1. Introduction
The brain is the vital organ responsible for receiving, processing and coordinating bodily activities. According to the Central Brain Tumor Registry of the United States (CBTRUS) report, over one million individuals in the USA suffer from primary brain tumors today, and another ~94,000 individuals will be diagnosed this year (1). The annual age-adjusted incidence rate (AAAIR) of all malignant and non-malignant brain and other central nervous system (CNS) tumors is 24.83 per 100,000 individuals. This includes a malignant AAAIR of 6.94 and a non-malignant AAAIR of 17.88. Overall, the incidence rate is higher in females than in males, with rates of 27.85 vs. 21.62 per 100,000 individuals, respectively. Patients with malignant brain tumors have an average survival rate of 35.7%, while those with nonmalignant brain tumors have an average survival rate of 91.8%. Brain tumors are the leading cause of cancer-related mortality among children and young adults aged ≤19 years (1). Additionally, hundreds of thousands of Americans are diagnosed with metastatic brain cancer each year, which presents unique challenges (1,2).
A brain tumor is the medical term used for an abnormal growth of brain cells (3). The World Health Organization (WHO) has categorized brain tumors into 120 types (4), based on the location, the type of tissue involved, the type of malignancy or benignity and additional contributing elements (4,5). In the CBTRUS report, the most common malignant brain and other CNS histopathologies was glioblastoma (14.2% of all tumors and 50.9% of all malignant tumors), and the most common predominantly non-malignant histopathology was meningioma (40.8% of all tumors and 56.2% of all non-malignant tumors) (1). The cerebellum tumors comprised the largest proportion of tumors, ~16.6% after tumors located in other brain (13.3%) and brain stem (12.6%) sites (1).
Medulloblastoma (MB) is the most common malignant childhood CNS (brain and spinal cord) tumor. MB comprises the largest percentage of embryonal tumors (70.2%) and almost 20% of all pediatric brain tumors (1). MB originates from the granule cell precursors in the external germinal layer of the developing cerebellum. MB tumor growth begins in the fourth ventricle and can completely occupy it. After filling up the fourth ventricle, MB spreads to the cerebellar vermis and the brainstem, seeding the craniospinal axis. MB is a highly malignant tumor with a propensity for local invasion and distant metastatic spread through the subarachnoid system (i.e., within the brain and along the spinal cord). Current treatment strategies involve a combination of maximal safe resection, chemotherapy and craniospinal irradiation (CSI), which has improved the long-term survival rates of patients to 70-80%. Despite aggressive multimodal therapy, ~30% of patients eventually succumb to this disease, and survivors cope with the long-term side effects of cognitive, neurologic and endocrinologic deficits, which markedly impact the quality of life of patients (6). The present review summarizes the currently available knowledge about epigenetic modifications in the upper layer, specifically focusing on DNA methylation, histone modifications and the regulation of gene expression by microRNAs in relation to MB. The epigenetic scenarios linked to various types of cancer, focusing on how different mechanisms interact to contribute to cancer development are introduced first, followed by a detailed discussion of the role of epigenetics in medulloblastoma.
Epigenetics was defined as ‘the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in the DNA sequence’ by Fincham (7). Immediately after DNA was experimentally discovered as the genetic material (8,9), in 1948, Hotchkiss (10) discovered 5-methylcytosine (5mC) in calf thymus DNA using paper chromatography (10,11), the first evidence of methylated DNA, the hallmark of epigenetics inheritance. DNA methylation, post-translational modifications of histones or histone variants, nucleosome positioning, maintenance of topologically associating domains, the regulation of heterochromatin silencing by RNA interference, or non-coding RNAs, all are the major mechanisms of epigenetics (12-15).
The discovery of epigenetics as the cause of cancers per se has changed the human cancer landscape (16). Cancer was typically considered a genetic disease; however, recent studies have provided a new perspective. Emerging research highlights the role of hypermethylation of proximal genomic regions, the hypomethylation of the genome, chromatin modeling aberrations, and a link among all these that contribute to the loss of function in essential and nonessential genes (17). Due to this complexity, defining cancer has become even more challenging. Notably, >50% of the genes mutated in familial cancers exhibit hypermethylation in sporadic cancers. In other words, the same gene candidates that are not mutated exhibit hypermethylated CpG islands in sporadic cancers. This represents a fascinating natural mechanism involving the interplay between DNA sequences (mainly CpG islands) and a wide range of proteins, particularly methyltransferases. An example of these mechanisms involves gene alterations from point mutations or deletions that lead to heterozygosity, followed by an event that can eliminate this mutation, a phenomenon known as loss of heterozygosity. In a number of tumor lines, despite the absence of loss of heterozygosity, genes are dysfunctional due to the hypermethylation of one of the alleles. For example, in 40% of hereditary non-polyposis colorectal tumors that contain hMLH1 germline mutations, but exhibit no loss of heterozygosity (LOH) at the hMLH1 gene, hypermethylation is present at hMLH1. By contrast, none of the eight hMLH1 germline mutant tumors with LOH display this hypermethylation (18). Similarly, other genes associated with familial forms of renal, breast and colon cancer, such as the von Hippel-Lindau syndrome, breast cancer 1 early onset and serine/threonine kinase 11 genes, have also been found to be silenced by hypermethylation in their sporadic cancer forms (19-22). There is a growing list of hypermethylated genes that are not mutated e.g., O6-methylguanine-DNA methyl-transferase (MGMT) encodes a DNA-repair protein (22,23); cyclin-dependent kinase inhibitor CDKN2B, which encodes cell cycle regulator p15 (22,24); and RASSF1A encodes a protein that binds to the RAS oncogene (22,25) (Fig. 1). Notably, the hypermethylation of MGMT predisposes mutations in key tumor suppressor gene p53 (TP53) and K-RAS (23). MGMT removes O6-methylguanine back to guanine; if this adduct is not removed, it results in conversion from a guanine-cytosine pair to an adenine-thymine pair, a critical step in the formation of mutations in cancer. There are also examples of human cancers where tumors contain hypomethylated non-promoter regions and structural elements such as centromeric DNA. For example, hypomethylation is found in pericentromeric regions of chromosomes 1, 9 and 16 due to a germ-line mutation in DNA methyltransferase (DNMT)3B. This DNMT3B mutation leads to immunodeficiency centromeric instability and facial abnormalities syndrome due to chromosomal structural changes (16,26,27) and enhanced genomic instability (27).
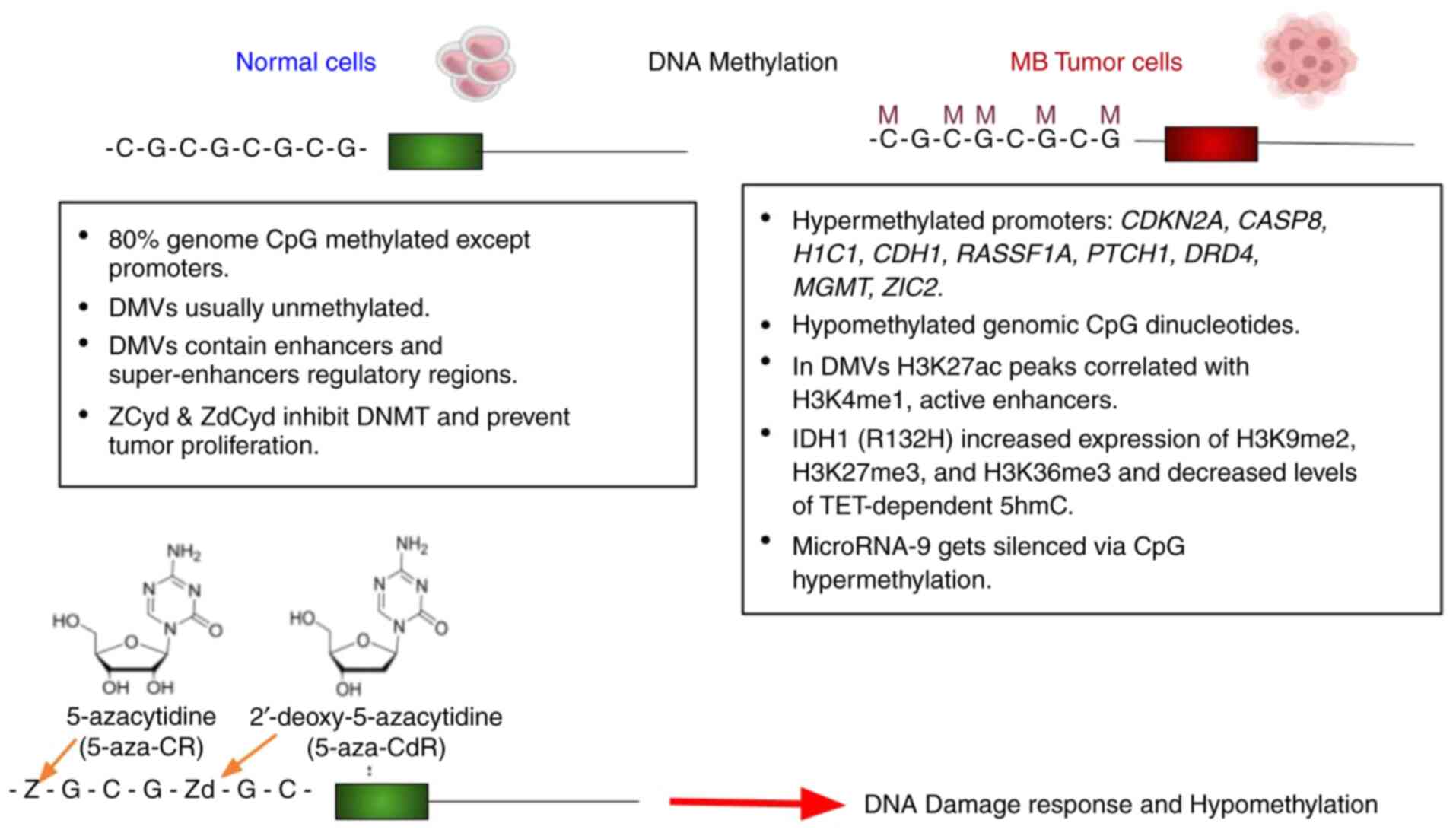 |
Figure 1
Methylation spectrum of normal cells vs. medulloblastoma cells. DMVs, DNA methylation valleys or regulatory genes devoid of DNA methylation. Zd is incorporated into DNA, while 80-90% of Z is incorporated into RNA, blocking mRNA and protein synthesis; 10-20% of Z is incorporated into DNA after its multiple enzymatic conversion into Zd. Z and Zd incorporation inhibits DNMT function, which causes the loss of methylation marks or hypomethylation (136,137). Z, 5-azacytidine; Zd, 2'deoxy-5-azacytidine; DNMT, DNA methyltransferase.
|
Histones were discovered by Kossel and Pringle (28) in 1884 at the laboratory of Felix Hoppe-Seyler from goose erythrocytes, following the serendipitous discovery of nucleic acids from neutrophils in the pus of wounded soldiers by Freidrich Meischer (29); both Kossel and Meischer were students of Felix Hoppe-Seyler. Several factors contribute to the development of cancer, including post-translational modifications of histones, mutations in histone genes (oncohistones) (30-32) and their interactions with chromatin regulatory complexes, chromatin-binding proteins, topologically associated domains, chromatin compaction and methylation enzymes. Histones are the subunits of a nucleosome, also known as beads on a string (Fig. 2). Histones undergo through a variety of post-translational modifications, such as lysine acetylation and lysine deacetylation, lysine methylation and arginine methylation, phosphorylation, propionylation and butyrylation, biotinylation, proline cis-trans isomerization, ubiquitylation, glycosylation, citrullination, SUMOylation, ADP ribosylation, deamination and crotonylation (Fig. 2). Histones play a crucial role in normal or cancer cell development (32-34) (Fig. 3). Of note, >150 histone-modifying proteins have been identified mainly from the inappropriate activation of proto-oncogenes or silencing of tumor suppressor genes (32). Histone-modifying enzymes are classified into three categories as follows: i1 |Writers: Histone acetyltransferases (HATs) and histone methyltransferases (HMTs); ii) erasers: Histone deacetylases (HDACs) and histone lysine demethylases (KDMs and Jumonji families); and iii) readers: Proteins that recognize these histone modifications (bromodomain, chromodomain and Tudor domain) (30).
