Introduction
Ischemia/reperfusion injury remains a leading cause
of mortality in developed countries, despite significant advances
in the medical treatment of heart failure (1). A number of investigators have
demonstrated that the transplantation of bone marrow (BM)-derived
mesenchymal stem cells (MSCs) represents a promising tool for the
repair and regeneration of cardiomyocytes and for the restoration
of heart function (2–4). However, the poor survival of
engrafted MSCs presents a major obstacle in MSC-based therapy, and
evidence suggests that the transplanted cells undergo apoptosis
(5,6). Previous studies have demonstrated
that hypoxia and serum deprivation (SD), which are both components
of ischemia, induce MSC apoptosis through the mitochondrial pathway
(7,8). Strategies that enhance tolerance to
apoptosis should thus significantly improve the efficacy of
MSC-based transplantation therapy.
C1q tumor necrosis factor-related proteins (CTRPs)
are members of the highly conserved family of adiponectins. Each of
the 10 known members (CTRP1-CTRP10) consists of 4 distinct domains,
including an N-terminal signal peptide, a short variable domain, a
collagen-like domain and a C-terminal C1q-like globular domain
(9,10). CTRP3 is expressed not only in
cartilage, where it was originally discovered, but also in adipose
tissue, preadipocyte cell lines, as well as in monocytes and
fibroblasts, and has been shown to be upregulated upon fasting and
inflammation (11–13). CTRP3, originally thought to
participate in lipid metabolism, has broad functions and regulates
various biological processes, including adipokine secretion, fatty
acid oxidation, inflammation, cell proliferation, differentiation
and apoptosis (14–17). CTRP3 protects cardiomyocytes from
apoptosis and plays a protective role in cardiac infarction
(17). Furthermore, recent
findings have suggested that CTRP3 acts via the phosphoinositide
3-kinase (PI3K)/Akt pathway to exert a marked anti-apoptotic effect
(17). The PI3K/Akt pathway is
involved in protecting numerous cell types, including MSCs, from
apoptosis. It is therefore possible that CTRP3 may be involved in
protecting MSCs against hypoxia/SD-induced apoptosis. CTRP3 and the
related downstream signaling pathway(s) may thus represent
promising targets for preventing hypoxia/SD-induced apoptosis.
Therefore, the present study aimed to examine the effects of CTRP3
on hypoxia/SD-induced MSC apoptosis and to investigate the related
signaling pathways.
Materials and methods
Animals
Male Sprague-Dawley rats, weighing 60–80 g, were
handled in accordance with the US National Institutes of Health
published guidelines. All procedures were approved by the
Institutional Animal Care and Use Committee of Harbin Medical
University. The study was conducted in compliance with the Guide
for the Care and Use of Laboratory Animals published by the
National Institutes of Health (NIH, revised in 1996).
Reagents
Dulbecco's modified Eagle's medium (DMEM)/F12 and
fetal bovine serum (FBS) were purchased from HyClone Laboratories
(Logan, UT, USA). The Annexin V-FITC apoptosis detection kit, and
anti-CD44, anti-CD29 and anti-CD90 antibodies were obtained from BD
Biosciences (San Diego, CA, USA). Anti-CD34 and anti-CD45
antibodies were obtained from eBioscience (San Diego, CA, USA).
Rabbit monoclonal antibodies against Akt, phospho-Akt (Tyr308,
Ser473), caspase-3, Bax, Bcl-2 and cytochrome c, and the
PI3K inhibitor, LY294002, were obtained from Cell Signaling
Technology (Danvers, MA, USA). Mouse polyclonal antibody against
β-actin (#TA-09) was purchased from Zhongshan Goldenbridge
Biotechnology (Beijing, China) and horseradish
peroxidase-conjugated secondary antibodies anti-mouse and -rabbit
were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). The JC-1 mitochondrial membrane potential assay kit was
purchased from the Beyotime Institute of Biotechnology (Nantong,
China), the Cell Counting kit-8 (CCK-8) assay kit was from HaiGene
Technology (Harbin, China) and recombinant human CTRP3 was from
Adipogen (San Diego, CA, USA).
