Introduction
Gangliocytic paraganglioma (GP) is a markedly rare
neuroendocrine tumor (NET) that was initially identified by Dahl
et al (1) in 1957. In
total, >260 cases of GP have been documented thus far worldwide.
GP can arise at any age. The median age at onset is 51.2 years and
a greater number of patients are male, with a male-to-female ratio
of 157:104(2). The majority of
documented cases were from the second and third sections of the
duodenum, particularly the duodenal ampulla around the abdomen
(3), The size of GP around the
ampulla ranges from 0.7 to 19.0 cm, with a median size of ~2.2 cm
(4). A few cases were detected
elsewhere, including the esophagus, mediastinum, pericardium,
thymus and lung (5-7).
GP consists of varying proportions of three types of tumor cells:
i) Epithelioid, ii) ganglion-shaped and iii) spindle-like cells,
and is identified by distinct histopathological and
immunohistochemical markers (8).
The proportion of the three tumor cell components varies from case
to case and misdiagnosis is common when one cell component
dominates in an individual. Duodenal GP (DGP) is typically
considered a benign tumor; however, there is a certain risk of
malignancy associated with it (9).
The present study describes the diagnosis and
treatment of a case of DGP that reappeared after endoscopic
resection, in an effort to improve the current understanding of
this disease and to provide a reference for future clinical
work.
Case report
The patient was a 39-year-old male with no previous
underlying disease, a heavy oily and spicy diet, a history of
smoking (10 cigarettes per day) and no history of alcohol intake.
The patient was admitted to the Second Affiliated Hospital of
Xuzhou Medical University (Xuzhou, China) in March 2020 with black
stools for 3 days. The patient had tarry stools, accompanied by
dizziness, palpitation and weakness of the limbs. The patient
vomited coffee-colored stomach contents, fainted once and regained
consciousness within a short period of time. Physical examination
revealed acute anemia, a soft belly with no pressure or rebound
pain and no abdominal mass.
After admission, hemoglobin was found to be 54.2 g/l
(normal, 150-175 g/l), with no abnormalities in coagulation, liver
or renal function, or tumor indicators. An enhanced computed
