Introduction
Diabetic nephropathy is one of the most prevalent
causes of end-stage renal disease. Increasing evidence has
indicated that diabetic nephropathy in experimental animals, as
well as in diabetic patients, induces fibrotic changes, such as
extracellular matrix (ECM) accumulation in glomerular mesangial
cells, which manifest as overt nephropathy (1). In vitro studies have also
revealed that high glucose levels induce the overt proliferation of
mesangial cells and ECM accumulation through a complex pathological
mechanism (2,3). Transforming growth factor-β (TGF-β)
has been shown to play a central role in the progression of
glomerular fibrosis and to transduce its signal through the type I
activin receptor-like kinase 5 (ALK5) (4). Additionally, TGF-β specifically
induces the expression of the large modular glycoprotein,
fibronectin (FN), and the antifibrinolytic agent, plasminogen
activator inhibitor type 1 (PAI-1), resulting in cellular
proliferation and ECM accumulation (5). A number of studies have shown that
the selective inhibition of ALK5 may be a potential therapeutic
strategy for the treatment of chronic renal disease secondary to
diabetic nephropathy (6,7).
Salt-inducible kinase 1 (SIK1), a serine/threonine
protein kinase, was originally found in the adrenal glands of rats
fed a high salt diet (8). SIK1 is
belongs to the family of 5′ adenosine monophosphate (AMP)-activated
protein kinases (AMPKs), since its amino acid sequence is closely
related to the catalytic α subunit of AMPK (8,9).
Additionally, the AMPK-related kinases share some functional
properties, several substrates and the upstream kinase,
serine/threonine kinase 11 (STK11, also termed LKB1). SIK1 is
predominantly localized in the nucleus in resting Y1 cells, where
it supresses cAMP-response element binding protein (CREB)-mediated
gene expression by phosphorylating the co-activator, CREB-regulated
transcription co-activator (CRTC). Following treatment with
adrenocorticotropic hormone (ACTH) and the subsequent
phosphorylation of the regulatory domain at Ser-577 by protein
kinase A (PKA), SIK1 translocates to the cytoplasm and loses its
repressive properties (10).
In contrast to the negative regulation by PKA, LKB1
phosphorylates SIK1 at Thr-182 in the activation loop (A-loop) of
the kinase domain, which is essential for catalytic activity. The
autophosphorylation at Ser-186, which is 4 amino acids downstream
from Thr-182, is also important for the sustained activity of SIK1
(11,12). Previous studies on CREB and LKB1
have revealed a number of physiological roles for SIK1, such as
cell cycle regulation (13),
muscle growth and differentiation (14) and tumor suppression (15). In diabetic states, SIK1 is subject
to hormonal control and constrains gluconeogenic and lipogenic gene
expression in the liver (16,17).
Cell biology has also identified SIK1 as a target of
the TGF-β/Smad signaling pathway. SIK1 forms complexes with ALK5,
Smad7, and Smurf2, which results in the downregulation of the
TGF-β-mediated activation of the ALK5 signaling pathway. Following
SIK1 knockdown by siRNA, endogenous ALK5 levels are significantly
higher, and the decreased repression of TGF-β signaling augments
the induction of the expression of FN and PAI-1 (18,19), both of which are involved in
fibrotic disorders, such as glomerulosclerosis. These findings
suggest that SIK1 may serve as a therapeutic target against
fibrosis through the specific downregulation of ALK5.
Although animal experiments have found the highest
SIK mRNA levels in the rat kidney (20), to our knowledge, there are no
studies available concerning the exact role of SIK1, which is
involved in the diabetic kidney. There is also limited knowledge
concerning the specific role of SIK1 in glucose metabolism,
cellular proliferation and ECM accumulation. In the current study,
we report that the protein level of SIK1 is downregulated by high
glucose in a rat glomerular mesangial cell line (HBZY-1 cells). By
contrast, the overexpression of SIK1 in HBZY-1 cells results in the
reduction of cell proliferation and ECM accumulation accompanied by
the inhibition of the ALK5 signaling pathway.
Materials and methods
Reagents and chemicals
The PCR primers and Lipofectamine 2000 were
purchased from Invitrogen (Carlsbad, CA, USA). The restriction
enzymes, BamHI, EcoRI and T4 DNA ligase, were
obtained from Takara Co. (Kyoto, Japan). Protein G Sepharose was
purchased from GE Healthcare (Piscataway, NJ, USA). Rabbit anti-rat
SIK1/PAI-1/FN polyclonal antibody was purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Rabbit polyclonal
antibody against phospho-SIK1 (phospho-Thr-182) and anti-SIK1
antiserum were a gift from Dr H. Takemori, Laboratory of Cell
Signaling and Metabolism, National Institute of Biomedical
Innovation, Osaka, Japan. Rabbit anti-rat ALK5 polyclonal antibody
was purchased from Bioworld Technology, Inc. (St. Louis Park, MN,
USA).
Expression plasmid construction
The expression plasmid, pCDF1-MCS2-EF1-copGFP, was
purchased from System Biosciences (Mountain View, CA, USA). The
plasmid vector of pEGFP and the E. coli strain, DH5α, were
preserved in our laboratory. The full-length cDNA coding for rat
SIK1 was amplified using the following primers:
5′-GCGGATCCGGTGCGGCCCGAAGCCATGGTGATCATGTCGGAGTTCAG-3′ and
5′-CCGGATCCATGGTGATCATGTCGGAGTTCAG-3′ and was cloned into the
BamHI/EcoRI site of pCDF1-MCS2; the full-length rat
SIK1 cDNA was subsequently subcloned into pEGFP-C1.
