Contents
Introduction
Chronic stressors act on the nervous system
Systemic pathways and neuroendocrine
transmitters
Modulation of the tumor microenvironment
Converging effects on cancer cells
Questions and perspective
Introduction
There is a long-standing hypothesis that
psychosocial factors can influence the incidence and progression of
cancers, and adequate psychotherapies may be beneficial to cancer
patients (1,2). Clinical and epidemiological studies
have also documented a prevalence of psychological stress among
cancer patients and the adverse effects on cancer outcomes.
However, stress-related disorders in cancer patients are not
adequately assessed nor treated due to the limited knowledge of the
psychological aspect of cancer and the ineffectiveness of
psychotherapies (3,4). Not until the recent past few years,
by using in vitro animal models and human clinical
perspective study approaches, have researchers begun to uncover the
complex relationship between psychosocial stress and cancer
progression at the systemic, biochemical and molecular levels.
Accumulating data indicate that the psychological stress caused by
chronic stressors is a major risk factor for cancer occurrence,
growth and metastasis (5–8). Psychological stress refers to the
emotional and physiological reactions experienced when an
individual confronts a situation in which the adaptation demands go
beyond their coping resources (9).
The effects of psychological stress on cancer cells are mediated by
the same key stress-related mediators and their corresponding
receptors of stress response in multifold pathways (2,6–8).
Understanding the action mechanisms of psychosocial stress in
promoting cancer progression is crucial for devising effective
interventions. In this article, we briefly review the recent theory
of the mechanisms involved from stressor to cancer progression.
Chronic stressors act on the nervous
system
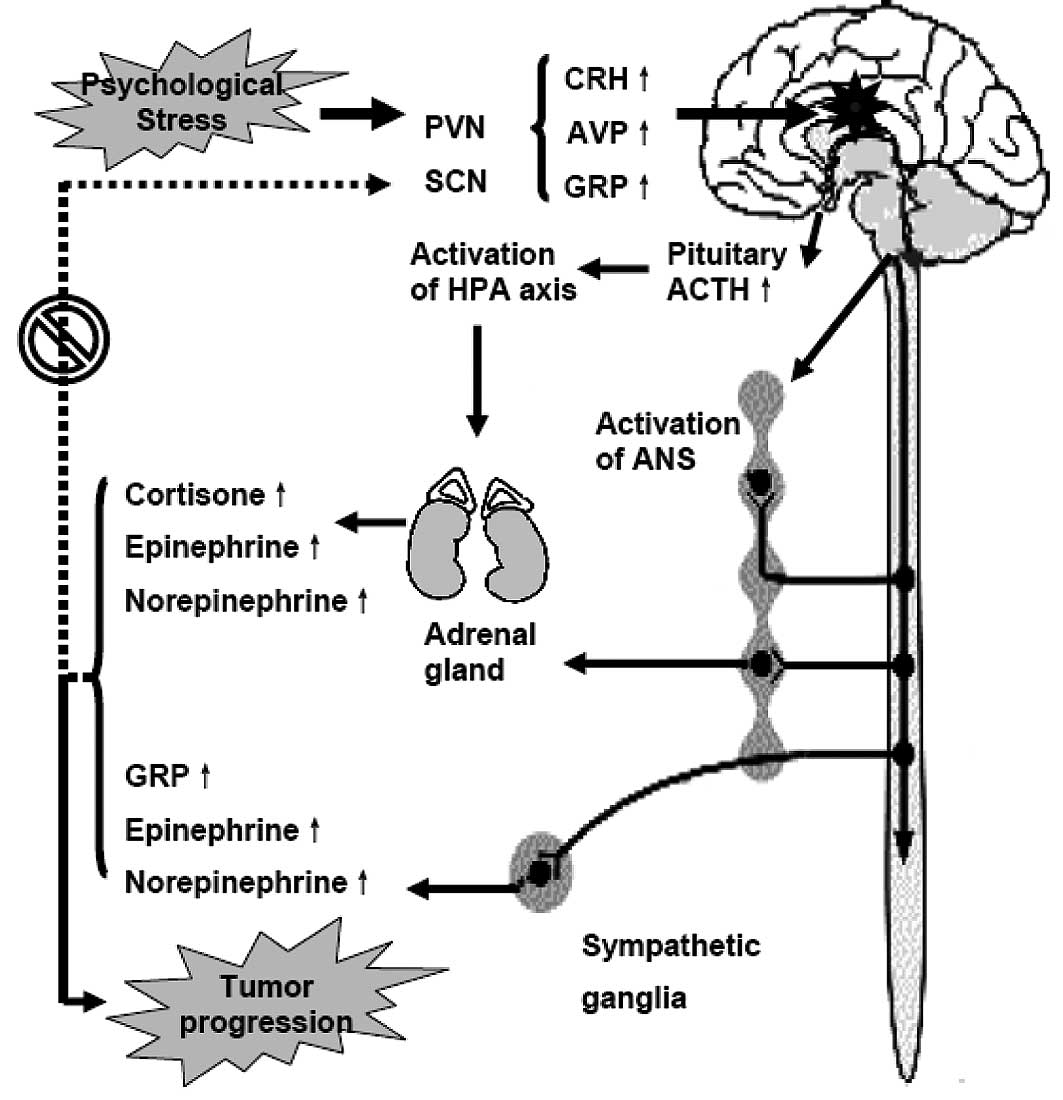
Chronic stressors first act on the central nervous
system, especially the paraventricular nucleus (PVN) and the
suprachiasmatic nuclei (SCN), causing an automatic process which
disrupts homeostasis (2,9–12).