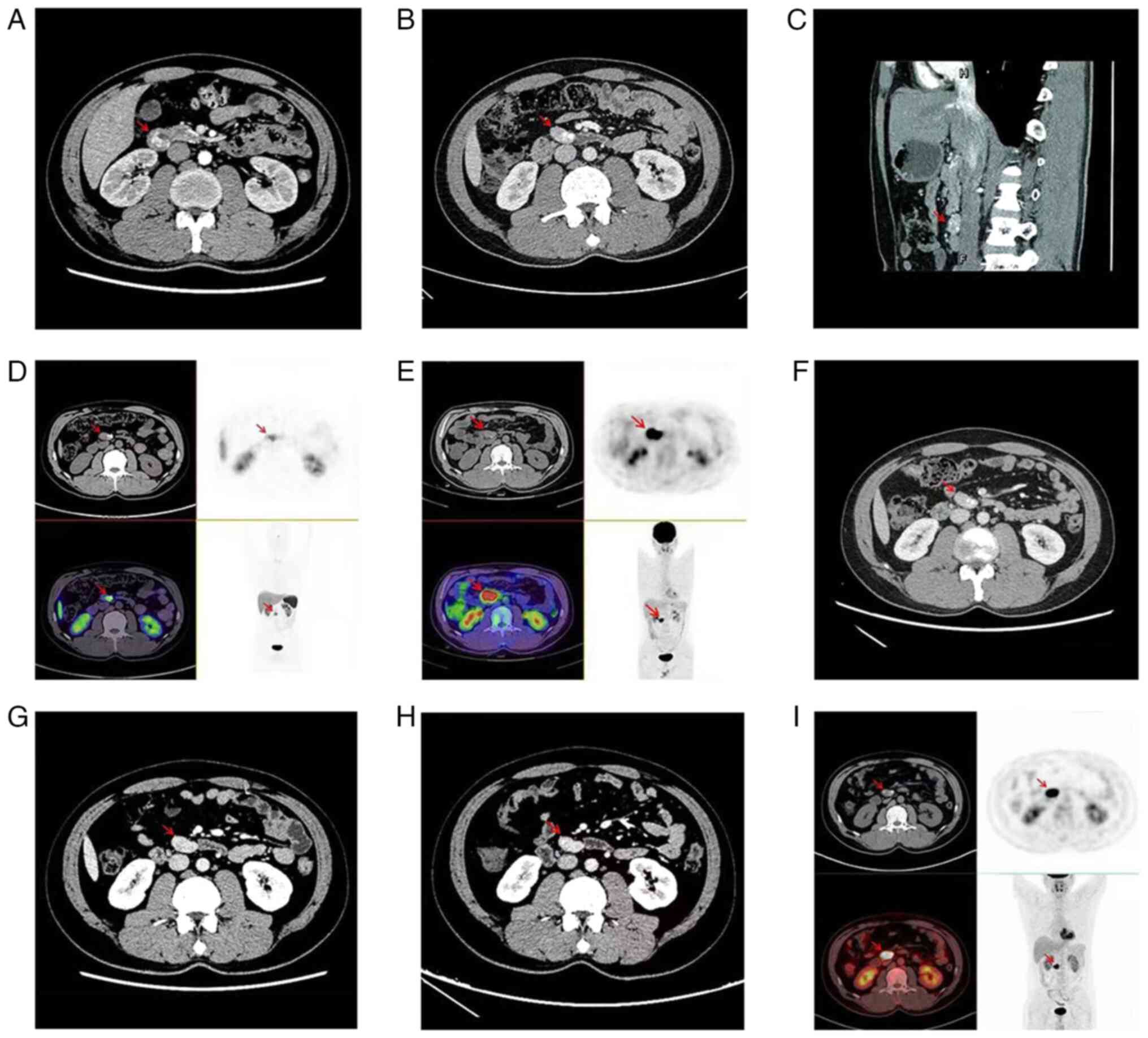
tomography (CT) scan of the whole abdomen revealed a soft tissue
mass, measuring ~2.7x2.4 cm, with a modest increase upon enhanced
scanning (Fig. 1A). During the
hospitalisation, the patient continuously vomited brilliant red
blood, destooled dark red blood, and appeared to be in hemorrhagic
shock; hemoglobin gradually decreased to 40 g/l. At 2-days
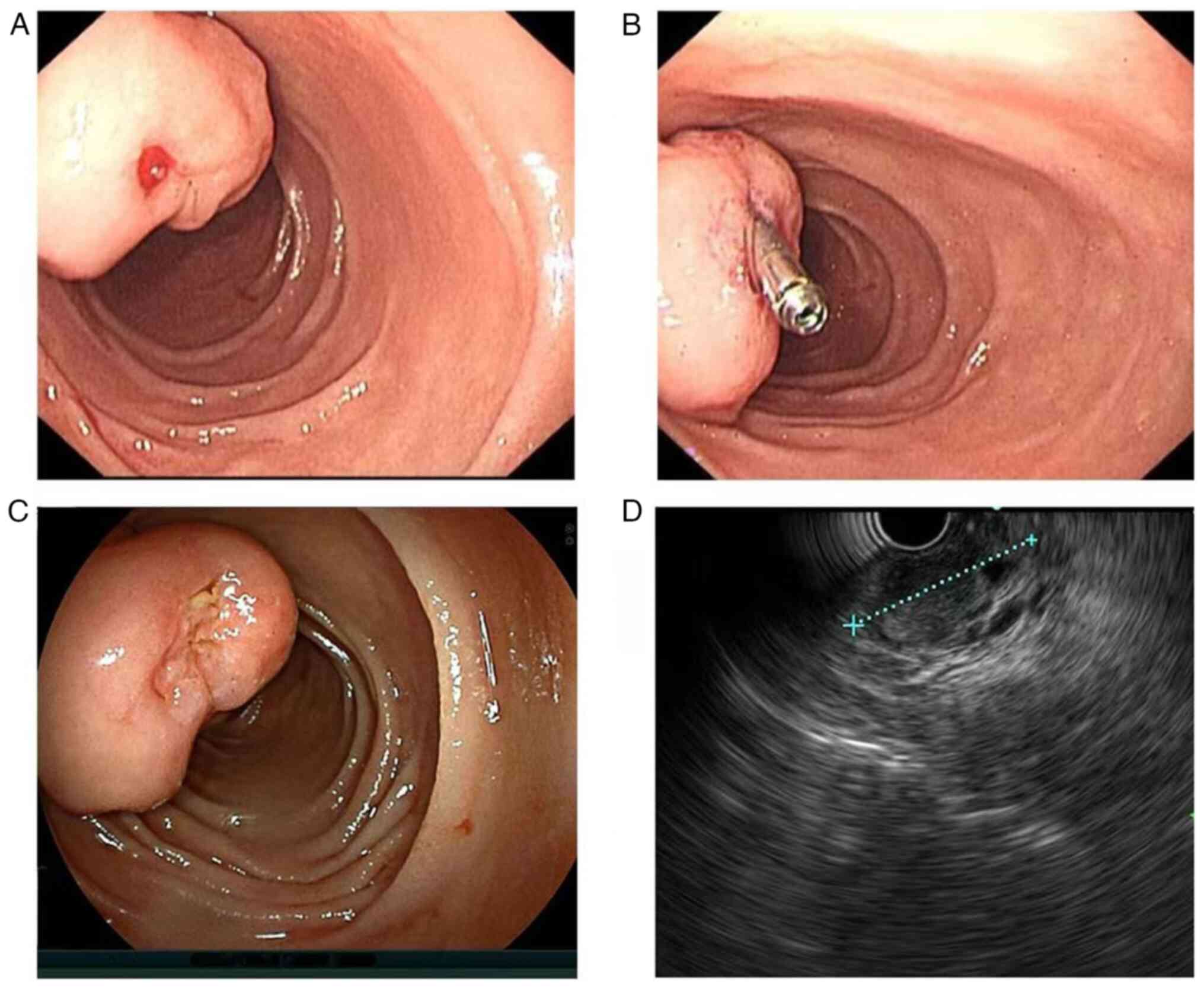
post-admission, an emergency gastroscopy revealed a large irregular
bulge in the duodenal papilla. The surface mucosa was smooth, the
papillary opening was not clearly visible and an ulcer and a red
thrombus head with limited bleeding were visible on the surface. A
titanium clip was attached to stop the bleeding (Fig. 2A and B). After 8 days of endoscopic
haemostasis, the patient's vital signs were stable, and the
bleeding was temporarily controlled. The duodenoscope and
endoscopic ultrasound revealed that the titanium clip had slipped
off. The duodenal papilla was large, with the white submucosal
tumour barely visible and bile outflowing from the papilla opening.
Ultrasonography indicated that the lesion was hypoechoic, with
well-defined borders and a generally uniform texture (Fig. 2C and D). Endoscopic treatment was advised in
the case of DGP.
A gastroenterologist suggested performing a major
surgery to remove the tumor. Following careful consideration, the
patient and his family requested endoscopic treatment for the
tumor. On March 20, 2020, endoscopic resection of a duodenal
papilloma was performed. The operation revealed a 2.7x2.5-cm firm
and smooth protrusion in the descending portion of the duodenum.
The root of the tumor was ligated with a snare, and most of the
tumor was mucous membrane. Electrocoagulation resection was
performed in stages and pulsatile haemorrhage was discovered in a
blood artery beneath the mucosa soon after cutting. To halt the
bleeding, electrocoagulation forceps were used and peripheral blood
vessels were treated. The procedure was successful. The wound was
found to be smooth and free of tumour residue, and it was rinsed to
ensure that there were no vascular residues or perforations on the
surface. The patient made a full recovery and was released 10 days
after the surgery.