Cell cultures and treatments
BM-MSCs were isolated from the femurs and tibias of
Sprague-Dawley rats, as previously described (18). Briefly, BM cells were flushed from
the femurs and tibias with 5 ml DMEM/F12. Red blood cells were
lysed and removed, and 5×105 cells were then plated in a
25-cm2 flask with 6 ml DMEM/F12 supplemented with 10%
FBS and 1% penicillin/streptomycin. After 3 days of culture at 37°C
under 5% CO2, the medium and non-adherent cells were
removed and replaced with fresh medium. Adherent MSCs were further
grown in medium, which was replaced every 3 days. When the cells
reached 80–90% confluence, the adherent cells were trypsinized and
expanded at 2:3 or 1:2 dilutions. All cells used were from passages
3–5.
The characteristics of the MSCs were determined by
immunophenotyping. Cells were harvested, washed with
phosphate-buffered saline (PBS), and labeled with phycoerythrin
(PE)-conjugated anti-CD45 and anti-CD90 antibodies, and fluorescein
isothiocyanate (FITC)-labeled anti-CD44, anti-CD29 and anti-CD34
antibodies. Labeled cells were assayed by flow cytometry and
analyzed using FACSDiva Pro software (BD Biosciences).
The in vitro induction of apoptosis by
hypoxia/SD was designed to mimic the in vivo conditions in
the ischemic myocardium, and was initiated as previously described
(19). Cells exposed to
hypoxia/SD alone were used as the apoptotic controls. Apoptosis was
induced by incubating the MSCs in serum-free medium in a controlled
(anaerobic)-atmosphere glove chamber (855-AC; Plas-Labs Inc.,
Lansing, MI, USA), so as to scavenge free oxygen. Cells cultured in
complete medium alone were used as the non-ischemic controls. Cells
were exposed to CTRP3 when subjected to hypoxia/SD. CTRP3
concentrations of 3–3,000 ng/ml were used to treat the cells
throughout the process.
To investigate the involvement of the PI3K/Akt
pathway, the cells were pre-incubated with the PI3K/Akt inhibitor,
LY294002 (25 μM), in complete medium for 90 min prior to exposure
to hypoxia/SD. CTRP3 was added in the presence of the inhibitor
during exposure to hypoxia/SD.
Analysis of cell apoptosis by flow
cytometry
Apoptotic rates were estimated by detecting
phosphatidylserine on the cell plasma membrane using the
fluorescent dye [propidium iodide (PI)] with the Annexin V-FITC
apoptosis detection kit (BD Biosciences), according to the
manufacturer's instructions. In brief, cells were harvested and
washed in ice-cold PBS, resuspended in 300 μl of binding buffer and
incubated with 5 μl of Annexin V-FITC solution for 30 min at 4°C in
the dark, followed by further incubation with 5 μl PI for 5 min.
Cells were then analyzed immediately by bivariate flow cytometry
using a BD FACSCanto cytometer equipped with FACSDiva Pro software.
Approximately 1–5×105 cells were analyzed in each
sample.
Cell proliferation assay
Cell proliferation was assessed using a CCK-8 assay
kit according to the manufacturer's instructions. Cells were
incubated with CCK-8 solution in 96-well plates for 1 h at 37°C.
The absorbance of each well was quantified at 450 nm.
Toxicity assay
The potential toxic effects of CTRP3 in cultured
MSCs were examined at various concentrations. The MSCs were
incubated in culture medium supplemented with CTRP3 for 24 h under
hypoxia/SD culture conditions, and for 3 days under normal culture
conditions. Trypan blue was added to the medium and the cells were
incubated at 37°C for 15 min. Trypan blue-positive cells were then
counted under a phase-contrast microscope. Five fields were
randomly selected for each dish and at least 3 dishes were counted
for each concentration.
