Introduction
Atherosclerosis forms the pathological basis of
atherosclerotic cardiovascular disease (ASCVD), which can lead to
ischemic heart disease, stroke and peripheral vascular disease
(1). Elevated levels of
low-density lipoprotein (LDL) cholesterol (LDL-C) have been
previously shown to increase the risk of ASCVD (2). By contrast, cholesterol homeostasis
is mainly regulated by the dynamic balance among its biosynthesis,
uptake, export and metabolism (3).
The liver is an important organ for cholesterol synthesis and
metabolism (4). The LDL receptor
(LDLR) is a cell surface protein that is mainly expressed in the
liver and mediates the clearance of >70% of plasma LDL-C through
endocytosis (5,6). Therefore, strategies to increase
cellular LDLR expression and stability to reduce plasma LDL-C
levels may be effective in preventing the development of ASCVD.
Recently, several drugs, such as proprotein convertase
subtilisin/Kexin type 9 (PCSK9) inhibitor and ezetimibe, which
target individual regulatory factors involved in the pathogenesis
of ASCVD, have been developed (7,8).
Anti-PCSK9 is a monoclonal antibody that functions as an inhibitor
to prevent PCSK9 binding to LDLR on the cell surface, which
inhibits LDLR degradation induced by PCSK9. Anti-PCSK9 can enhance
the clearance of LDL particles from the circulation mediated by
LDLR to subsequently reduce plasma LDL-C levels (9,10).
At the transcriptional level, LDLR gene expression
is controlled by a cholesterol-responsive negative feedback
mechanism through sterol regulatory element (SRE)-binding proteins
(SREBPs), which bind to the SRE region of the LDLR promoter and
enhance mRNA transcription, thereby increasing the expression of
hepatocyte surface LDLR (11-13).
Post-translational regulation of LDLR can be achieved through two
major pathways, namely PCSK9 and inducible degrader of the LDLR
(IDOL) (14). PCSK9 is a plasma
protein that is mainly produced and secreted by the liver (15). It is transcriptionally regulated by
SREBP-2 and hepatocyte nuclear factor 1α (HNF1α) (16). Inflammatory pathological stimuli
can increase PCSK9 expression (16,17).
PCSK9 binds to the extracellular structural domain of LDLR on the
cell surface, interfering with LDLR endocytosis, followed by the
intracellular degradation of endocytosed LDLR by lysosomes, which
results in reduced LDLR recycling back to the cell surface
(18,19). IDOL is an E3 ubiquitin ligase that
triggers the ubiquitination of LDLR cytoplasmic domain and promotes
lysosomal degradation, reducing the abundance of LDLR on the cell
surface (14). Unlike PCSK9, IDOL
gene expression is induced only by the liver X receptor (LXR),
which belongs to a key family of nuclear receptors required for the
transcription of genes involved in cholesterol and lipid metabolism
(20). These regulators
collectively function to control the abundance of LDLR on
hepatocytes (21,22).
Dendrobium nobile Lindl. alkaloids (DNLA) are
active ingredients that can be extracted from Dendrobium
nobile Lindl., a traditional Chinese herbal medicine with a
long history of use in China (23). A previous study has shown that DNLA
reduced carbon tetrachloride-induced liver injury in mice by
reducing mitochondrial oxidative stress, which was evidenced by the
decrease in mitochondrial malondialdehyde production and a marked
increase in manganese superoxide dismutase activity (24). Another recent study reported that
DNLA exerted neuroprotective effects against lipopolysaccharide
(LPS)-induced neuronal damage and cognitive impairment by
attenuating NOD-, LRR- and pyrin domain-containing protein
3-mediated pyroptosis in mice (25). In addition, DNLA has also been
documented to effectively ameliorate streptozotocin (STZ)-induced
elevation of blood glucose and lipid levels to protect against
STZ-induced fatty liver degeneration (26). DNLA has been suggested to exert a
beneficial effect on hepatic lipid homeostasis (27). Treatment of diabetic mice with DNLA
has been reported to confer beneficial effects on glucose and lipid
metabolism (28). However, the
mechanisms underlying the DNLA-mediated improvement of lipid
metabolism remain to be fully elucidated. To address this, the
present study investigated the effects of DNLA on lipid metabolism
and the associated mechanisms in HepG2 cells following treatment
with LPS.
Materials and methods
Chemicals
Dendrobium Nobile Lindl. (DNL) was purchased
from Xintian Traditional Chinese Medicine Industry Development Co.,
Ltd. DNLA was isolated from DNL based on our previous research
methods (29,30), and analyzed by Thermo Fisher
Q-Exactive UPLC-Q/Orbitrap MS (Thermo Fisher Science, Inc.).
Alkaloids accounted for 79.8% of DNLA, with 92.6% dendrobium
(C16H25O2N) as the major compound
based on liquid chromatography-mass spectrometry/mass spectrometry
analysis as described previously (31-33).
Minimum Essential Medium (MEM) was purchased from Procell Life
Science & Technology Co., Ltd., and FBS was obtained from
Gibco; Thermo Fisher Scientific, Inc.
Cell culture
The HepG2 human liver cancer cell line was used. The
cell line was provided by China Infrastructure of Cell Line
Resources, Institute of Basic Medical Sciences, Chinese Academy of
Medical Sciences (Beijing, China) with a statement of
authentication using the short tandem repeat profiling method.
HepG2 cells were incubated in MEM containing 10% FBS, 100 units/ml
penicillin and 100 mg/ml streptomycin at 37˚C in a 5%
CO2 incubator.
In LPS (Beijing Solarbio Science & Technology
Co., Ltd.) stimulation experiments, HepG2 cells were grown to 80%
confluence and the medium without FBS was subsequently replaced.
The HepG2 cells were then co-incubated with both LPS (5 µg/ml) and
different concentrations of DNLA for 48 h at 37˚C. For LXR agonist
experiments, following replacement with 0% FBS medium, the cells
were treated with 5 µM T0901317 (Selleck Chemicals) with or without
3.5 µg/ml DNLA for 48 h at 37˚C. For statin experiments, the medium
was replaced with medium without FBS and the cells were treated
with LPS alone or LPS and 1 µM rosuvastatin calcium (Beijing
Solarbio Science & Technology Co., Ltd.) and/or 3.5 µg/ml DNLA
for 48 h at 37˚C. The HepG2 cells in the control group were treated
with PBS of the same volume as DNLA or LPS in the other groups. The
reagents were dissolved in DMSO and diluted with medium to a
maximum final DMSO concentration of ≤0.025%.
Cell viability assay
The cytotoxic effects of DNLA and LPS were evaluated
in HepG2 cells using the Cell Counting Kit-8 (CCK-8) assay.
HepG2 cells were seeded into 96-well plates
(1x104 cells/well) for 24 h. Following the incubation
period at 37˚C, the cells were treated with or without DNLA (0.035,
0.35 and 3.5 µg/ml) for 24 and 48 h at 37˚C, respectively.
HepG2 cells were seeded into 96-well plates
(1x104 cells/well) for 24 h at 37˚C. Following the
incubation period, the cells were treated with or without LPS (1,
2, 5 and 10 µg/ml) for 48 h at 37˚C.
Following the treatment period, 10 µl CCK-8 (Dojindo
Molecular Technologies, Inc.) was added for incubation at 37˚C for
2 h before the absorbance values were measured at 450 nm using a
microplate reader.
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
A total of 5x105 HepG2 cells per well
were seeded into 12-well plates and then treated with DNLA (0.035,
0.35 and 3.5 µg/ml) at 37˚C for 48 h in the presence of LPS (5
µg/ml) . To quantify gene expression, the total RNA was extracted
from HepG2 cells using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) and then converted to cDNA using
the PrimeScript™ RT Master Mix (cat. no. RR036A; Takara Bio, Inc.).
Reverse transcription was performed at 37˚C for 15 min and then
85˚C for 5 sec, holding at 4˚C. qPCR was performed in triplicate
using SYBR Green qPCR master mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and the cDNA was amplified using a Vii7 Real-Time
PCR System. The qPCR thermocycling conditions were as follows: 95˚C
for 3 min, followed by 40 cycles of 95˚C for 15 secs and 60˚C for
60 sec, with a 4˚C hold. The expression of the target genes in each
group were compared using the
2-ΔΔCq method (34) and normalized using statistical
analysis. The primers for the reactions were as follows: LDLR
forward, 5'-CAGCTACCCCTCGAGACAGA-3' and reverse,
5'-GCAGGCAATGCTTTGGTCTT-3'; and GAPDH forward,
5'-CATGAGAAGTATGACAACAGCC-3' and reverse,
5'-AGTCCTTCCACGATACCAAAG-3'.