Cell culture and gene transfer
HBZY-1 cells, a rat glomerular mesangial cell line,
were purchased from the China Center for Type Culture Collection
(Wuhan, China), maintained at 37°C in a humidified atmosphere of 5%
CO2 and cultured in Dulbecco’s modified Eagle’s medium
(DMEM; Gibco-BRL, Gaithersburg, MD, USA) supplemented with 10%
fetal bovine serum, 100 U/ml penicillin and 100 μg/ml
streptomycin. The cells were grown to ~80% confluence and then
incubated in serum-free medium for 24 h. To investigate the effects
of glucose on the protein expression of SIK1 and pT182-SIK1,
serum-deprived HBZY-1 cells were cultured in DMEM containing 30 mM
glucose (high glucose) for 0, 12, 24 and 48 h. At the end of each
time period, SIK1, pT182-SIK1, ALK5, FN and PAI-1 expression levels
were examined. To determine the direct effects of high glucose on
the specific cellular localization of SIK1, the HBZY-1 cells were
transfected with the expression vectors for green fluorescence
protein (GFP)-fused full-length SIK1 (pEGFP-SIK1), cultured with
high glucose in glass-bottomed culture dishes (GBD-35-20; Nest
Biotechnology Co., Ltd., Shanghai, China) for 0 and 48 h, and then
observed under an Olympus confocal microscope. To examine how SIK1
negatively regulates the ALK5 signaling pathway, HBZY-1 cells were
randomly divided into three groups: a non-transfected normal group,
an empty vector control group and a pCDF1-SIK1 group. At 48 h after
transfection, the HBZY-1 cells were collected and analyzed for SIK1
expression. The cells were cultured with high glucose for a further
48 h to analyze SIK1, ALK5, FN and PAI-1 expression by western blot
and immunocytochemistry analyses. The transient transfection of
HBZY-1 cells was carried out using Lipofectamine 2000 according to
the manufacturer’s instructions. At 48 h after transfection, a
fluorescent microscope was used to examine GFP expression, and the
cells were then collected for protein extraction.
Immunocytochemistry and
immunocytofluorescent analysis
Cells were cultured on glass coverslips in 12-well
plates for 0, 12, 24 and 48 h, gently washed twice with
phosphate-buffered saline (PBS) and fixed with 4% polyformaldehyde
for 20 min at room temperature. The cells were then washed three
times with PBS, and the coverslips with the cells were made
permeable with 0.3% Triton X-100 in PBS for 15 min. The cells were
then blocked by pre-treatment with 3% H2O2
for 10 min at 37°C. Finally, the HBZY-1 cells were incubated with
rabbit anti-PAI-1/FN polyclonal antibody (1:100) overnight at 4°C.
The following day, the cells were washed with PBS three times and
incubated with polymer helper for 30 min at 37°C. Thereafter, the
HBZY-1 cells were washed three times with PBS and then treated for
30 min at 37°C with polyperoxidase-anti-rabbit IgG. After being
washed with PBS, the coverslips were stained with DAB. Negative
controls were obtained by replacing specific antibody with
non-immune serum. Images were taken using a light Olympus
microscope. For indirect immunocytofluorescence, HBZY-1 cells on
glass coverslips were fixed with 4% polyformaldehyde in 0.1% Triton
X-100 and blocked in PBS containing 5% goat serum. An antibody
against SIK1 was used at a dilution of 1:100. AlexaFluor
488-conjugated secondary antibodies were also used at a dilution of
1:200. Counterstaining of the nuclei was performed by the addition
of 1 mM 4’,6-diamidino-2-phenylindole (DAPI) (Boster Biological
Technology Ltd., Wuhan, Hubei, China) for 15 min. Images were taken
using an Olympus confocal microscope.
Reverse transcription-polymerase chain
reaction (RT-PCR)
The cells were washed twice with PBS. RT-PCR was
performed as previously described (21). The primers used were: SIK1 sense,
5′-AGCACCACTCAGCCGTCTCATCT-3′ and antisense,
5′-CAGGTCCTCCATCTCACAATCCC-3′; and β-actin sense,
5′-CGTTGACATCCGTAAAGACCTC-3′ and antisense,
5′-TAGGAGCCAGGGCAGTAATCT-3′. After electrophoresis the gels were
recorded using a digital recorder, and the mRNA expression levels
were semi-quantified using ImageJ software (National Institutes of
Health, USA).
Western blot analysis and
immunoprecipitation
Western blot analysis was carried out as previously
described (22). For
immunoprecipitation, the proteins (500 μg/sample) extracted
from the cells were incubated with anti-SIK1 antiserum for 16 h at
4°C with rotation. Protein G agarose beads (20 μl/sample)
were then added to each sample and incubated for an additional 2 h
at 4°C with rotation. The complexes were spun down, and the pellet
was washed three times with immunoprecipitation buffer. The
immunoprecipitates were immobilized on a polyvinylidene difluoride
membrane after separation on 12% SDS-PAGE gels. The membrane was
immunoblotted with anti-pT182-SIK1 antibody by enhanced
chemiluminescence (ECL) (Pierce Chemical, Co., Rockford, IL, USA)
method. The same membrane was subsequently reblotted with anti-SIK1
antibody after stripping to verify that equivalent amounts of SIK1
protein were loaded onto each lane, and the protein expression
levels were semi-quantified using ImageJ software.
Cell proliferation measurements
Cell proliferation was measured using a
dimethylthiazol-diphenyltetrazolium bromide (MTT) assay in 96-well
microplates. HBZY-1 cells were exposed to high glucose for
different periods of time with or without gene transfection. Medium
(180 μl) was added to each well. After 48 h, 20 μl of
5 mg/ml MTT were added to each well. Four hours later, the medium
was replaced with 200 μl of dimethyl sulfoxide, and the
96-well microplate was shaken gently. The absorbance was then
measured at 570 nm, and these data were transformed into a variable
representing the number of cells in each well using a standard
curve that correlated the absorbance to the number of HBZY-1
cells.