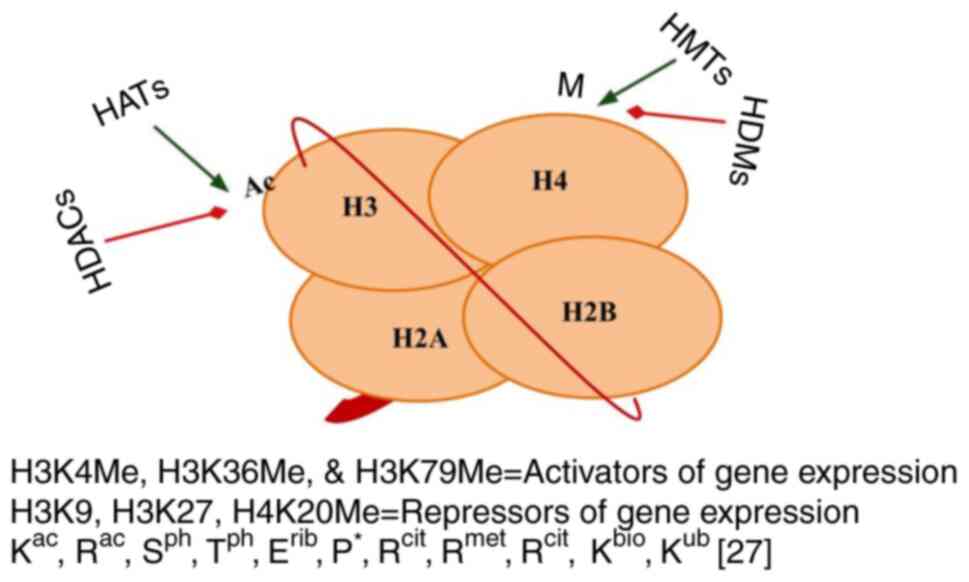 |
Figure 2
Structure of a nucleosome. A 146-bp segment of DNA wrapped around an octamer of histones, which consists of two H2A-H2B dimers and one H3-H4 tetramer. Modifications at different histones' N- and C-termini and globular domains, ac, acetyl; ph, phosphoryl; rib, ADP-ribosyl; met, methyl; cit, citrullyl, bio, biotinyl; asterisks above proline show cis-trans isomerization. bp, base pair; HDACs, histone deacetylases; HATs, histone acetyltransferases; HMTs, histone methyltransferases; HDMs, histone demethylases.
|
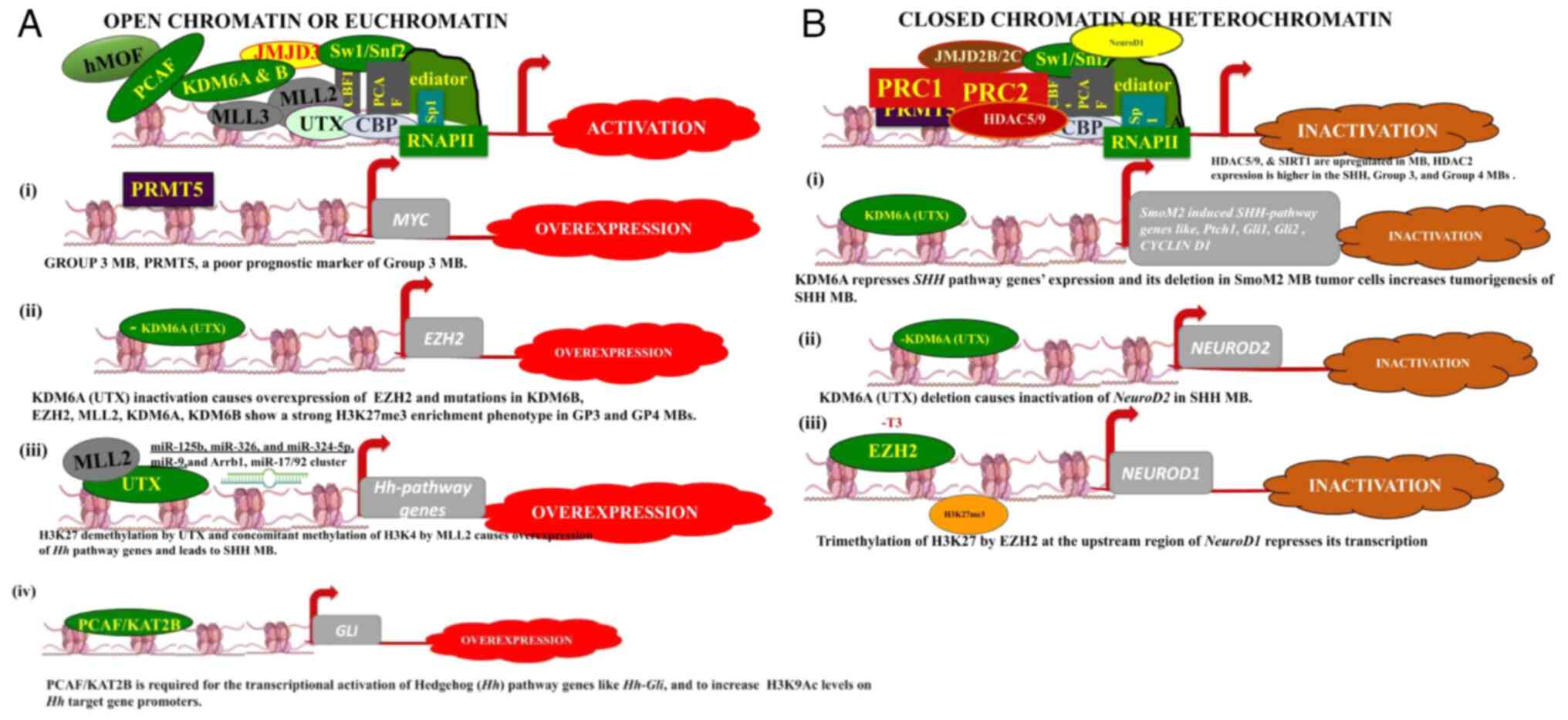 |
Figure 3
(A and B) Schematic diagram outlining the general modes of open and closed chromatin. The diagram illustrates how PCAF, KDM6A, KDM6B, MLL2 and MLL3 contribute to transcriptional activation, while the PRC complex is involved in gene repression. Each side of the panels illustrates how specific histone modifications can influence the overexpression or inactivation of particular genes in MB cells. In the absence of thyroid hormone, particularly its active form T3, EZH3 trimethylates H3K27 and suppresses the expression of NeuroD1, a key transcription factor promoting cell differentiation.
|
Role of histone modifications
The histone modification complexes, their particular subunits and their targets or recognition sites exhibit loss-of-function, gain-of-function mutations, or up/downregulation in various types of cancer. For example, class I, II and IV HDACs are upregulated in breast and colorectal cancers, and in hematological malignancies. Polycomb group (PcG) repressive complexes 1 and 2 (PRC1 and PRC2) that catalyze H3K27me3 and H2AK119ub (35) modifications (Fig. 3B), exhibit loss-of-function mutations in myeloid disorders and malignant peripheral nerve sheath tumors. The enhancer of Zeste homologs 1 and 2 (EZH1/2) are the mutually exclusive catalytic subunits of the PRC2 complex and work in concert with EED, SUZ12, and additional subunits. Notably, the allosteric activation site of EZH2, which is influenced by EED, is mutated in various types of cancer and is also linked to Weaver's syndrome (31,32,36). BMI1, also known as PCGF4, is part of the PRC1 complex and can form homodimers and heterodimers with RING1 or PHC subunits, these dimers are important for chromosome compaction. BMI1, the specific biomarker of AML, is frequently upregulated in AML and is necessary for the self-renewal and maintenance of healthy and leukemic stem cells; its depletion reduces proliferation or results in the apoptosis of epithelial and leukemic cells and also in murine colorectal cancer xenograft models (32,37-41).
Role of methylation and histone complexes
As aforementioned, histone modifications, chromatin modeling complexes, mega-nucleosomal complexes and methylation marks are interconnected. While most of the genome is methylated, CpG islands, which are typically 1 kb in length and often include promoter or RNA polymerase II binding sites, are usually unmethylated in the majority of somatic cells. Unmethylated CpG islands, found in euchromatin, are associated with actively transcribed genes, whereas methylated CpG islands in heterochromatin are linked to transcriptionally repressed genes. Unmethylated chromatin is characterized by hyperacetylated histones, which aid in recruiting transcriptional machinery necessary for gene expression. For example, the 5S rRNA gene is only expressed if histone H4 is acetylated in its nucleosomes (12,42) and transcription factor IIIA cannot bind to it. Similarly, the GAL4A activator cannot transcribe DNA that does not contain acetylated histones, such as H4. The hyperacetylated histones are present on unmethylated CpG islands and these chromatin domains are preferentially accessible to nucleases (43,44). The mechanisms which trigger histone acetylation on unmethylated CpG islands and the mechanisms through which this histone acetylation occurs, remain unknown. In addition, it is unclear whether histone deacetylation or hypermethylation occur first. However, it is known that the fragile X mental retardation gene 1 (FMR1) promoter, the methylation of the CGG repeat sequence, increases its affinity for the histone octamer and nucleosome assembly, which can alter the functional organization of chromatin and causes abnormal expression of the FMR1 gene and its phenotype (12,45). Transcriptionally inactive genes also exhibit an abundance of linker histones H1 near methylated and heterochromatin regions (12,46,47). The direct evidence of DNA methylation in the formation of heterochromatin is derived from the role of methyl-CpG binding domain (MDB) proteins in the methylation and recruitment of histone deacetylase complex (48). The MDB-containing proteins or MBD family are the readers of DNA methylation and recruit methylases, histone deacetylases and chromatin compacters or remodelers at the sites (48). For example, the Mi-2 complex is a well-known model to used to elucidate the role of MBDs in DNA methylation and chromatin repression by histone deacetylation. The Mi-2 complex is conserved among invertebrates. For example, in Drosophila melanogaster and Caenorhabditis elegans, the Mi-2 complex is made up of methyl-CpG binding protein (MBDs) MBD3, histone binding protein RbAp48, the histone deacetylase, along with three other polypeptides that together deacetylate histones (12,48,49).