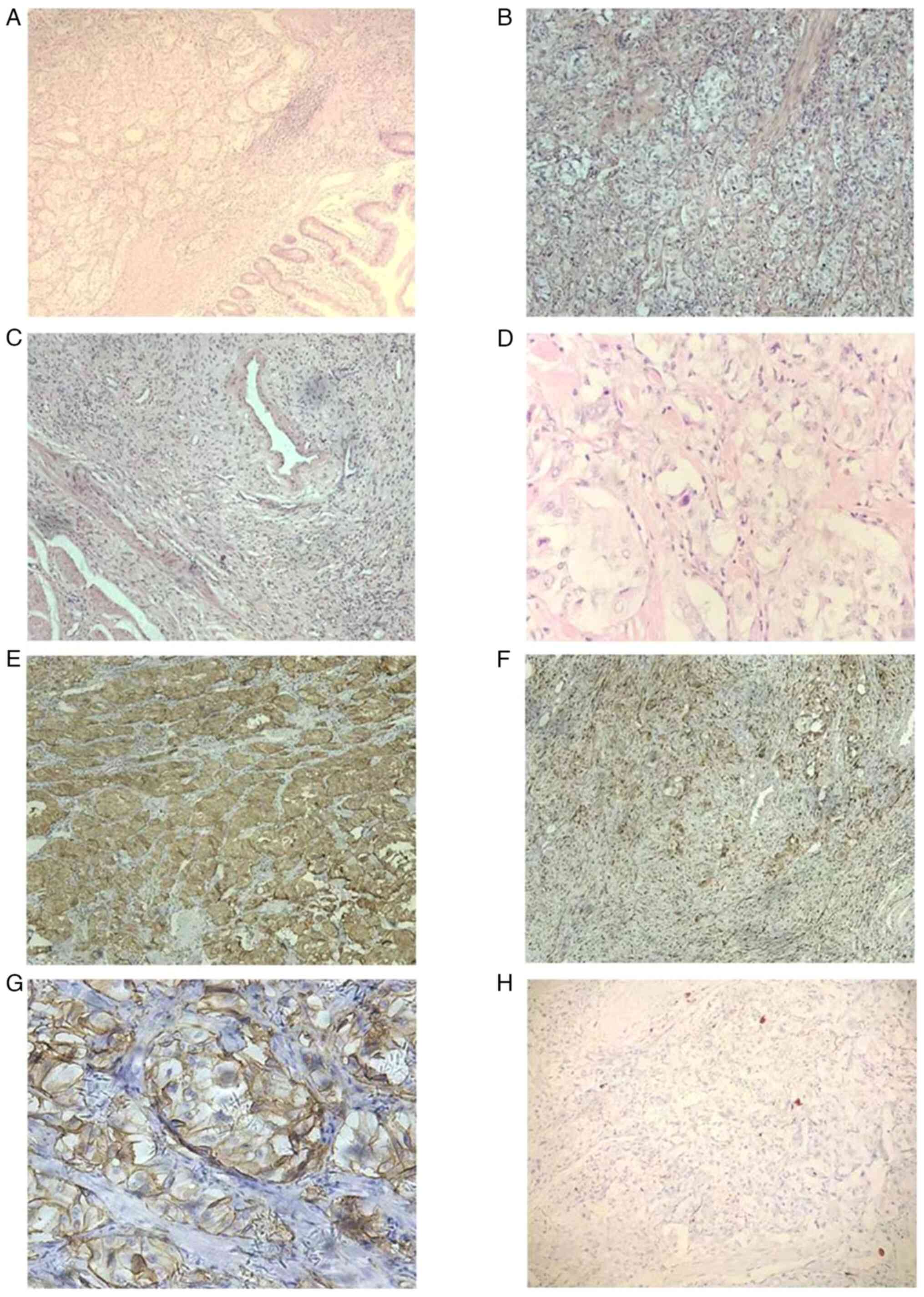
Regarding the findings of the histopathological
analyses (duodenum), which were performed according to standard
procedures, tumor tissue was found in the mucous membrane, mucous
membrane muscular and submucous membrane layers (Fig. 3A). The tumor was a single piece of
grayish-white nodular tissue that measured ~2.7x2.4x1.9 cm. The
tumor tissue was composed of three different types of cells, namely
epithelioid, ganglion-shaped and spindle-like cells. The majority
of the tumour cells were arranged in nests; the cells were oval,
with abundant and light-stained cytoplasm and oval nuclei, and
nuclear mitotic figures were difficult to discern. There were
visible short spindle cells arranged in sheets or bundles and
nodule-like cells dispersed throughout the area (Fig. 3B-D).
Immunohistochemical staining was performed according
to the standard percedures (10).
Regarding the immunohistochemical findings, the tumor cells
expressed somatostatin receptor (SSTR)2(+) (cat. no. 704011;
1:1,000 dilution), the epithelioid cells exhibited the following
characteristics: Synaptophysin(+) (Syn; cat. no. MA5-14532; 1:1,000
dilution) (Fig. 3E), chromogranin
A(+) (CgA; cat. no. MA1-25038; 1:100 dilution), CD56(+) (cat. no.
MA5-11563; 1:500 dilution) (Fig.
3G), cytokeratin(+) (CK; cat. no. MA5-32118; 1:500 dilution),
melan-A(+) (cat. no. MA5-14168; 1:200 dilution) and epithelial
membrane antigen(-) (EMA; cat. no. MA5-11202; 1:100 dilution). The
spindle-like cells showed the following characteristics: S100(+)
(cat. no. MA5-12969; 1:100 dilution) (Fig. 3F), neurofilament dispersed(+) (NF;
cat. no. PA5-78668; 1:500 dilution) and partly CD34(+) (cat. no.
MA1-10202; 1:100 dilution). The ganglion-shaped cells exhibited the
following characteristics: NF(+) (Fig.