Statistical analysis
Data analysis was conducted using GraphPad Prism 5.0
software, and the data are expressed as the means ± SEM.
Comparisons among multiple groups were performed using one-way
ANOVA followed by a Newman-Keuls test. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
High glucose levels suppress the activity
and decrease the expression of SIK1 and regulate the specific
localization and distribution of SIK1 in HBZY-1 cells
HBZY-1 cells were stimulated with high glucose, and
the activity resulting from the Thr-182 phosphorylation of the SIK1
gene was monitored. The HBZY-1 cells were cultured in medium
containing 30 mM glucose for 0, 12, 24, or 48 h.
Immunoprecipitation followed by western blot analysis revealed that
SIK1 activity decreased following exposure with glucose in a
time-dependent manner (Fig. 1A).
The levels of Thr-182 phosphorylation at 12, 24 and 48 h were 24.8,
58 and 58.4%, respectively, compared to 0 h. The level of
phospho-Thr-182 is important to maintain the SIK1 protein level
(12), which was also evident in
the HBZY-1 cells treated with high glucose (Fig. 1B). The density ratios of
SIK1/β-actin at 12, 24 and 48 h were 18.2, 61.2 and 72%,
respectively, compared to 0 h. Further semi-quantitative RT-PCR
analysis also demonstrated equivalent results at the
transcriptional level under high glucose conditions at each time
point (Fig. 1C). These results
demonstrate that high glucose concentrations suppress the activity
and decrease the expression of SIK1.
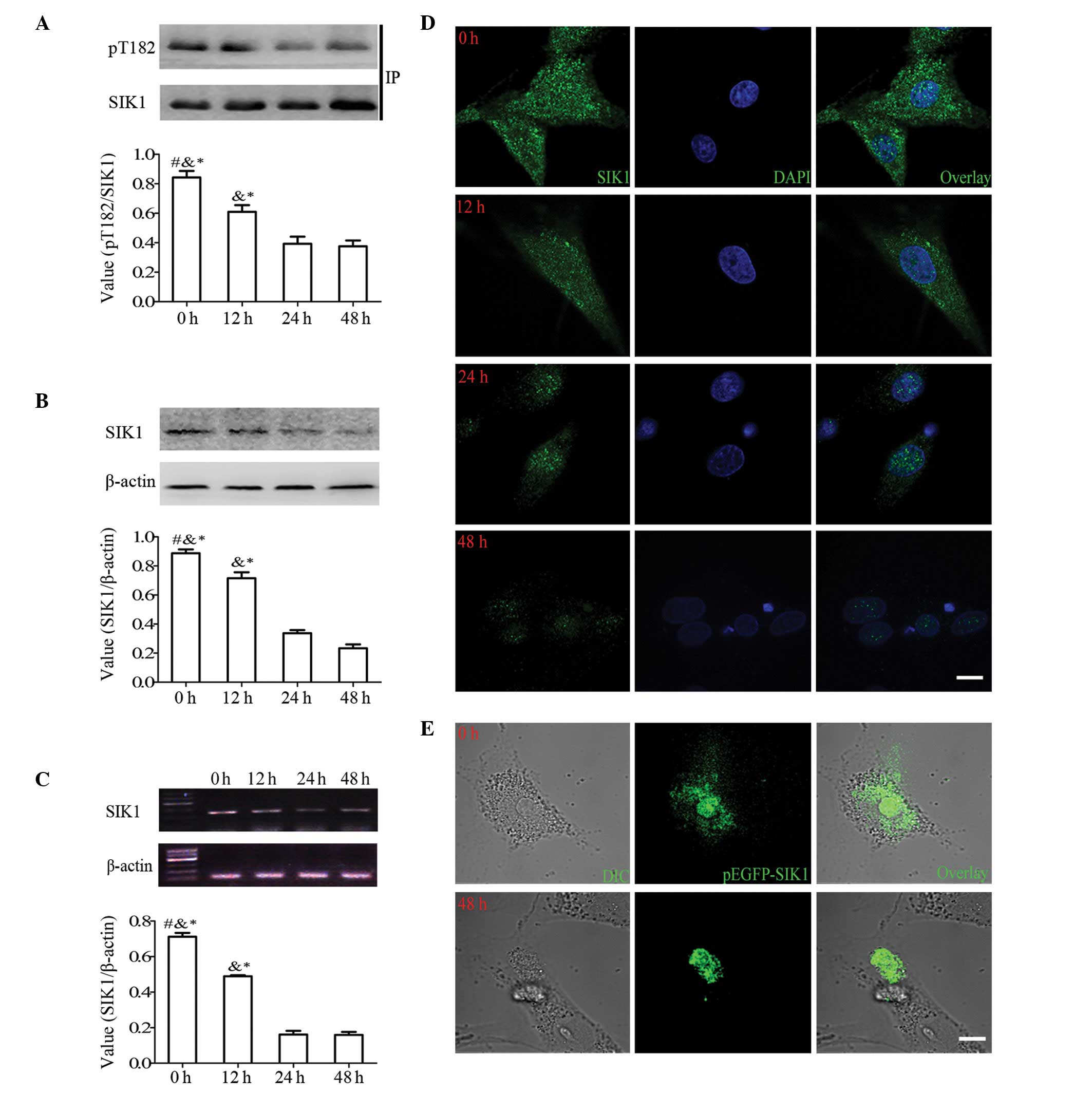 | Figure 1.High glucose levels significantly
suppress the expression and activity of salt-inducible kinase 1
(SIK1) and regulate the nuclear redistribution of SIK1 in HBZY-1
cells. (A) Immunoprecipitation and western blot analysis showing
the levels of pT182-SIK1 and the SIK1 control in HBZY-1 cells
cultured in high glucose (30 mM) at 0, 12, 24, and 48 h. (B)
Western blot analysis showing the levels of SIK1 protein and the
β-actin control at different culture times. (C) RT-PCR showing the
levels of SIK1 mRNA and the β-actin control in HBZY-1 cells at
different culture times. (D) Representative confocal images of
HBZY-1 cells that were cultured on coverslips in high glucose for
0, 12, 24, 48 h for immunocytofluorescent analysis. Green
fluorescent signals of endogenous SIK1 (left panel), blue
fluorescent signals representing nuclear staining with DAPI (middle
panel), and merged images of both (right panel). Scale bar, 10
μm. (E) Representative confocal images of HBZY-1 cells
cultured in glass-bottomed culture dishes and transfected with
overexpression vectors for green fluorescence protein (GFP)-fused
full-length SIK1 (pEGFP-SIK1) protein stimulated with high glucose
for 0 and 48 h. Confocal DIC images of HBZY-1 cells (left panel),
green fluorescent signals of GFP-SIK1 (middle panel), and overlaid
images of both (right panel). Scale bar, 10 μm.