Western blot analysis
A total of 1x106 HepG2 cells were seeded
into 6-well plates and treated as aforementioned. The cells were
lysed in RIPA buffer (Thermo Fisher Scientific, Inc.) containing 1
mM phenylmethylsulfonyl fluoride, homogenized on ice, allowed to
stand for 30 min and centrifuged at 13,500 x g for 15 min at 4˚C.
The protein concentrations were determined using a bicinchoninic
assay kit (Thermo Fisher Scientific, Inc.). Total proteins (30 µg
from each sample) were then separated using 10% SDS-PAGE and
transferred onto polyvinylidene difluoride membranes. Subsequently,
the membranes were blocked with 5% skimmed dry milk in Tris-buffer
solution with 0.1% Tween-20 (TBST) at room temperature for 2 h
before being incubated overnight at 4˚C with the following primary
antibodies (1:1,000): Rabbit anti-LDLR (cat. no. ab30532; Abcam),
rabbit anti-PCSK9 (cat. no. ab185194; Abcam), rabbit anti-IDOL
(cat. no. bs-9674R; BIOSS), rabbit anti-SREBP2 (cat. no. ab30682;
Abcam), rabbit anti-LXRα (cat. no. bs-10311R; BIOSS), rabbit
anti-HNF1α (cat. no. 89670S; Cell Signaling Technology, Inc.),
rabbit anti-3-hydroxy-3-methyl glutaryl-coenzyme A reductase
(HMGCR; cat. no. ab242315; Abcam), rabbit anti-cytochrome P450
(CYP) 7A1 (cat. no. bs-21430R; BIOSS) and mouse anti-GAPDH (cat.
no. 97166S; Cell Signaling Technology, Inc.). The membranes were
then washed with TBST three times and subsequently probed with the
appropriate HRP-conjugated secondary antibodies (1:2,000) (cat.
nos. SA00001-1 abd SA00001-2; Proteintech Group, Inc.) for 1 h at
room temperature. All protein bands were visualized using an
electrochemiluminescence kit (Thermo Scientific SuperSignal West
Pico PLUS Chemiluminescent Substrate; cat. no. 34577; Thermo Fisher
Scientific, Inc.) and semi-quantified by ImageJ 1.48j software
(National Institutes of Health).
1,1'-dioctadecyl-3,3,3',3'-tetramethyl-indocarbocyanideperchlorate
(Dil)-LDL uptake assay
LDL labeling with Dil-LDL represents the cellular
cholesterol uptake ability (35).
A total of 5x105 HepG2 cells were cultured, inoculated
into confocal dishes and subjected to different treatments with
both LPS (5 µg/ml) and different concentrations of DNLA (0.035,
0.35 and 3.5 µg/ml) for 48 h at 37˚C. Following the removal of
culture medium and replacement with serum-free medium, the cells
were incubated with 10 µg/ml Dil-LDL (Beijing Solarbio Science
& Technology Co., Ltd.) for 4-5 h at 37˚C in the dark.
Subsequently, the cells were washed with PBS and fixed in 4%
paraformaldehyde (Beijing Zhongshan Jinqiao Biotechnology Co.,
Ltd.) at room temperature for 15 min. The nuclei of the cells were
then stained with DAPI (Beyotime Institute of Biotechnology) for 10
min. The cells were examined by confocal microscope (Leica SP8
laser-scanning confocal microscope) and subsequently analyzed using
ImageJ 1.48j software (National Institutes of Health). In each
group, six fields of view were used per well for
quantification.
LDLR immunofluorescence
A total of 5x105 HepG2 cells were seeded
into 12-well plates lined with cell slides and treated as
aforementioned. Following three washes with PBS, HepG2 cells were
fixed with 4% paraformaldehyde for 15 min at room temperature and
0.5% Triton X-100 in PBS was applied for 10 min. Subsequently, the
cells were blocked in 10% sheep serum blocking solution (Beijing
Zhongshan Jinqiao Biotechnology Co., Ltd.) for 1 h at room
temperature and incubated with primary anti-LDLR rabbit (1:1,000;
cat. no. SA00001-2; Proteintech Group, Inc.) antibody at 4˚C
overnight, followed by incubation with Alexa Fluor 488-conjugated
goat anti-rabbit IgG (1:1,000; cat. no. ZF-0511; Beijing Zhongshan
Jinqiao Biotechnology Co., Ltd.) at room temperature for 1 h.
Excess antibody was removed by washing with PBS. The cells were
subsequently stained with DAPI (5 µg/ml; Beyotime Institute of
Biotechnology) at room temperature for 10 min before being imaged
using a laser-scanning confocal microscope (Leica SP8). The cells
were analyzed using the ImageJ 1.48j software (National Institutes
of Health). In each group, six fields of view used per well for
quantification.
Statistical analysis
All experiments were repeated at least three times.
The data are presented as the mean ± standard deviation. The
significance of the differences was evaluated using one-way ANOVA
followed by the Bonferroni post hoc test. Statistical analysis was
performed using SPSS 19.0 (IBM Corp.) and GraphPad Prism 8.0
software (Dotmatics). P<0.05 was considered to indicate a
statistically significant difference.
Results
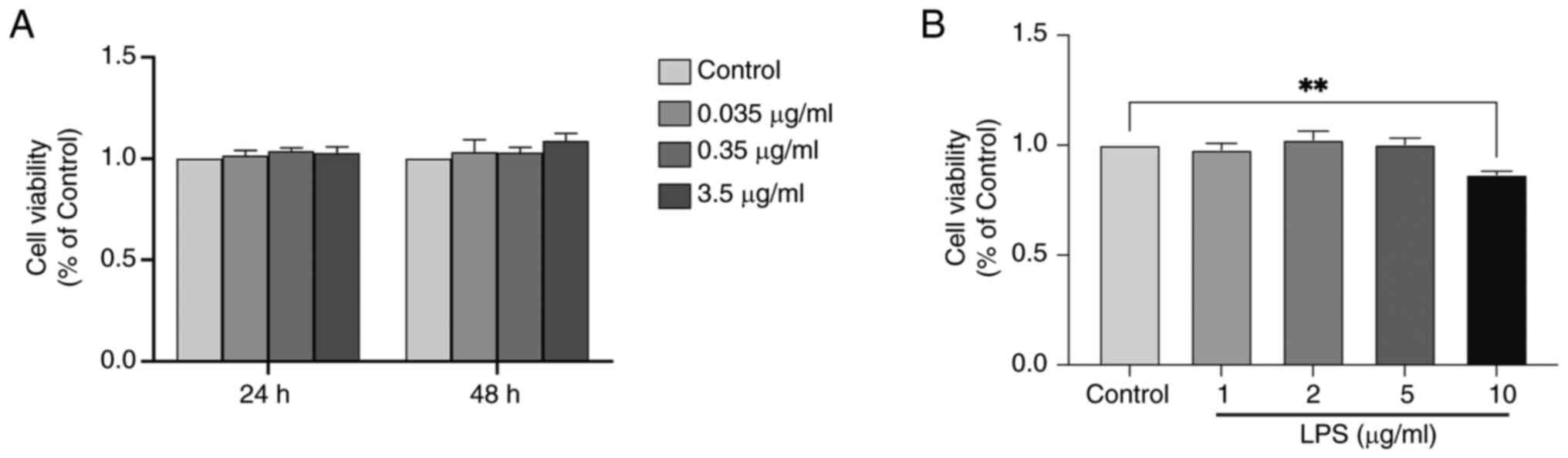
Effects of different concentrations of
DNLA and LPS on HepG2 cell viability
To assess the cytotoxic effects of DNLA and LPS,
HepG2 cells were treated with various concentrations of DNLA
(0.035, 0.35 and 3.5 µg/ml) for 24 or 48 h, followed by evaluation
of cell viability with the CCK-8 assay. None of the concentrations
of DNLA tested exerted significant cytotoxic effects on HepG2 cells
at the 24 and 48 h time points (Fig.
1A). Examination of the viability of HepG2 cells treated with
different concentrations of LPS (1, 2, 5 and 10 µg/ml) for 48 h
revealed that 10 µg/ml LPS exerted a significant cytotoxic effect
at this time point compared with the control group (Fig. 1B). Based on these findings, 5 µg/ml
LPS and DNLA (0.035, 0.35 and 3.5 µg/ml) were selected for 48 h
treatment under basal conditions for subsequent experiments.
DNLA restores LDL uptake by HepG2
cells under LPS stimulation
To investigate the effect of DNLA on the uptake of
LDL, treated HepG2 cells were incubated with Dil-LDL particles for
4-5 h, before LDL uptake was visualized by confocal microscope. As
shown in Fig. 2A and B, LPS significantly reduced LDL uptake.