Western blot analysis
Western blot analysis was carried out as previously
described (20). Briefly, cells
were washed twice with ice-cold PBS and ruptured with lysis buffer
containing 20 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, protease
and phosphatase inhibitors. Cell extracts were centrifuged for 5
min at 12,000 × g and supernatants were collected. A total of 20 μg
of protein was resolved by SDS-PAGE and transferred onto PVDF
membranes. Membranes were blocked for 1 h with 5% skim milk in
Tris-buffered saline solution containing 0.1% Tween-20 and were
incubated with primary antibodies [rabbit monoclonal antibodies
against Akt, phospho-Akt (Tyr308, Ser473), caspase-3, Bax, Bcl-2,
cytochrome c and mouse polyclonal antibody against β-actin]
at 4°C overnight. The membranes were washed, incubated for 1 h with
the appropriate horseradish peroxidase-conjugated secondary
antibodies (anti-mouse and anti-rabbit), developed using
chemiluminescent substrates, photographed using ChemiDoc XRS
equipment (Bio-Rad, Hercules, CA, USA) and quantified using
Quantity One software (Bio-Rad).
Mitochondrial membrane potential
assay
Mitochondrial membrane potential was determined
using the JC-1 mitochondrial membrane potential assay kit. Briefly,
the MSCs were seeded in 6-well plates. Following treatment, the
cells were washed twice with PBS, and 1 ml staining dye/well was
added (culture medium:JC-1 working dye, 1:1) and incubated at 37°C
for 20 min. The cells were then washed twice with cold JC-1
staining buffer and examined under a fluorescence microscope.
Statistical analysis
Data are expressed as the means ± standard deviation
(SD). Differences among groups were tested by a one-way ANOVA.
Comparisons between groups were evaluated using the Student's
t-test. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
Cells isolated from rat BM show MSC
characteristics
MSCs were uniformly spindle-like. FACS analysis
revealed that the majority of cells from passage 3 expressed the
common MSC surface markers, CD29 (98.98±0.55%), CD90 (99.06±0.63%)
and CD44 (25.30±5.80%), but were found negative for CD34
(0.97±0.03%) and CD45 (1.38±1.30%) (Fig. 1). Cells cultured in vitro
for 3–5 passages were therefore used in the subsequent
experiments.
CTRP3 protects MSCs from
hypoxia/SD-induced apoptosis
In preliminary experiments, the maximal induction of
early apoptosis by hypoxia/SD in the MSCs occurred at 24 h. We
investigated whether CTRP3 protects MSCs from this process. MSCs
were exposed to increasing concentrations of CTRP3 (3–3,000 ng/ml)
followed by exposure to hypoxia/SD for 24 h, and cell apoptosis was
determined by FACS analysis. CTRP3 (3–3,000 ng/ml) effectively
blocked the apoptotic process, and the ratio of apoptotic cells
decreased with a pronounced effect at 300 ng/ml [treated cells,
2.60±0.23 vs. apoptotic control (untreated cells), 6.95±0.37]
(Fig. 2A and B).
Caspase-3 is a key mediator of apoptosis. We further
evaluated the anti-apoptotic effects of CTRP3 by western blot
analysis using a caspase-3 monoclonal antibody, and confirmed that
CRTP3 significantly inhibited the cleavage of caspase-3 in a
concentration-dependent manner following exposure to hypoxia/SD
(apoptotic control, 6.17±0.17 vs. treated cells, 2.63±0.34)
(Fig. 2C and D).
