Introduction
Non-alcoholic fatty liver disease (NAFLD)
encompasses a spectrum of liver diseases pertaining to fat
accumulation in the liver without significant alcohol consumption.
It has become the leading cause of chronic liver injury in the
Western world (1). Currently, no
effective treatments are available to treat NAFLD; thus, lifestyle
modifications via diet and exercise are most commonly recommended
(2). Therefore, there is an
urgent need to develop novel treatment options for preventing
NAFLD. NAFLD is a clinicohistopathological entity characterized by
lipid accumulation in the liver (simple steatosis) (3). In hepatic steatosis, an excess
amount of triglycerides (TGs) are acquired and accumulate in
hepatocytes. The main sources of TGs are from fatty acids stored in
adipose tissue and de novo lipogenesis of fatty acids within
the liver (4). Therefore, the
role of hepatic glycerol uptake in the development of NAFLD
warrants investigation.
Aquaporins (AQPs) are a family of membrane-bound,
homotetrameric water channel proteins that allow the movement of
water through cell membranes (5).
Thus far, 13 AQP subtypes have been identified in human (6). Among these, AQP3, 7, 9 and 10 are
subcategorized as aquaglyceroporins which permeabilize glycerol, as
well as water (7). AQP9 is
expressed in a number of organs, but it is highly expressed in the
liver (8). It is thought to be
the sole aquaglyceroporin subfamily expressed glycerol channel in
liver cells and is localized at the sinusoidal plasma membrane that
faces the portal vein (9).
However, the role of the AQP9 protein in the pathogenesis of NAFLD
remains to be fully elucidated.
In this study, we constructed pEGFP-N1-AQP9 and
pGenesil-1-AQP9-short hairpin RNA (shRNA) recombinant eukaryotic
expression vectors, which were then transfected into cell models of
oleic acid-induced NAFLD, then the expression of AQP9 was detected
by RT-PCR and western blot analysis. We demonstrate that
transfection with these plasmids can effectively increase and
decrease AQP9 expression, respectively. We also demonstrate that
high levels of AQP9 protein increase steatosis and that the
silencing of AQP9 reverses this effect in cell models of oleic
acid-induced NAFLD. Taken together, our results reveal that the
downregulation of AQP9 represents a novel potential molecular
target for therapeutic intervention in NAFLD.
Materials and methods
Construction of recombinant pEGFP-N1-AQP9
eukaryotic expression vector
Total RNA was extracted from the LO2 cells using
RNAiso Plus (Takara, Dalian, China) and first-strand DNA was
synthesized using the RT reagent kit (Takara) with random hexamer
primers. The full length AQP9 cDNA was obtained by RT-PCR. The AQP9
nested PCR primer (forward, 5′-GATTTCGGGTTCTAAGTCGC-3′ and reverse
primer, 5′-GAGAATCCCAAACTGACTGC-3′; nested forward,
5′-GGAAGATCTGATGCAGCCTGAGGGAG-3′ and reverse,
5′-CGGGGTACCCTGAGTTCATATTTCTC TGG-3′) and β-actin (forward,
5′-ACTGTGCCCATCTAC GAGG-3′ and reverse primer, 5′-GAAAGGGTGTAACG
CAACTA-3′) were synthesized by Takara. The human full-length
AQP9-cDNA was retrieved following the procedures of the E.Z.N.A.
Gel Extraction kit (Omega Bio-Tek Inc., Norcross, GA, USA) and
ligated to the multi-clony sites of pEGFP-N1 which was digested by
BglII and KpnI at a ratio of 10:1 with T4 DNA ligase
(Takara) and incubated at 16°C overnight. The ligated product was
transformed into competent E. coli DH5α. The recombinant
plasmid pEGFP-N1-AQP9 was extracted from a single positive clone
which was selected with kanamycin, then identified using single
digested with BglII and double digested with BglII
and KpnI and the products were evaluated with 1% agarose gel
electrophoresis. The sequence of pEGFP-N1-AQP9 was confirmed by
Sangon Biotech Co., Ltd. (Shanghai, China).
Construction of recombinant
pGenesil-1-AQP9-shRNA eukaryotic expression vector
According to the Homo sapien AQP9 gene
sequence (GenBank Accession no. NM_182966) and the principles of
shRNA design, a shRNA site targeting the AQP9 gene was selected.
The oligonucleotide sequences encoding AQP9 shRNA (forward,
5′-GATCCGCTGTGTC TTTAGCAATGTGTTTCGACACATTGCTAAAGACACAG CTTTTTTA-3′
and reverse, 5′-AGCTTAAAAAAGCTGTGT
CTTTAGCAATGTGTCGAAACACATTGCTAAAGACACA GCG-3′); and scrambled shRNA
(forward, 5′-GATCCG AATCCGCACTACTCCTTACATTCGTGTAAGGAGTAGTG
CGGATTCTTTTTTA-3′ and reverse, 5′-AGCTTAAAAA
AGAATCCGCACTACTCCTTACACGAATGTAAGGAGTA GTGCGGATTCG-3′) were
synthesized by Invitrogen (Carlsbad, CA, USA); these two pairs of
oligonucleotides were annealed into double-stranded DNA, then
cloned into the pGenesil-1 vector which was double digested with
BamHI and HindIII. These recombinant plasmids were
double digested with EcoRI and HindIII for enzyme
digestion analysis and sequenced by Sangon Biotech Co., Ltd.