3H), <2% Ki-67(+) (cat. no. MA5-14520; 1:200 dilution),
HMB45(-) (cat. no. MA5-13232; 1:80 dilution), desmin(-) (cat. no.
MA5-13259; 1:100 dilution), smooth muscle actin(-) (SMA; cat. no.
14-9760-82; 1:500 dilution), gastrointestinal stromal tumor 1(-)
(Dog-1; cat. no. MA5-16358; 1:100 dilution) and CD117 (cat. no.
MA5-15894; 1:500 dilution) (all Thermo Fisher Scientific, Inc.).
DGP was considered when combining the morphological findings
revealed by hematoxylin and eosin staining with the
immunohistochemical findings.
In June 2020, the patient was examined by an
abdominal enhanced CT scan, which revealed a mass of ~2.9x1.9 cm in
the horizontal segment of the duodenum, and a nodular cell
paraganglioma recurrence was considered (Fig. 1B and C). The next day, 68Ga-Dotatate
(Target Molecule Corp.) and 18F-FDG (Target Molecule
Corp.) positron emission tomography (PET)-CT were further performed
for refinement to evaluate metastasis or recurrence, which also
identified a retroperitoneal duodenal nodule above the horizontal
portion; there was positive growth inhibitory receptor imaging and
considerably active glucose metabolism (Fig. 1D and E). It was suggested that the patient may
undergo endoscopic, radical surgical or biological treatments.
After consideration, the patient requested biotherapy and received
octreotide (Novartis Corp.) at 30 mg/28 days for 12 consecutive
cycles. After 47 months of postoperative follow-up, the patient did
not have any further symptoms of gastrointestinal bleeding and was
re-examined in September 2020, February 2021 and February 2024,
with hemoglobin and gastrointestinal tumor markers within the
normal ranges. The lesions at the horizontal duodenum appeared to
be unremarkable compared with the previous imaging changes and no
new lesions or metastases were observed (Fig. 1F-I).
Discussion
GP is now classified as composite
gangliocytoma/neuroma and neuroendocrine tumor according to the
2022 World Health Organization (WHO) Classification of
Neuroendocrine Neoplasms (11,12).
NETs frequently overexpress SSTRs, particularly of type 2(13). 68Ga-Dotatate is a
selective SSTR-2 PET tracer with a strong affinity for
SSTR-2-expressing NETs; thus, it is recommended as the imaging
modality of choice for the early diagnosis of GP (14). The final diagnosis of GP is based
on its distinct histopathological features, which include a mixture
of three cell types, namely epithelioid, spindle-like and
ganglion-shaped, in varying proportions (8). Epithelioid cells in GPs are similar
to those that compose paragangliomas and/or carcinoid tumors, with
polygonal cells, abundant cytoplasm, ovoid nuclei, indistinct
nucleoli and dense core granules visible on electron microscopy,
and are arranged in dense nests and trabeculae. Epithelioid cells
are mainly positively immunoreactive for Syn, neuron-specific
enolase (NSE), CgA, CD56, growth inhibitors and pancreatic
polypeptide (PP) (15).
Spindle-like cells show a neurofibromatous structure with elongated
nuclei in bundles surrounding nests of epithelioid cells and
ganglion-shaped cells, and are mainly positive for S100 and NSE
(16). Ganglion-shaped cells are
spread singly or in nests amid epithelioid and spindle-like cells,
with infrequent mitotic cell divisions and no evident anisotropy or
necrosis. They proliferate in a manner similar to that of
ganglioneuromas, with big nuclei containing large quantities of
eosinophilic cytoplasm. Ganglion-shaped cells show positive
reactivity for Syn, NSE and CD56(17).
The tumor proliferation index or Ki-67 is used
clinically to assess tumor cell division and proliferation
activities. The higher the value, the greater the proliferation
activity of tumor cells (18). In
the present study, positive expression of Ki-67 was <2%,
indicating that tumor cell proliferation activity was weak and
prognosis was good. The proportion of the three tumor cell
components varies from case to case and misdiagnosis is common when
one cell component dominates in an individual.
GP is notably rare and typically appears clinically
in the second and third sections of the duodenum, particularly in
the area surrounding the duodenal jugular abdomen (3), although it can also be present in the
biliary tract, pancreas, esophagus, stomach, jejunum, cecum,
thymus, mediastinum, lungs, bladder and other areas of the body
(5-7,19-22).
GP can arise at any age. It has been recorded in patients aged 15
to 92 years. The median age at onset is 51.2 years and the majority
of them are males, with a male-to-female ratio of 157:104(2). Previous research also indicated that
there was no gender difference in terms of occurrence of GP and the
size of GP around the ampulla ranges from 0.7 to 19.0 cm, with a
median size of ~2.2 cm (4). The
2019 WHO Classification of Tumors of the Digestive System (5th
edition) (23) indicated that GP
was a NET with a fair prognosis that is not likely to experience
recurrence or metastasis; however, there is still a risk of
malignancy. The 1-, 3- and 5-year survival rates of GP around the
duodenal jugular abdomen were reported to be 100, 83.3 and 55.6%,
respectively, according to a study by Chiang et al (4). An increasing number of case reports
of GP with distant metastasis, recurrence and lymph node metastasis
have been published (9,24). The most common type of metastasis
is lymph node metastasis, followed by liver, lung, bone and other
organ metastases. Li et al (25) reported a case of lethal GP with
multifocal metastasis. The present study reports a case of DGP with
short-term recurrence. No lymph node or distant metastases were
identified during 4 years of follow-up.
The primary factor influencing the clinical
presentation of GP is the tumor growth site. While gastrointestinal
bleeding, abdominal pain, anemia, diarrhea, wasting and other
symptoms are typical of DGP, a few patients may experience biliary
obstruction symptoms (26) or even
no symptoms at all that are unintentionally identified during
physical examination. The main clinical manifestations of DGP
reported in the present study were black stool and anemia, which
could easily be misdiagnosed as other digestive diseases.