Role of RNA interference (RNAi)
When cancer became enigmatic or sporadic cancers, such as colon, lung, lymphoid, brain and other tumors were found without genomic mutations (22,24,25), the epigenetics of cancers was discovered. The most notable discovery is the role of RNAi in targeting and repressing histones or nucleosome complexes. In ciliate Tetrahymena thermophile, small RNA together with an Argonaute family protein generates H3K9me (gene repression mark) at histones along with its normal role of DNA silencing or degradation (50,51). The role of dicer, Argonaute and RNA-dependent polymerase in heterochromatin formation was first discovered in S. pombe lysine methyltransferase (KMT) for H3K9me and the HP1 homologue Swi6(50). Small RNAs modify chromatin structure and silence transcription by guiding Argonaute-containing complexes to complementary nascent RNA scaffolds and then recruit histone and DNA methyltransferases. In addition, recent advances in genomics, ChIP-seq, and RNA-seq assays suggest that chromatin-associated long non-coding RNA scaffolds also recruit chromatin-modifying complexes independently of small RNAs. These co-transcriptional silencing mechanisms form powerful RNA surveillance systems that detect and silence inappropriate transcription events and provide a memory of these events via self-reinforcing epigenetic loops (50,52,53). Understanding these RNA surveillance systems may be potent therapies for cancers. The section below focuses mainly on the epigenetic landscape of medulloblastoma and explores currently available treatments.
2. The epigenetic hallmarks of medulloblastoma
Medulloblastoma (MB) is one of the most common forms of brain cancer in children, affecting the cerebellum located in the posterior fossa, also referred to as an infratentorial tumor. It mainly arises from disruptive cerebellar development, which may be related to extracellular cues and aberrant developmental signaling pathways of neurogenesis. These unregulated signaling pathways, such as hedgehog (Hh) and wingless (WNT) alter the pathway of granule neuron precursors (GNPs) from differentiation or maturation to proliferation or tumor formation (54-57). The environmental cues include parental exposure to various solvents and chemicals, such as benzene, chlorinated and polycyclic hydrocarbons during the 5-year time frame prior to conception (58). MB exhibits a higher propensity for children than adults and affects 1.58-fold more male children than female children, which could vary in different types of MBs (59). Based on the CBTRUS report, the most common MB subtype is sonic hedgehog (SHH)-activated and TP53-wildtype (included in the SHH group), with an incidence rate of 0.03 per 100,000 population and a median age of diagnosis of 19 years. The second most prevalent subtype is the non-WNT/non-SHH MB, which includes both group 3 (group C) and group 4 (group D). This subtype has an incidence rate of 0.02 cases per 100,000 and a median age at diagnosis of 8 years. The WNT-activated MB subtype has a lower incidence rate of 0.01 cases per 100,000 individuals and a median age at diagnosis of 10 years (1,2).
Based on histopathological studies, MB was classified into four major groups: Classic, desmoplastic nodular, MB with extensive nodularity and large cell/anaplastic (55). Furthermore, MB is subdivided into four subgroups based on transcriptional and epigenetic features: WNT, SHH, group 3 and group 4(60). Advanced high-throughput techniques, such as RNA sequencing (RNA-seq), next-generation sequencing (NGS), chromatin immunoprecipitation followed by sequencing (ChIP-seq), and DNA methylation analysis have refined the classification of MB into 7 to 12 subtypes. These detailed classifications provide valuable insights into genetic signatures, mutations, histone modifications, methylation patterns and signaling pathways associated with these subtypes (55,56,59-63). The majority of MB types originate in the cerebellum, with the exception of the WNT group, which arises outside the cerebellum. The WNT tumors are distributed within the fourth ventricle and infiltrate the dorsal surface of the brainstem (57). WNT and SHH subtypes can be traced back to mutations that cause the constitutive activation of the WNT and SHH pathways; however, groups 3 and 4 are difficult to characterize. The main player of WNT MB in 85-90% of cases is the mutated β-catenin encoding gene, CTNNB1 but its phosphorylation-dependent ubiquitination partner complex APC (associated with familial adenomatous polyposis syndrome) shows mutations in cases where β-catenin is normal. WNT MB tumors also contain many recurrently mutated genes, DDX3X, SMARCA4, TP53, CSNK2B and PIK3CA (60). SHH-MB tumors are known for the loss-of-function or heterozygous mutations in patched-1 homolog (PTCH1), activating mutations in smoothened (Smo), and glioma-associated oncogene homolog (GLI)1 or GLI2 and MYCN amplifications (60,61,64). In addition, recurrent focal copy-number alterations in the p53 and PI3K genes are present in patients with SHH-MB. Those in the SHH-MB group who carry p53 mutations belong to a particularly high-risk category (65). Recent advancements in various genomic techniques have helped to provide a better understanding of group 3 and 4 MB. Patients with group 3 MB have a high-level amplification of MYC in 17% of cases, MYCN in 5%, OTX2 in 3% and GFI1 and GFI1B in 15-20% of cases. Group 4 MB tumors exhibit an MYCN amplification of 6%; BRACA1, BRACA2 mutations; in-frame insertions in KBTBD4; the aberrant induction of GFI1 or GFI1B by enhancer hijacking; a markedly enhanced expression of PRDM6 by the hijacking of a super-enhancer of SNCAIP gene; the SNCAIP gene is 600 kb upstream of PRDM6; due to a duplication event it becomes closer to PRDM6 in a topologically associated domain and super enhances its expression in patients with group 4 MB (61,66). In summary, among several recurrent mutations, certain genes, such as KMT2D, KMT2C, DDX3X and SMARCA4 are common among all four MB types (60). Even though there ample mutational data in MB, the epigenetic mechanisms of MB remain unclear. In the sections below, epigenetic mechanisms, such as DNA methylation, histone modifications and RNAi in medulloblastoma are discussed.
Evidence from DNA methylation
The evidence of hypermethylation of CpG islands, known as CGI methylator phenotype (CIMP) was first described in colorectal tumors (67). However, the first CIMP studies in brain cancer came from isocitrate dehydrogenase (IDH1) mutations in patients with glioma. The mutation at arginine (R132) of the IDH1 active site changes the whole methylation landscape of certain genes; some of these genes are de novo methylated, and some hypomethylated genes exhibit a higher methylation (Fig. 1) (59,68-70). In a previous study, the expression of the IDH1 mutation arginine into histidine (R132H) increased the levels of H3K9me2, H3K27me3 and H3K36me3, and decreased the levels of TET2-dependent 5-hydroxymethylcytosine (5hmC) in immortalized primary human astrocytes. Since 5mC and 5hmC are oxidized to 5-carboxylcytosine (5caC) by Tet dioxygenase, the pathway for active DNA demethylation (71); a decrease in TET2 promoter activity by IDH1 R132H mutation led to extensive DNA methylation, ultimately leading to a CIMP pattern (70). These samples also revealed the methylation of the differentiation marker, neuronal differentiation 1 (NeuroD1) (70), which has been found as a signature gene of differentiating GNPs (56). Previously, IDH1 mutations confirmed the molecular basis of CIMP in patients with SHH-MB (59,61,72). Hypermethylation profiling assays have revealed that CDKN2A, CASP8, H1C1, CDH1 and RASSF1 (tumor suppressor genes), MGMT (DNA repair gene) (73,74), PTCH1 (the negative regulator of SHH signaling), the SFRP family (inhibitors of the WNT signaling), DRD4 (brain development) and ZIC2 (transcriptional repressor) in MBs are silenced by promoter CGI methylation (59) (Table I). MGMT promoter hypermethylation has been linked with G:C to A:T transition mutations in K-Ras and p53 in colorectal tumors (20,23). Patients with SHH-MB with Tp53 mutations form an extremely high-risk group of patients (60,65). DNA methylation profiling has also revealed DNA methylation valleys (DMVs) or canyons of regulatory genes devoid of DNA methylation located in large genomic domains (75). DMVs are not only visible in the ChIP-seq analysis of samples of patients with MB (e.g., H3K27ac or H3K4me3, active enhancer marks), but these core regulatory circuits are MB subgroup-specific. These DMVs exhibit hypermethylation and the repressed transcription of important markers, as well as hypomethylation and increased gene expression of other markers. The methylated domains affect up to one-third of the genome with increased mutation rates and gene silencing in an MB subgroup-specific manner (59,75-77). These DMVs are well known as enhancers and super-enhancers, and their role in topologically associated domain formation, chromosome compaction, nucleosome formation and transcription (76-79). Using H3K27ac and BRD4 ChIP-seq analysis from 28 MB specimens, 78,516 enhancers were previously found. The H3K27ac peaks correlated with H3K4me1 (activation mark), but not with DNA methylation and repressive H3K27me3 histone marks (76), and also bidirectional RNA transcripts or enhancer RNAs (eRNAs) were overlapping with these peaks. These findings revealed enhancer-promoter interactions for TGFBR1 and SMAD9 in group 3 MB cell line HD-MB03. These studies also confirmed enhancers regulating ALK, a receptor tyrosine kinase that is altered in other human cancers, are highly active in the WNT subgroup. In light of these new findings of CIMP, DMVs, methylation spectrums, and enhancers and super-enhancers in MB samples, improved treatment strategies and prognoses for patients with MB may now be possible.
 |
Table I
List of different epigenetic modifications, their signature markers, and drugs that target these.
|
Table I
List of different epigenetic modifications, their signature markers, and drugs that target these.
| Epigenetic modification type |
Signature markers |
Drugs |
| DNA methylation |
IDH1, CDKN2A, CASP8, H1C1, CDH1, RASSF1, PTCH1, DRD4, MGMT, ZIC2. |
5-azacytidine (5-aza-CR) or 5-aza-2'deoxycytidine (5-aza-CdR) or ZdCyd, zebularine, SG-1027 |
| Histone modifications |
|
|
| Histone acetyltransferases |
hMOF, PCAF/KAT2B, CBP and P300, MYST3 |
JQ-1 (Thieno-triazolo-1,4-diazepine), |
| Histone deacetylases |
HDAC1, HDAC2, HDAC5, HDAC9, & SIRT1 |
Trichostatin A (TSA), vorinostat, romidepsin, belinostat, panobinostat, curcumin, MAZ1863 and MAZ1866 |
| Histone methyltransferases |
PRMT5, EZH2, MLL2/KMT2D and MLL3/KMT2C, EHMT1, SMYD4 |
EPZ015666, UNC0638 |
| Histone demethylases |
KDM6A, KDM6B |
SP2509 |
| MicroRNAs |
miR-10, miR-9/9*, miR-20a, miR-19b, miR-130a, miR-29c and miR-29b, miR-124, miR-125a, miR-125b, miR-138, miR-181a, let-7, miR-29b, and miR-26a, miR-326, and miR-324-5p, miR-218, miR-17/92 cluster and miR-4521 |
miR-124, miR-9 and miR-125a |
Evidence from histone modifications
Almost every category of histone modifiers, whether writers, erasers, or readers are involved in cancer. The post-translational modifications of histone tails play an essential role in cell cycle, cell development, proliferation, maturation and differentiation; a number of these pathways have been found aberrant in numerous types of cancer, including MB. For example, the first histone lysine methyltransferase (HKMT) identified was SUV39H1 that targets H3K9 and has been shown to be mutated in MB (63). Histone methylation can occur either at lysine or arginine of H3 or H4 and also at the core globular domains of histones (80). Histone H3 lysine methylation occurs at K4, K9, K27, K36 and K79, and histone H4 lysine methylation occurs at K20. The H3K4, H3K36 and H3K79 methylation occurs on transcriptionally active genes, while H3K9, H3K27 and H4K20 methylation causes gene suppression (59) (Fig. 2).