However, treatment with DNLA (at concentrations of 0.35 and 3.5
µg/ml) significantly restored the uptake of LDL by HepG2 cells in
the presence of LPS (Fig. 2).
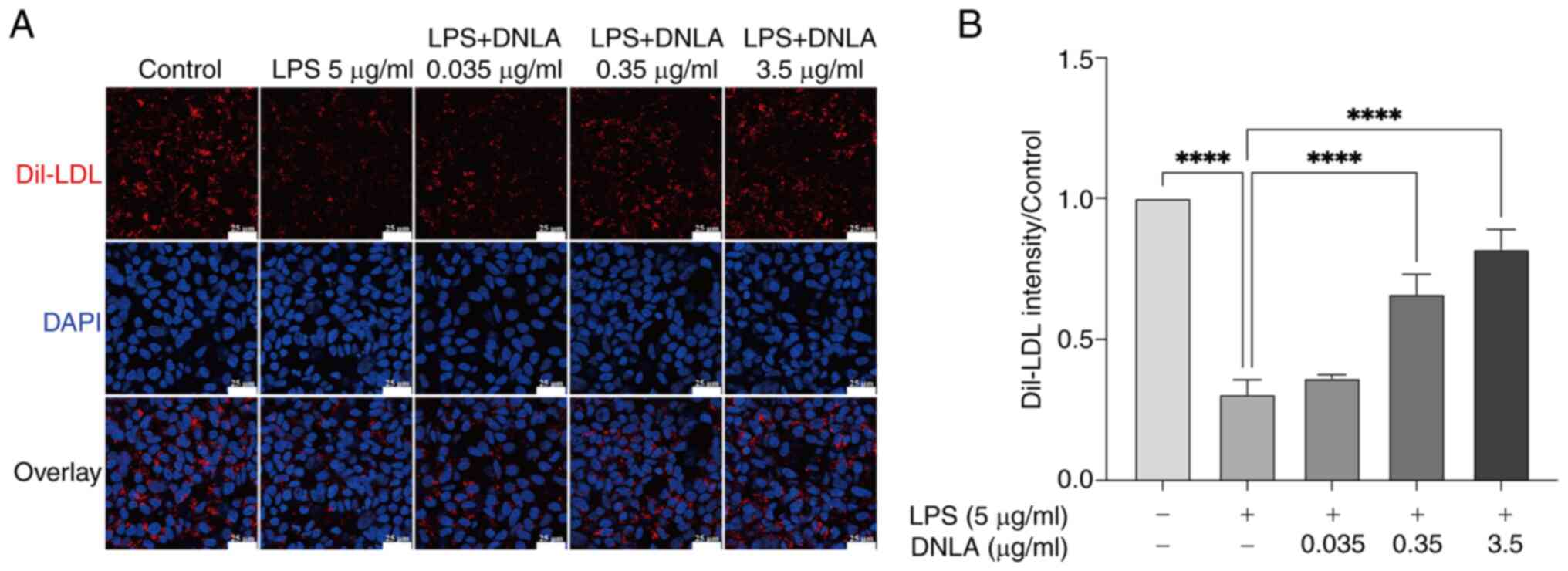 | Figure 2DNLA increases LDL uptake inhibited
by LPS in HepG2 cells as determined using confocal microscopy. (A)
Dil-LDL staining (red), DAPI staining (blue) and overlay.
Magnification, x400. Scale bar, 25 µm. (B) Semi-quantification of
(A). Data are presented as the mean ± SD of three independent
experiments. ****P<0.0001. DNLA, Dendrobium
nobile Lindl. alkaloids; LPS, lipopolysaccharides; LDL,
low-density lipoprotein; Dil,
1,1'-dioctadecyl-3,3,3',3'-tetramethyl-indocarbocyanideperchlorate. |
DNLA causes an upregulation in the
protein and mRNA expression levels of LDLR in HepG2 cells
LDL uptake is associated with the amount of LDLR on
the cell surface (20).
Immunofluorescence and western blotting results revealed a
significant decrease in LDLR protein expression following LPS
stimulation compared with that in the control group (Fig. 3A-D). DNLA treatment (at
concentrations of 0.35 and 3.5 µg/ml) led to a marked increase in
the LDLR protein expression in HepG2 cells in the presence of LPS
(Fig. 3A-D). To investigate
whether LDLR expression could be regulated by DNLA at the
transcriptional level, LDLR mRNA expression was then examined. The
results of RT-qPCR demonstrated that DNLA significantly increased
the mRNA expression levels of LDLR in HepG2 cells in the presence
of LPS (Fig. 3E). Therefore, these
findings suggested that DNLA could increase the expression levels
of LDLR at both transcriptional and post-translation levels in the
presence of LPS.
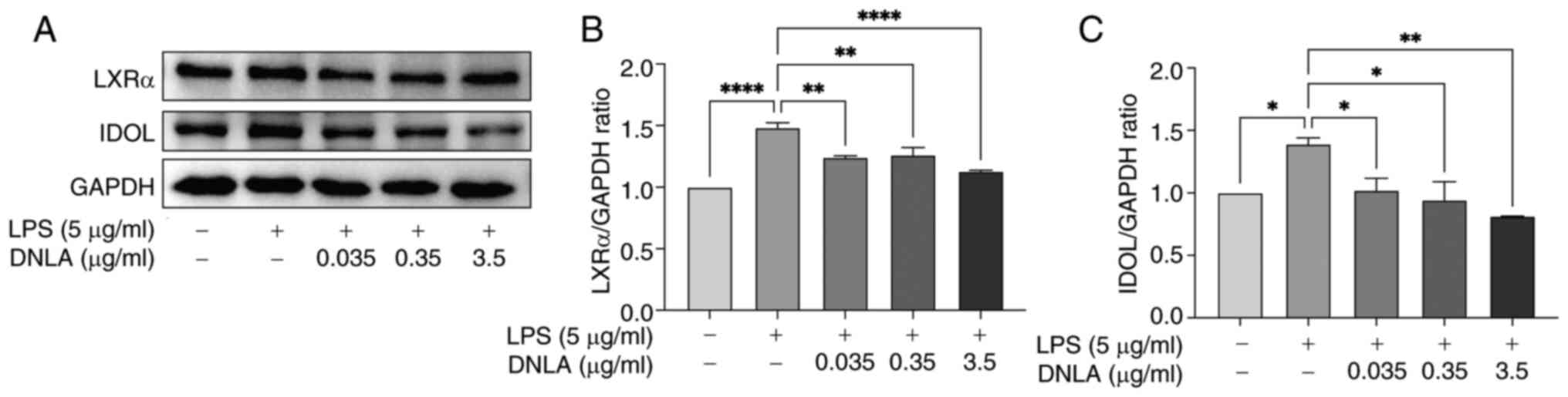
DNLA downregulates IDOL and LXRα
protein expression in LPS-stimulated HepG2 cells
IDOL is an important factor involved in
post-transcriptional regulation of LDLR, promoting its
ubiquitination and lysosomal degradation, and IDOL expression is
regulated by LXRα (36). Western
blot analysis was used to examine IDOL and LXRα protein expression
in treated cells. As shown in Fig.
4, LPS stimulation significantly enhanced the protein
expression levels of IDOL and LXRα compared with those in the
control group. By contrast, treatment with DNLA induced a
significant reversal of the increase in IDOL and LXRα protein
expression caused by LPS (Fig. 4).
These results suggested that DNLA could serve a role in
ameliorating lipid metabolism disorder by activating the
LXRα/IDOL/LDLR signal pathway.
DNLA increases LDLR content in
LPS-stimulated HepG2 cells through regulation of the LXRα/IDOL
axis
LXRs are cholesterol-sensitive transcription factors
activated in response to excess intracellular cholesterol to induce
the expression of key genes involved in the regulation of
cholesterol homeostasis, including IDOL (37). To further investigate the potential
mechanism underlying the therapeutic effects of DNLA on lipid
metabolism disorders, the LXR agonist T0901317 was next utilized.
Before the protein expression levels of LDLR, IDOL and LXRα were
detected by western blot analysis. Treatment with T0901317
significantly reversed the DNLA-mediated reduction of IDOL and LXRα
protein expression, whilst significantly decreasing LDLR expression
in HepG2 cells (Fig. 5). Based on
these findings, it was proposed that DNLA increased LDLR expression
by regulating the LXRα/IDOL signaling pathway in HepG2 cells.