#P<0.05 vs. at time of 12 h;
&P<0.05 vs. at time of 24 h;
*P<0.05 vs. at time of 48 h. |
Since the intracellular distribution of SIK1
reflects its functional activity (19), we decided to examine the
localization of SIK1 protein in the HBZY-1 cells. As shown in
Fig. 1D, immuno-positive signals
for SIK1 were detected in the cytoplasm and nucleus of the
unstimulated HBZY-1 cells. Followign stimulation with high glucose,
the SIK1 signals were gradually detected in the nucleus with
decreased intensity.
To confirm our results from immunocytochemical
analysis, green fluorescence protein (GFP)-tagged SIK1 protein was
transfected into the HBZY-1 cells and the subcellular translocation
of GFP signals was monitored before and after exposure to high
glucose. In the unstimulated cells, the GFP-SIK1 signal was
observed in both the nuclear and cytoplasmic compartments (Fig. 1E). When the cells were treated
with high glucose, the green fluorescence signal was subsequently
translocated into the nucleus from the cytoplasm. When the GFP-tag
alone was introduced into the HBZY-1 cells, the fluorescence signal
was not localized at any precise subcellular area, and its
distribution did not change after exposure to high glucose (data
not shown). These results indicate that high glucose levels induce
the nuclear import of SIK1, which may result from the reduction in
SIK1 kinase activity.
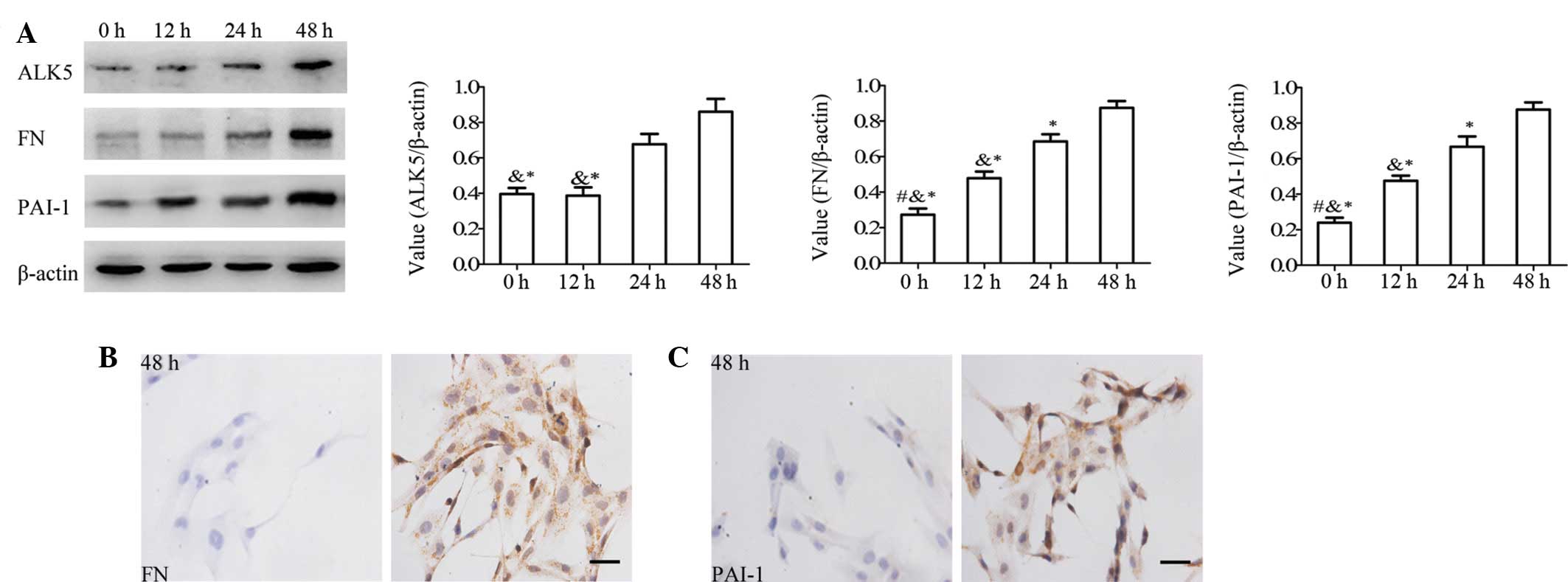
High glucose induces the activation of
the ALK5 signaling pathway in HBZY-1 cells
The ALK5 signaling pathway has been shown to be
induced by high glucose levels and promotes cell proliferation with
ECM accumulation (23,24). Western blot analysis using HBZY-1
cells also showed that the level of ALK5 was upregulated by high
glucose levels (Fig. 2A) with
1.82- and 2.92-fold inductions at 24 and 48 h, respectively
(normalized with β-actin), whereas no significant difference in the
ALK5 level was observed between 0 and 12 h. In contrast to ALK5,
the protein levels of FN and PAI-1, downstream targets of ALK5,
were also increased by high glucose levels in a time-dependent
manner. Immunocytochemical analysis of FN (Fig. 2B) and PAI-1 (Fig. 2C) protein in the HBZY-1 cells also
provided consistent results at 48 h, suggesting that high glucose
levels induce the activation of the ALK5 signaling pathway, which
may be attributed to the downregulation of SIK1.