To maintain homeostasis, two strictly controlled
information-processing cascades precede in similar ways to properly
maintain the stress response and to keep the central pacemaker in
position: the sympathetic nervous system (SNS)-adrenal medullary
axis and the hypothalamic-pituitary-adrenal (HPA) axis. Activation
of the SNS causes increased release of norepinephrine (NEPI) from
sympathetic nerve terminals, and NEPI and epinephrine (EPI) from
the adrenal medulla. Activation of the HPA axis causes increased
release of corticotrophin-releasing hormone (CRH) from the PVN of
the hypothalamus, resulting in increased secretion of
adrenocorticopic hormone (ACTH) in the anterior pituitary. ACTH, in
turn, stimulates secretion of glucocorticoid (GC) hormones from the
adrenal cortex. GCs modulate the activity of the HPA axis via
negative feedback effects on glucocorticoid receptors (GRs) in the
hippocampus. However, the information-processing cascade is out of
control, and stress-related mediators can not be supressed during
psychological stress, resulting in constant abnormalities in
hormone levels and behavior (2,9,10,13).
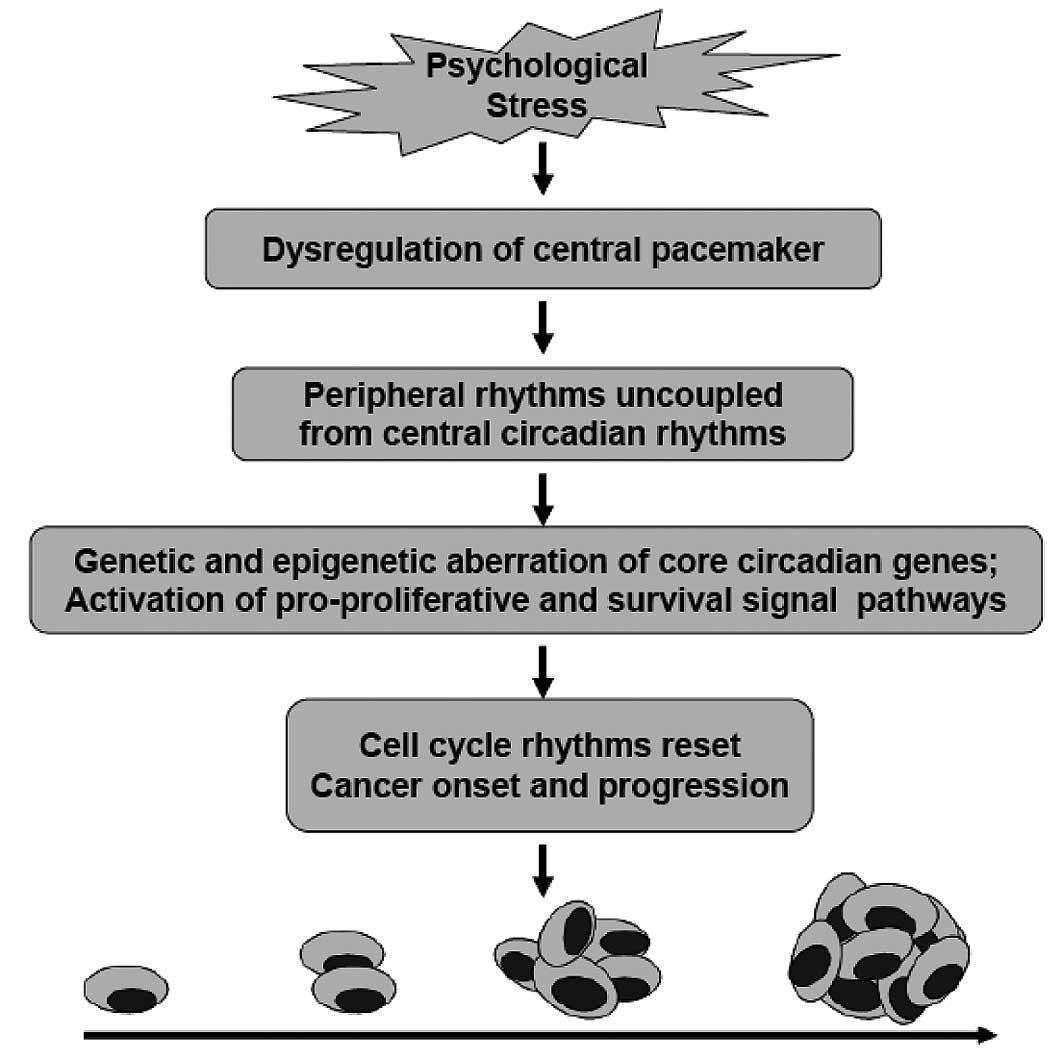
Meanwhile, the central circadian rhythms are disrupted for the
dysfunction of the central pacemaker located in the SCN (11,12).
Circadian rhythms are 24-h oscillations in behavior and physiology,
which provide organisms a survival advantage when confronted with
cyclic changes in the environment. Circadian rhythms are driven by
an internal timing machine – the ‘circadian clock’ operated by
numerous gene products in the central pacemaker as well as in most
peripheral tissues. Peripheral clocks exist in almost every cell
type to regulate daily changes in cell biology. To guarantee normal
growth and avoid tumor formation, the timing of cell division must
be under strict control. The central pacemaker controls peripheral
clocks mainly through the HPA axes and SNS. GCs and melatonin
(N-acetyl-5-methoxytryptamine) are in feedback control of CRH
release from SCN or HPA axes. The function of the circadian
pacemaker is disrupted due to two major mechanisms: loss of
feedback control of CRH and up-regulation of the operating genes of
the circadian pacemaker such as gastrin-releasing peptide (GRP) and
GRP receptors (GRPRs) in SCN (Fig.