Establishment of oleic acid-induced NAFLD
cell models
The 3rd passage LO2 cells (1×104
cells/well) were inoculated into a 96-well plate and cultured in
RPMI-1640 supplemented with 10% fetal bovine serum in 5%
CO2 at 37°C. The following day, cells were treated with
0, 10, 20, 30, 40 or 50 μg/ml of oleic acid (Sigma-Aldrich Corp.,
St. Louis, MO, USA) for 72 h, then the culture medium was discarded
and the cells were washed three times in phosphate buffer solution,
and 20 μl of MTT labeling reagent (0.5 mg/ml) (Sigma-Aldrich Corp.)
were added to each well. The plates were then incubated for 4 h and
150 μl of dimethylsulfoxide was added. Optical density values were
detected at the 490 nm wavelength. A cell model of NAFLD was
induced by oleic acid. Each assay was performed in octuplicate.
Transfection of the recombinant
pEGFP-N1-AQP9 and pGenesil-1-AQP9-shRNA eukaryotic expression
vector into cell models of NAFLD
Endotoxin-free plasmid was extracted using the
E.Z.N.A. Endo-Free Plasmid Mini kit I (Omega Bio-Tek Inc.). Cell
models of Oleic acid-induced NAFLD were cultured in RPMI-1640
containing 10% fetal bovine serum at 37°C in a humidified
atmosphere of 5% CO2. Cells were passaged and
transfected with endotoxin-free plasmid at 80–90% confluence using
Lipofectamine 2000 reagent (Invitrogen) according to the
manufacturer’s instructions. Cells were divided into eight groups:
untreated, oleic acid, pEGFP-N1-AQP9, pEGFP-N1,
pGenesil-1-AQP9-shRNA, pGenesil-1-scrambled-shRNA, pGenesil-1 and
double-distilled water (ddH2O) group. Seventy-two hours
after transfection, the expression of GFP was observed under an
inverted fluorescence microscope and the cells were collected to
conduct subsequent experiments.
RT-PCR analysis
Total RNA was isolated from each group by RNAiso
Plus according to the manufacturer’s instructions (Takara). cDNA
was prepared using the RT reagent kit and RT-PCR was performed
using AQP9 primer (forward, 5′-CTTTGGACGGATGAAATGGTT-3′ and
reverse, 5′-GAG TCAGGCTCTGGATGGTG-3′) and β-actin was detected as
an internal reference. Each assay was performed in duplicate.
Western blot analysis
Total protein was isolated from each group and the
protein concentration was detected using the BCA protein assay kit
(Beyotime Institute of Biotechnology, Hangzhou, China). Western
blot analysis was performed using rabbit anti-human AQP9 polyclonal
antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and
rabbit anti-human β-actin polyclonal antibody (Biosynthesis
Biotechnology Co., Ltd., Beijing, China), followed by incubation
with HRP-conjugated goat anti-rabbit IgG (Biosynthesis
Biotechnology Co., Ltd.). The proteins of interest were detected
using BeyoECL plus reagent (Beyotime Institute of Biotechnology)
following the manufacturer’s instructions. Each assay was performed
in duplicate.
Oil red O staining
Briefly, the culture medium was discarded and the
cells were washed were washed three times in phosphate buffer
solution, then fixed with 4% paraformaldehyde for 30 min and
stained with 5% oil red O solution for 30 min. They were then
washed with 60% isopropanol for 30 sec and then rinsed with
ddH2O for 30 sec, counterstained with hematoxylin for 3
min, rinsed with ddH2O for 5 min, fixed with neutral
balata, and then observed under an upright microscope. Each assay
was performed in triplicate.
Determination of TG, free fatty acid
(FFA) and glycerol levels
Briefly, the cells were washed with phosphate buffer
solution three times and lysed by repeated freezing and thawing.
Intracellular lipid content was determined using the TG assay kit
(Beihua Kangtai, Beijing, China), FFA assay kit (Jiancheng
Bioengineering Institute, Nanjing, China) and glycerol GPO-POD
assay kit (Applygen, Beijing, China) according to the
manufacturer’s instructions. Each assay was performed in
quintuplicate.
Statistical analysis
Statistical analyses were conducted using SPSS 17.0
software. Data are presented as the means ± standard deviation.
One-way ANOVA was conducted to assess differences among groups. A
P-value ≤0.05 was considered to indicate a statistically
significant difference.