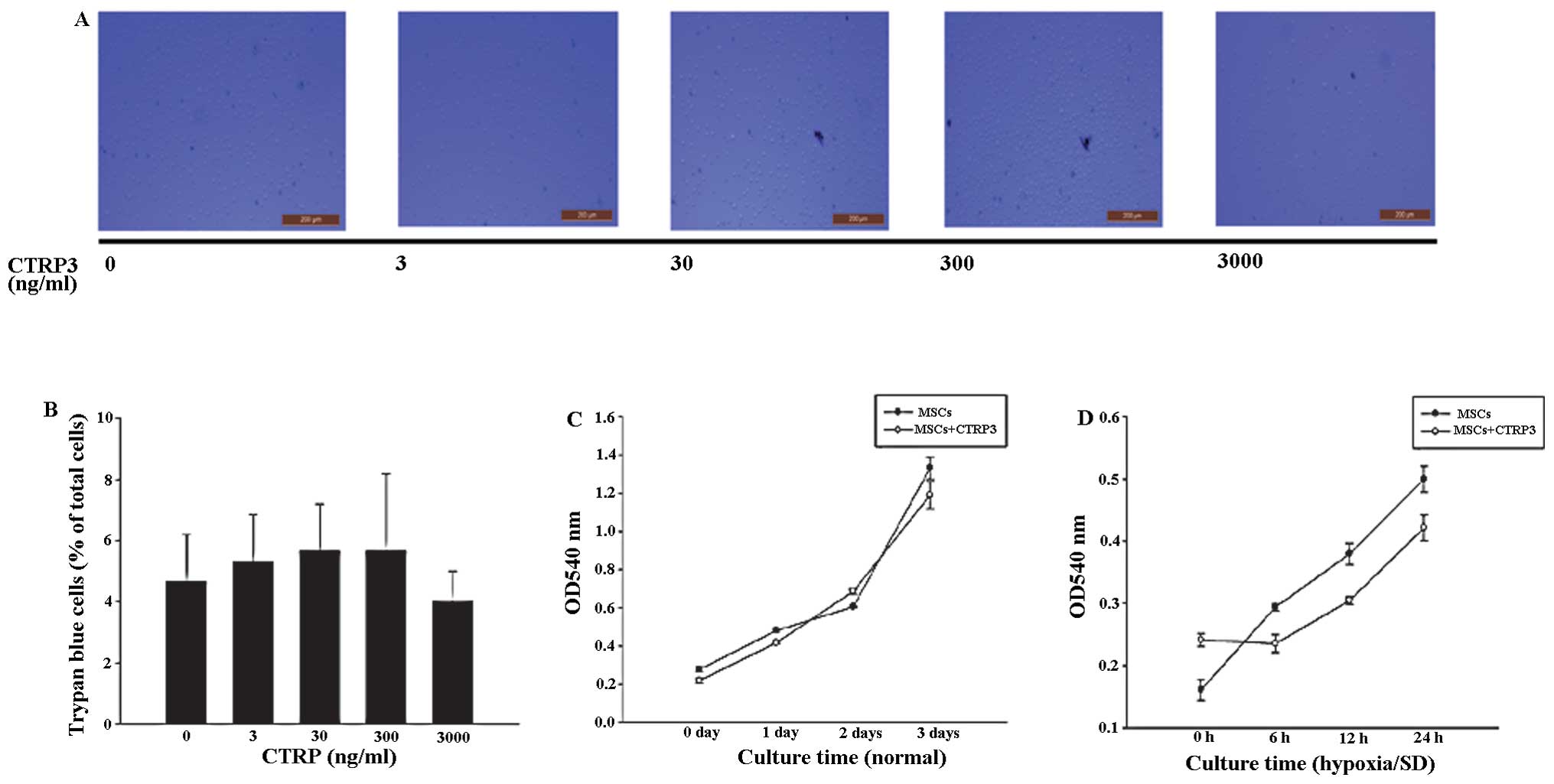
To the best of our knowledge, the potential toxicity
of CTRP3 to MSCs at the tested concentrations has not been
previously addressed. We therefore examined the effects of CTRP3 on
MSC viability. Trypan blue assays indicated that up to 3,000 ng/ml
of CTRP3 had no adverse effects on the viability of MSCs (Fig. 3A and B).
Apoptosis occurred over several hours, and we
therefore measured cell proliferation, which could interfere with
the measurements of the apoptotic process. The cell proliferation
rate, as assessed by CCK-8 assay, showed no significant change
during the 3 days of treatment with 300 ng/ml CTRP3, either under
normal culture or under 24-h hypoxia/SD culture conditions
(Fig. 3C and D).