HMTs
There are two major types of HMTs, lysine-specific, which can be SET (Su(var)3-9, enhancer of zeste and trithorax) domain or non-SET domain-containing HKMTs, and arginine specific, also known as protein arginine methyltransferases (PRMTs) (81-83). Group 3 MBs account for ~25% of MBs and are associated with the worst prognosis, with a 5-year survival rate of 50% (83). In recent studies on 491 MB samples, the evidence of PRMT5 mRNA overexpression was found to be significantly high as compared to nine normal cerebellar controls (84) (Table I). This was found to be linked with MYC amplification in patients with group 3 MB (Fig. 3A) (84). The high levels of PRMT5 expression were associated with the poor survival of patients with MB and hence, PRMT5 has been considered as a poor prognostic marker of group 3 MB. Furthermore, the knockdown of PRMT5 resulted in a decreased MYC expression and a statistically significant reduction in cell proliferation among MB cell lines (84). Co-immunoprecipitation assays from MB cells also indicated that PRMT5 was present in Myc-immunoprecipitated complexes (84). Based upon these results, the dose-dependent efficacy of the PRMT5 inhibitor, EPZ015666, was observed, as evidenced by MB cell growth suppression and the induction of the apoptosis of MYC-driven MB cells (84) (Table I). Using high-resolution SNP genotyping, whole-genome sequencing and ChIP assay, landmark mutations of histones were found in pediatric MB tumor samples. For example, inactivating mutations of HKMTs MLL2/KMT2D and MLL3/KMT2C; the homozygous deletion of the SET domain-containing euchromatic histone-lysine N-methyltransferase 1 gene (EHMT1), SET and MYND domain-containing histone lysine methyltransferase 4 gene (SMYD4), PcG genes (L3MBTL2 and L3MBTL3 and SCML2), the amplification of histone lysine demethylase (JMJD2B and JMJD2C) genes and the recurrent amplification of histone lysine acetyltransferase gene MYST3 in human pediatric MB tumor specimens were identified (59,83,85,86). Furthermore, the restoration of genes controlling H3K9 methylation was shown to significantly diminish the proliferation of MB cell lines in vitro (86), which suggests the further exploration of H3K9 methylation in patients with MB for targeted therapy.
Histone-modifier MLL2, an H3K4me3 methylase is a transcriptional activator; KDM6A or UTX cooperates with MLL2 by demethylating H3K27me3 (Fig. 3A); hence, KDM6A is also a potent activator; however, both of these genes are mutated in numerous types of cancer, including MB (85). KDM6A and KDM6B contain the Jumonji C (JmJC) domain at their C termini, specialized for the lysine demethylation of H3K27me2/me3. In MB tumors, MLL2 and KDM6A mutations are associated with the loss of H3K4me3 and H3K27me3 marks. The KDM6A inactivation causes the overexpression of EZH2 (subunit of PRC2 complex), an H3K27-methyltransferase, and mutations in another H3K27 demethylase KDM6B (Fig. 3A). This spatiotemporal expression of EZH2 (H3K27MT), KDM6A (H3K27DM), KDM6B (H3K27DM) and MLL2 (H3K4MT) exhibits a strong H3K27me-3 enrichment phenotype in groups 3 and 4 tumors as compared with the WNT and SHH subgroups (Fig. 3A) (85). Another example is the trimethylation of H3K27 by EZH2 on the upstream region of a basic helix-loop-helix transcription activator NeuroD1 shown by ChIP-PCR in the study by Cheng et al (54). H3K27me3 is a potent transcriptional repression marker and causes the repression of NeuroD1, which results in MB tumorigenesis (Fig. 3B); however, Cheng et al (54) demonstrated that the overexpression of NeuroD1 or the application of EZH2 inhibitors shifted the path of the cells to differentiation. These differentiated MB cells resemble mature GNPs (54,64,87,88). The results of these studies provide a novel therapeutic approach for inhibiting MB tumor proliferation using EZH2 inhibitors along with NeuroD1 induction.
KDMs
KDMs are enzymes that remove methyl groups from histone proteins and these are involved in the regulation of chromatin structure and transcription. Until 2002, histone methylation was considered a stable, static and irreversible modification. At first, it was considered that the conversion of arginine to citrulline via a deamination reaction was the only strategy for reversing arginine methylation; however, in 2004, lysine-specific demethylase 1 (LSD1) that removes methyl groups from H3K4me1/2 (89,90) and jumonji domain-containing protein JMJD6 that demethylates H3R2 and H4R3 (91,92) were discovered. Histone demethylases or erasers are classified into two protein families: i) KDMs that include LSD1 or KDM1A and LSD2 or KDM1B; and ii) the JmJC domain-containing large KDM family comprises ~30 enzymes grouped into KDM2-7 subfamilies in humans (59,85,92) (Table I). The X-linked H3K27me2/3 eraser UTX on chromosome X (also known as KDM6A; lysine-specific demethylase 6A) is a member of the MLL2 histone H3K4 methyltransferase complex. It erases di-and tri-methyl groups from lysine 27 of H3. The UTX/KDM6 is known for correct reprogramming, embryonic development, and tissue-specific differentiation, and contributes to animal body patterning by regulation of homeobox (HOX) genes (92-94). The recruitment of the UTX complex to HOX genes results in H3K27 demethylation and concomitant methylation of H3K4(93). Group 4 MBs have been found to contain the UTX/KDM6A mutations as afore mentioned (59,85,92). Recently, the generation of a UTX-deleted SmoM2-mouse model of SHH-MB revealed extensive proliferation by BrdU staining. UTX appeared to suppress SHH-MB initiation and proliferation, exhibiting tumor suppressor function (94) (Fig. 3B). This tumor suppressor function was challenged/reversed by the deletion of another H3K27me demethylase JMJD3/KDM6B also. In both mouse MB models and human MB cells, UTX activated Th1-type chemokines that attracted T-cell migration, which is critical for targeting MB cells (94). Another proneuronal gene, NeuroD2, also requires UTX for its expression in precancerous progenitors. The overexpression of NeuroD2 decreases GNPs proliferation and increases their differentiation; UTX deletion leads to the repression of NeuroD2 and impaired neural differentiation (94); thus, UTX deletion increases MB proliferation due to impaired NeuroD2 (Fig. 3B); hence UTX and NeuroD2 are the next in line for MB therapeutics.
HATs
HATs are enzymes that modify histone proteins by the addition of acetyl groups to lysine residues, and in general, histone acetylation is a gene activation mark. Based upon the sequence conservation with the catalytic HAT domain of a general control non-derepressible-5 (Gcn5), a transcriptional adaptor from yeast, HATs are divided into three major families: The GCN5-related N-acetyltransferase family, the MYST family (MOZ, Ybf2, Sas2, TIP60) and the orphan family (CBP/P300 and nuclear receptors) (95,96). Research on MB tumors has demonstrated the downregulation of human MOF (hMOF) of the MYST family, a HAT for H4K16 acetylation (Table I) (97). Another key HAT, PCAF/KAT2B (CBP/P300 family), is required for the transcriptional activation of Hedgehog (Hh) Hh-Gli target genes; PCAF binds to glioma-associated oncogene (Gli), and both proteins are required for the increased H3K9Ac levels on Hh target gene promoters (Fig. 3A) (Table I). The Hh-Gli pathway is deregulated in glioblastomas and MBs and PCAF is a key player. The depletion of PCAF causes an impairment in cell proliferation and the induction of apoptosis in glioblastomas and MB cell lines Therefore, targeting PCAF activity presents an attractive strategy for treating Hh/Gli-dependent SHH MB tumors (98). Apart from H3K9, the orphan family members CBP and P300 that acetylate H3K27, a marker for strong and active enhancers, have also been found to be mutated in MB tumors (59). The enzymes that remove these modifications include HDACs, phosphatases, demethylases and deubiquitinases (99).
HDACs
HDACs are a class of enzymes that remove acetyl groups from an ε-N-acetyl lysine amino acid on both histone and non-histone proteins. HDACs help histones wrap tightly to DNA. This is crucial as DNA is wrapped around histones, and DNA expression is regulated by acetylation and deacetylation (100). HDACs are classified into four classes based upon their sequence identity and domain organization. Class I, II and IV are zinc-dependent HDACs, but class III/Sirtuins are NAD-dependent HDACs. The class I HDACs are homologous to yeast repressor reduced potassium dependency 3 (Rpd3) (HDAC1, HDAC2, HDAC3 and HDAC8), class IIs are homologous to yeast histone deacetylase 1 (Hda1) (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 and HDAC10), class III or Sirtuins have catalytic domain similar to yeast NAD+-dependent deacetylase silent information regulator 2 (Sir2) (SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6 and SIRT7), class IV (HDAC11) (99). Several HDACs, such as HDAC5, HDAC9, and SIRT1 are upregulated in MB (Table I) and cause an aberrant cell cycle; for example, SIRT1 siRNA arrests 65% of UW228-3 MB cells in the G1 phase and induced 14.5% of cells to apoptosis at the 48-h time point (101,102). SIRTI is a potential therapeutic target of MBs. Similarly, the expression HDAC2 is higher in SHH, group 3 and group 4 MBs (103). Other examples of histone phosphorylation and ubiquitination include WEE1, a tyrosine kinase, and a regulator of the S/G2 checkpoint, which impairs MB cell growth. WEE1 phosphorylates H2B at Tyr37 and inhibits the transcription of multiple histone genes (59,104-107). The resultant change in the DNA/histone ratio affects mitotic entry. A component of the Cullin E3 ubiquitin ligase reduces the expression and the allelic deletion of REN (KSCTD11) in SHH-MBs; it ubiquitinates and promotes the degradation of HDAC1, resulting in the suppression of SHH signaling and MB growth (59,108). From the aforementioned findings, the key MB targets can be denoted as follows: NeuroD1, PRMT5, MYC, MLL2/KMT2D and MLL3/KMT2C, EHMT1, SMYD4, L3MBTL2 and L3MBTL3 and SCML2, JMJD2B, JMJD2C, KDM6A or UTX, hMOF, PCAF, HDAC5, HDAC9, and SIRT1, WEE1, and REN (KSCTD11).
Evidence from RNAi
For a very long time, until the turn of the 21st century, it was deemed that protein-coding genes constituted only a small portion of the mammalian genome and that the remainder of the genome was merely non-coding or transcriptional noise (109,110). As regards DNA methylation and histone modifications, as aforementioned herein, both of these processes can become highly aberrant if the transcriptional noise (non-coding RNA) becomes even slightly low. These non-coding RNAs are the key players, from fertilized egg development, cell proliferation and cell differentiation, to the whole function of the organism. The cerebellum or the ‘little brain’ comprises only 10% of the total mass, but contains 80% of all neurons (111).