Results
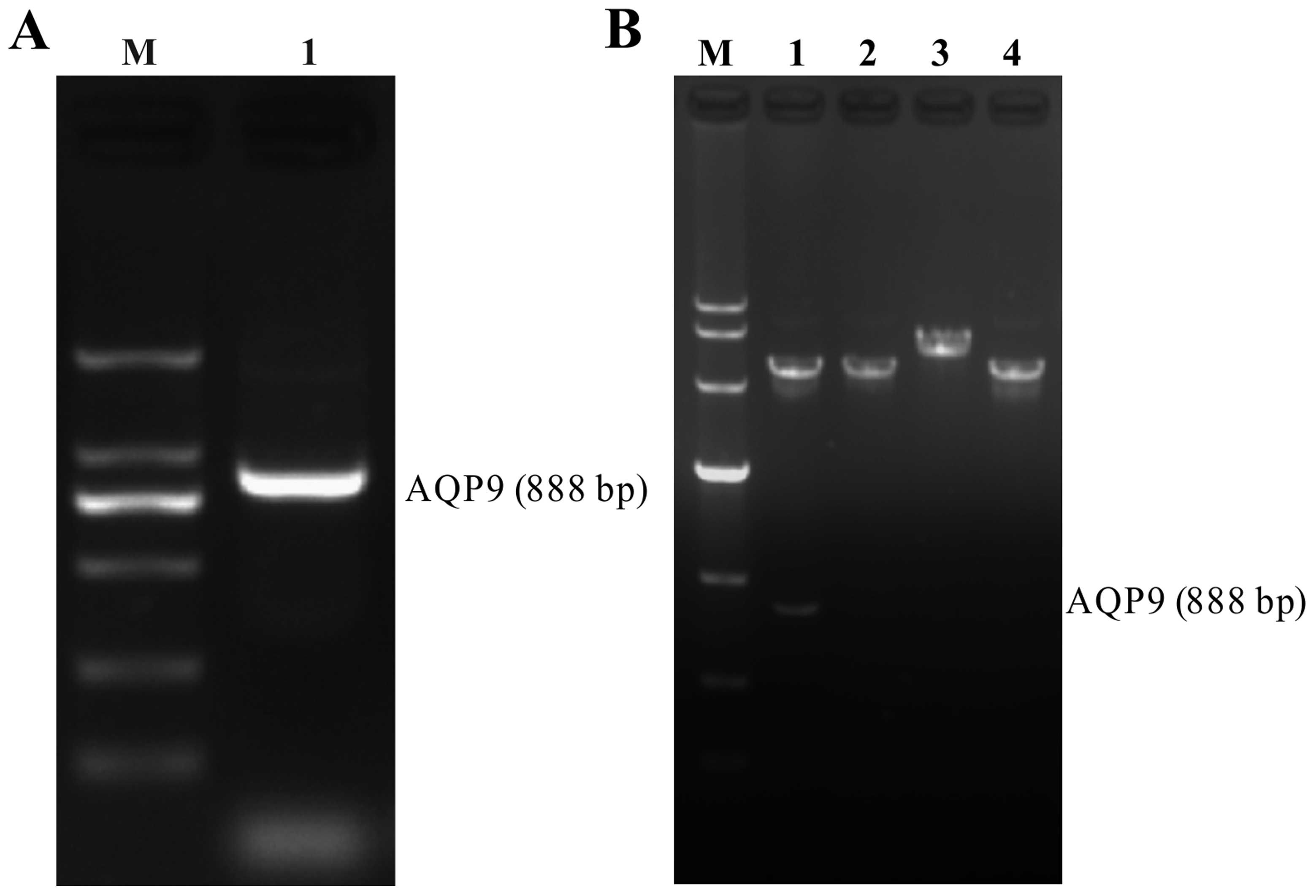
Evaluation of RT-PCR product and
recombinant pEGFP-N1-AQP9 eukaryotic expression vector
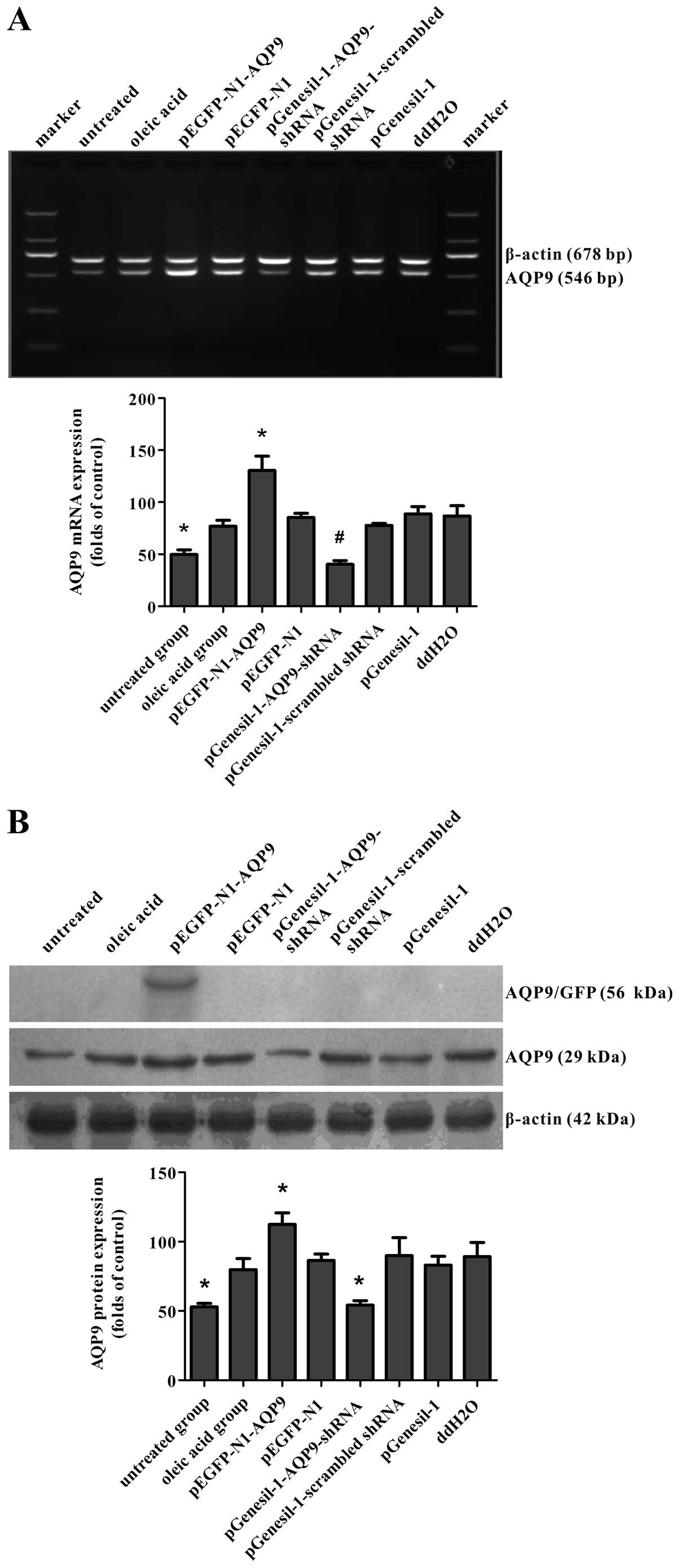
The RT-PCR products were loaded on 1.5% agarose
gels, and the band for full-length AQP9 cDNA was located at 888 bp
(Fig. 1A). After the AQP9 cDNA
fragment was inserted into the pEGFP-N1 plasmid (5428 bp),
pEGFP-N1-AQP9 recombinant plasmids were digested by restriction
enzymes and separated on a 1% agarose gel. The positive plasmid was
confirmed by DNA sequencing. Our data demonstrated that after both
pEGFP-N1-AQP9 and pEGFP-N1 were single and double digested,
separately, the AQP9 band was detected in the double digested