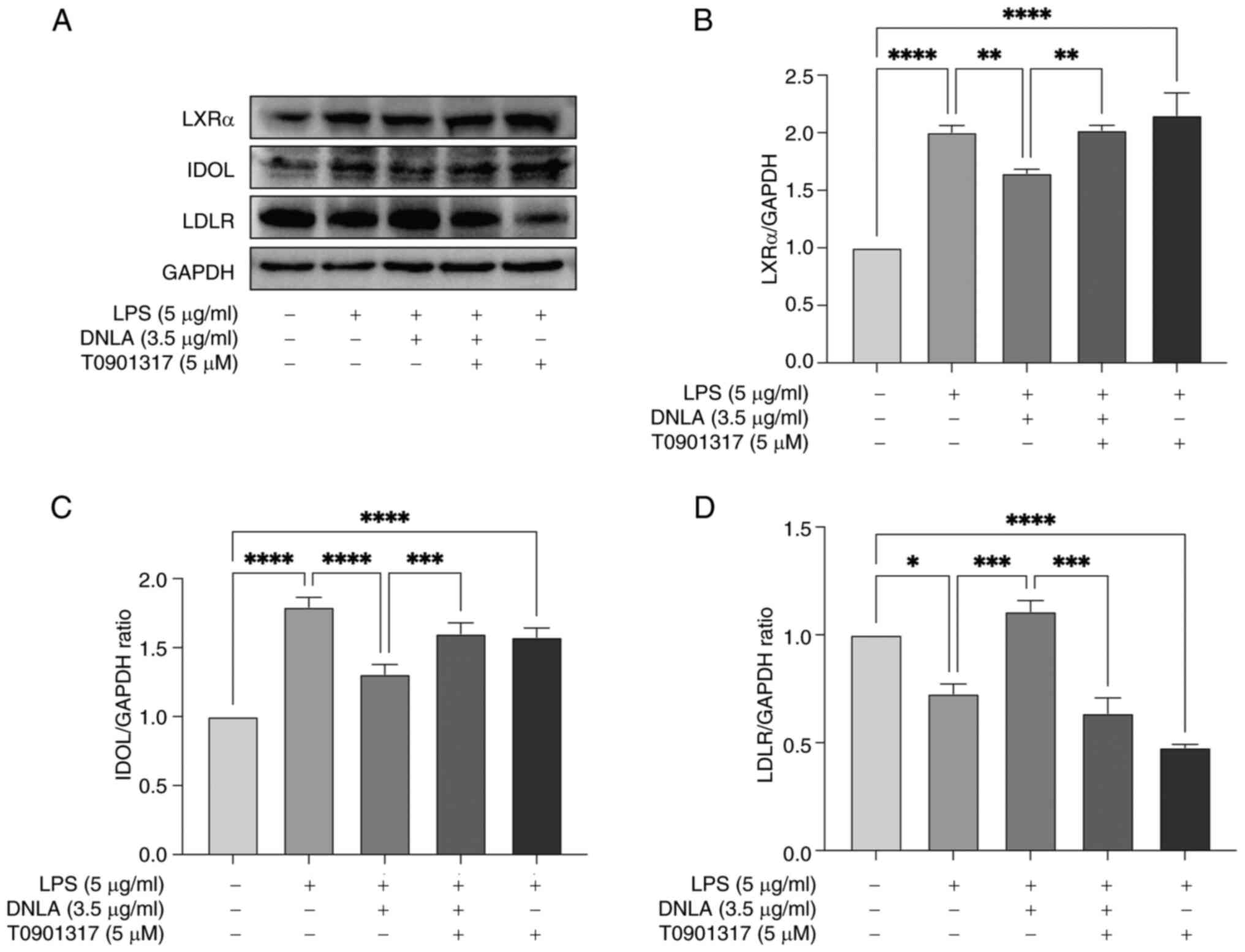 | Figure 5T0901317, an LXRα agonist, reverses
the decrease in IDOL and LXRα expression induced by DNLA and
reduces the LDLR content in HepG2 cells. (A) Protein expression of
LXRα, IDOL and LDLR in HepG2 cells assessed by western blotting.
Normalized intensity of (B) LXRα, (C) IDOL and (D) LDLR vs. GAPDH,
presented as the mean ± SD of three independent experiments.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. DNLA,
Dendrobium nobile Lindl. alkaloids; LPS,
lipopolysaccharides; LXRα, liver X receptor α; IDOL, inducible
degrader of the low-density lipoprotein receptor; LDLR, low-density
lipoprotein receptor. |
DNLA suppresses PCSK9, SREBP2 and
HNF1α protein expression in LPS-stimulated HepG2 cells
PCSK9 is a post-transcriptional regulator that can
promote the degradation of LDLR and reduce its recycling to the
cell membrane (38). As shown in
Fig. 6D, LPS stimulation led to
significant upregulation of PCSK9 protein expression, which was
significantly reversed following DNLA intervention. PCSK9
expression is further regulated by SREBP2 and HNF1α (39). LPS was found to induce a
significant increase in SREBP2 and HNF1α expression, which was
markedly reversed by DNLA treatment (Fig. 6A and B). Taken together, these data suggested
that DNLA may improve lipid metabolism disorders by regulating the
expression levels of PCSK9, SREBP2 and HNF1α.
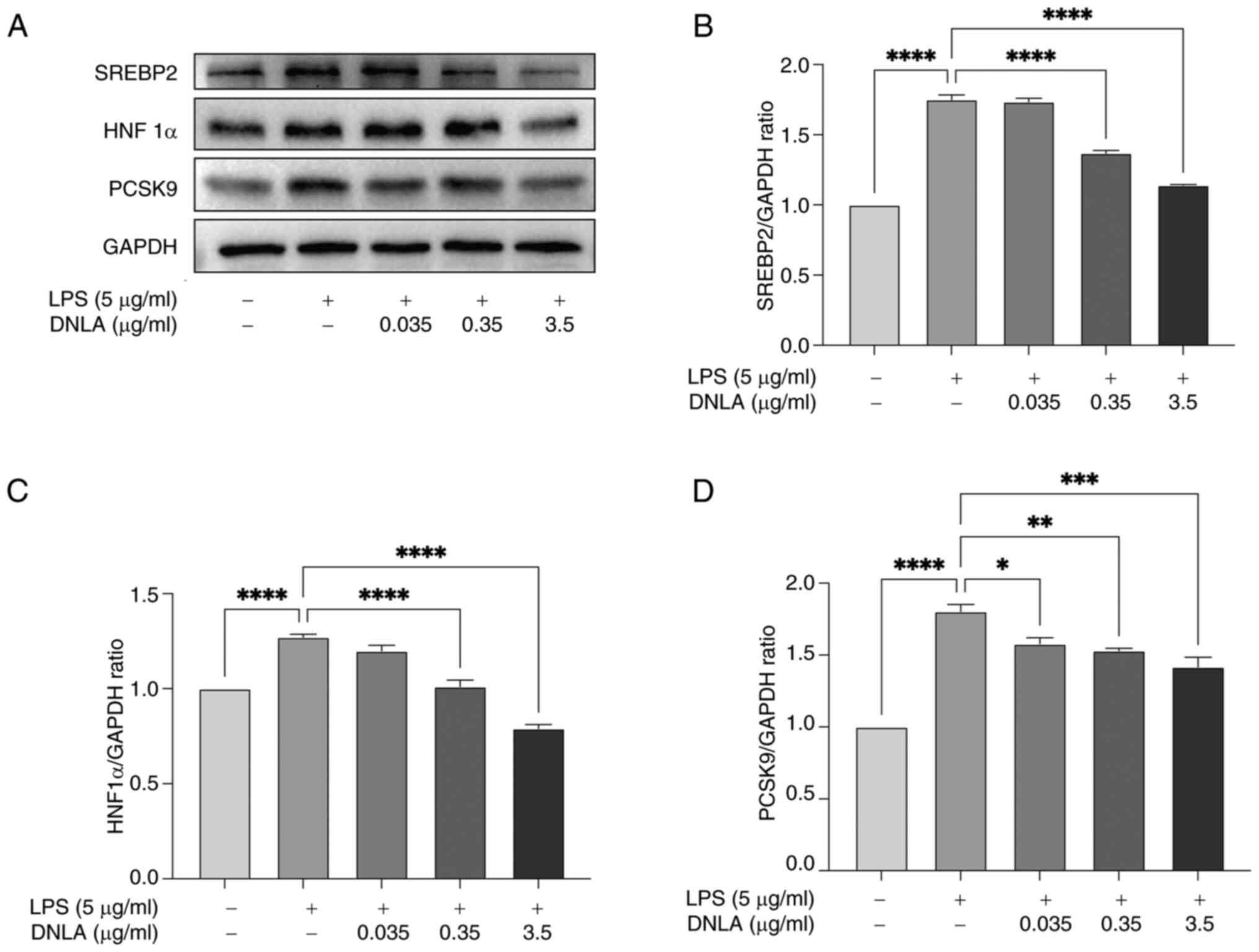 | Figure 6DNLA suppresses PCSK9, SREBP2 and
HNF1α protein expression in HepG2 cells. HepG2 cells were treated
with varying concentrations of DNLA (0.035, 0.35 and 3.5 µg/ml)
and/or LPS (5 µg/ml) for 48 h. (A) Protein expression of SREBP2,
HNF1α and PCSK9 in HepG2 cells assessed through western blotting.