The journey begins with microRNA (miRNA/miR)-10, a major player in cerebellar development. Two important homeodomain transcription factors, Otx2 and Gbx2, regulate the formation of the midbrain-hindbrain boundary or isthmic organizer and miR-10 controls the expression of both. MiR-10 downregulates Otx2 and upregulates Gbx2 in human neural progenitor cells (NPCs) (112-114). MiR-10 displays a highly specific regionalized expression pattern in the developing human brain, suggesting a role for miR-10 in the caudalization of hNPCs (112,113). The loss of miR-10 expands the Gbx2 domain that affects the proper development of the cerebellum (113). The miRNA map of cerebellar development is almost complete; for example, in Bergmann glia cells, miR-9 under Dicer1 specifically guides transcriptional signature genes, such as brain lipid binding protein of the Notch pathway for cerebellar morphology (115). Purkinje cells express miR-124, miR-125b, miR-138, miR-181a, let-7, miR-29b and miR-26a (113) (Table I). miRNA-20a, miRNA-19b, and miRNA-130a exhibit a significant downregulation, while miRNA-29c and miRNA-29b exhibit a significant upregulation at 30 days postnatally, as compared to 9 days postnatally, in adult rat Purkinje cells (116) (Fig. 4) (Table I), miRNAs also protect Purkinje neurons from degeneration (113). miR-124 is a highly expressed brain-specific miRNA in neuronal cells; it is a neuronal marker that regulates neurogenesis and cerebellar development (117) and is the first identified miRNA linked with MB (113). High throughput miRNA expression profiling in human MB specimens has revealed the downregulated expression of numerous miRNAs, including miR-9 and miR-125a (113) (Fig. 4). These findings were validated by the rescued expression of miR-9 and miR-125a, which promoted MB cell growth arrest and apoptosis by targeting the neurotrophin receptor, truncated tyrosine kinase isoform (tTrkC) (118,119). In parallel with these findings, miR-125b, miR-326 and miR-324-5p have been found to antagonize the SHH proliferation pathway during cerebellar granular neuronal precursor cell (GNPC) differentiation by targeting the activator, Smo, and the effector Gli1 (113) (Figs. 3A and 4). In addition, miR-326 targets Arrb1; Arrb1 encodes for an adaptor and scaffold protein that regulates several signaling pathways in cell development and cancer, both miR-326 and Arrb1 are downregulated in SHH-MBs (Fig. 4) (120). In granule cells, Dicer-dependent pathways of miRNAs sustain SHH signaling and DNA damage response and control GC proliferation and differentiation (113). miRNAs can alter the trajectory of signaling pathways and the fate of cells. miR-124 and miR-9* can transform human fibroblasts into neurons through SW1/SNF-like BAF chromatin-remodeling complexes (121-123). These studies demonstrate how miR9/9* maintains the genetic circuitry in human fibroblasts per se. Both miR-9* and miR-124 repress the BAF53a subunit of the neural-progenitor BAF remodeling complex; BAF53a is replaced by BAF53b, BAF45a by BAF45b and BAF45c for post-mitotic functions. This process is facilitated by NEUROD2, and the addition of neurogenic transcription factors (ASCL1 and MYT1L1) enhances the rate of conversion and the maturation of the converted neurons. miR-9/9* and miR124 play an instructive role in neural fate determination (121-123). miR9/9* exhibits a versatile expression in human cancers, such as breast cancer, cervical cancer, squamous cell carcinoma of the skin and oral cavity, hematological malignancies, and glioblastoma (122). In glioblastoma multiforme brain tumors, that are resistant to alkylating agents, miR-9 is highly expressed and miR-9* is downregulated. miR-9 contributes to chemoresistance by directly targeting PTCH1 and the subsequent activation of the SHH pathway (122). miR-9 is a critical neural developmental marker, and it has been found to be silenced by CpG hypermethylation in MB samples, as well as in MB-derived cell lines. miR-9 binds at the 3'UTR of hairy and enhancer of split 1 (HES1) and decreases its protein expression; the low expression of miR-9 increases HES1 and causes MB tumor proliferation (124,125). The low expression of miR-9 is a predictive marker for MB poor prognosis (124). miR-9/9* and miR-124 play a crucial instructive role in neural maturational genetic circuits, and hence are the potential target of future miRNA-based therapies against MB and other human malignancies.
 |
Figure 4
Key microRNAs that are significantly upregulated and downregulated in various groups of medulloblastoma.
|
Numerous roles of miRNAs in cerebellar development, the neural axis and neuronal maturation process came from miRNA biogenesis machinery knock-out studies. One of the genes disrupted by the 22q11.2 microdeletion is the DGCR8, a component of the ‘microprocessor’ complex that is essential for miRNA production. The DGCR8 deletion results in the abnormal processing of specific brain miRNAs and working memory deficits, Dgcr8+/- mutant mice display significant behavioral and cognitive defects (104,126). The deletion of Dicer1 expression in different neuronal cells shows missing targeted cell population or dedifferentiation (proliferation). One of the most extensively studied regulatory loops involved in neural stem cell differentiation is the repressor element 1-silencing transcription factor (REST) complex/small C-terminal domain phosphatase (SCP1) pathway. The REST complex is also known as the neuron-restrictive silencer factor or NRSF. This complex inhibits neuronal genes in non-neuronal cells by suppressing miR-9 and miR-124. In neurons, however, the negative feedback loop via miR-124 suppresses the SCP1 and REST that leads to neuronal differentiation. The deletion of miR-9 in the hindbrain increases cell proliferation due to the indirect downregulation of the cell cycle inhibitor p27(108). Cyclin-dependent kinase-6 (CDK6) is regulated by miR-124 and is inversely related means if CDK6 is overexpressed then miR-124 is low in MB cell lines and when miR-124 is restored it decreases CDK6 expression (127). The ectopic expression of miR-124 by intracerebellar or subcutaneous transplantation in immunosuppressed mice results in MB growth arrest (128). As another example, miR-218 also modulates CDK6 expression (129). As regards the miR-17/92 polycistron, the first validated oncogenic miRNA clusters, the first discovery in MB was derived from deep sequencing and comparative expression analysis of proliferating GNPCs and MBs relative to the mature mouse cerebellum. Among the 26 overexpressed miRNAs, nine belong to the miR-17/92 cluster family; three of the miR-17/92 cluster miRNAs, miR-92, miR-19a, and miR-20, are overexpressed with a constitutively activated SHH pathway (Fig. 4) (130-133). This miR-17/92 cluster in SHH-MB samples is induced by MYCN/c-Myc amplification; the surge of these transcription factors is associated with SHH pathways that result in the loss of GNPC differentiation and hence, medulloblastoma (130-134). In tissue samples obtained from patients with MB, the significant upregulation of the transcription factor Forkhead Box M1 (FOXM1) was found to be associated with a loss of its putative regulating microRNA, miR-4521(135). In all of the MB types, SHH, WNT, group 3 and 4, miR-4521 mRNA expression is low; however, group 4 exhibits a highly significant reduced miR-4521 expression (135) (Fig. 4). mir-4521 directly targets the 3'UTR of FOXM1. miR-4521 overexpression reduces the proliferation and invasion of MB cell lines and induces programmed cell death through the activation of caspase-3/7(135); hence, miR-4521 plays a critical role in MB and is another target for MB therapies.
3. Therapies for medulloblastoma, status quo
Some of the first epigenetic drugs proposed as anticancer therapeutics were DNA methylation inhibitors, such as 5-azacytidine (5-aza-CR) or Zcyd and 5-aza-2'deoxycytidine (5-aza-CdR) or ZdCyd that stimulate the differentiation of tumor cells (136,137). DNMT inhibitors are the pioneer agents used to target gene silencing by hypomethylation. When (cytosine-C5) methyltransferase [DNA (C5) MTase, DNMT] binds at these cytidine analogs, it is trapped and becomes inactivated due to covalent linkage to ZCyd residues in CpG sites of DNA (138). This trapping of DNMT depletes the availability and activity of DNMT in the cell, hence restoring the expression of tumor suppressor genes (Fig. 1). Other nucleoside inhibitors, such as zebularine, an inhibitor of SHH pathway, non-nucleoside inhibitors compound 5, and compound 2, the structural isomers of SG-1027 have been tested in MB mouse stem cells expressing higher levels of DNMT1; however, these findings require validation (139) (Table I).
The first report of the deacetylation of histones came from calf thymus extract and the role of acetylation and deacetylation interactions between histones and chromatin was proposed in 1969 based upon the effects of their interactions (140). As HDACs exist in complexes with other HDACs, histone modifiers and transcriptional factors, the first report of HDAC purification came after over two decades from Taunton et al in 1996(141). While performing a deep biochemical analysis of HDAC activity in nuclei, n-butyrate was found to induce the accumulation of acetylated histones (141). Subsequently, in 1990, trichostatin A (TSA) and HDAC (HDACi) from a Streptomyces strain was isolated, originally identified as an antifungal antibiotic and potent inducer of murine erythroleukemia cell differentiation (100,141). Following the discovery of TSA a number of HDACis that can induce morphological changes in transformed cells were designed or isolated, as for example: Trapoxin, a fungal cyclic peptide and an irreversible HDAC inhibitor (142); SAHA or vorinostat, the first HDAC inhibitor approved for chemotherapy in 2006(143); following SAHA, FK228 or istodax in 2009 for cutaneous and peripheral T-cell lymphoma, and then benzamides such as CI-994 and MS-275 (Entinostat) also entered trials. Based upon the chemical structure, HDACis have been categorized into four classes as follows: Class A, hydroxamic acid (TSA, vorinostat, belinostat, panobinostat and pracinostat); class B, short-chain fatty acid (valproic acid, butyric acid, and pivanex); class C, benzamide (CI-994, entiostat, MGCD0103); class D, cyclic peptide (romidepsin, largazole, CHAP31, trapoxin, apicidin). HDACis are still under investigation or trials for various types of cancer, such as leukemias, lymphomas and MBs; however, these have still been unsuccessful, particularly for MB. For example, vorinostat, romidepsin and belinostat are FDA-approved for T-cell lymphomas and are under clinical phase trials for MB (Table I). Recently, the pan-HDACi, panobinostat was found to suppress leptomeningeal seeding, a rare complication in MB spreading, causing brain and spinal cord inflammation in mouse models. The other examples of HDACis are the HDAC1/2-selective inhibitors, HDiA and HDiB, that block Gli1 and Gli2 activity through acetylation and SHH MB cell growth in several SHH MB lines (143). The natural compound, curcumin, has been found to inhibit HDAC and HDAC4 depletion and it reduces tumor growth; however, further studies are required (143). The Class IIa-selective HDACis, MAZ1863 and MAZ1866, in group 3 MB cells had only very minimal effects on MYC-MB cells, whereas vorinostat and entinostat efficiently reduce metabolic activity in MYC-MB cells (139). HDAC2 depletion promotes MB cell death and pre-clinical research has revealed that MYC-amplified group 3 MB cell lines exhibit sensitivity to class I HDAC inhibitors (139). The repression of the class III/Sirtuins HDAC, SIRT1, leads to MB growth arrest and apoptosis (101). Since SIRT1 is part of the BLC6/BCOR/SIRT1 complex, its downregulation attenuates the activation of the SHH pathway in mouse MB models; thus, SIRT1 inhibitors may also be next in line for MB therapy (144).
One of the critical epigenetic readers is bromodomain and extra-terminal domain proteins (BETs) that recognize the acetyl-lysine residues or acetylated chromatin, which are enhancers and super-enhancer markers. These high levels of H3K27Ac marks regulate key genes of cancer growth and are sensitive to BET inhibition; hence, they have been extensively studied in MB tumors. The BET inhibitor, JQ-1, a novel thieno-triazolo-1,4-diazepine, decreases MB tumor proliferation by lowering Gli1 and Gli2 expression and inhibits the SHH pathway (Table I). JQ-1 also downregulates MYC in group 3 MB; JQ-1 may provide a future treatment strategy for MB if its pharmacokinetic and pharmacodynamic properties can be improved (116,145). The lysine trimethylase EHMT2 G9a represses deubiquitinase USP37 in MB; its inhibition by UNC0638 leads to arrest of the MB cell proliferation (146) (Table I). the first lysine demethylase 1 discovered was LSD1 or KDM1A; SP2509 inhibits the LSD1, and blocks the MB tumor growth of various MB cell lines such as DAOY, D283med and ONS-76(147) (Table I). The dose-dependent efficacy of the PRMT5 inhibitor, EPZ015666, has also been shown; it suppresses MB tumor growth with the subsequent induction of apoptosis in MYC-driven MB cells (84). The inhibition of the SHH pathway by vismodegib (GDC-0449) has been shown; this is an FDA-approved SMO inhibitor for SHH-driven cancers, but is still under trials for MB (84,148).