pEGFP-N1-AQP9, but not in pEGFP-N1, and the band of single digested
pEGFP-N1-AQP9 was higher than that of pEGFP-N1 (Fig. 1B). From sequencing analysis, there
was a nonsense mutation at site 45: A→G. The sequencing map was as
shown in Fig. 2.
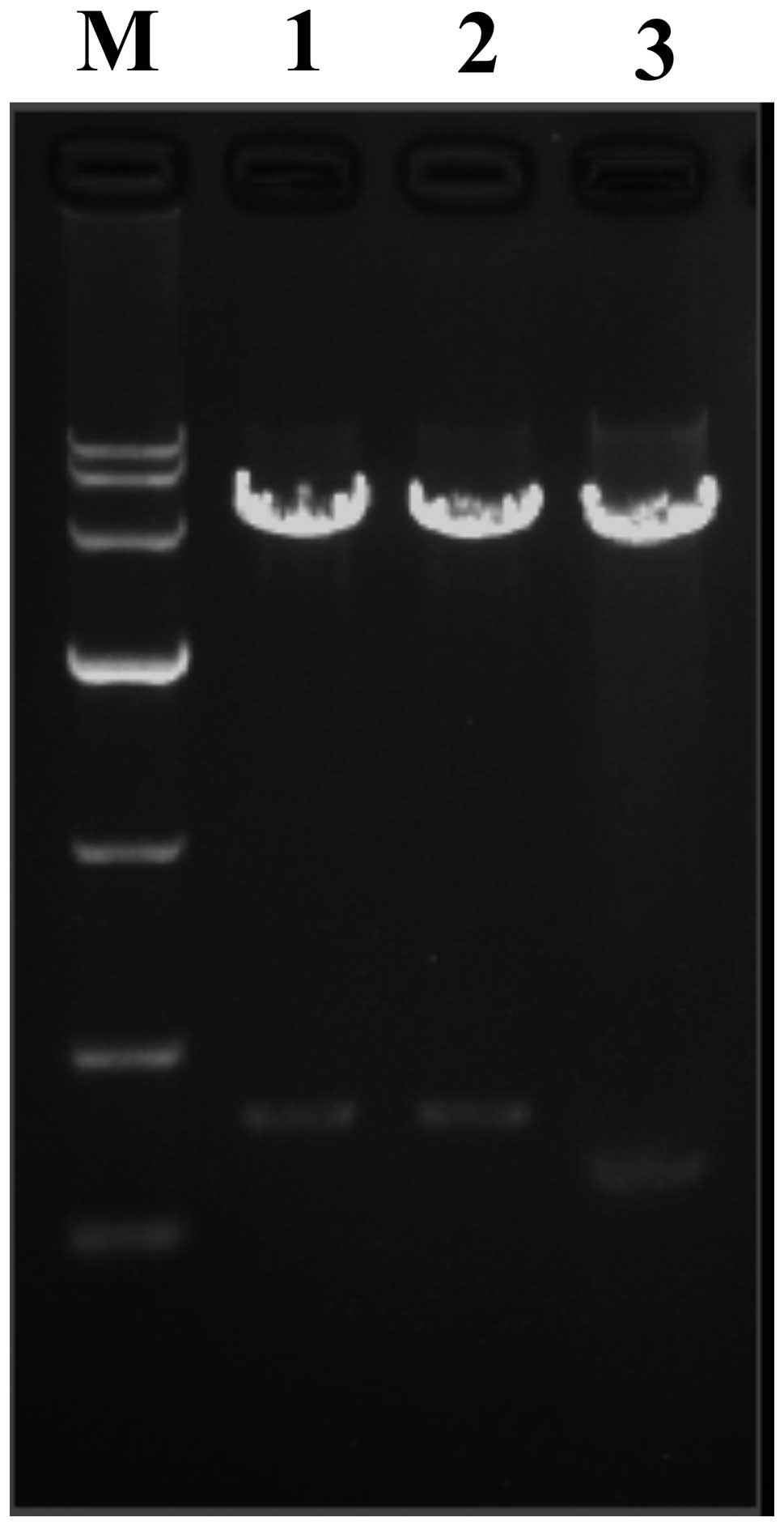
Evaluation of recombinant
pGenesil-1-AQP9-shRNA eukaryotic expression vector
pGenesil-1-AQP9-shRNA recombinant plasmids were
digested by restriction enzymes and evaluated with 1% agarose gel
electrophoresis. The positive plasmid was confirmed by DNA
sequencing. The band of double digested and pGenesil-1-scrambled
shRNA was higher than that of pGenesil-1 (Fig. 3). Sequencing analysis confirmed
that the inserted sequence was consistent with the expected
sequence (Fig. 4).