Normalized intensity of (B) SREBP2, (C) HNF1α and (D) PCSK9 vs.
GAPDH, presented as the mean ± SD of three independent experiments.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. DNLA,
Dendrobium nobile Lindl. alkaloids; LPS,
lipopolysaccharides; PCSK9, proprotein convertase subtilisin/Kexin
type 9; SREBP2, sterol regulatory element-binding protein 2; HNF1α,
hepatocyte nuclear factor 1α. |
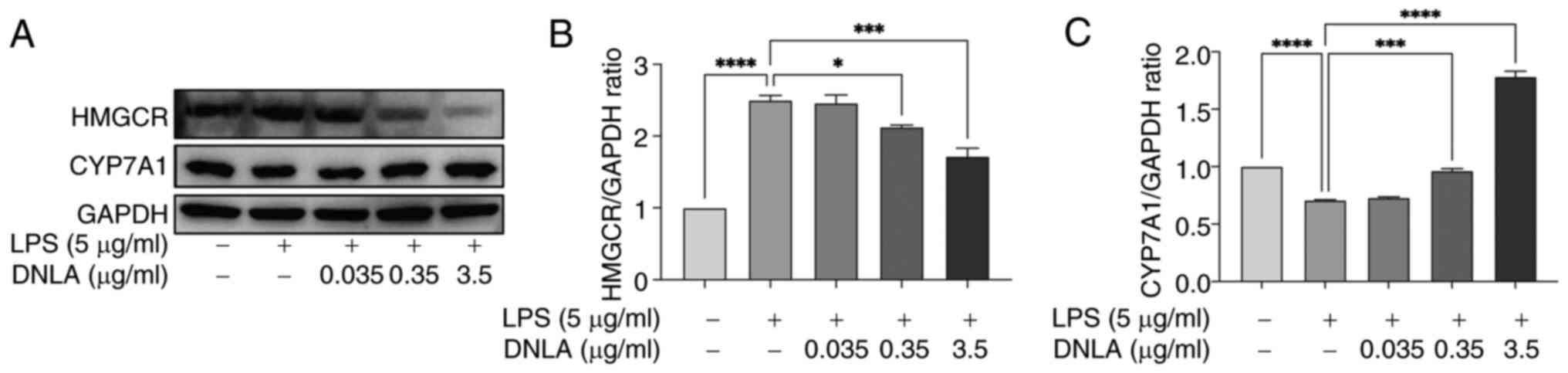
DNLA enhances CYP7A1 expression and
suppresses HMGCR expression in LPS-stimulated HepG2 cells
DNLA appeared to enhance the cellular uptake of LDL.
To determine whether DNLA serves a role in maintaining
intracellular cholesterol homeostasis, its effects on the
expression levels of HMGCR, which is the rate-limiting enzyme in
the cholesterol synthesis pathway, were examined (40). In addition, the effects of DNLA on
the expression levels of CYP7A1, which is the rate-limiting enzyme
in the classical pathway of cholesterol conversion to bile acids,
were examined (41). DNLA
effectively decreased HMGCR and increased CYP7A1 expression at the
protein level in HepG2 cells following LPS stimulation (Fig. 7).
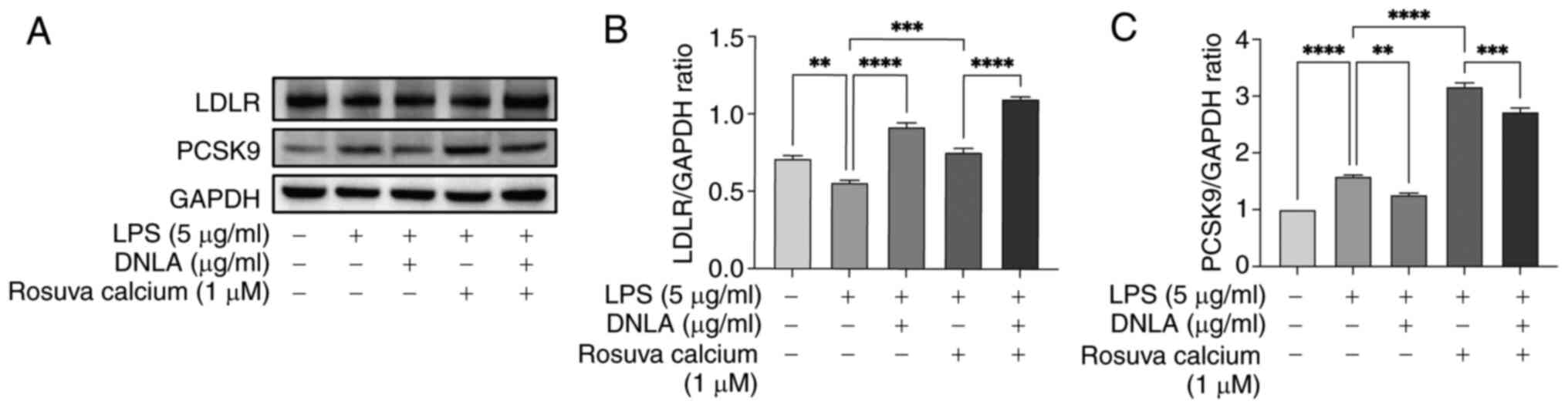
DNLA reduces PCSK9 expression in
rosuvastatin calcium-treated HepG2 cells
Statins are widely used to lower plasma LDL-C levels
due to their ability to upregulate hepatic LDLR expression whilst
enhancing the subsequent uptake of LDL-C from the blood, in
addition to their inhibitory effects on HMGCR activity (42). In the present study, the effects of
DNLA in combination with statins on the expression levels of LDLR
and PCSK9 were examined. LDLR and PCSK9 protein levels were
determined by western blot analysis. As shown in Fig. 8, rosuvastatin calcium induced the
upregulation of PCSK9 expression compared with the LPS stimulation
group. Administration of DNLA led to a significant decrease in
rosuvastatin calcium-induced PCSK9 expression (Fig. 8). The combined treatment with DNLA
and rosuvastatin calcium resulted in an overall increase in LDLR
expression compared with treatment with rosuvastatin in HepG2 cells
under LPS stimulation. These findings support the hypothesis that
DNLA could inhibit statin-induced PCSK9 expression and increase the
intracellular LDLR content, thereby potentially improving the
efficacy of statin therapy.
Discussion
The present study focused on the effects of DNLA on
LPS-induced lipid metabolism disorders in HepG2 cells. The results
suggest that DNLA effectively increased the expression levels of
LDLR, enhanced the uptake of Dil-LDL, and inhibited IDOL, LXRα,
PCSK9, SREBP2 and HNF1α protein expression in HepG2 cells.
Simultaneously, DNLA inhibited the expression of HMGCR, the
rate-limiting enzyme in the cholesterol synthesis pathway whilst
promoting that of CYP7A1, the rate-limiting enzyme in the classical
metabolic pathway of cholesterol to bile acids. In addition, DNLA
in combination with statins was found to enhance LDLR expression in
HepG2 cells, and DNLA inhibited the statin-induced increase in
PCSK9 expression. These findings suggested that DNLA alleviated
lipid metabolism disorders by regulating the LXRα/IDOL/LDLR pathway
in HepG2 cells.
Elevated plasma LDL-C is an important risk factor
for atherosclerosis, where inflammation serves a key role (43). In the absence of stimulation by
other inflammatory factors, excessive free cholesterol can induce
an inflammatory response, which then further promotes the uptake
and accumulation of lipids by cells to inhibit the outflow of
cellular lipids, increasing the risk of lipid metabolism disorders
to accelerate the process of atherosclerosis (44). Disorders in lipid metabolism
eventually trigger an inflammatory response (45). Atherosclerosis is a complex
pathological process, where abnormal lipid metabolism is typically
accompanied by an inflammatory response (46,47).
Previous studies have shown that LPS stimulation could suppress
LDLR expression, and increase SREBP2 and PCSK9 expression, whilst
decreasing Dil-LDL uptake by HepG2 cells (48,49).
In mice, LPS-induced systemic inflammation was observed to increase
PCSK9 mRNA expression, decrease hepatic LDLR expression and
increase plasma LDL-C levels (50). In the present study, stimulation
with LPS consistently suppressed the protein levels of LDLR and the
uptake of LDL in HepG2 cells.