1).
Systemic pathways and neuroendocrine
transmitters
The question of how psychological stress affects
cancer cells has intrigued both researchers and patients. Recent
studies have confirmed that the effects of psychological stress on
cancer cells are mediated by key stress hormones and their
corresponding receptors through similar processes observed in the
stress response (2,6–8,11–14).
At least three systemic pathways are involved (Fig. 1): the SNS and the
sympathetic-adrenal medullary axis, the HPA axis and an altered
pattern of coping. First, the sympathetic fibers that descend from
the brain into peripheral tissues can release a wide variety of
neurotransmitters that influence cancer growth by binding to
receptors on cancer cells. Some of the most significant
neurotransmitters recently identified by a series of studies
include cortisol, EPI, NEPI and GRP. Sympathetic signals increase
the release of EPI, NEPI and GRP from the sympathetic terminal
fibers or even tumor cells. Second, the
sympathetic-adrenal-medullary axis and the
hypothalamic-pituitary-adrenal/ovarian axis can increase the
release of EPI, NEPI, cortisol, GRP, prolactin, growth hormone,
melatonin, β-endorphin and enkephalin from the brain, pituitary and
adrenal glands. The unchecked release of these stress-related
neurotransmitters results in their chronic-flattened elevations in
both blood and tissues. Finally, the altered pattern of coping may
have an equal importance in linking stressors to cancer cells. The
altered pattern of coping refers to the unhealthy behavior of
individuals who suffer from psychological stress developed when
coping with an adverse situation. Commonly observed unhealthy
behaviors include overeating, alcoholism, sleeplessness, smoking,
reduction in socialization and non-compliance to medical
treatments. Through the three main pathways and numerous mediators,
the effects of psychological stress are amplified and transmitted
as humoral and behavioral functions to the blood, tissues and the
coping mechanism.
Modulation of the tumor
microenvironment
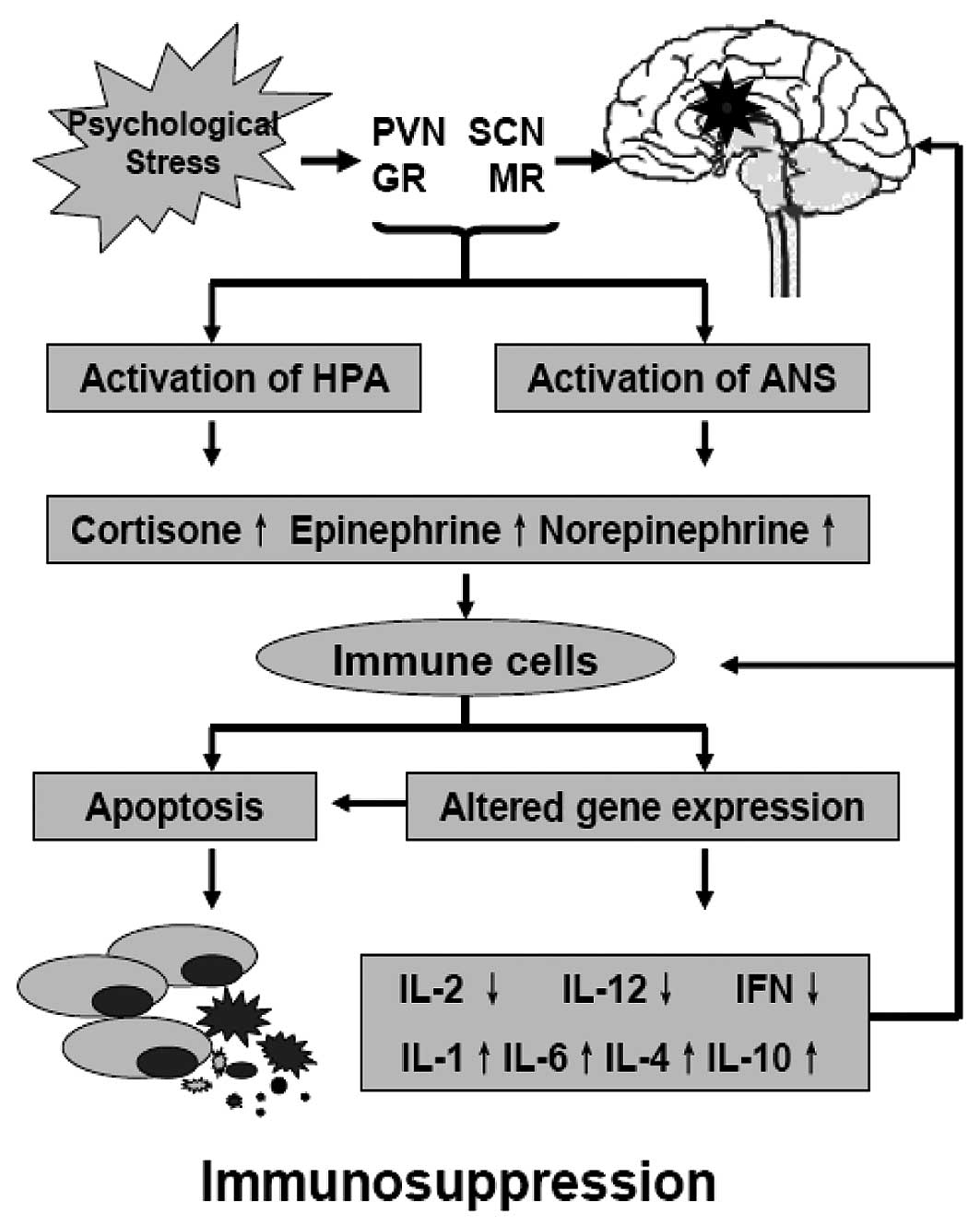
Changes in stress-related neuroendocrine
transmitters during psychological stress lead to a modulation of
the tumor microenvironment and immune cells. The first most
important mechanism is the modulation of the immune function
against tumor cells. Recent studies indicate that the impact of
stress on the immune response is mediated by a bidirectional signal
network between the nervous, endocrine and immune systems, and
chronic stressors are associated with suppression of both cellular
and humoral immune functions (Fig.