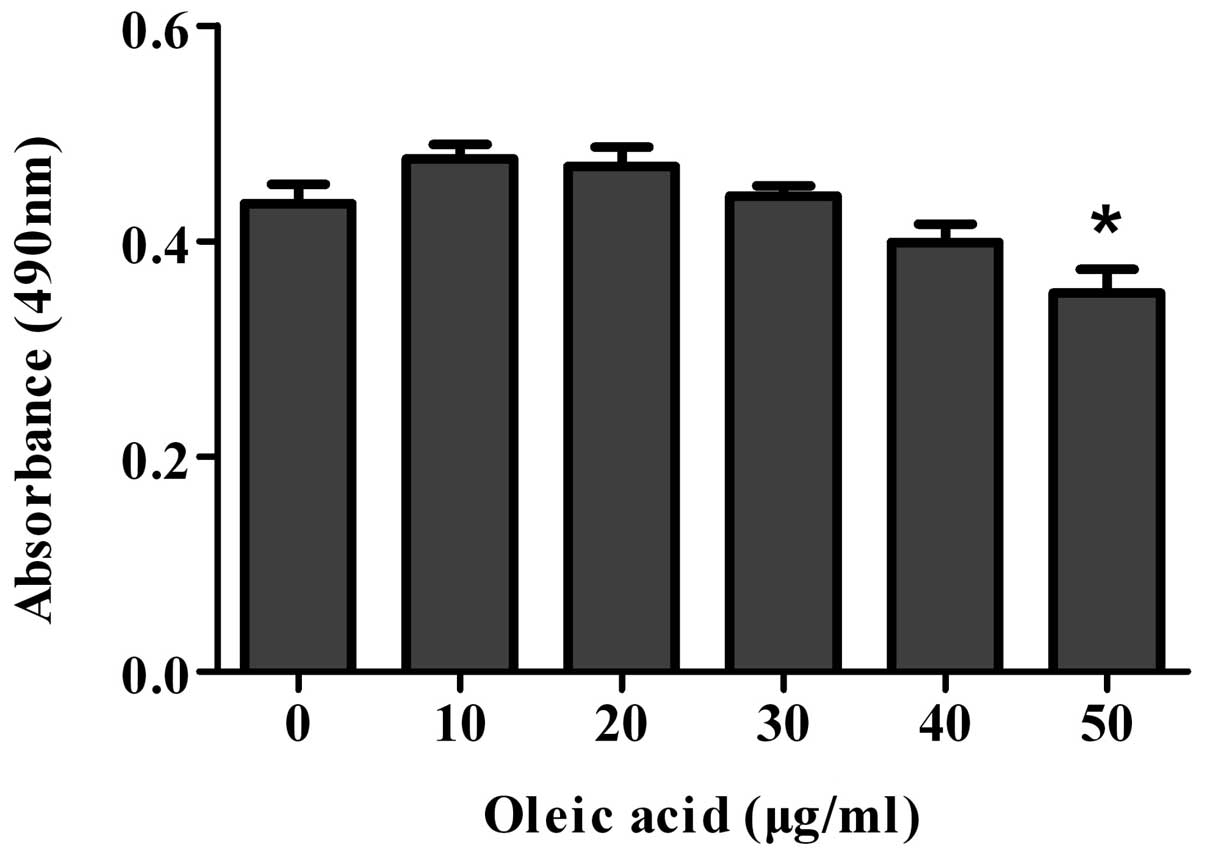
Optimizing the concentration of oleic
acid by MTT assay
To determine the optimal concentration of oleic acid
for the induction of steatosis, the cell viability of the cell
models of oleic acid-induced NAFLD was determined by MTT assay
following treatment with oleic acid (0, 10, 20, 30, 40 or 50 μg/ml)
for 72 h. The cell viability of the cells treated with >20 μg/ml
of oleic acid decreased and significantly decreased when the cells
were treated with 50 μg/ml of oleic acid (Fig. 5). Therefore, according to our
results, 20 μg/ml of oleic acid was the optimal concentration for
inducing steatosis; thus, we selected this dose for the following
experiments.
mRNA and protein expression of AQP9 in
cell models of oleic acid-induced NAFLD
After the cell models of NAFLD were transfected with
recombinant plasmids for 72 h, GFP protein expression was observed
under an inverted fluorescence microscope. GFP was observed in the
pEGFP-N1-AQP9, pEGFP-N1, pGenesil-1-AQP9-shRNA,
pGenesil-1-scrambled shRNA and pGenesil-1 group, but not in the
ddH2O, oleic acid or untreated group (data not shown).