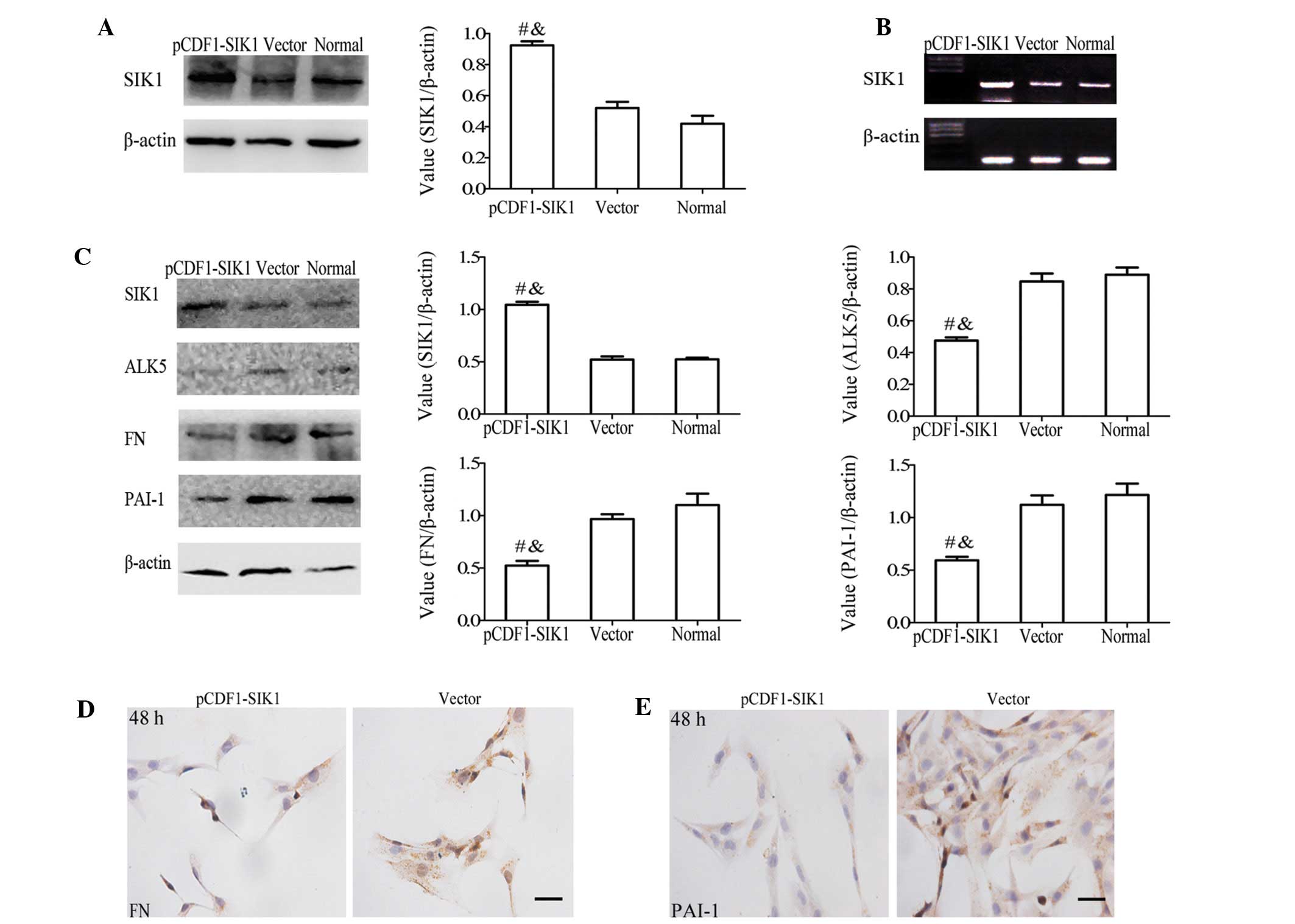
Forced expression of SIK1 induces the
degradation of ALK5 and reduces ECM production in HBZY-1 cells
To determine the involvement of the reduced levels
of SIK1 in the upregulation of ALK5 signaling in glomerular
mesangial cells, we performed paradoxical assays through the
enforced expression of SIK1 in HBZY-1 cells. The transformation
efficiency, monitored by the control copGFP-expression, was ~60%
(data not shown). When the SIK1 expression plasmid was transformed
into the HBZY-1 cells, the SIK1 protein (Fig. 3A) and mRNA (Fig. 3B) levels increased by ~1.8- and
1.9-fold, respectively, compared to the non- or mock-transfected
cells. Following the stimulation of the cells with high glucose for
48 h, a negative correlation between the levels of SIK1 protein and
ALK5, FN and PAI-1 protein leels was observed (Fig. 3C). When the SIK1 expression
plasmid was transformed into the HBZY-1 cells, the ALK5 protein
expression decreased by ~38% in the pCDF1-SIK1 transfected cells
compared to the control cells. Immunocytochemical analysis of FN
(Fig. 3D) and PAI-I (Fig. 3E) also supported the negative
correlation between SIK1 and ALK5 signaling.