Even though numerous DNA methyltransferase inhibitors, DNA methylase inhibitors, HDACis and BETis have been mentoined, current MB therapies include a combination of surgical resection followed by CSI, chemotherapy and immunotherapy with severe treatment-related toxicity and secondary or refractory tumors, and a maximum 5-year survival rate. Thus, more molecular-targeted therapeutic approaches need to be explored, in order to reduce the toxic side-effects and increase the life expectancy of patients. There are novel therapeutic approaches on the horizon for MB treatment. These directly involve the inhibition of MB tumor proliferation by downregulating the basic developmental pathways such as Hh and by promoting tumor cell differentiation. For example, the overexpression of a helix-loop-helix transcription factor, NeuroD1 promotes the differentiation of MB cells. By using EZH2 inhibitors, the H3K27me3 modification of NeuroD1 can be prevented, thus also enhancing the differentiation of MB cells (56). Recently, the new role of the active form of thyroid hormone, T3, in suppressing the EZH2 methylation of H3K27 on NeuroD1 that leads to the differentiation of MB cells has been demonstrated in both SHH-MB and group-3 MB cells, suggesting new promising differentiation-based strategies not only for MB, but also for other cancers (148) (Fig. 3B). The neuroepithelial stem cell protein (Nestin), largely present in nerve cells, is a type VI intermediate filament protein. Nestin drives embryonic cell development and upon cell differentiation, it becomes downregulated and replaced by tissue-specific intermediate filament proteins. However, in MB tumors, the continuous expression of nestin inhibits the GLi3 negative feedback loop that keeps the SHH proliferation pathway. Therefore, identifying a novel method with which to inhibit nestin could be a viable approach to combat MB tumor proliferation (149). The further exploration of these signaling pathway candidates in conjunction with existing treatments is needed.
miRNAs can also be possibly used as therapeutic targets, or as pre-screening and prognosis markers for MB tumors. Current MB treatments still have limited efficacy and severe side-effects. First and foremost, the miRNA expression analysis, microarray and NanoString (a variation of microarray technology) have identified strong candidates, such as miR-10, miR-9/miR-9*, miR-124, miR-218a, miR125a, miR-326, miR-324-5p, the miR17/92 cluster and miR-4521 that can be used to classify, diagnose and treat MB tumors in combined approaches with radiation and chemotherapy or immunotherapy. For example, miR-124 inhibits the overexpression of CDK6 and prevents the proliferation of glioblastoma and MB cells in vitro and in vivo. miR-124 also potently inhibits the growth of MB xenograft tumors in mice (128) (Table I). miR-9 and miR-125a overexpression promotes MB cell growth arrest and apoptosis by targeting the neurotrophin receptor, tTrkC in MB cell lines (Table I). However, there are no miRNA inhibitors for MB yet available. Combined approaches may enhance MB tumor differentiation; for example, anti-miR-21 and anti-miR-10b co-delivery using poly lactic-co-glycolic acid nanoparticles has been shown to enhance glioblastoma cell killing by temozolomide (124). The development of combined approaches including miRNA inhibitors or miRNAs that enhance MB apoptosis and prevent the chemoresistance of MB may be acheivable in the near future.
4. Conclusions and future perspectives
The journey began with James Wright's discovery of undifferentiated nerve cell tumors, which he termed neurocytes or neuroblasts (Wright, 1910) (150) to Bailey and Cushing in 1926, when they postulated that these tumors derived from primitive embryonic neuroepithelial cells as medulloblasts (151). Subsequently, in 1995, in the study by Hamilton et al (152), brain tumors in Turcot's syndrome were described as MB (152); in 2010, MBs were first classified into four molecular groups (153); finally, in 2017, molecular signatures categorized MBs were classified into 12 subgroups (55). The development of a versatile set of techniques, such as next-generation sequencing, chromatin profiling, DNA methylation assay, miRNA profiling, chromatin immunoprecipitation followed by sequencing (ChIP-seq), RNA sequencing (RNA-seq), assay for transposase-accessible chromatin using sequencing (ATAC-seq), Hi-ChIP, Hi-Seq, etc. has led to a new era in molecular-targeted therapeutic approaches for these MB tumors. One of the MB types, WNT-MB is associated with an improved survival rate, as WNT-MBs have a weak blood-brain barrier, which facilitates systemic chemo- or immunotherapy access and contributes to better prognosis and survival response (154). The major challenge and focus lie in the improved understanding of the elusive molecular biology of group 3 and 4 tumors to identify better markers, and targeted approaches to improve the clinical outcomes of patients. With the continuing surge of epigenomic therapeutics, such as DNMT inhibitors (Zcyd and ZdCyd), HDACis (vorinostat in combination with temozolomide or with bortezomib), HDAC/PI3Kinase inhibitor (fimepinostat), EZH2 inhibitors, BET inhibitor (JQ-1), drugs targeting EHMT1/EHMT2 and LSD1, PRMT5 inhibitor (EPZ015666), HAT inhibitors, and as many of these are already in trials, improved treatments for MB can be foreseen. A number of non-coding RNAs such as miR-9, miR-10, miR-124, miR125a, miR-125b and miR-324-5p protect GNPCs from constant proliferation and trigger differentiation. miR-124, miR-9/9*, miR-17/92, miR-218, miR-326, miR-34a, miR-128a, miR125a and miR-125b are only a few strong candidates for further MB studies. miR inhibitors may be part of the combined regimen for future MB therapies.
The clarity of epigenetics mechanisms, such as DNA methylation or CIMP, histones methylation and demethylation, histones acetylation and deacetylation, miRNA networking, or RNA surveillance may aid in the development of strategies with which to inhibit MB tumorigenicity via inducing tumor differentiation and finding small molecules to induce MB differentiation. However, the main unanswered question is what causes these epigenetic changes, the mechanisms behind the changes in miRNA expression, and abrupt histone modifications that result in cell proliferation or tumor formation rather than cell differentiation. The processes through which chromatin domains regulate these mechanisms and what controls the nuclear chromatin also remain to be elucidated. By understanding these epigenetic mechanisms, a better understanding of MB and other cancers may be achieved. In addition, the understanding of these epigenetic mechanisms may also solve neurodegenerative disorders, such as Parkinson's disease, Alzheimer's disease and amyotrophic lateral sclerosis.
Acknowledgements
The author is deeply grateful to Dr Harvey Rubin and his laboratory (Department of Medicine, University of Pennsylvania, Philadelphia, PA, USA) for providing invaluable scientific expertise and mentorship. The author would also like to extend his heartfelt thanks to Dr Zengji Yang and his laboratory (Fox Chase Cancer Center, Philadelphia, PA, USA) for providing the opportunity to delve into the study of medulloblastoma. The author would also like to thank Dr Vijay Juneja (USDA-Agricultural Research Service, Wyndmoor, PA, USA) for his invaluable assistance in editing the present review.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Author's contributions
The author (KKA) wrote and edited the manuscript. Data authentication is not applicable. The author has read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that he has no competing interests.
References
|
1
|
Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2016-2020. Neuro Oncol. 25 (12 Suppl 2):iv1–iv99. 2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Neff C, Price M, Cioffi G, Kruchko C, Waite KA, Barnholtz-Sloan JS and Ostrom QT: Complete prevalence of primary malignant and nonmalignant brain tumors in comparison to other cancers in the United States. Cancer. 129:2514–2521. 2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Johnson DR, Guerin JB, Giannini C, Morris JM, Eckel LJ and Kaufmann TJ: 2016 updates to the WHO brain tumor classification system: What the radiologist needs to know. Radiographics. 37:2164–2180. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kaifi R: A review of recent advances in brain tumor diagnosis based on AI-based classification. Diagnostics (Basel). 13(3007)2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Buckner JC, Brown PD, O'Neill BP, Meyer FB, Wetmore CJ and Uhm JH: Central nervous system tumors. Mayo Clin Proc. 82:1271–1286. 2007.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mahapatra S and Amsbaugh MJ: Medulloblastoma. In: StatPearls [Internet]. StatPearls Publishing, Treasure Island, FL, 2024.
|
|
7
|
Fincham JRS: Epigenetic mechanisms of gene regulation. Edited by V. E. A. Russo, R. A. Martienssen and A. D. Riggs. Cold Spring Harbor Laboratory Press, 1996. 693+xii pages. Price $125. ISBN 0 87969 490 4. Genetical Res 69: 159-162, 1997.