To determine the AQP9 mRNA and protein levels in the cell models of
oleic acid-induced NAFLD, RT-PCR and western blot analysis were
performed. RT-PCR revealed that the delivery of pEGFP-N1-AQP9 and
pGenesil-1-AQP9-shRNA resulted in an approximately 70%
overexpression and a 45% knockdown of mRNA levels compared with the
empty vector group, respectively (Fig. 6A). Western blot analysis revealed
a corresponding change in AQP9 protein expression (Fig. 6B). These results indicated that
the recombinant plasmids were successfully transfected into the
cell models of NAFLD and that the vectors, pEGFP-N1-AQP9 and
pGenesil-1-AQP9-shRNA, were effective in increasing and suppressing
endogenous AQP9 expression, respectively.
Intracellular lipid accumulation is
dependent on AQP9 expression in cell models of NAFLD
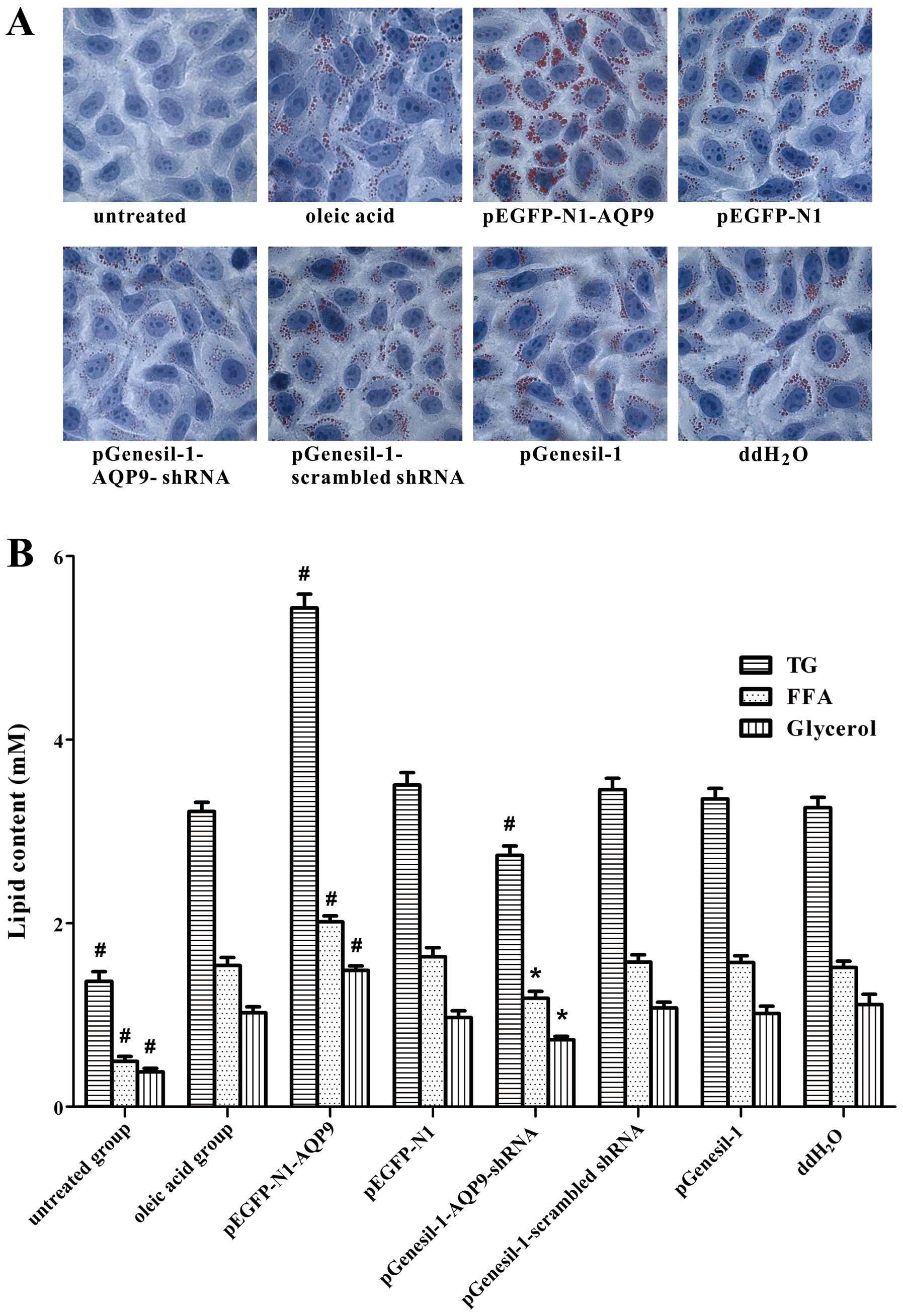
To detect intracellular lipid accumulation, oil red
O staining was performed. A large number of lipid droplets appeared
jacinth with oil red O staining accompanied by lipid droplet fusion
in the pEGFP-N1-AQP9 group; a small number of lipid droplets
appeared jacinth and lipid droplet fusion was not as evident in the
pGenesil-1-AQP9-shRNA group; in the oleic acid group, we also
observed lipid droplet fusion, but no lipid droplets were observed
in the untreated group (Fig. 7A).
These results indicate that oleic acid induces intracellular lipid
accumulation in LO2 cells. AQP9 overexpression significantly
aggravates intracellular lipid accumulation; however, the silencing
of AQP9 alleviates this effect.