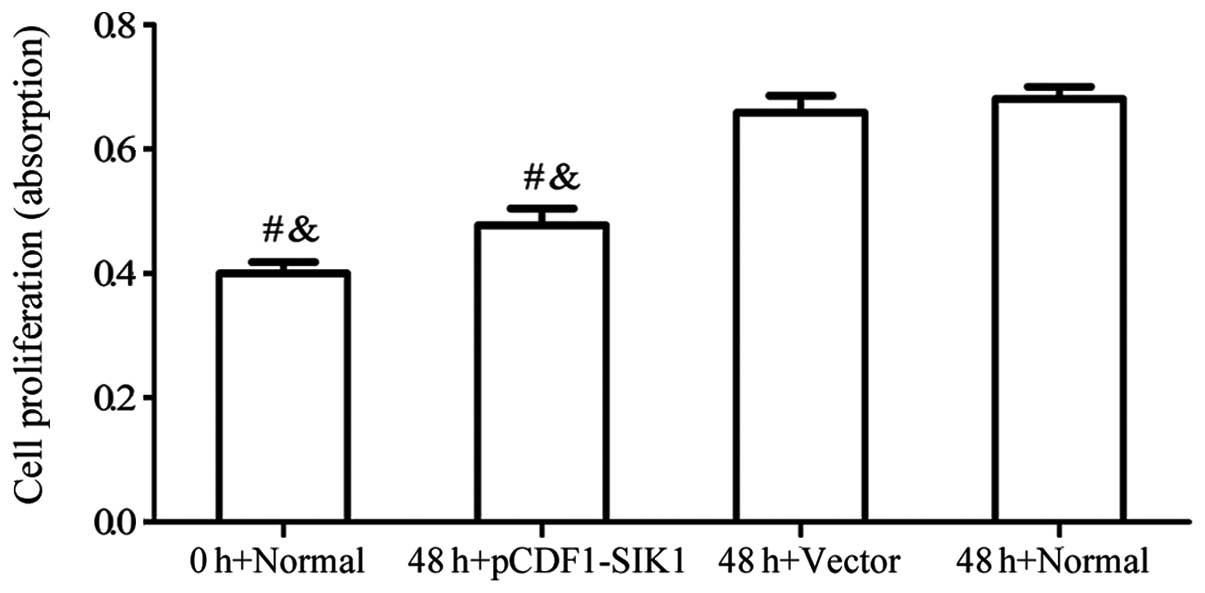
Upregulation of SIK1 reverses the high
glucose-induced over-proliferation of HBZY-1 cells
To determine whether SIK1 exerts antiproliferative
effects on HBZY-1 cells, we analyzed HBZY-1 cell proliferation
using MTT assays. The HBZY-1 cells were transfected with or without
pCDF1-SIK1 and subjected to high glucose levels for 48 h. As shown
in Fig. 4, a significant
reduction in the proliferation of HBZY-1 cells due to the
overexpression of SIK1 was observed.
Discussion
The progression of diabetic nephropathy to end-stage
kidney disease is manifested by the gradual, inexorable process of
renal glomerular fibrosis. Several studies have attempted to
develop effective therapies and preventive strategies for diabetic
nephropathy by elucidating the molecular mechanisms that lead to
chronic glomerular fibrosis. SIK1 may be a therapeutic target for
diabetic nephropathy as it is a negative regulator of the ALK5
signaling pathway and is abundantly expressed in the rat
kidney.
In the present study, we demonstrate that high
glucose levels suppress SIK1 activity in HBZY-1 cells. This result
is consistent with evidence showing that AMPK-related kinases, such
as AMPK, SIK2 (an isoform of SIK1) and SNF1/AMPK-related kinase
(SNARK), are activated by glucose deprivation (25). A proposed mechanism for high
glucose-induced SIK1 suppression could be traceable to its upstream
kinase, LKB1. Suppressed LKB1 signaling by high glucose may
contribute to the decreased activity of SIK1 (11,26). Similar to the kinase activity
levels of SIK1, the intracellular distribution of SIK1 has also
been shown to be a marker of the LKB1-mediated regulation of SIK1
activity.
Since Ser-577, a determinant of the intracellular
distribution of SIK1, is also an autophosphorylation site, inactive
SIK1 as well as CREB-repressing active SIK1 are present as
dephospho-forms at Ser-577 and are localized in the nucleus
(27). Therefore, SIK1 is
localized only in the nucleus in LKB1-defective HeLa cells, and
LKB1 overexpression isolates a portion of SIK1 into the cytoplasm,
whereas the combination of LKB1 and PKA completely induces the
cytoplasmic localization of SIK1 (28).
In this study, indirect immunocytofluorescence and
GFP-tagged SIK1 revealed the nuclear redistribution of SIK1
following stimulation with high glucose, suggesting that high
glucose levels decreased the activity of SIK1, most likely
inactivating the upstream kinase, LKB1. The correlation of the
decreased activity of SIK1 with its protein level in HBZY-1 cells
under high glucose conditions is also consistent with findings in
HeLa cells in which the level of phospho-Thr-182/Ser-186 correlated
with the SIK1 protein level (11). By contrast, the elevated levels of
SIK1 mRNA and protein in Y1 cells have been shown to be accompanied
by an increase in SIK1 kinase activity (27). This suggests that the reduction in
SIK1 protein levels in HBZY-1 cells following exposure to high
glucose may result from inactive SIK1 and decreased SIK1 mRNA
levels.