2) (12–16). For example, stress hormones affect
the immune function through receptors present on immune cells, and
the immune cells in tern modulate the activity of the hypothalamus
by producing cytokines. GRs expressed on a variety of immune cells,
which bind cortisol interfere with the function of nuclear
factor-κB (NF-κB), which regulates the activity of
cytokine-producing immune cells. Adrenergic receptors (ARs) bind
EPI and NEPI to activate the cAMP response element-binding protein
(CREB); CREB induces the transcription of genes encoding for a
variety of cytokines. Changes in gene expression result in a shift
of immune response from T-helper lymphocyte type 1 cells (Th1) to
T-helper lymphocyte type 2 cells (Th2), which impairs the immune
responses against tumor cells (12–16).
The second important mechanism is the increased generation of
mitogenic factors in the tumor microenvironment. Most of the
stress-related transmitters and cytokines are also mitogenic for
tumor cells and can promote the growth and invasion in endocrine,
paracrine and autocrine manners (2,6–8,17).
The third mechanism is the resettling of the tumor cell circadian
clock that, decoupled from the control of central circadian rhythms
via the effects of altered hormone levels on tumor cells, impacts
the tumor vs. host metabolism and neuroimmune effects resulting in
cancer-related immunosuppression (11,12).
Converging effects on cancer cells
The total effects of psychological stress converging
on cancer cells are mediated by more complicated processes, and the
mechanisms remain largely unknown. However, the recognition of the
direct influences of stress hormones and GC on cancer cells may be
the most important advance in exploring the mechanisms from
stressor to cancer progression. Using mouse model and in
vitro approaches, researchers have demonstrated that the
effects of psychological stress on cancer cells are mainly mediated
by the key stress hormones (EPI and NEPI) and β-adrenergic
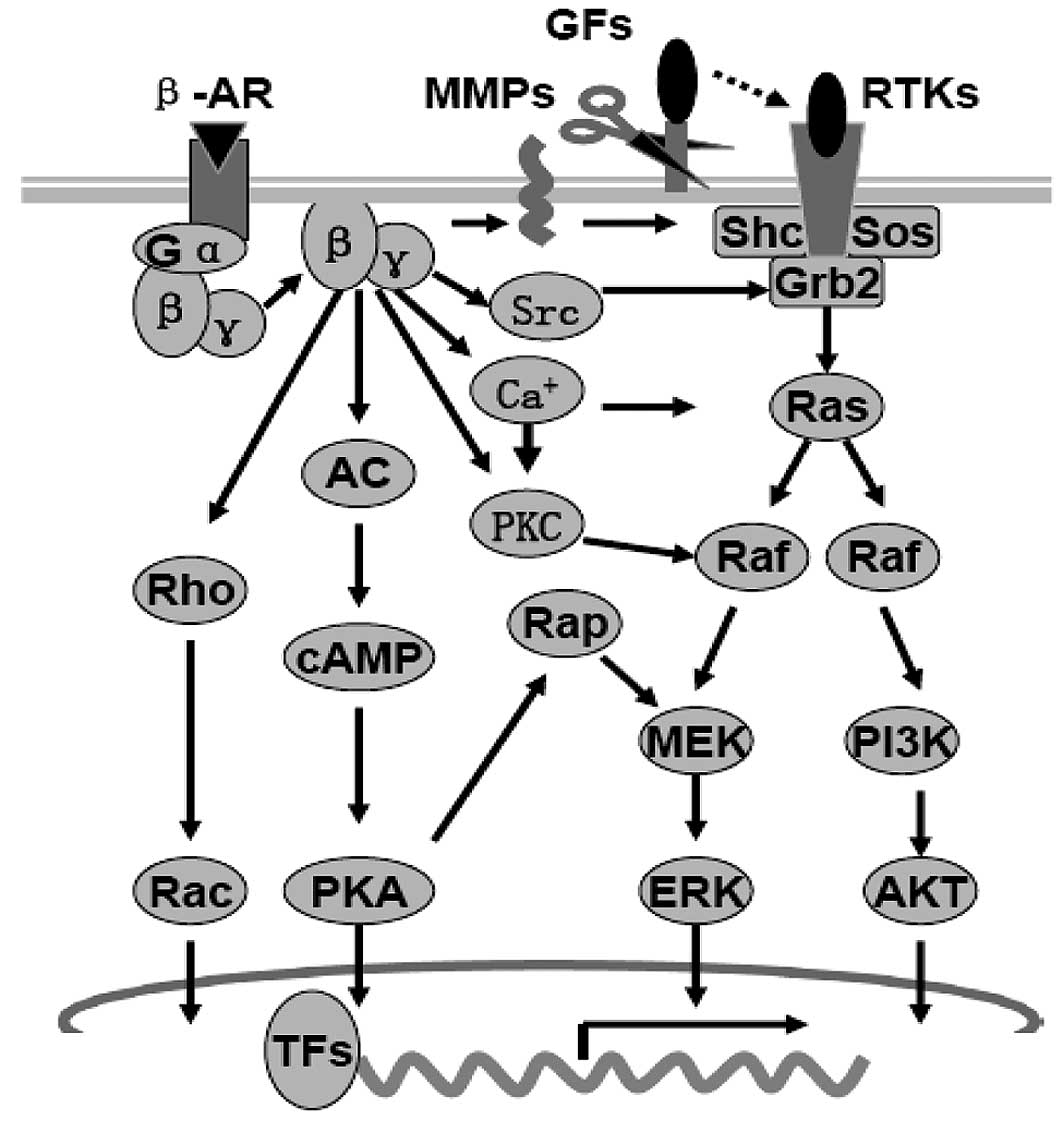
receptors (β-AR), especially the β2-AR (2,7,8,17).