Dendrobium species, which have been documented to
confer beneficial therapeutic effects, have been widely used as a
traditional Chinese medicine (23). The use of Dendrobium nobile
Lindl. as a herbal medicine is particularly common in Guizhou,
China (25,26), which is included in the Chinese
Pharmacopoeia. To date, ≥82 active ingredients have been isolated
from Dendrobium nobile Lindl., including alkaloids,
glycosides, polysaccharides, phenanthrene and dibenzyl compounds
(51,52). DNLA appears to be the main active
compound, and its pharmacological effects are relatively complex
(53). To the best of our
knowledge, its effects on LDL have not been previously studied. In
total, >70% of plasma LDL-C is cleared by cell surface
LDLR-mediated endocytosis (8).
LDLR is a receptor that is mainly expressed in the liver.
Therefore, the liver is an important site for cholesterol
metabolism (4,5). Increasing hepatic LDLR expression or
its activity will likely accelerate the clearance of circulating
LDL particles, serving as a potential strategy for regulating
cholesterol metabolism. Changes in LDLR expression are driven by a
combination of transcriptional and post-translational regulation
processes (54-56).
At the transcriptional level, LDLR expression is mainly regulated
by SREBP, which binds to the SRE region of the LDLR promoter to
promote transcription, thereby increasing the expression of LDLR on
the cell membrane (54). IDOL and
PCSK9 are the two main regulators of LDLR stability during the
post-translational phase (14).
Both of these aforementioned proteins can induce the lysosomal
degradation of LDLR through different pathways. Extracellular
mature PCSK9 interacts with cell surface LDLR to trigger
receptor-mediated endocytosis, leading to lysosomal LDLR
degradation and reducing its localization to the cell membrane
(19). By contrast, intracellular
IDOL binds to the intracellular structural domain of LDLR,
promoting ubiquitination of this region and protein localization to
lysosomes for degradation (51,57).
In the present study, DNLA was found to increase LDLR protein
expression and enhance the uptake of LDL in LPS-stimulated HepG2
cells.
IDOL expression is mainly regulated by the LXR,
which is activated by a number of LXR ligands, such as oxysterols
and synthetic agonists (18,58).
Previous genome-wide association studies have identified genetic
variants at the IDOL locus that can affect serum LDL-C levels
(59-61).
Furthermore, knockdown of IDOL expression with short interference
RNA has been previously associated with elevated LDLR levels in
HepG2 cells (62,63). Therefore, inhibition of
IDOL-mediated LDLR degradation may provide a therapeutic direction
to improve hepatic clearance of LDL-C. In the present study, DNLA
suppressed IDOL and LXRα protein expression in LPS-stimulated HepG2
cells. To further investigate the mechanisms by which DNLA can
improve lipid metabolism disorders, the LXR synthetic agonist
T0901317 was used. T0901317 was able to reverse the inhibitory
effects of DNLA on IDOL and LXRα protein expression to reduce the
expression of LDLR in HepG2 cells. These findings suggested that
DNLA could promote LDLR expression in HepG2 cells in association
with the inhibition of IDOL and LXRα expression. Specifically, DNLA
reduced LXRα expression, which in turn reduced IDOL expression.
Therefore, it can be proposed that DNLA functioned as an inhibitor
of LXRα rather than antagonizing the activity of LXRα. Accordingly,
DNLA may exert beneficial effects on lipid metabolism in HepG2
cells by regulating the LXRα/IDOL/LDLR pathway.
PCSK9 is a plasma protein regulated mainly by SREBP2
and HNF1α (64). Deficiency of
PCSK9 has been previously associated with an increase in cell
surface LDLR expression and a decrease in the plasma cholesterol
concentration (65). PCSK9 has
been identified to be a therapeutic target for cardiovascular
diseases, where its inhibitors compete with LDLR to interact with
PCSK9. This suppresses endocytosis and degradation of LDLR to
increase the LDLR content on the hepatocyte membrane, thereby
promoting the metabolism of LDL-C by the liver and reducing plasma
LDL-C levels (66). The results of
the present study indicated that DNLA downregulated PCSK9, SREBP2
and HNF1α protein expression in LPS-stimulated HepG2 cells,
suggesting that its activity against lipid metabolism disorders may
also be associated with regulation of the PCSK9-related pathway.
However, the present study only focused on the LXRα/IDOL/LDLR
pathway using the agonist of LXR, and the results demonstrated that
DNLA regulated LDLR expression. Further studies are necessary to
establish the underlying mechanisms.
Statins are the most widely used
cholesterol-lowering drugs. They act as potent inhibitors of HMGCR,
the rate-limiting enzyme for ab initio cholesterol synthesis
(67,68). Statins upregulate LDLR and PCSK9
expression whilst increasing LDL-C uptake by activating the SREBP2
pathway (69). Concomitant PCSK9
expression attenuates LDLR protein activity and is considered to
limit the efficacy of statins in lowering cholesterol levels
(70). In the present study, DNLA
intervention suppressed rosuvastatin calcium-induced PCSK9
expression and further increased LDLR levels compared with those
mediated by rosuvastatin calcium alone. However, the molecular
mechanism of action underlying the effects of this DNLA-statin
combination remain to be fully established. The present study
showed that DNLA reduced statin-induced PCSK9 expression and
increased LDLR protein stability. Therefore, combinations of these
drugs with different mechanisms of action may exert a synergistic
effect, thereby improving the efficacy of statin therapy.
Intracellular cholesterol homeostasis is maintained
by cholesterol biosynthesis and export in addition to relying on
uptake (3). In total, ~30 reaction
steps are involved in cholesterol biosynthesis. HMGCR, the
rate-limiting enzyme involved in this pathway, is regulated by
SREBP2(71). Conversion of
cholesterol to bile acids and biliary excretion of cholesterol are
critical mechanisms for cholesterol removal in vivo
(72). CYP7A1 is involved in the
initial and rate-limiting steps in the classical pathway of
cholesterol conversion to bile acids (73). In the present study, DNLA treatment
was found to inhibit HMGCR expression and enhance CYP7A1 expression
in LPS-stimulated HepG2 cells. One suggested theory to explain
these findings is that DNLA maintains intracellular cholesterol
homeostasis with regard to synthesis and metabolism whilst
increasing LDL uptake by HepG2 cells.
In conclusion, the results of the present study
suggested that DNLA could serve a role in ameliorating lipid
metabolism disorders by regulating the LXRα/IDOL/LDLR pathway to
increase LDLR expression in HepG2 cells. In addition, the effects
of DNLA on LDLR are potentially associated with inhibition of
SREBP2, HNF1α and PCSK9 protein expression, and the detailed
mechanism of the effects of the SREBP2/HNF1α/PCSK9 pathway on LDLR
expression should be examined in future studies. In combination
with statins, DNLA could increase LDLR expression whilst inhibiting
the statin-induced upregulation of PCSK9 protein expression in
HepG2 cells. Simultaneously, DNLA could regulate the expression of
HMGCR and CYP7A1 proteins, which are rate-limiting enzymes in
cholesterol synthesis and metabolism. The results of the present
study provided insights into the pathways regulated by DNLA in
terms of lipid metabolism. However, further in vivo studies
are necessary to comprehensively establish the pharmacological
actions of DNLA and evaluate its utility as a combination drug for
the prevention and treatment of ASCVD.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Peking Union Medical
College Youth Fund (grant no. 3332018200), National Natural Science
Foundation of China (grant no. 82060750) and Guizhou Provincial
Department of Education 125 Major Special Projects (grant no.
2012-012).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JS, HRL and YXZ completed the project, analyzed the
data and wrote the manuscript. QW and RXX established the study,
interpreted the data, and reviewed and edited the manuscript. WZ
contributed to analyzing the data. JSS contributed to establishing
the study, interpreting the data and reviewing the manuscript. QW
and RXX confirm the authenticity of all the raw data. All authors
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kobiyama K and Ley K: Atherosclerosis.
Circ Res. 123:1118–1120. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ridker PM: LDL cholesterol: Controversies
and future therapeutic directions. Lancet. 384:607–617.
2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Luo J, Yang H and Song BL: Mechanisms and
regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol.
21:225–245. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Barale C, Melchionda E, Morotti A and
Russo I: PCSK9 biology and its role in atherothrombosis. Int J Mol
Sci. 22(5880)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li H, Yu XH, Ou X, Ouyang XP and Tang CK:
Hepatic cholesterol transport and its role in non-alcoholic fatty
liver disease and atherosclerosis. Prog Lipid Res.
83(101109)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Brown MS and Goldstein JL: A
receptor-mediated pathway for cholesterol homeostasis. Science.