Glucose induces Ca2+ signaling, which
activates Ca2+ calmodulin-dependent kinases (CaMKs).
CaMK I/IV phosphorylates SIK2 at Thr-484 in the motif of RRHT
(29), which is conserved in
SIK1. Subsequently, phospho-SIK2 is degraded by the proteasome,
which restores the signaling cascades that have been suppressed by
SIK2, such as CREB-TORC1. The fact that glucose toxicity induces
the activation of proteasomes (30) and CaMK I/IV (31), along with the findings that high
glucose levels induce TGF-β/ALK5 signaling followed by cell
proliferation and ECM accumulation in mesangial cells (4,32),
suggests that glucose-mediated signaling pathways may suppress SIK1
function. This suppression may result in the activation of the ALK5
signaling pathway. However, there is a small discrepancy between
the induction of ALK5 and the induction of its downstream targets.
High glucose levels gradually upregulate ALK5 expression but
rapidly induce FN and PAI-I expression, suggesting that an
alternate pathway that is completely independent of ALK5 may induce
ALK5 downstream. The delayed induction of ALK5 is consistent with
previous studies showing that high glucose levels do not induce
rapid changes in ALK5 mRNA or protein levels (23,32), but that ALK5 is markedly induced
after prolonged stimulation under high glucose conditions (23).
TGF-β plays a central role in the progression of
diabetic nephropathy. However, TGF-β is a pleiotropic factor
involved in a variety of biological processes, including modulation
of cell growth, angiogenesis, ECM deposition and various immune
responses (33). Many of these
actions may be important to balance the response to chronic kidney
injury. However, the appropriate levels and long-term effect of
TGF-β inhibition has always been controversial (34). From the many studies on the TGF-β
signaling pathway, it is becoming clear that ALK5 mediates many of
the actions of TGF-β. Therefore, the direct inhibition of ALK5
represents an alternative and attractive mechanism for the
prevention of the detrimental profibrotic effects of TGF-β. In this
study, in vitro experimental data clearly demonstrated that
high glucose levels activated the ALK5 signaling pathway and
downregulated SIK1. In other words, upregulating SIK1 may be a
beneficial method to reduce high glucose-induced proliferation and
fibrosis by enhancing ALK5 degradation. To support this hypothesis,
a specific plasmid for SIK1 was transfected into HBZY-1 cells using
Lipofectamine 2000. As a result, there was a marked downregulation
in ALK5 protein expression, resulting in the inhibition of FN and
PAI-1 expression and cellular proliferation in addition to the
antagonism of fibrosis processing mechanisms. Taken together, these
findings indicate that SIK1 protects mesangial cells against high
glucose-induced cellular proliferation and ECM accumulation through
the inhibition of the activation of the ALK5 signaling pathway.
In conclusion, our results indicate that high
glucose levels decrease SIK1 expression and activity in glomerular
mesangial cells. The decrease in SIK1 expression reduces the
pathogenesis of mesangial cell proliferation and ECM accumulation
through the activation of the ALK5 signaling pathway. Given the
importance of glucose in the pathogenesis of mesangial cell
proliferation, the SIK1-mediated regulation of ALK5 may provide
innovative strategies for the treatment of diseases involving
mesangial cells, such as glomerular fibrosis.
Acknowledgments
This study was supported by the Ministry of
Education Specialized Research Fund for the Doctoral Program of
Education (no. 20110142110016) and the Innovation Fund of Huazhong
University of Science and Technology Ph.D. Dissertation
(2011–2012).
References
|
1.
|
Border WA and Noble NA: Evidence that
TGF-β should be a therapeutic target in diabetic nephropathy.
Kidney Int. 54:1390–1391. 1998.
|
|
2.
|
Ayo SH, Radnik RA, Glass W, et al:
Increased extracellular matrix synthesis and mRNA in mesangial
cells grown in high-glucose medium. Am J Physiol. 260:F185–F191.
1991.PubMed/NCBI
|
|
3.
|
Paradis V, Perlemuter G, Bonvoust F, et
al: High glucose and hyperinsulinemia stimulate connective tissue
growth factor expression: a potential mechanism involved in
progression to fibrosis in nonalcoholic steatohepatitis.
Hepatology. 34:738–744. 2001. View Article : Google Scholar
|
|
4.
|
Schnaper HW, Hayashida T, Hubchak SC and
Poncelet AC: TGF-β signal transduction and mesangial cell
fibrogenesis. Am J Physiol Renal Physiol. 284:F243–F252. 2003.
|
|
5.
|
Lebrin F, Deckers M, Bertolino P and Ten
Dijke P: TGF-β receptor function in the endothelium. Cardiovasc
Res. 65:599–608. 2005.
|
|
6.
|
Moon J, Kim H, Cho I, Sheen Y and Kim D:
IN-1130, a novel transforming growth factor-β type I receptor
kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive
nephropathy. Kidney Int. 70:1234–1243. 2006.
|
|
7.
|
Laping NJ: ALK5 inhibition in renal
disease. Curr Opin Pharmacol. 3:204–208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Wang Z, Takemori H, Halder SK, Nonaka Y
and Okamoto M: Cloning of a novel kinase (SIK) of the SNF1/AMPK
family from high salt diet-treated rat adrenal. FEBS Lett.