β-AR signals can activate several common intracellular
pro-proliferative and pro-migratory signaling pathways, such as the
cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA), the
mitogen-activated protein kinase (MAPK)/extracellular
signal-regulated kinase (ERK1/2) and phosphatidylinositol-3-kinase
(PI3K)/AKT (protein kinase B) signaling pathways by (i) the ‘G
protein switching’ mechanism; (ii) the homodimerization and
heterodimerization mechanism; and (iii) the MMP mechanism (18,19)
(Fig. 3). Activation of the
cAMP-PKA signaling pathway is suggested to be the central aspect of
β-AR signaling in tumor cells. Through PKA or Epac (the exchange
protein directly activated by cAMP), cAMP can further activate
small GTPases such as RhoA, Rac, Rap1 and Rap2 to activate
MAPK/ERK1/2, PI3K/AKT and Rho/Rac/Cdc42 signaling pathways
(18–24). Through homodimerization and
heterodimerization with other ARs, G-protein-coupled receptors
(GPCRs) or receptor tyrosine kinases (RTKs), β-AR signals directly
activate the RTKs, or increase in matrix metalloproteinases (MMPs),
which can release growth factors to activate the RTKs indirectly
(25,26).
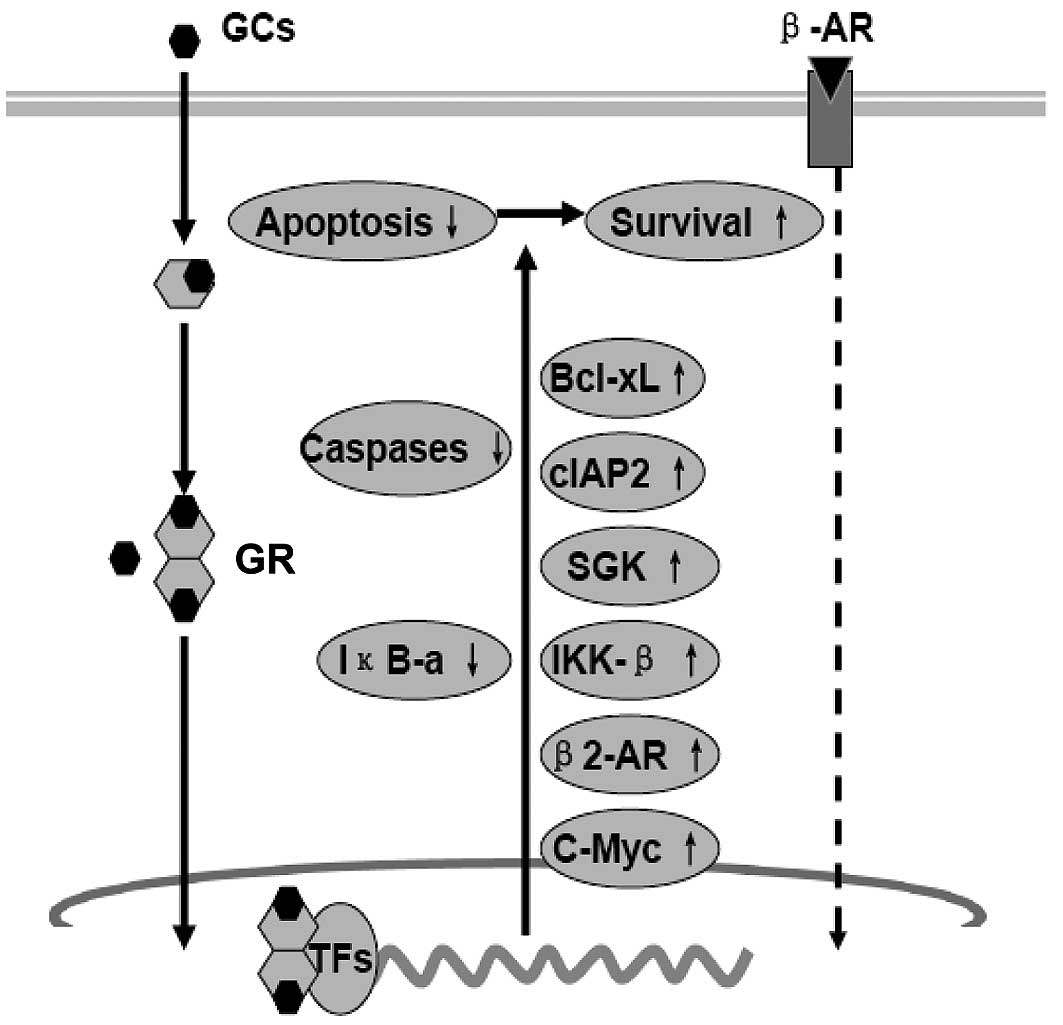
GCs can function in synergy with stress hormones to
promote cancer progression. Dexamethasone, prednisone and
cortisone, for example, can promote cell survival and
chemoresistance in a number of solid tumors (27,28).