232:34–47. 1986.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Burnett JR and Hooper AJ: MK-0616: An oral
PCSK9 inhibitor for hypercholesterolemia treatment. Expert Opin
Investig Drugs. 32:873–878. 2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Raschi E, Casula M, Cicero AFG, Corsini A,
Borghi C and Catapano A: Beyond statins: New pharmacological
targets to decrease LDL-cholesterol and cardiovascular events.
Pharmacol Ther. 250(108507)2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
O'Donoghue ML, Fazio S, Giugliano RP,
Stroes ESG, Kanevsky E, Gouni-Berthold I, Im K, Pineda AL,
Wasserman SM, Češka R, et al: Lipoprotein(a), PCSK9 inhibition, and
cardiovascular risk. Circulation. 139:1483–1492. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Rosenson RS, Hegele RA, Fazio S and Cannon
CP: The evolving future of PCSK9 inhibitors. J Am Coll Cardiol.
72:314–329. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Goldstein JL, DeBose-Boyd RA and Brown MS:
Protein sensors for membrane sterols. Cell. 124:35–46.
2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hua X, Yokoyama C, Wu J, Briggs MR, Brown
MS, Goldstein JL and Wang X: SREBP-2, a second
basic-helix-loop-helix-leucine zipper protein that stimulates
transcription by binding to a sterol regulatory element. Proc Natl
Acad Sci USA. 90:11603–11607. 1993.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shimano H, Shimomura I, Hammer RE, Herz J,
Goldstein JL, Brown MS and Horton JD: Elevated levels of SREBP-2
and cholesterol synthesis in livers of mice homozygous for a
targeted disruption of the SREBP-1 gene. J Clin Invest.
100:2115–2124. 1997.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yang HX, Zhang M, Long SY, Tuo QH, Tian Y,
Chen JX, Zhang CP and Liao DF: Cholesterol in LDL receptor
recycling and degradation. Clin Chim Acta. 500:81–86.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bartolomei M, Bollati C, Li J, Arnoldi A
and Lammi C: Assessment of the cholesterol-lowering effect of
MOMAST®: Biochemical and cellular studies. Nutrients.
14(493)2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ding Z, Pothineni NVK, Goel A, Lüscher TF
and Mehta JL: PCSK9 and inflammation: Role of shear stress,
pro-inflammatory cytokines, and LOX-1. Cardiovasc Res. 116:908–915.
2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jia Q, Cao H, Shen D, Li S, Yan L, Chen C,
Xing S and Dou F: Quercetin protects against atherosclerosis by
regulating the expression of PCSK9, CD36, PPARγ, LXRα and ABCA1.
Int J Mol Med. 44:893–902. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Olsson PA, Korhonen L, Mercer EA and
Lindholm D: MIR is a novel ERM-like protein that interacts with
myosin regulatory light chain and inhibits neurite outgrowth. J
Biol Chem. 274:36288–36292. 1999.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lagace TA: PCSK9 and LDLR degradation:
Regulatory mechanisms in circulation and in cells. Curr Opin
Lipidol. 25:387–393. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
van Loon NM, van Wouw SAE, Ottenhoff R,
Nelson JK, Kingma J, Scheij S, Moeton M and Zelcer N: Regulation of
intestinal LDLR by the LXR-IDOL axis. Atherosclerosis. 315:1–9.
2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zelcer N, Hong C, Boyadjian R and Tontonoz
P: LXR regulates cholesterol uptake through Idol-dependent
ubiquitination of the LDL receptor. Science. 325:100–104.
2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang L, Reue K, Fong LG, Young SG and
Tontonoz P: Feedback regulation of cholesterol uptake by the
LXR-IDOL-LDLR axis. Arterioscler Thromb Vasc Biol. 32:2541–2546.
2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
da Silva JA and Ng TB: The medicinal and
pharmaceutical importance of Dendrobium species. Appl Microbiol
Biotechnol. 101:2227–2239. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhou J, Zhang Y, Li S, Zhou Q, Lu Y, Shi
J, Liu J, Wu Q and Zhou S: Dendrobium nobile Lindl.
alkaloids-mediated protection against CCl4-induced liver
mitochondrial oxidative damage is dependent on the activation of
Nrf2 signaling pathway. Biomed Pharmacother.
129(110351)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li DD, Fan HX, Yang R, Li YY, Zhang F and
Shi JS: Dendrobium Nobile Lindl. Alkaloid suppresses NLRP3-mediated
pyroptosis to alleviate LPS-induced neurotoxicity. Front Pharmacol.
13(846541)2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Huang Q, Liao X, Wu Q, Li F, Wang LY and
Shi JS: Effects of Dendrobium nobile Lindl. alkaloids on blood
glucose and liver fatty degeneration in diabetic rats. Chin J New
Drugs Clin Rem. 32:490–493. 2013.
|
|
27
|
Xu YY, Xu YS, Wang Y, Wu Q, Lu YF, Liu J
and Shi JS: Dendrobium nobile Lindl. alkaloids regulate metabolism
gene expression in livers of mice. J Pharm Pharmacol. 69:1409–1417.
2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Huang S, Wu Q, Liu H, Ling H, He Y, Wang
C, Wang Z and Lu Y and Lu Y: Alkaloids of dendrobium nobile lindl.
Altered hepatic lipid homeostasis via regulation of bile acids. J
Ethnopharmacol. 241(111976)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhou J, Zhang Y, Li S, Zhou Q, Lu Y, Shi
J, Liu J, Wu Q and Zhou S: Dendrobium nobile Lindl.
alkaloids-mediated protection against CCl4-induced liver
mitochondrial oxidative damage is dependent on the activation of
Nrf2 signaling pathway. Biomed Pharmacother.
129(110351)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Xian S, Yang Y, Nan N, Fu X, Shi J, Wu Q
and Zhou S: Inhibition of mitochondrial ROS-mediated necroptosis by
Dendrobium nobile Lindl. alkaloids in carbon tetrachloride induced
acute liver injury. J Ethnopharmacol. 330(118253)2024.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Nie J, Tian Y, Zhang Y, Lu YL, Li LS and
Shi JS: Dendrobium alkaloids prevent Aβ25-35-induced neuronal and
synaptic loss via promoting neurotrophic factors expression in
mice. PeerJ. 4(e2739)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Li LS, Lu YL, Nie J, Xu YY, Zhang W, Yang
WJ, Gong QH, Lu YF, Lu Y and Shi JS: Dendrobium nobile Lindl
alkaloid, a novel autophagy inducer, protects against axonal
degeneration induced by Aβ25-35 in hippocampus neurons in vitro.
CNS Neurosci Ther. 23:329–340. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Huang J, Huang N, Zhang M, Nie J, Xu YY,
Wu Q and Shi J: Dendrobium alkaloids decrease Aβ by regulating α-
and β-secretases in hippocampal neurons of SD rats. PeerJ.
7(e7627)2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Emmanuel UO: TNFα-Induced LDL cholesterol
accumulation involve elevated LDLR cell surface levels and SR-B1
downregulation in human arterial endothelial cells. Int J Mol Sci.
22(6236)2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Scotti E, Calamai M, Goulbourne CN, Zhang
L, Hong C, Lin RR, Choi J, Pilch PF, Fong LG, Zou P, et al: IDOL
stimulates clathrin-independent endocytosis and multivesicular
body-mediated lysosomal degradation of the low-density lipoprotein
receptor. Mol Cell Biol. 33:1503–1514. 2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Nazih H and Bard JM: Cholesterol,
oxysterols and LXRs in breast cancer pathophysiology. Int J Mol
Sci. 21(1356)2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Seidah NG and Prat A: The multifaceted
biology of PCSK9. Endocr Rev. 43:558–582. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Nourse JL, Leung VM, Abuwarda H, Evans EL,
Izquierdo-Ortiz E, Ly AT, Truong N, Smith S, Bhavsar H, Bertaccini
G, et al: Piezo1 regulates cholesterol biosynthesis to influence
neural stem cell fate during brain development. J Gen Physiol.
154(e202213084)2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yang T, Wang Y, Cao X, Peng Y, Huang J,
Chen L, Pang J, Jiang Z, Qian S, Liu Y, et al: Targeting mTOR/YY1
signaling pathway by quercetin through CYP7A1-mediated
cholesterol-to-bile acids conversion alleviated type 2 diabetes
mellitus induced hepatic lipid accumulation. Phytomedicine.