453:135–139. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Steinberg GR and Kemp BE: AMPK in health
and disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Takemori H, Katoh Y, Horike N, Doi J and
Okamoto M: ACTH-induced nucleocytoplasmic translocation of
salt-inducible kinase. J Biol Chem. 277:42334–42343. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Hashimoto YK, Satoh T, Okamoto M and
Takemori H: Importance of autophosphorylation at Ser186 in the
A-loop of salt inducible kinase 1 for its sustained kinase
activity. J Cell Biochem. 104:1724–1739. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Al-Hakim AK, Goransson O, Deak M, et al:
14-3-3 cooperates with LKB1 to regulate the activity and
localization of QSK and SIK. J Cell Sci. 118:5661–5673. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Romito A, Lonardo E, Roma G, Minchiotti G,
Ballabio A and Cobellis G: Lack of sik1 in mouse embryonic stem
cells impairs cardiomyogenesis by down-regulating the
cyclin-dependent kinase inhibitor p57kip2. PloS One. 5:e90292010.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Berdeaux R, Goebel N, Banaszynski L, et
al: SIK1 is a class II HDAC kinase that promotes survival of
skeletal myocytes. Nat Med. 13:597–603. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Cheng H, Liu P, Wang ZC, et al: SIK1
couples LKB1 to p53-dependent anoikis and suppresses metastasis.
Sci Signal. 2:ra352009.PubMed/NCBI
|
|
16.
|
Koo SH, Flechner L, Qi L, et al: The CREB
coactivator TORC2 is a key regulator of fasting glucose metabolism.
Nature. 437:1109–1111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Yoon YS, Seo WY, Lee MW, Kim ST and Koo
SH: Salt-inducible kinase regulates hepatic lipogenesis by
controlling SREBP-1c phosphorylation. J Biol Chem. 284:10446–10452.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Kowanetz M, Lönn P, Vanlandewijck M,
Kowanetz K, Heldin CH and Moustakas A: TGFβ induces SIK to
negatively regulate type I receptor kinase signaling. J Cell Biol.
182:655–662. 2008.
|
|
19.
|
Lönn P, Vanlandewijck M, Raja E, et al:
Transcriptional induction of salt-inducible kinase 1 by
transforming growth factor β leads to negative regulation of type I
receptor signaling in cooperation with the Smurf2 ubiquitin ligase.
J Biol Chem. 287:12867–12878. 2012.PubMed/NCBI
|
|
20.
|
Feldman JD, Vician L, Crispino M, Hoe W,
Baudry M and Herschman HR: The Salt-inducible kinase, SIK, is
induced by depolarization in brain. J Neurochem. 74:2227–2238.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Wen X, Zeng Y, Liu L, et al: Zhenqing
recipe alleviates diabetic nephropathy in experimental type 2
diabetic rats through suppression of SREBP-1c. J Ethnopharmacol.
142:144–150. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Huang W, Yu J, Jia X, Xiong L, Li N and
Wen X: Zhenqing recipe improves glucose metabolism and insulin
sensitivity by repressing hepatic FOXO1 in type 2 diabetic rats. Am
J Chin Med. 40:721–733. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Wang S and Hirschberg R: Diabetes-relevant
regulation of cultured blood outgrowth endothelial cells. Microvasc
Res. 78:174–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Hills CE, Al-Rasheed N, Willars GB and
Brunskill NJ: C-peptide reverses TGF-β1-induced changes in renal
proximal tubular cells: implications for treatment of diabetic
nephropathy. Am J Physiol Renal Physiol. 296:F614–F621.
2009.PubMed/NCBI
|
|
25.
|
Du J, Chen Q, Takemori H and Xu H: SIK2
can be activated by deprivation of nutrition and it inhibits
expression of lipogenic genes in adipocytes. Obesity (Silver
Spring). 16:531–538. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Lee MJ, Feliers D, Sataranatarajan K, et
al: Resveratrol ameliorates high glucose-induced protein synthesis
in glomerular epithelial cells. Cell Signal. 22:65–70. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Lin X, Takemori H, Katoh Y, et al:
Salt-inducible kinase is involved in the ACTH/cAMP-dependent
protein kinase signaling in Y1 mouse adrenocortical tumor cells.
Mol Endocrinol. 15:1264–1276. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Katoh Y, Takemori H, Lin X, et al:
Silencing the constitutive active transcription factor CREB by the
LKB1-SIK signaling cascade. FEBS J. 273:2730–2748. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Sasaki T, Takemori H, Yagita Y, et al:
SIK2 is a key regulator for neuronal survival after ischemia via
TORC1-CREB. Neuron. 69:106–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Xu J, Wu Y, Song P, Zhang M, Wang S and
Zou MH: Proteasome-dependent degradation of guanosine
5′-triphosphate cyclohydrolase I causes tetrahydrobiopterin
deficiency in diabetes mellitus. Circulation. 116:944–953.
2007.
|
|
31.
|
Trumper A, Trumper K and Horsch D:
Mechanisms of mitogenic and anti-apoptotic signaling by
glucose-dependent insulinotropic polypeptide in beta (INS-1)-cells.
J Endocrinol. 174:233–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Wu L and Derynck R: Essential role of
TGF-β signaling in glucose-induced cell hypertrophy. Dev Cell.
17:35–48. 2009.
|
|
33.
|
Gouville AC, Boullay V, Krysa G, et al:
Inhibition of TGF-β signaling by an ALK5 inhibitor protects rats
from dimethylnitrosamine-induced liver fibrosis. Br J Pharmacol.
145:166–177. 2009.
|
|
34.
|
Yata Y, Gotwals P, Koteliansky V and
Rockey DC: Dose-dependent inhibition of hepatic fibrosis in mice by
a TGF-β soluble receptor: implications for antifibrotic therapy.
Hepatology. 35:1022–1030. 2002.
|