The mechanism involves direct stimulation of cell growth and
survival, as well as immunosuppression (Fig. 4). GRs are present in a number of
human malignancies (29).
Activated-GR acts as a transcription factor, either through binding
to GC response elements (GRE) in the regulatory sequences of target
genes or through cross-talk and/or interference with other
transcription factors such as activator protein-1 (AP-1), signal
transducers and activators of transcription 5 (STAT5) and NF-κB to
promote the growth and survival of cancer cells. GCs can also
support growth and metastasis of cancer cells through the
up-regulation of proto-oncogene c-Myc, anti-apoptotic protein
Bcl-xL, cytosolic caspase inhibitor cIAP2 and β2-AR (30–32).
Activity of NF-κB has been shown to play an important role in
preventing apoptosis of cancer cells. Activation of NF-κB requires
activation of the IκB (inhibitor of NF-κB) kinase-β (IKK-β). IKK-β
activation is preceded by phosphorylation by upstream kinases such
as mitogen-activated protein kinase kinase-1 (MAPKK1 or MEK1),
NF-κB-inducing kinase, protein kinase C (PKC) and PI3K. GCs can
also activate the IKK-NF-κB signal pathway by activation of the
serum- and glucocorticoid-regulated kinase-1 (SGK1) and
inactivation of the forkhead transcription factor 3a (FOXO3a)
(33–35).
The changed tumor microenvironment also impinges on
the internal cell timers or molecular clocks in tumor cells. The
circadian clock functions in vivo as a tumor suppressor at
the systemic, cellular and molecular levels (36). Ablation of SCN in mice resulted in
an accelerated growth of implanted malignant tumors. Overexpression
of either the circadian clock gene Per1 or Per2 in cancer cells
inhibits growth and increases apoptosis (11,12,36–38).
Repeated stress-response activation disrupts the circadian rhythms
at both the central and peripheral levels (Figs. 1 and 5). There are several mechanisms by which
circadian disruption might hasten tumor growth: the increased
stress-related mediators and cytokines in the tumor
microenvironment induce immunosuppression, influence the metabolic
pathways and the circadian clock gene expression in tumor cells,
and reduce the sensitivity of tumor cells to treatment (11,12,39,40).
For example, the flattened elevation of cortisone can cooperate
with cell timers to establish circadian cell cycle rhythms by
regulating the expression of cell cycle genes. Recent studies have
revealed that expression of cell cycle genes such as Wee1, Cyclins
and c-Myc is directly under the regulation of the circadian
transcriptional complex (CTC). Circadian clock genes regulate
cell-cycle progression and apoptosis through cAMP/PKA, MAPK/ERK1/2
and β-catenin/the T-cell factor/lymphoid enhancer binding factor
(TCF/LEF) pathways (11,12). Hence, the disruption of circadian
rhythms, in a sense, is also a mediator of psychological stress.
However, the detailed mechanisms that accounts for the effects of
psychological stress on central circadian rhythms and the molecular
clock remain to be clarified.
Questions and perspective
As described above, psychological stress is closely
associated with tumorigenesis and cancer progression. The
mechanisms for mediating the effects of psychological stress can be
outlined. Chronic stressors act on the PVN and SCN. The effects are
then transmitted through SNS and the HPA axis, amplified by the
unchecked release of stress-related mediators and altered
behaviors. All these mediators act as immunosuppressors or mitogens
in the tumor microenvironment. The converging effects of
psychological stress on cancer cells finally signal through
receptors of the stress mediators and cytokines to activate the
intracellular pro-proliferative and pro-migratory signaling
pathways and reset the molecular clock in tumor cells (Figs. 1–5). There emerges an opportunity to devise
effective interventions targeting the signaling pathways from
stressor to cancer progression for anticancer treatment; for
example, psychosocial and the pharmacological interventions. Recent
studies have demonstrated that social support and massage
therapies, especially cognitive-behavioral stress management (CBSM,
a structured, manualized group intervention that emphasizes skill
learning, cognitive behavioral modification and relaxation
training) have potent effects of improving the outcome of cancer
patients (41–43). Some drugs such as antidepressants
and β-AR antagonist also exhibit certain effectiveness in
attenuating the effect of psychological stress on cancer cells
(44–47). However, it is not so simple; the
in vivo mechanisms involved in the influence of
psychological stress on cancer cells and most of the details remain
unclear (48). Some questions must
be resolved before devising more effective interventions. The first
question involves how chronic stressors act on the PVN and SCN.
This is important for devising adequate interventions to initially
protect cancer patients from psychological stress. The second
question must answer how many key effectors are in tumor cells. As
there are several known key stress-related mediators responsible
for the influence of psychological stress on cancer cells and each
acts on the cell in synergy with others through different
processes, it is impossible to develop one blocker for all. The
third question must addres how psychological stress affects the
molecular clock. Since the circadian clock functions in vivo
as a tumor suppressor, it is logical to assume that the effect of
psychological stress can be suppressed by the up-regulation of a
certain key circadian gene. By exploring the cascades linking the
psychological stress to cancer progression, some useful targets
will be found for anticancer treatment in the future.
References
|
1.
|
Riley V: Mouse mammary tumors: alteration
of incidence as apparent function of stress. Science. 189:465–467.