GP is uncommon in clinical practice and clinicians
have limited knowledge of this condition. GP is formed by the
combination of epithelioid, spindle-like and ganglion-shaped cells
in different ratios. It is usually dominated by epithelioid and
spindle-like cells, with ganglion-shaped cells dispersed
throughout. In clinical practice, it is easy to cause missed
diagnosis and misinterpretation when a sample is acquired
incompletely or a specific cell component is prominent.
It is necessary to differentiate this condition from
the following illnesses when epithelioid cells predominate over GP:
i) NET grade 1 (NET G1): In 2022, the WHO classified NET into G1,
G2 and G3 according to their mitotic count or Ki67 index. The
diagnostic criteria for NET include: G1, <2 mitoses/2
mm2 and/or Ki67 <3%; G2, 2-20 mitoses/2
mm2 and/or Ki67 3-20%; and G3, >20 mitoses/2
mm2 and/or Ki67 >20% (11). NET G1 is a carcinoid that accounts
for 50% of all gastrointestinal and pancreatic NETs. Clinical
symptoms include abdominal discomfort, abdominal pain, black stool,
weight loss and other symptoms. Tumor cells can be arranged in an
island-like, trabecular or adenoid pattern on pathology.
Immunohistochemistry shows that Syn, CgA, NSE, carcinoembryonic
antigen (CEA), CD56 and Ki-67 are all positively expressed, similar
to GP. However, GP epithelioid cells were previously reported to be
positive for PP and progesterone receptor, while NET G1 are
negative for these, which is helpful to differentiate them
(2); and ii) poorly-differentiated
adenocarcinoma: When GP epithelioid cells exhibit invasive
proliferation, they must be distinguished from
poorly-differentiated adenocarcinoma. The immunophenotype of GP
epithelioid cells is often positive in Syn and CgA expression,
while negative for EMA and CEA expression, whereas the
immunophenotype of poorly differentiated adenocarcinoma cells is
simple in composition and apparent in atypia.
When GP is dominated by spindle-like cells, it
should be distinguished from the following diseases: i)
Gastrointestinal stromal tumor (GIST), which is a type of
gastrointestinal mesenchymal tumor that often affects the stomach
and small intestine. It is more frequent in middle and old age, and
there are no evident clinical signs in the early stage. Patients
with mid- and late-stage GIST may experience gastrointestinal
bleeding, abdominal pain, abdominal mass, anemia, emaciation and
other symptoms (27). The
pathology of GIST is mainly composed of spindle and/or epithelioid
cells, which may be mistaken for GP dominated by spindle-like
cells. The immunohistochemistry of GIST is characterized by CD117
(c-KIT), CD34 and DOG-1 expression, but negative Syn and S100
expression, whereas GP often shows positive SSTR2 and Syn
expression but negative CD117, CD34 and DOG-1 expression (28). In the present study, the expression
of SSTR2 and Syn in DGP was positive, while CD117 and DOG-1 were
negative. Accordingly, the difference in immunohistochemical
phenotype is helpful to distinguish DGP from GIST; and ii)
gastrointestinal leiomyoma, which is a benign tumor caused by
aberrant smooth muscle hyperplasia that typically develops in the
esophagus and colon, but rarely in the stomach or small intestine.
In total, 90% of tumors are solitary, round or oval, with no
genuine envelope, clear boundary, hard, smooth surface and core
ulcer development. Histologically, the tumor cells are organized in
a cross bundle, with no or limited mitogenic signals and limited
malignant signs. Leiomyoma has a positive SMA and desmin
immunohistochemical phenotype, but exhibits negative S100, CD117
and DOG-1 expression (29). In the
present case report, the tumor was positive for S100 but negative
for SMA and desmin. As a result, S100, SMA and desmin can help to
distinguish between gastrointestinal leiomyoma and GP. Furthermore,
the characteristics in terms of expression of CD117, DOG-1, CD34,
SMA and desmin allow to distinguish GIST from leiomyoma.
GP should also be distinguished from ganglioneuroma
(GNs) when it is dominated by ganglion-shaped cells. GN is a benign
tumor that develops from the neural crest of the sympathetic
nervous system and is common in young patients.
Histopathologically, GNS is composed of ganglion-shaped cells and
nerve fibers, with the fundamental distinction from GP being the
absence of epithelioid cells (30). Thus, identifying epithelioid cells
is key to distinguishing between GP and GNS.
At present, there are no defined diagnostic or
therapeutic standards for GP; thus, local or radical resection is
the primary treatment for GP in clinical practice. Endoscopic
submucosal dissection (ESD) offers advantages such as reduced
surgical trauma, less bleeding, shorter surgical time and faster
recuperation compared to surgical operation. ESD has become the
recommended treatment technique for DGP when the tumor diameter is
<2.0 cm and if the tumor is restricted to the mucosal or
submucosal layers, with no local invasion or distant metastases
(31), and a study has relaxed the
tumor diameter to 3.0 cm (32). In
a systematic review by Okubo (33), 27 individuals with GP who received
endoscopic treatment had positive outcomes. When GP is >2.0 cm
in diameter, it infiltrates into the intrinsic muscle layer, and if
it has no local invasion or distant metastases, it can be removed
locally using laparoscopic or open surgery (34,35).
Cathcart et al (36)
coupled duodenoscopy with laparoscopy and employed laparoscopy with
endoscopic-assisted localization to completely resect the DGP and
adjacent duodenal wall, resulting in a successful outcome and safe
discharge from the hospital 10 days later. When infiltrative growth
is detected with vascular and lymph node invasion or distant
metastasis, pancreaticoduodenectomy and local lymph node dissection
are advised as palliative surgical treatments (24).