|
|
8
|
Avery OT, Macleod CM and McCarty M: Studies on the chemical nature of the substance inducing transformation of pneumococcal types: Induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J Exp Med. 79:137–158. 1944.PubMed/NCBI View Article : Google Scholar
|
|
9
|
McCarty M and Avery OT: Studies on the chemical nature of the substance inducing transformation on pneumococcal types; an improved method for the isolation of the transforming substance and its application to Pneumococcus types II, III, and VI. J Exp Med. 83:97–104. 1946.PubMed/NCBI
|
|
10
|
Hotchkiss RD: The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem. 175:315–332. 1948.PubMed/NCBI
|
|
11
|
Moore LD, Le T and Fan G: DNA methylation and its basic function. Neuropsychopharmacology. 38:23–38. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
El-Osta A and Wolffe AP: DNA methylation and histone deacetylation in the control of gene expression: Basic biochemistry to human development and disease. Gene Expr. 9:63–75. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lazar NH, Nevonen KA, O'Connell B, McCann C, O'Neill RJ, Green RE, Meyer TJ, Okhovat M and Carbone L: Epigenetic maintenance of topological domains in the highly rearranged gibbon genome. Genome Res. 28:983–997. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lu Y, Chan YT, Tan HY, Li S, Wang N and Feng Y: Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol Cancer. 19(79)2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI and Martienssen RA: Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 297:1833–1837. 2002.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Xu GL, Bestor TH, Bourc'his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ and Viegas-Péquignot E: Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 402:187–191. 1999.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Flavahan WA, Gaskell E and Bernstein BE: Epigenetic plasticity and the hallmarks of cancer. Science. 357(eaal2380)2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Esteller M, Fraga MF, Guo M, Garcia-Foncillas J, Hedenfalk I, Godwin AK, Trojan J, Vaurs-Barrière C, Bignon YJ, Ramus S, et al: DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet. 10:3001–3007. 2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Esteller M, Avizienyte E, Corn PG, Lothe RA, Baylin SB, Aaltonen LA and Herman JG: Epigenetic inactivation of LKB1 in primary tumors associated with the Peutz-Jeghers syndrome. Oncogene. 19:164–168. 2000.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Esteller M, Risques RA, Toyota M, Capella G, Moreno V, Peinado MA, Baylin SB and Herman JG: Promoter hypermethylation of the DNA repair gene O(6)-methylguanine-DNA methyltransferase is associated with the presence of G:C to A:T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res. 61:4689–4692. 2001.PubMed/NCBI
|
|
21
|
Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, et al: Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA. 91:9700–9704. 1994.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Jones PA and Baylin SB: The fundamental role of epigenetic events in cancer. Nat Rev Genet. 3:415–428. 2002.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Esteller M, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Watkins DN, Issa JP, Sidransky D, Baylin SB and Herman JG: Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res. 60:2368–2371. 2000.PubMed/NCBI
|
|
24
|
Herman JG, Civin CI, Issa JP, Collector MI, Sharkis SJ and Baylin SB: Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res. 57:837–841. 1997.PubMed/NCBI
|
|
25
|
Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, et al: Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 93:691–699. 2001.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang Q: Cancer predisposition genes: Molecular mechanisms and clinical impact on personalized cancer care: Examples of Lynch and HBOC syndromes. Acta Pharmacol Sin. 37:143–149. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Weemaes CM, van Tol MJ, Wang J, van Ostaijen-ten Dam MM, van Eggermond MC, Thijssen PE, Aytekin C, Brunetti-Pierri N, van der Burg M, Graham Davies E, et al: Heterogeneous clinical presentation in ICF syndrome: Correlation with underlying gene defects. Eur J Hum Genet. 21:1219–1225. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kossel A and Pringle H: Über Protamine und Histone. J Biol Chemistry. 49:301–321. 1906.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Dahm R: Discovering DNA: Friedrich Miescher and the early years of nucleic acid research. Hum Genet. 122:565–581. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hyun K, Jeon J, Park K and Kim J: Writing, erasing and reading histone lysine methylations. Exp Mol Med. 49(e324)2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ III, Voigt P, Martin SR, Taylor WR, De Marco V, et al: Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 461:762–767. 2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Valencia AM and Kadoch C: Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat Cell Biol. 21:152–161. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Audia JE and Campbell RM: Histone modifications and cancer. Cold Spring Harb Perspect Biol. 8(a019521)2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Dhall A and Chatterjee C: Chemical approaches to understand the language of histone modifications. ACS Chem Biol. 6:987–999. 2011.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Angrand PO: Structure and function of the polycomb repressive complexes PRC1 and PRC2. Int J Mol Sci. 23(5971)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lee CH, Yu JR, Kumar S, Jin Y, LeRoy G, Bhanu N, Kaneko S, Garcia BA, Hamilton AD and Reinberg D: Allosteric activation dictates PRC2 activity independent of its recruitment to chromatin. Mol Cell. 70:422–434.e6. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Abdouh M, Hanna R, El Hajjar J, Flamier A and Bernier G: The polycomb repressive complex 1 protein BMI1 is required for constitutive heterochromatin formation and silencing in mammalian somatic cells. J Biol Chem. 291:182–197. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Gray F, Cho HJ, Shukla S, He S, Harris A, Boytsov B, Jaremko Ł, Jaremko M, Demeler B, Lawlor ER, et al: BMI1 regulates PRC1 architecture and activity through homo- and hetero-oligomerization. Nature Commun. 7(13343)2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lessard J and Sauvageau G: Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 423:255–260. 2003.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ and Clarke MF: Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 423:302–305. 2003.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yuan J, Takeuchi M, Negishi M, Oguro H, Ichikawa H and Iwama A: Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia. 25:1335–1343. 2011.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wakamori M, Okabe K, Ura K, Funatsu T, Takinoue M and Umehara T: Quantification of the effect of site-specific histone acetylation on chromatin transcription rate. Nucleic Acids Res. 48:12648–12659. 2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Hebbes TR, Clayton AL, Thorne AW and Crane-Robinson C: Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. EMBO J. 13:1823–1830. 1994.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Krajewski WA and Becker PB: Reconstitution of hyperacetylated, DNase I-sensitive chromatin characterized by high conformational flexibility of nucleosomal DNA. Proc Natl Acad Sci USA. 95:1540–1545. 1998.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Godde JS, Kass SU, Hirst MC and Wolffe AP: Nucleosome assembly on methylated CGG triplet repeats in the fragile X mental retardation gene 1 promoter. J Biol Chem. 271:24325–24338. 1996.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ball DJ, Gross DS and Garrard WT: 5-methylcytosine is localized in nucleosomes that contain histone H1. Proc Natl Acad Sci USA. 80:5490–5494. 1983.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Steinbach OC, Wolffe AP and Rupp RA: Somatic linker histones cause loss of mesodermal competence in Xenopus. Nature. 389:395–399. 1997.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Du Q, Luu PL, Stirzaker C and Clark SJ: Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics. 7:1051–1073. 2015.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F and Wolffe AP: Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 23:62–66. 1999.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Djupedal I and Ekwall K: Epigenetics: Heterochromatin meets RNAi. Cell Res. 19:282–295. 2009.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Taverna SD, Coyne RS and Allis CD: Methylation of histone h3 at lysine 9 targets programmed DNA elimination in tetrahymena. Cell. 110:701–711. 2002.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Holoch D and Moazed D: RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 16:71–84. 2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J and Wolffe AP: Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 19:187–191. 1998.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Cheng Y, Liao S, Xu G, Hu J, Guo D, Du F, Contreras A, Cai KQ, Peri S, Wang Y, et al: NeuroD1 dictates tumor cell differentiation in medulloblastoma. Cell Rep. 31(107782)2020.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Doussouki M, Gajjar A and Chamdine O: Molecular genetics of medulloblastoma in children: Diagnostic, therapeutic and prognostic implications. Future Neurol. 14:2019.
|
|
56
|
Guo D, Qu Y, Yang Y and Yang ZJ: Medulloblastoma cells resemble neuronal progenitors in their differentiation. Mol Cell Oncol. 7(1810514)2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Roussel MF and Hatten ME: Cerebellum development and medulloblastoma. Curr Top Dev Biol. 94:235–282. 2011.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Rawal ZD, Upadhyay VA, Patel DD and Trivedi TI: Medulloblastoma under siege: Genetic and molecular dissection concerning recent advances in therapeutic strategies. J Pediatr Neurosci. 15:175–182. 2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Haltom AR, Toll SA, Cheng D, Maegawa S, Gopalakrishnan V and Khatua S: Medulloblastoma epigenetics and the path to clinical innovation. J Neurooncol. 150:35–46. 2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, Rutkowski S, Ellison DW, Malkin D, Taylor MD, et al: Medulloblastoma. Nat Rev Dis Primers. 5(11)2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Gröbner S, Segura-Wang M, Zichner T, Rudneva VA, et al: The whole-genome landscape of medulloblastoma subtypes. Nature. 547:311–317. 2017.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Northcott PA, Dubuc AM, Pfister S and Taylor MD: Molecular subgroups of medulloblastoma. Expert Rev Neurother. 12:871–884. 2012.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Smoll NR and Drummond KJ: The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J Clin Neurosci. 19:1541–1544. 2012.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Tian Z, Yu T, Liu J, Wang T and Higuchi A: Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 14:135–145. 2008.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Ramaswamy V, Nör C and Taylor MD: Taylor, p53 and meduloblastoma. Cold Spring Harb Perspect Med. 6(a026278)2015.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Shiraishi R and Kawauchi D: Epigenetic regulation in medulloblastoma pathogenesis revealed by genetically engineered mouse models. Cancer Sci. 112:2948–2957. 2021.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB and Issa JP: CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 96:8681–8686. 1999.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al: Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 164:550–563. 2016.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al: Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 17:510–522. 2010.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al: IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 483:479–483. 2012.PubMed/NCBI View Article : Google Scholar
|
|
71
|
He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al: Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 333:1303–1307. 2011.PubMed/NCBI View Article : Google Scholar
|
|
72
|
El-Ayadi M, Egervari K, Merkler D, McKee TA, Gumy-Pause F, Stichel D, Capper D, Pietsch T, Ansari M, von Bueren AO, et al: Concurrent IDH1 and SMARCB1 mutations in pediatric medulloblastoma: A case report. Front Neurol. 9(398)2018.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Kurimoto T, Kondo A, Ogino I, Fujimura J, Arakawa A, Arai H and Shimizu T: Effect of O6-methylguanine-DNA methyltransferase methylation in medulloblastoma. Mol Clin Oncol. 7:1107–1111. 2017.PubMed/NCBI View Article : Google Scholar
|
|
74
|