Content of TG, FFA and glycerol is
depenentd on AQP9 expression in cell models of NAFLD
The content of TG, FFA and glycerol was
significantly increased in the oleic acid group, compared with the
untreated group (Fig. 7B). Our
data also demonstrated that the content of TG, FFA and glycerol was
significant increased in the pEGFP-N1-AQP9 group compared with the
oleic acid group, while it was significantly decreased in the
pGenesil-1-AQP9-shRNA group compared with the oleic acid group.
These results suggested that oleic acid increased the content of
TG, FFA and glycerol. AQP9 overexpression induced a significant
increase in TG, FFA and glycerol content; however, the silencing of
AQP9 significantly reversed this effect. Taken together, these
results suggest that AQP9 plays an important role in the
progression of hepatic steatosis.
Discussion
NAFLD consists of a spectrum of pathological states
ranging from the hepatic accumulation of lipids known as steatosis
to non-alcoholic steatohepatitis, cirrhosis and ultimately, liver
failure (10). It occurs in
children and adults of all age groups and both genders (11). Its key feature is steatosis in the
absence of pathologies, such as viral hepatitis or alcohol abuse
(12). By current estimations,
NAFLD is perhaps the most common liver disease as it has a
prevalence of 6–35% with a median of 20% according to a study on
populations in several countries (13). NAFLD is particularly associated
with obesity, since the prevalence of steatosis is 57.5–74% in
obese subjects from Japan and Italy (14).
Concerning the pathogenesis of NAFLD, the most
accepted theory is the ‘two hit’ hypothesis, in which the first hit
involves the accumulation of hepatic TG, which in turn triggers the
second hit, inflammation and oxidative stress (15). An extended hypothesis proposed a
decrease capacity of hepatic regeneration and the adverse effects
of FFA lipotoxicity as the third hit (16,17). These data suggest that alleviating
hepatocyte lipid accumulation is an effective therapeutic strategy
to prevent NAFLD.
AQP belongs to the major intrinsic protein (MIP)
family which can be classified as two types. One type is aquaporin
which accounts for the majority of MIPs, including AQP1, AQP2,
AQP4, AQP5, AQP6, AQP8, AQP10, AQP11 and AQP12, only permeable to
water. The other type is aquaglyceroporin, including AQP3, AQP7,
AQP9 and AQP10, permeable to not only water, but also urea,
glycerol and even some inorganic ions (18), which play an important role in
regulating glycerol transportation and lipid metabolism. AQP9 is a
protein channel highly expressed in the liver and is localized at
the sinusoidal membrane (19). It
is thought to be the sole aquaglyceroporin subfamily expressed
glycerol channel in liver cells (7,9).
In this study, we used gain- and loss-of-function
approaches to determine the role of AQP9 in steatosis in cell
models of oleic acid-induced NAFLD. Our results revealed that AQP9
overexpression significantly increased intracellular lipid content;
however, the silencing of AQP9 had the opposite effect. These
results are consistent with those from previous studies, showing
that an abnormal increase in AQP9 expression leads to a large
amount of glycerol entering hepatocytes and increased fat synthesis
and accumulation, resulting in the formation of fatty liver
(20,21). Glycerol uptake is significantly
decreased in cultured astrocytes following transfection with
AQP9-small interfering RNA (22).
The decreased expression of AQP9 in hepatocytes can effectively
block the entry of glycerol into hepatocytes; therefore, targeting
AQP9 may be used to prevent and treat NAFLD, which may provide a
promising therapeutic strategy for NAFLD. Furthermore, AQP9 acts as
an absorption channel for certain drugs. AQP9 has been shown to aid
the transfer of the chemotherapeutic drug, arsenic trioxide
(23,24). The enforced expression of AQP9 in
the tumor cell membrane can effectively enhance the
chemotherapeutic efficacy, reducing drug dosage and toxic effects;
thus it may provide a novel treatment strategy for tumors (25,26). It has been reported that insulin
suppresses AQP9 expression in H4IIE hepatocytes (27). The treatment of rats with
streptozotocin (STZ) has been shown to result in a 20-fold increase
in AQP9 expression in the liver, which was blocked by the
administration of insulin (28).
Moreover, insulin resistance is the key factor in the pathogenesis
and potential evolution of hepatic steatosis, which is associated
with increased peripheral lipolysis with the release of FFA from
visceral fat, the hepatic uptake of FFA and hepatocellular TG
synthesis and accumulation (29,30). Thus, the knockout of AQP9 may play
an important role against insulin resistance.
In conclusion, in this study, we successfully
constructed the pEGFP-N1-AQP9 and pGenesil-1-AQP9-shRNA recombinant
eukaryotic expression vectors, which were then transfected into
cell models (derived from LO2 cells) of oleic acid-induced NAFLD.
Thus, AQP9 expression was effectively increased and suppressed,
respectively. In addition, the overexpression of AQP9 significantly
increased intracellular lipid content. By contrast, the knockdown
of AQP9 exerted the opposite effect. Our findings provide evidence
of the potential therapeutic effectiveness of AQP9 in NAFLD.
Further studies are required to clarity the molecular mechanisms of
action of AQP9 in lipid accumulation in NAFLD.
Acknowledgements
This study was supported by grants (81070318) from
the National Natural Science Foundation of China (NSFC81070318) and
the Medical Research Project from Chongqing Board of Health
(YuWeiKeJiao 2010-2-100).