GP is a type of NET; thus, the pharmacological
treatment options for NETs are also useful for GP, including
biotherapy, nuclide therapy, targeted therapy, chemotherapy and
immunotherapy. Biotherapy is recommended for individuals with Ki-67
<10%, which includes long-acting somatostatin analogues (SSAs)
and interferon (IFN)-α. IFN-α is less frequently utilized in
clinical settings and is mostly used for refractory functional
NETs. The most widely used long-acting SSAs exert
antiproliferative/antitumor effects via the SSTR2 (37,38),
including octretide, lanreotide and pasireotide, among which
octreotide is mainly used for gastrointestinal NETs, while
lanreotide is also used for pancreatic NETs. According to Nesti
et al (39), the
progression-free overall survival (PFS) of patients with NET in the
octreotide group was noticeably higher than that of patients in the
placebo group. Lanotide significantly extended the PFS of patients
with gastroenteropancreatic (GEP)-NET (40). According to a study by Caplin et
al (41), patients with
advanced enteropancreatic NETs who were in the lanreotide group had
a significantly higher PFS than patients in the control group. In
addition, patients in the lanreotide group experienced a
significantly lower incidence of treatment-related adverse events,
as well as an overall decrease in adverse events. In the present
study, after surgical recurrence, the patient, who exhibited Ki67
<2%, received biotherapy with octreotide, and, during routine
follow-up assessment, no metastasis was observed in the lesion.
Currently, NETs can be treated with nuclear therapy as a second or
third line of treatment. Lutetium-177 (177Lu) is a
commonly used radioactive nucleotide that frequently binds to
somatostatin analogues to form 177Lu-dotatate, which
targets SSTR-positive NETs, inducing DNA breaks in tumor cells,
leading to apoptosis (42). In
September 2017 and January 2018, 177Lu-dotatate was
licensed in Europe and the US, respectively, for the treatment of
SSTR-positive GEP NETs (43). In a
phase III clinical trial, patients with advanced mid-gut NETs
treated with 177Lu-Dotatate in conjunction with
long-acting octreotide exhibited a significant improvement in PFS
compared to those treated with high-dose long-acting octreotide
alone (44). Chemotherapy is the
first line of treatment for patients with high-grade (G3) NETs and
has an antitumor effect by preventing tumor cells from entering the
mitotic cycle. It is not usually utilized for patients in the G1 or
G2 stages of cancer (45),
including alkylating agents (cisplatin, temozolomide),
topoisomerase inhibitors (etoposide) and thymidylate synthase
inhibitors (capecitabine). Molecularly targeted drugs are divided
into two categories, namely i) mammalian target of rapamycin
inhibitors, with the most representative drug being everolimus; and
ii) tyrosine kinase inhibitors, including sunitinib and sofantinib
(46). Everolimus plus
temozolomide may be the first-line treatment for metastatic
high-grade GEP NETs, according to a prospective multicenter phase
II trial (47). Immunotherapy
drugs include programmed cell death protein 1 inhibitors, such as
pabolizumab, and programmed cell death ligand 1 inhibitors, such as
dovalizumab (48). The efficacy of
single immune checkpoint inhibitors in the treatment of advanced
neuroendocrine neoplasms is limited. At present, dual immune
checkpoint therapy or a combination of other drugs is typically
employed to treat them. In a non-randomized controlled multicohort
phase II clinical trial, tremelimumab in combination with
durvalumab demonstrated good antitumor effectiveness and safety in
GEP and lung NETs (49). The
selection of initial and post-progression treatment options for GEP
NETs must be evaluated in various aspects according to the tumor's
SSTR expression, stage, primary characteristic, hormonal status and
other characteristics. When the tumor is SSTR (-), chemotherapy and
targeted drugs are recommended for patients with stage G1 to G2;
when the tumor is SSTR (+), long-acting SSAs are first recommended
for patients. Systemic chemotherapy is recommended for patients
with G3 stage tumors independently of SSTR expression by the tumor
(50).
In conclusion, DGP is a clinically atypical NET,
which lacks specific clinical manifestations. The diagnosis of DGP
is primarily based on its distinct histopathological
manifestations, which dictate the treatment modality based on the
tumor's size, depth of infiltration and metastasis. In our opinion,
for gastrointestinal tumors, when the preoperative diagnosis is
unknown, endoscopic local excision can be selected first to
maximize the guarantee of negative margins and reduce the risk of
secondary surgery, and, if the pathology exists in the presence of
positive margins and plexus nerve invasion, additional surgical
treatment may be performed. In the present case, the tumor
originated from the mucosal layer and the depth of infiltration was
shallow, therefore ESD therapy was used to totally remove the
tumor. The patient required further biological therapy after the
tumor recurred following surgery. There was no lymph node
metastases or distant metastasis discovered during the ~4-year
follow-up period. GP is a low-grade cancer with a low risk of
metastasis and a favorable prognosis. ESD or surgical resection is
preferred, and for patients with postoperative recurrence,
metastasis or those unsuitable for surgery, biotherapy, nuclear
therapy, chemotherapy, targeted therapy and immunotherapy can be
selected independently based on factors including hormone receptor
expression and tumor grading.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
NX and LHS made substantial contributions to the
conception or design of the work, and confirm the authenticity of
all the raw data. SMZ, HL and LFL gathered, analyzed and
interpreted the data. NX, LHS and SMZ wrote the original
manuscript, and reviewed and edited the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Second Affiliated Hospital of Xuzhou Medical
University (approval no. 2024031205).
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of the present case report and the
accompanying associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dahl EV, Waugh JM and Dahlin DC:
Gastrointestinal ganglioneuromas; brief review with report of a
duodenal ganglioneuroma. Am J Pathol. 33:953–965. 1957.PubMed/NCBI
|
|
2
|
Okubo Y, Yoshioka E, Suzuki M, Washimi K,
Kawachi K, Kameda Y and Yokose T: Diagnosis, pathological findings,
and clinical management of gangliocytic paraganglioma: A systematic
review. Front Oncol. 8(291)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Matsubayashi H, Ishiwatari H, Matsui T,
Fujie S, Uesaka K, Sugiura T, Okamura Y, Yamamoto Y, Ashida R, Ito
T, et al: Gangliocytic paraganglioma of the minor papilla of the
duodenum. Intern Med. 56:1029–1035. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chiang CS, Shyr BU, Chen SC, Shyr YM and
Wang SE: Periampullary gangliocytic paraganglioma. J Gastrointest
Surg. 23:2247–2254. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Weinrach DM, Wang KL, Blum MG, Yeldandi AV
and Laskin WB: Multifocal presentation of gangliocytic
paraganglioma in the mediastinum and esophagus. Hum Pathol.