von Bueren AO, Bacolod MD, Hagel C, Heinimann K, Fedier A, Kordes U, Pietsch T, Koster J, Grotzer MA, Friedman HS, et al: Mismatch repair deficiency: A temozolomide resistance factor in medulloblastoma cell lines that is uncommon in primary medulloblastoma tumours. Br J Cancer. 107:1399–1408. 2012.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al: Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 153:1134–1148. 2013.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, et al: Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 510:537–541. 2014.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz HJ, et al: Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 530:57–62. 2016.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Batora NV, Sturm D, Jones DT, Kool M, Pfister SM and Northcott PA: Transitioning from genotypes to epigenotypes: Why the time has come for medulloblastoma epigenomics. Neuroscience. 264:171–185. 2014.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Fraser J, Williamson I, Bickmore WA and Dostie J: An overview of genome organization and how we got there: From FISH to Hi-C. Microbiol Mol Biol Rev. 79:347–372. 2015.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Tropberger P and Schneider R: Going global: Novel histone modifications in the globular domain of H3. Epigenetics. 5:112–117. 2010.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Bryant JP, Heiss J and Banasavadi-Siddegowda YK: Arginine methylation in brain tumors: Tumor biology and therapeutic strategies. Cells. 10(124)2021.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K and Zhang Y: Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 12:1052–1058. 2002.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, et al: The genetic landscape of the childhood cancer medulloblastoma. Science. 331:435–439. 2011.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Chaturvedi NK, Mahapatra S, Kesherwani V, Kling MJ, Shukla M, Ray S, Kanchan R, Perumal N, McGuire TR, Sharp JG, et al: Role of protein arginine methyltransferase 5 in group 3 (MYC-driven) Medulloblastoma. BMC Cancer. 19(1056)2019.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez-Lago M, Kool M, Jones DT, Unterberger A, Morrissy AS, et al: Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 125:373–384. 2013.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, et al: Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 41:465–472. 2009.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Guo D, Wang Y, Cheng Y, Liao S, Hu J, Du F, Xu G, Liu Y, Cai KQ, Cheung M, et al: Tumor cells generate astrocyte-like cells that contribute to SHH-driven medulloblastoma relapse. J Exp Med. 218(e20202350)2021.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Liu Y, Yuelling LW, Wang Y, Du F, Gordon RE, O'Brien JA, Ng JMY, Robins S, Lee EH, Liu H, et al: Astrocytes promote medulloblastoma progression through hedgehog secretion. Cancer Res. 77:6692–6703. 2017.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Bannister AJ and Kouzarides T: Regulation of chromatin by histone modifications. Cell Res. 21:381–395. 2011.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA and Shi Y: Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 119:941–953. 2004.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Chang B, Chen Y, Zhao Y and Bruick RK: JMJD6 is a histone arginine demethylase. Science. 318:444–447. 2007.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Van der Meulen J, Speleman F and Van Vlierberghe P: The H3K27me3 demethylase UTX in normal development and disease. Epigenetics. 9:658–668. 2014.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE and Helin K: UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 449:731–734. 2007.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Yi J, Shi X, Xuan Z and Wu J: Histone demethylase UTX/KDM6A enhances tumor immune cell recruitment, promotes differentiation and suppresses medulloblastoma. Cancer Lett. 499:188–200. 2021.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Marmorstein R and Zhou MM: Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol. 6(a018762)2014.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Sterner DE and Berger SL: Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 64:435–459. 2000.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Pfister S, Rea S, Taipale M, Mendrzyk F, Straub B, Ittrich C, Thuerigen O, Sinn HP, Akhtar A and Lichter P: The histone acetyltransferase hMOF is frequently downregulated in primary breast carcinoma and medulloblastoma and constitutes a biomarker for clinical outcome in medulloblastoma. Int J Cancer. 122:1207–1213. 2008.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Malatesta M, Steinhauer C, Mohammad F, Pandey DP, Squatrito M and Helin K: Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res. 73:6323–6333. 2013.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Biswas S and Rao CM: Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharmacol. 837:8–24. 2018.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Seto E and Yoshida M: Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 6(a018713)2014.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Ma JX, Li H, Chen XM, Yang XH, Wang Q, Wu ML, Kong QY, Li ZX and Liu J: Expression patterns and potential roles of SIRT1 in human medulloblastoma cells in vivo and in vitro. Neuropathology. 33:7–16. 2013.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P, Deubzer HE, Lodrini M, Taylor MD, von Deimling A, et al: HDAC5 and HDAC9 in Medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res. 16:3240–3252. 2010.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Ecker J, Oehme I, Mazitschek R, Korshunov A, Kool M, Hielscher T, Kiss J, Selt F, Konrad C, Lodrini M, et al: Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol Commun. 3(22)2015.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Wang X, Holgado BL, Ramaswamy V, Mack S, Zayne K, Remke M, Wu X, Garzia L, Daniels C, Kenney AM and Taylor MD: miR miR on the wall, who's the most malignant medulloblastoma miR of them all? Neuro Oncol. 20:313–323. 2018.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Harris PS, Venkataraman S, Alimova I, Birks DK, Balakrishnan I, Cristiano B, Donson AM, Dubuc AM, Taylor MD, Foreman NK, et al: Integrated genomic analysis identifies the mitotic checkpoint kinase WEE1 as a novel therapeutic target in medulloblastoma. Mol Cancer. 13(72)2014.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Mahajan K, Fang B, Koomen JM and Mahajan NP: H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nat Struct Mol Biol. 19:930–937. 2012.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Mahajan K and Mahajan NP: WEE1 tyrosine kinase, a novel epigenetic modifier. Trends Genet. 29:394–402. 2013.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Wang J, Qiu Z and Wu Y: Ubiquitin regulation: The histone modifying Enzyme's story. Cells. 7(118)2018.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al: Landscape of transcription in human cells. Nature. 489:101–108. 2012.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Pennisi E: Genomics. ENCODE project writes eulogy for junk DNA. Science. 337:1159–1161. 2012.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Butts T, Green MJ and Wingate RJ: Development of the cerebellum: Simple steps to make a ‘little brain’. Development. 141:4031–4041. 2014.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Jönsson ME, Nelander Wahlestedt J, Åkerblom M, Kirkeby A, Malmevik J, Brattaas PL, Jakobsson J and Parmar M: Comprehensive analysis of microRNA expression in regionalized human neural progenitor cells reveals microRNA-10 as a caudalizing factor. Development. 142:3166–3177. 2015.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Laneve P and Caffarelli E: The Non-coding side of medulloblastoma. Front Cell Dev Biol. 8(275)2020.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Roussel MF and Stripay JL: Epigenetic drivers in pediatric medulloblastoma. Cerebellum. 17:28–36. 2018.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Kuang Y, Liu Q, Shu X, Zhang C, Huang N, Li J, Jiang M and Li H: Dicer1 and MiR-9 are required for proper Notch1 signaling and the Bergmann glial phenotype in the developing mouse cerebellum. Glia. 60:1734–1746. 2012.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Pieczora L, Stracke L, Vorgerd M, Hahn S, Theiss C and Theis V: Unveiling of miRNA expression patterns in purkinje cells during development. Cerebellum. 16:376–387. 2017.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Han D, Dong X, Zheng D and Nao J: MiR-124 and the underlying therapeutic promise of neurodegenerative disorders. Front Pharmacol. 10(1555)2019.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Ferretti E, De Smaele E, Po A, Di Marcotullio L, Tosi E, Espinola MS, Di Rocco C, Riccardi R, Giangaspero F, Farcomeni A, et al: MicroRNA profiling in human medulloblastoma. Int J Cancer. 124:568–577. 2009.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Laneve P, Di Marcotullio L, Gioia U, Fiori ME, Ferretti E, Gulino A, Bozzoni I and Caffarelli E: The interplay between microRNAs and the neurotrophin receptor tropomyosin-related kinase C controls proliferation of human neuroblastoma cells. Proc Natl Acad Sci USA. 104:7957–7962. 2007.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Miele E, Po A, Begalli F, Antonucci L, Mastronuzzi A, Marras CE, Carai A, Cucchi D, Abballe L, Besharat ZM, et al: β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer. 17(488)2017.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Cheng LC, Pastrana E, Tavazoie M and Doetsch F: miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci. 12:399–408. 2009.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Nowek K, Wiemer EAC and Jongen-Lavrencic M: The versatile nature of miR-9/9* in human cancer. Oncotarget. 9:20838–20854. 2018.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW and Crabtree GR: MicroRNA-mediated conversion of human fibroblasts to neurons. Nature. 476:228–231. 2011.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Fiaschetti G, Abela L, Nonoguchi N, Dubuc AM, Remke M, Boro A, Grunder E, Siler U, Ohgaki H, Taylor MD, et al: Epigenetic silencing of miRNA-9 is associated with HES1 oncogenic activity and poor prognosis of medulloblastoma. Br J Cancer. 110:636–647. 2014.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Ingram WJ, McCue KI, Tran TH, Hallahan AR and Wainwright BJ: Sonic Hedgehog regulates Hes1 through a novel mechanism that is independent of canonical Notch pathway signalling. Oncogene. 27:1489–1500. 2008.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Fénelon K, Mukai J, Xu B, Hsu PK, Drew LJ, Karayiorgou M, Fischbach GD, Macdermott AB and Gogos JA: Deficiency of Dgcr8, a gene disrupted by the 22q11.2 microdeletion, results in altered short-term plasticity in the prefrontal cortex. Proc Natl Acad Sci USA. 108:4447–4452. 2011.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Pierson J, Hostager B, Fan R and Vibhakar R: Regulation of cyclin dependent kinase 6 by microRNA 124 in medulloblastoma. J Neurooncol. 90:1–7. 2008.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Silber J, Hashizume R, Felix T, Hariono S, Yu M, Berger MS, Huse JT, VandenBerg SR, James CD, Hodgson JG and Gupta N: Expression of miR-124 inhibits growth of medulloblastoma cells. Neuro Oncol. 15:83–90. 2013.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Venkataraman S, Birks DK, Balakrishnan I, Alimova I, Harris PS, Patel PR, Handler MH, Dubuc A, Taylor MD, Foreman NK and Vibhakar R: MicroRNA 218 acts as a tumor suppressor by targeting multiple cancer phenotype-associated genes in medulloblastoma. J Biol Chem. 288:1918–1928. 2013.PubMed/NCBI View Article : Google Scholar
|
|
130
|
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ and Hammond SM: A microRNA polycistron as a potential human oncogene. Nature. 435:828–833. 2005.PubMed/NCBI View Article : Google Scholar
|
|
131
|
O'Donnell KA, Wentzel EA, Zeller KI, Dang CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 435:839–843. 2005.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L, Buettner R, et al: MYCN regulates oncogenic MicroRNAs in neuroblastoma. Int J Cancer. 122:699–704. 2008.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Uziel T, Karginov FV, Xie S, Parker JS, Wang YD, Gajjar A, He L, Ellison D, Gilbertson RJ, Hannon G and Roussel MF: The miR-17~92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastoma. Proc Natl Acad Sci USA. 106:2812–2817. 2009.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Mollashahi B, Aghamaleki FS and Movafagh A: The roles of miRNAs in medulloblastoma: A systematic review. J Cancer Prev. 24:79–90. 2019.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Senfter D, Samadaei M, Mader RM, Gojo J, Peyrl A, Krupitza G, Kool M, Sill M, Haberler C, Ricken G, et al: High impact of miRNA-4521 on FOXM1 expression in medulloblastoma. Cell Death Dis. 10(696)2019.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Agrawal K, Das V, Vyas P and Hajdúch M: Nucleosidic DNA demethylating epigenetic drugs-A comprehensive review from discovery to clinic. Pharmacol Ther. 188:45–79. 2018.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Diesch J, Zwick A, Garz AK, Palau A, Buschbeck M and Götze KS: A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin Epigenetics. 8(71)2016.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Christman JK: 5-Azacytidine and 5-aza-2'-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene. 21:5483–5495. 2002.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Zwergel C, Romanelli A, Stazi G, Besharat ZM, Catanzaro G, Tafani M, Valente S and Mai A: Application of small epigenetic modulators in pediatric medulloblastoma. Front Pediatr. 6(370)2018.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Inoue A and Fujimoto D: Enzymatic deacetylation of histone. Biochem Biophys Res Commun. 36:146–150. 1969.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Taunton J, Hassig CA and Schreiber SL: A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 272:408–411. 1996.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Kijima M, Yoshida M, Sugita K, Horinouchi S and Beppu T: Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem. 268:22429–22435. 1993.PubMed/NCBI
|
|
143
|
Bolden JE, Peart MJ and Johnstone RW: Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 5:769–784. 2006.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Tiberi L, Bonnefont J, van den Ameele J, Le Bon SD, Herpoel A, Bilheu A, Baron BW and Vanderhaeghen P: A BCL6/BCOR/SIRT1 complex triggers neurogenesis and suppresses medulloblastoma by repressing Sonic Hedgehog signaling. Cancer Cell. 26:797–812. 2014.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Tang Y, Gholamin S, Schubert S, Willardson MI, Lee A, Bandopadhayay P, Bergthold G, Masoud S, Nguyen B, Vue N, et al: Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 20:732–740. 2014.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Dobson THW, Hatcher RJ, Swaminathan J, Das CM, Shaik S, Tao RH, Milite C, Castellano S, Taylor PH, Sbardella G and Gopalakrishnan V: Regulation of USP37 expression by REST-Associated G9a-Dependent histone methylation. Mol Cancer Res. 5:1073–1084. 2017.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Inui K, Zhao Z, Yuan J, Jayaprakash S, Le LTM, Drakulic S, Sander B and Golas MM: Stepwise assembly of functional C-terminal REST/NRSF transcriptional repressor complexes as a drug target. Protein Sci. 26:997–1011. 2017.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Maier H, Dalianis T and Kostopoulou ON: New approaches in targeted therapy for medulloblastoma in children. Anticancer Res. 41:1715–1726. 2021.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Gordon RE, Zhang L and Yang ZJ: Restore the brake on tumor progression. Biochem Pharmacol. 138:1–6. 2017.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Wright JH: Neurocytoma or neuroblastoma, a kind of tumor not generally recognized. J Exp Med. 12:556–561. 1910.PubMed/NCBI View Article : Google Scholar
|
|
151
|
MacKenzie DJ: A classification of the tumours of the glioma group on a histogenetic basis with a correlated study of prognosis. Can Med Assoc J. 16(872)1926.
|
|
152
|
Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, et al: The molecular basis of Turcot's syndrome. N Engl J Med. 332:839–847. 1995.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, et al: Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 29:1408–1414. 2011.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J and McMahon AP: Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 322:1247–1250. 2008.PubMed/NCBI View Article : Google Scholar
|