1975. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Antoni MH, Lutgendorf SK, Cole SW, Dhabhar
FS, Sephton SE, McDonald PG, Stefanek M and Sood AK: The influence
of bio-behavioural factors on tumour biology: pathways and
mechanisms. Nat Rev Cancer. 6:240–248. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Massie MJ: Prevalence of depression in
patients with Cancer. J Natl Cancer Inst Monogr. 32:57–71. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Carlson LE, Angen M, Cullum J, Goodey E,
Koopmans J, Lamont L, MacRae JH, Martin M, Pelletier G, Robinson J,
Simpson JS, Speca M, Tillotson L and Bultz BD: High levels of
untreated distress and fatigue in cancer patients. Br J Cancer.
90:2297–2304. 2004.PubMed/NCBI
|
|
5.
|
Garssen B: Psychological factors and
cancer development: evidence after 30 years of research. Clin
Psychol Rev. 24:315–338. 2004.PubMed/NCBI
|
|
6.
|
Chida Y, Hamer M, Wardle J and Steptoe A:
Do stress-related psychosocial factors contribute to cancer
incidence and survival? Nat Clin Pract Oncol. 5:466–475. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Thaker PH, Han LY, Kamat AA, Arevalo JM,
Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori
M, Merritt WM, Lin YG, Mangala LS, Kim TJ, Coleman RL, Landen CN,
Li Y, Felix E, Sanguino AM, Newman RA, Lloyd M, Gershenson DM,
Kundra V, Lopez-Berestein G, Lutgendorf SK, Cole SW and Sood AK:
Chronic stress promotes tumor growth and angiogenesis in a mouse
model of ovarian carcinoma. Nat Med. 12:939–944. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Sood AK, Bhatty R, Kamat AA, Landen CN,
Han L, Thaker PH, Li Y, Gershenson DM, Lutgendorf S and Cole SW:
Stress hormone-mediated invasion of ovarian cancer cells. Clin
Cancer Res. 12:369–375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
McEwen BS: Mood disorders and allostatic
load. Biol Psychiatry. 54:200–207. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Vanitallie TB: Stress: a risk factor for
serious illness. Metabolism. 51:40–45. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Fu L and Lee CC: The circadian clock:
pacemaker and tumour suppressor. Nat Rev Cancer. 3:350–361. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Sephton S and Spiegel D: Circadian
disruption in cancer: a neuroendocrine-immune pathway from stress
to disease? Brain Behav Immun. 17:321–328. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Glaser R and Glaser JK: Stress-induced
immune dysfunction: implications for health. Nat Rev Immunol.
5:243–251. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Reiche EM, Nunes SO and Morimoto HK:
Stress, depression, the immune system and cancer. Lancet Oncol.
5:617–625. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Padgett DA and Glaser R: How stress
influences the immune response. Trends Immunol. 24:444–448. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Segerstrom SC and Miller GE: Psychological
stress and the human immune system: A meta-analytic study of 30
years of inquiry. Psychol Bull. 130:601–630. 2004.PubMed/NCBI
|
|
17.
|
Lutgendorf SK, Cole S, Costanzo E, Bradley
S, Coffin J, Jabbari S, Rainwater K, Ritchie JM, Yang M and Sood
AK: Stress-related mediators stimulate vascular endothelial growth
factor secretion by two ovarian cancer cell lines. Clin Cancer Res.
9:4514–4521. 2003.PubMed/NCBI
|
|
18.
|
Hall RA: Beta-adrenergic receptors and
their interacting proteins. Semin Cell Dev Biol. 15:281–288. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Dorsam RT and Gutkind JS:
G-protein-coupled receptors and cancer. Nat Rev Cancer. 7:79–94.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
20.
|
O’Connor KL and Mercurio AM: Protein
kinase A regulates Rac and is required for the growth
factor-stimulated migration of carcinoma cells. J Biol Chem.
276:47895–47900. 2001.PubMed/NCBI
|
|
21.
|
Rangarajan S, Enserink JM, Kuiperij HB,
Rooij J, Price LS, Schwede F and Bos JL: Cyclic AMP induces
integrin-mediated cell adhesion through Epac and Rap1 upon
stimulation of the β2-adrenergic receptor. J Cell Biol.
160:487–493. 2003.PubMed/NCBI
|
|
22.
|
Schmitt JM and Stork JS: β2-Adrenergic
receptor activates extracellular signal-regulated kinases (ERKs)
via the small G protein Rap1 and the serine/threonine kinase B-Raf.
J Biol Chem. 275:25342–25350. 2000.
|
|
23.
|
Shenoy SK, Drake MT, Nelson CD, Houtz DA,
Kunhong X, Madabushi S, Reiter E, Premont RT, Lichtarge O and
Lefkowitz RJ: β-arrestin-dependent, G protein-independent ERK1/2
activation by the β2-adrenergic receptor. J Biol Chem.
281:1261–1273. 2006.
|
|
24.
|
Tsurutani J, Castillo SS, Brognard J,
Granville CA, Chunyu Z, Gills JJ, Sayyah J and Dennis PA: Tobacco
components stimulate Akt-dependent proliferation and
NF-κB-dependent survival in lung cancer cells. Carcinogenesis.
26:1182–1195. 2005.PubMed/NCBI
|
|
25.
|
Maudsley S, Pierce KL, Zamah AM, Miller
WE, Ahn S, Daaka Y, Lefkowitz RJ and Luttrell LM: The β2-Adrenergic
receptor mediates extracellular signal-regulated kinase activation
via assembly of a multi-receptor complex with the epidermal growth
factor receptor. J Biol Chem. 275:9560–9572. 2000.
|
|
26.
|
Drube S, Stirnweiss J, Valkova C and
Liebmann C: Ligand-independent and EGF receptor-supported
transactivation: lessons from beta2-adrenergic receptor signalling.