35:1288–1291. 2004.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Palacio D, Jo N, Del Pilar Gutierrez M,
Shponka V and Betancourt S: Multimodality imaging appearance of
intrapericardial paragangliomas. Clin Radiol. 77:952–959.
2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Naso JR, Wang D, Romero AO, Leclair T,
Smit P, Boland JM, Folpe AL and Bois MC: Pulmonary gangliocytic
paraganglioma: An under-recognized mimic of carcinoid tumor. Hum
Pathol. 146:23–27. 2024.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mete O, Asa SL, Gill AJ, Kimura N, de
Krijger RR and Tischler A: Overview of the 2022 WHO classification
of paragangliomas and pheochromocytomas. Endocr Pathol. 33:90–114.
2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Park HK and Han HS: Duodenal gangliocytic
paraganglioma with lymph node metastasis. Arch Pathol Lab Med.
140:94–98. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ramos-Vara JA: Principles and methods of
immunohistochemistry. Methods Mol Biol. 1641:115–128.
2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Rindi G, Mete O, Uccella S, Basturk O, La
Rosa S, Brosens LAA, Ezzat S, de Herder WW, Klimstra DS, Papotti M
and Asa SL: Overview of the 2022 WHO classification of
neuroendocrine neoplasms. Endocr Pathol. 33:115–154.
2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Helderman NC, Suerink M, Kilinç G, van den
Berg JG, Nielsen M and Tesselaar MET: Relation between WHO
classification and location- and functionality-based
classifications of neuroendocrine neoplasms of the digestive tract.
Neuroendocrinology. 114:120–133. 2024.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Oronsky B, Ma PC, Morgensztern D and
Carter CA: Nothing but NET: A review of neuroendocrine tumors and
carcinomas. Neoplasia. 19:991–1002. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sanli Y, Garg I, Kandathil A, Kendi T,
Zanetti MJB, Kuyumcu S and Subramaniam RM: Neuroendocrine tumor
diagnosis and management: 68Ga-DOTATATE PET/CT. AJR Am J
Roentgenol. 211:267–277. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Li J, Wang LP and Zhu PS: Is gangliocytic
paraganglioma designated as a subtype of composite paragangliomas
and originated from pancreas islet? A case report and review of
literature. Front Endocrinol (Lausanne). 13(847632)2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang B, Zou Y, Zhang H, Xu L, Jiang X and
Sun K: Duodenal gangliocytic paraganglioma: Report of two cases and
review of literature. Int J Clin Exp Pathol. 8:9752–9759.
2015.PubMed/NCBI
|
|
17
|
Furihata M, Sonobe H, Iwata J, Ido E,
Ohtsuki Y and Ohnishi S: Immunohistochemical characterization of a
case of duodenal gangliocytic paraganglioma. Pathol Int.
46:610–613. 1996.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Rubisz P, Ciebiera M, Hirnle L,
Zgliczyńska M, Łoziński T, Dzięgiel P and Kobierzycki C: The
usefulness of immunohistochemistry in the differential diagnosis of
lesions originating from the myometrium. Int J Mol Sci.
20(1136)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Harries K, Nunn T, Shah V, Richards D and
Manson JM: First reported case of esophageal paraganglioma. A
review of the literature of gastrointestinal tract paraganglioma
including gangliocytic paraganglioma. Dis Esophagus. 17:191–195.
2004.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Nonaka K, Matsuda Y, Okaniwa A, Kasajima
A, Sasano H and Arai T: Pancreatic gangliocytic paraganglioma
harboring lymph node metastasis: A case report and literature
review. Diagn Pathol. 12(57)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Seo G, Park J, Lee E, Han J, Kim D, Kim D,
Park J, Kwag M and Jung S: Gangliocytic paraganglioma of the
gastrointestinal tract: A case report of cecal origin. Curr Med
Imaging. 18:95–98. 2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Usuda H and Emura I: Composite
paraganglioma-ganglioneuroma of the urinary bladder. Pathol Int.
55:596–601. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Nagtegaal ID, Odze RD, Klimstra D, Paradis
V, Rugge M, Schirmacher P, Washington KM, Carneiro F and Cree IA:
WHO Classification of Tumours Editorial Board. The 2019 WHO
classification of tumours of the digestive system. Histopathology.
76:182–188. 2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Choi H, Choi JW, Ryu DH, Park S, Kim MJ,
Yoo KC and Woo CG: Ampullary gangliocytic paraganglioma with lymph
node metastasis: A case report with literature review. Medicine
(Baltimore). 101(e29138)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li B, Li Y, Tian XY, Luo BN and Li Z:
Malignant gangliocytic paraganglioma of the duodenum with distant
metastases and a lethal course. World J Gastroenterol.
20:15454–15461. 2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sharma S, Gaspar BL, Kumar P, Yadav TD and
Vasishta RK: Gangliocytic paraganglioma with atypical
immunohistochemical features presenting as extrahepatic biliary
obstruction. Int J Surg Pathol. 23:561–566. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Serrano C, Martin-Broto J, Asencio-Pascual
JM, Lopez-Guerrero JA, Rubio-Casadevall J, Bague S, Garcia-Del-Muro
X, Fernandez-Hernandez JA, Herrero L, Lopez-Pousa A, et al: 2023
GEIS guidelines for gastrointestinal stromal tumors. Ther Adv Med
Oncol. 15(17588359231192388)2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Munteanu A, Patrascu S, Bordu S, Laskou S,
Surlin V and Radu P: Clinical and morphological characteristics of
gastrointestinal stromal tumor. Chirurgia (Bucur). 118:618–623.