References
|
1
|
Browning JD, Szczepaniak LS, Dobbins R, et
al: Prevalence of hepatic steatosis in an urban population in the
United States: Impact of ethnicity. Hepatology. 40:1387–1395. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rodriguez B, Torres DM and Harrison SA:
Physical activity: an essential component of lifestyle modification
in NAFLD. Nat Rev Gastroenterol Hepatol. 9:726–731. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fabbrini E, Sullivan S and Klein S:
Obesity and nonalcoholic fatty liver disease: biochemical,
metabolic, and clinical implications. Hepatology. 51:679–689. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krawczyk M, Bonfrate L and Portincasa P:
Nonalcoholic fatty liver disease. Best Pract Res Clin
Gastroenterol. 24:695–708. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang HF, He RH, Sun CC, Zhang Y, Meng QX
and Ma YY: Function of aquaporins in female and male reproductive
systems. Hum Reprod Update. 12:785–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu H, Zheng Z and Wintour EM: Aquaporins
and fetal fluid balance. Placenta. 29:840–847. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maeda N: Implications of aquaglyceroporins
7 and 9 in glycerol metabolism and metabolic syndrome. Mol Aspects
Med. 33:665–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elkjaer M, Vajda Z, Nejsum LN, et al:
Immunolocalization of AQP9 in liver, epididymis, testis, spleen,
and brain. Biochem Biophys Res Commun. 276:1118–1128. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maeda N, Funahashi T and Shimomura I:
Metabolic impact of adipose and hepatic glycerol channels aquaporin
7 and aquaporin 9. Nat Clin Pract Endocrinol Metab. 4:627–634.
2008.PubMed/NCBI
|
|
10
|
Chalasani N, Younossi Z, Lavine JE, et al:
The diagnosis and management of non-alcoholic fatty liver disease:
practice Guideline by the American Association for the Study of
Liver Diseases, American College of Gastroenterology, and the
American Gastroenterological Association. Hepatology. 55:2005–2023.
2012. View Article : Google Scholar
|
|
11
|
Roberts EA: Pediatric nonalcoholic fatty
liver disease (NAFLD): A ‘growing’ problem? J Hepatol.
46:1133–1142. 2007.
|
|
12
|
Angulo P and Lindor KD: Non-alcoholic
fatty liver disease. J Gastroenterol Hepatol. 17(Suppl): S186–S190.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vernon G, Baranova A and Younossi ZM:
Systematic review: the epidemiology and natural history of
non-alcoholic fatty liver disease and non-alcoholic steatohepatitis
in adults. Aliment Pharmacol Ther. 34:274–285. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tailleux A, Wouters K and Staels B: Roles
of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys
Acta. 1821:809–818. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Day CP and James OF: Steatohepatitis: a
tale of two ‘hits’? Gastroenterology. 114:842–845. 1998.
|
|
16
|
Feldstein AE, Werneburg NW, Canbay A, et
al: Free fatty acids promote hepatic lipotoxicity by stimulating
TNF-alpha expression via a lysosomal pathway. Hepatology.
40:185–194. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Day CP: From fat to inflammation.
Gastroenterology. 130:207–210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gomes D, Agasse A, Thiebaud P, Delrot S,
Geros H and Chaumont F: Aquaporins are multifunctional water and
solute transporters highly divergent in living organisms. Biochim
Biophys Acta. 1788:1213–1228. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Carbrey JM, Gorelick-Feldman DA, Kozono D,
Praetorius J, Nielsen S and Agre P: Aquaglyceroporin AQP9: solute
permeation and metabolic control of expression in liver. Proc Natl
Acad Sci USA. 100:2945–2950. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hara-Chikuma M and Verkman AS:
Physiological roles of glycerol-transporting aquaporins: the
aquaglyceroporins. Cell Mol Life Sci. 63:1386–1392. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maeda N, Hibuse T and Funahashi T: Role of
aquaporin-7 and aquaporin-9 in glycerol metabolism; involvement in
obesity. Handb Exp Pharmacol. 190:233–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Badaut J, Brunet JF, Guerin C, Regli L and
Pellerin L: Alteration of glucose metabolism in cultured astrocytes
after AQP9-small interference RNA application. Brain Res.
1473:19–24. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hamdi M, Sanchez MA, Beene LC, et al:
Arsenic transport by zebrafish aquaglyceroporins. BMC Mol Biol.
10:1042009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Drobna Z, Walton FS, Paul DS, Xing W,
Thomas DJ and Styblo M: Metabolism of arsenic in human liver: the
role of membrane transporters. Arch Toxicol. 84:3–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leung J, Pang A, Yuen WH, Kwong YL and Tse
EWC: Relationship of expression of aquaglyceroporin 9 with arsenic
uptake and sensitivity in leukemia cells. Blood. 109:740–746. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao L, Gao Y, Li X, et al: Aquaporins
mediate the chemoresistance of human melanoma cells to arsenite.
Mol Oncol. 6:81–87. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hibuse T, Maeda N, Nagasawa A and
Funahashi T: Aquaporins and glycerol metabolism. Biochim Biophys
Acta. 1758:1004–1011. 2006. View Article : Google Scholar
|
|
28
|
King LS, Kozono D and Agre P: From
structure to disease: the evolving tale of aquaporin biology. Nat
Rev Mol Cell Biol. 5:687–698. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Parekh S and Anania FA: Abnormal lipid and
glucose metabolism in obesity: implications for nonalcoholic fatty
liver disease. Gastroenterology. 132:2191–2207. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shoelson SE, Herrero L and Naaz A:
Obesity, inflammation, and insulin resistance. Gastroenterology.
132:2169–2180. 2007. View Article : Google Scholar : PubMed/NCBI
|