Cell Signal. 18:1633–1646. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Herr I, Ucur E, Herzer K, Okouoyo S,
Ridder R, Krammer PH, Doeberitz MK and Debatin KM: Glucocorticoid
cotreatment induces apoptosis resistance toward cancer therapy in
carcinomas. Cancer Res. 63:3112–3120. 2003.PubMed/NCBI
|
|
28.
|
Herr I and Pfitzenmaier J: Glucocorticoid
use in prostate cancer and other solid tumours: implications for
effectiveness of cytotoxic treatment and metastases. Lancet Oncol.
7:425–430. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
NØrgaard P and Poulsen HS: Glucocorticoid
receptors in human malignancies: a review. Ann Oncol. 2:541–557.
1991.PubMed/NCBI
|
|
30.
|
Petrella A, Ercolino SF, Festa M,
Gentilella A, Tosco A, Conzen SD and Parente L: Dexamethasone
inhibits TRAIL-induced apoptosis of thyroid cancer cells via Bcl-xL
induction. Eur J Cancer. 42:3287–3293. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Runnebaum IB and Brüning A:
Glucocorticoids inhibit cell death in ovarian cancer and
up-regulate caspase inhibitor cIAP2. Clin Cancer Res. 11:6325–6332.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Hadcock JR, Wang HY and Malbon CC:
Agonist-induced destabilization of beta-adrenergic receptor mRNA.
Attenuation of glucocorticoid-induced up-regulation of
beta-adrenergic receptors. J Biol Chem. 264:19928–19933.
1989.PubMed/NCBI
|
|
33.
|
Landen CN Jr, Lin YG, Armaiz Pena GN, Das
PD, Arevalo JM, Kamat AA, Han LY, Jennings NB, Spannuth WA, Thaker
PH, Lutgendorf SK, Savary CA, Sanguino AM, Lopez-Berestein G, Cole
SW and Sood AK: Neuroendocrine modulation of signal transducer and
activator of transcription-3 in ovarian cancer. Cancer Res.
67:10389–10396. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Liping Z, Cui R, Xiaodong CH and Jie D:
Antiapoptotic effect of serum and glucocorticoid-inducible protein
kinase is mediated by novel mechanism activating IκB kinase. Cancer
Res. 65:457–464. 2005.PubMed/NCBI
|
|
35.
|
Wei W, Min Z, Brickley DR, Pew T and
Conzen SD: Glucocorticoid receptor activation signals through
forkhead transcription factor 3a in breast cancer cells. Mol
Endocrinol. 20:2304–2314. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Goodspeed MC and Lee CC: Tumor suppression
and circadian function. J Biol Rhythms. 22:291–298. 2007.
View Article : Google Scholar
|
|
37.
|
Hastings M, O’Neill JS and Maywood ES:
Circadian clocks: regulators of endocrine and metabolic rhythms. J
Endocrinol. 195:187–198. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ
and Chang JG: Deregulated expression of the PER1, -PER2 and PER3
genes in breast cancers. Carcinogenesis. 26:1241–1246. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Rasmuson T, Ljungberg B, Grankvist K,
Jacobsen J and Olsson T: Increased serum cortisol levels are
associated with high tumour grade in patients with renal cell
carcinoma. Acta Oncol. 40:83–87. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Dickmeis T: Glucocorticoids and the
circadian clock. J Endocrinol. 200:3–22. 2009. View Article : Google Scholar
|
|
41.
|
Andersen BL, Farrar WB, Golden-Kreutz DM,
Glaser R, Emery CF, Crespin TR, Shapiro CL and Carson WE:
Psychological, behavioral and immune changes after a psychological
intervention: a clinical trial. J Clin Oncol. 22:3570–3580. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Jacobsen PB and Jim HS: Psychosocial
interventions for anxiety and depression in adult cancer patients:
achievements and challenges. CA Cancer J Clin. 58:214–230. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Küchler T, Bestmann B, Rappat S,
Henne-Bruns D and Wood-Dauphinee S: Impact of psychotherapeutic
support for patients with gastrointestinal cancer undergoing
surgery: 10-year survival results of a randomized trial. J Clin
Oncol. 25:2702–2708. 2007.PubMed/NCBI
|
|
44.
|
Steingart AB and Cotterchio M: Do
antidepressants cause, promote, or inhibit cancers? J Clin
Epidemiol. 48:1407–1412. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Brandes LJ: Hormetic effects of hormones,
antihormones and antidepressants on cancer cell growth in culture:
in vivo correlates. Crit Rev Toxicol. 35:587–592. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Algazi M, Plu-Bureau G, Flahault A, Dondon
MG and Le MG: Is beta-blocker treatment associated with a decrease
in the risk of cancer? Drug Des Discov. 3:653–661. 2006. View Article : Google Scholar
|
|
47.
|
Benish M, Bartal I, Goldfarb Y, Levi1 B,
Avraham1 R, Raz A and Ben-Eliyahu S: Perioperative use of
β-blockers and COX-2 inhibitors may improve immune competence and
reduce the risk of tumor metastasis. Ann Surg Oncol. 15:2042–2052.
2008.
|
|
48.
|
Boesen EH and Johansen C: Impact of
psychotherapy on cancer survival: time to move on? Curr Opin Oncol.
20:372–377. 2008. View Article : Google Scholar : PubMed/NCBI
|