2023.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Virani N, Pang J and Lew M: Cytologic and
immunohistochemical evaluation of low-grade spindle cell lesions of
the gastrointestinal tract. Arch Pathol Lab Med. 140:1038–1044.
2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Goldberg JL, Tong J and McGrath LB Jr:
Spinal ganglioneuroma. World Neurosurg. 162:15–16. 2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Barret M, Rahmi G, van Huyen JP, Landi B,
Cellier C and Berger A: Duodenal gangliocytic paraganglioma with
lymph node metastasis and an 8-year follow-up: A case report. Eur J
Gastroenterol Hepatol. 24:90–94. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hernandez AG, Lanuza ED, Matias AC,
Huertas RP, Rodriguez KM, Perez PG and Mompean FO: Large
gangliocytic paraganglioma of the duodenum: A rare entity. World J
Gastrointest Surg. 7:170–173. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Okubo Y: Gangliocytic paraganglioma: An
overview and future perspective. World J Clin Oncol. 10:300–302.
2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhang Z, Tu Z, Lv Z, Luo Y and Yuan J:
Case report: Totally laparoscopic resection of retroperitoneal
paraganglioma masquerading as a duodenal gastrointestinal stromal
tumor. Front Surg. 8(586503)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Moriyama H, Asai K, Watanabe R, Nagata T,
Futawatari N, Enomoto T, Nagao S, Watanabe M, Oharazeki T and Saida
Y: Laparoscopic tumor resection for asymptomatic paraganglioma-a
case study. Gan To Kagaku Ryoho. 50:1924–1927. 2023.PubMed/NCBI(In Japanese).
|
|
36
|
Cathcart SJ, Sasson AR, Kozel JA, Oliveto
JM and Ly QP: Duodenal gangliocytic paraganglioma with lymph node
metastases: A case report and comparative review of 31 cases. World
J Clin Cases. 5:222–233. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lamberts SW, Hofland LJ and Nobels FR:
Neuroendocrine tumor markers. Front Neuroendocrinol. 22:309–339.
2001.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Stueven AK, Kayser A, Wetz C, Amthauer H,
Wree A, Tacke F, Wiedenmann B, Roderburg C and Jann H: Somatostatin
analogues in the treatment of neuroendocrine tumors: Past, present
and future. Int J Mol Sci. 20(3049)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Nesti C, Bräutigam K, Benavent M, Bernal
L, Boharoon H, Botling J, Bouroumeau A, Brcic I, Brunner M, Cadiot
G, et al: Hemicolectomy versus appendectomy for patients with
appendiceal neuroendocrine tumours 1-2 cm in size: A retrospective,
Europe-wide, pooled cohort study. Lancet Oncol. 24:187–194.
2023.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Saif MW: Lanreotide for the treatment of
gastroenteropancreatic neuroendocrine tumors. Expert Opin
Pharmacother. 17:443–456. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Caplin ME, Pavel M, Phan AT, Ćwikła JB,
Sedláčková E, Thanh XT, Wolin EM and Ruszniewski P: CLARINET
Investigators. Lanreotide autogel/depot in advanced
enteropancreatic neuroendocrine tumours: Final results of the
CLARINET open-label extension study. Endocrine. 71:502–513.
2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Becx MN, Minczeles NS, Brabander T, de
Herder WW, Nonnekens J and Hofland J: A clinical guide to peptide
receptor radionuclide therapy with 177Lu-DOTATATE in
neuroendocrine tumor patients. Cancers (Basel).
14(5792)2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Mittra ES: Neuroendocrine tumor therapy:
177Lu-DOTATATE. AJR Am J Roentgenol. 211:278–285.
2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Strosberg JR, Caplin ME, Kunz PL,
Ruszniewski PB, Bodei L, Hendifar A, Mittra E, Wolin EM, Yao JC,
Pavel ME, et al: 177Lu-Dotatate plus long-acting
octreotide versus high-dose long-acting octreotide in patients with
midgut neuroendocrine tumours (NETTER-1): Final overall survival
and long-term safety results from an open-label, randomised,
controlled, phase 3 trial. Lancet Oncol. 22:1752–1763.
2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Chan DL and Singh S: Current chemotherapy
use in neuroendocrine tumors. Endocrinol Metab Clin North Am.
47:603–614. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hijioka S, Morizane C, Ikeda M, Ishii H,
Okusaka T and Furuse J: Current status of medical treatment for
gastroenteropancreatic neuroendocrine neoplasms and future
perspectives. Jpn J Clin Oncol. 51:1185–1196. 2021.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Morken S, Langer SW, Sundlöv A, Vestermark
LW, Ladekarl M, Hjortland GO, Svensson JB, Tabaksblat EM, Haslerud
TM, Assmus J, et al: Phase II study of everolimus and temozolomide
as first-line treatment in metastatic high-grade
gastroenteropancreatic neuroendocrine neoplasms. Br J Cancer.
129:1930–1939. 2023.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Ooki A, Osumi H, Fukuda K and Yamaguchi K:
Potent molecular-targeted therapies for gastro-entero-pancreatic
neuroendocrine carcinoma. Cancer Metastasis Rev. 42:1021–1054.
2023.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Capdevila J, Hernando J, Teule A, Lopez C,
Garcia-Carbonero R, Benavent M, Custodio A, Garcia-Alvarez A,
Cubillo A, Alonso V, et al: Durvalumab plus tremelimumab for the
treatment of advanced neuroendocrine neoplasms of
gastroenteropancreatic and lung origin. Nat Commun.
14(2973)2023.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Del Rivero J, Perez K, Kennedy EB, Mittra
ES, Vijayvergia N, Arshad J, Basu S, Chauhan A, Dasari AN, Bellizzi
AM, et al: Systemic therapy for tumor control in metastatic
well-differentiated gastroenteropancreatic neuroendocrine tumors:
ASCO guideline. J Clin Oncol. 41:5049–5067. 2023.PubMed/NCBI View Article : Google Scholar
|