113(154703)2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Li W, Li H, Zha C, Che B, Yu Y, Yang J and
Li T: Lipids, lipid-modified drug target genes, and the risk of
male infertility: A Mendelian randomization study. Front Endocrinol
(Lausanne). 15(1392533)2024.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Gormley M, Yarmolinsky J, Dudding T,
Burrows K, Martin RM, Thomas S, Tyrrell J, Brennan P, Pring M,
Boccia S, et al: Using genetic variants to evaluate the causal
effect of cholesterol lowering on head and neck cancer risk: A
Mendelian randomization study. PLoS Genet.
17(e1009525)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Navarese EP, Robinson JG, Kowalewski M,
Kolodziejczak M, Andreotti F, Bliden K, Tantry U, Kubica J, Raggi P
and Gurbel PA: Association between baseline LDL-C level and total
and cardiovascular mortality after LDL-C lowering: A systematic
review and meta-analysis. JAMA. 319:1566–1579. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Lan R, Luo H, Wu F, Wang Y and Zhao Z:
Chitosan oligosaccharides alleviate heat-stress-induced lipid
metabolism disorders by suppressing the oxidative stress and
inflammatory response in the liver of broilers. Antioxidants
(Basel). 12(1497)2023.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sun Y, Ishibashi M, Seimon T, Lee M,
Sharma SM, Fitzgerald KA, Samokhin AO, Wang Y, Sayers S, Aikawa M,
et al: Free cholesterol accumulation in macrophage membranes
activates Toll-like receptors and p38 mitogen-activated protein
kinase and induces cathepsin K. Circ Res. 104:455–465.
2009.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Wadström BN, Pedersen KM, Wulff AB and
Nordestgaard BG: Inflammation compared to low-density lipoprotein
cholesterol: Two different causes of atherosclerotic cardiovascular
disease. Curr Opin Lipidol. 34:96–104. 2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Hussain A and Ballantyne CM: New
approaches for the prevention and treatment of cardiovascular
disease: Focus on lipoproteins and inflammation. Annu Rev Med.
72:431–446. 2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Wu YR, Li L, Sun XC, Wang J, Ma CY, Zhang
Y, Qu HL, Xu RX and Li JJ: Diallyl disulfide improves lipid
metabolism by inhibiting PCSK9 expression and increasing LDL uptake
via PI3K/Akt-SREBP2 pathway in HepG2 cells. Nutr Metab Cardiovasc
Dis. 31:322–332. 2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Gu B, Jiang Y, Huang Z, Li H, Yu W, Li T,
Liu C, Wang P, Chen J, Sun L, et al: MRG15 aggravates
sepsis-related liver injury by promoting PCSK9 synthesis and
secretion. Int Immunopharmacol. 140(112898)2024.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Feingold KR, Moser AH, Shigenaga JK,
Patzek SM and Grunfeld C: Inflammation stimulates the expression of
PCSK9. Biochem Biophys Res Commun. 374:341–344. 2008.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Nie X, Chen Y, Li W and Lu Y: Anti-aging
properties of Dendrobium nobile Lindl.: From molecular mechanisms
to potential treatments. J Ethnopharmacol.
257(112839)2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Fan C, Sun X, Wang X and Yu H: Therapeutic
potential of the chemical composition of Dendrobium nobile Lindl.
Front Pharmacol. 14(1163830)2023.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Mou Z, Zhao Y, Ye F, Shi Y, Kennelly EJ,
Chen S and Zhao D: Identification, Biological activities and
biosynthetic pathway of dendrobium alkaloids. Front Pharmacol.
12(605994)2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Horton JD, Goldstein JL and Brown MS:
SREBPs: Activators of the complete program of cholesterol and fatty
acid synthesis in the liver. J Clin Invest. 109:1125–1131.
2002.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Sorrentino V and Zelcer N:
Post-transcriptional regulation of lipoprotein receptors by the
E3-ubiquitin ligase inducible degrader of the low-density
lipoprotein receptor. Curr Opin Lipidol. 23:213–219.
2012.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Lin XL, Xiao LL, Tang ZH, Jiang ZS and Liu
MH: Role of PCSK9 in lipid metabolism and atherosclerosis. Biomed
Pharmacother. 104:36–44. 2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Yu Q, Zheng H and Zhang Y: Inducible
degrader of LDLR: A potential novel therapeutic target and emerging
treatment for hyperlipidemia. Vascul Pharmacol.
140(106878)2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Scotti E, Hong C, Yoshinaga Y, Tu Y, Hu Y,
Zelcer N, Boyadjian R, de Jong PJ, Young SG, Fong LG and Tontonoz
P: Targeted disruption of the idol gene alters cellular regulation
of the low-density lipoprotein receptor by sterols and liver x
receptor agonists. Mol Cell Biol. 31:1885–1893. 2011.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Teslovich TM, Musunuru K, Smith AV,
Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S,
Chasman DI, Willer CJ, et al: Biological, clinical and population
relevance of 95 loci for blood lipids. Nature. 466:707–713.
2010.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Chasman DI, Paré G, Mora S, Hopewell JC,
Peloso G, Clarke R, Cupples LA, Hamsten A, Kathiresan S, Mälarstig
A, et al: Forty-three loci associated with plasma lipoprotein size,
concentration, and cholesterol content in genome-wide analysis.
PLoS Genet. 5(e1000730)2009.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Isaacs A, Willems SM, Bos D, Dehghan A,
Hofman A, Ikram MA, Uitterlinden AG, Oostra BA, Franco OH, Witteman
JC and van Duijn CM: Risk scores of common genetic variants for
lipid levels influence atherosclerosis and incident coronary heart
disease. Arterioscler Thromb Vasc Biol. 33:2233–2239.
2013.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Dong B, Wu M, Cao A, Li H and Liu J:
Suppression of Idol expression is an additional mechanism
underlying statin-induced up-regulation of hepatic LDL receptor
expression. Int J Mol Med. 27:103–110. 2011.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Hong C, Marshall SM, McDaniel AL, Graham
M, Layne JD, Cai L, Scotti E, Boyadjian R, Kim J, Chamberlain BT,
et al: The LXR-Idol axis differentially regulates plasma LDL levels
in primates and mice. Cell Metab. 20:910–918. 2014.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Lin YK, Yeh CT, Kuo KT, Yadav VK, Fong IH,
Kounis NG, Hu P and Hung MY: Pterostilbene increases LDL metabolism
in HL-1 cardiomyocytes by modulating the PCSK9/HNF1α/SREBP2/LDLR
signaling cascade, upregulating epigenetic hsa-miR-335 and
hsa-miR-6825, and LDL receptor expression. Antioxidants (Basel).
10(1280)2021.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Lebeau PF, Byun JH, Platko K, Saliba P,
Sguazzin M, MacDonald ME, Paré G, Steinberg GR, Janssen LJ, Igdoura
SA, et al: Caffeine blocks SREBP2-induced hepatic PCSK9 expression
to enhance LDLR-mediated cholesterol clearance. Nat Commun.
13(770)2022.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Sabatine MS: PCSK9 inhibitors: Clinical
evidence and implementation. Nat Rev Cardiol. 16:155–165.
2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D
and Park SW: Sterol-dependent regulation of proprotein convertase
subtilisin/kexin type 9 expression by sterol-regulatory element
binding protein-2. J Lipid Res. 49:399–409. 2008.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Rashid S, Curtis DE, Garuti R, Anderson
NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA and Horton JD: Decreased
plasma cholesterol and hypersensitivity to statins in mice lacking
Pcsk9. Proc Natl Acad Sci USA. 102:5374–5379. 2005.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Liang J, Li W, Liu H, Li X, Yuan C, Zou W
and Qu L: Di'ao Xinxuekang capsule improves the
anti-atherosclerotic effect of atorvastatin by downregulating the
SREBP2/PCSK9 signalling pathway. Front Pharmacol.
13(857092)2022.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Careskey HE, Davis RA, Alborn WE, Troutt
JS, Cao G and Konrad RJ: Atorvastatin increases human serum levels
of proprotein convertase subtilisin/kexin type 9. J Lipid Res.
49:394–398. 2008.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Shi Q, Chen J, Zou X and Tang X:
Intracellular cholesterol synthesis and transport. Front Cell Dev
Biol. 10(819281)2022.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Roca-Agujetas V, Barbero-Camps E, de Dios
C, Podlesniy P, Abadin X, Morales A, Marí M, Trullàs R and Colell
A: Cholesterol alters mitophagy by impairing optineurin recruitment
and lysosomal clearance in Alzheimer's disease. Mol Neurodegener.
16(15)2021.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Rizzolo D, Buckley K, Kong B, Zhan L, Shen
J, Stofan M, Brinker A, Goedken M, Buckley B and Guo GL: Bile acid
homeostasis in a cholesterol 7α-Hydroxylase and sterol
27-Hydroxylase double knockout mouse model. Hepatology. 70:389–402.
2019.PubMed/NCBI View Article : Google Scholar
|