CTRP3 protects MSCs from
hypoxia/SD-induced apoptosis through the PI3K/Akt pathway
The PI3K/Akt signaling pathway is considered to be
important in promoting survival in numerous cell types (21–23). We therefore investigated the
potential involvement of this pathway in the anti-apoptotic effects
of CTRP3 in MSCs. Western blot analysis revealed low detectable
levels of phospho-Akt in the untreated control cells. Akt
phosphorylation was increased by CTRP3 in a time-dependent manner,
peaking at 60 min [Akt (Ser473) 60 min, 4.57±0.16 vs. 0 min,
1.00±0.00; Akt (Tyr308) 60 min, 4.47±0.33 vs. 0 min, 1.00±0.00] and
remained detectable for up to 90 min (Fig. 4A and B). To determine the role of
the PI3K/Akt signaling pathway in the CTRP3-induced protective
effects against hypoxia/SD, MSCs were treated with the specific
PI3K/Akt inhibitor, LY294002. The CTRP3-induced phosphorylation of
Akt was inhibited by LY294002 (25 μM) [Akt (Ser473) with LY294002,
22.65±2.51 vs. without LY294002, 73.20±3.02%; Akt (Tyr308) with
LY294002, 18.79±1.09 vs. without LY294002, 63.47±3.21%] (Fig. 4C and D). In addition, FACS
analysis revealed that LY294002 (25 μM) completely blocked the
anti-apoptotic effects of CTRP3 (with LY294002, 6.56±0.11 vs.
without LY294002, 3.13±0.56) (Fig. 4E
and F).
CTRP3 exerts anti-apoptotic effects by
inhibiting the activation of the mitochondrial pathway
To determine whether CTRP3 exerts its anti-apoptotic
effects by inhibiting the activation of the mitochondrial pathway
under hypoxia/SD conditions, we examined its effects on the
Bcl-2/Bax ratio, the release of cytochrome c, caspase-3
activation and the mitochondrial membrane potential. CTRP3
increased the Bcl-2/Bax ratio (with treatment, 3.07±0.12 vs.
without treatment, 0.43±0.09) (Fig.
5A), inhibited the release of cytochrome c (without
treatment, 0.51±0.04 vs. with treatment, 2.95±0.06) (Fig. 5B) and inhibited the activation of
caspase-3 (with treatment, 2.44±0.18 vs. without treatment,
5.81±0.28) (Fig. 5C). In a
parallel experiment, CTRP3 reversed the decrease in mitochondrial
membrane potential that was induced by hypoxia/SD (Fig. 5D). The PI3K/Akt inhibitor,
LY294002 (25 μM), blocked the inhibitory effects of CTRP3 on the
mitochondrial pathway (Fig.
5).
Discussion
Autologous MSCs can be easily prepared from adult
patients and are immunologically safe. They therefore offer great
advantages for regenerating and repopulating the injured myocardium
and restoring its function when transplanted into ischemic or
infarcted hearts (24). However,
despite the implantation of large numbers of cells, the ratio of
MSC engraftment remains low due to poor cell survival within the
ischemic environment that they are introduced into (6,21).
The results of the present study suggest that CTRP3 represents a
good candidate for protecting MSCs from apoptosis induced by
hypoxia/SD; this protective effect is mediated by the activation of
the PI3K/Akt signaling pathway and the inhibition of the
mitochondrial apoptotic pathway.
CTRP3 is a key member of the family of adipokines
and has broad functions, not only in adipokine secretion and
metabolism, but also in inflammation, cell proliferation,
differentiation, apoptosis and cardiac protection (13–17). Recent studies have found that the
plasma levels of CTRP3 were decreased in patients with cardiac
infarction. In addition, the replenishment of CTRP3 has been shown
to attenuate cardiac remodeling and dysfunction and to protect
cardiomyocytes from apoptosis in a model of cardiac infarction
(13–17). It is possible that the
replenishment of CTRP3 in the infarcted myocardium may offer
cardioprotection through its ability to influence cell survival,
and could potentially enhance MSC survival through the activation
of pro-survival signaling pathways. As regards apoptosis, CTRPs
have been confirmed to exert strong anti-apoptotic effects in
various types of cells (17,25), and have been shown to protect
cardiomyocytes from apoptosis in cardiac infarction through the
PI3K/Akt pathway (17). The
PI3K/Akt pathway further displays significant anti-apoptotic
effects in MSCs (20,22,23). We thus hypothesized that CTRP3 may
protect MSCs from apoptosis induced by hypoxia/SD.
In preliminary experiments, early apoptosis in MSCs
induced by hypoxia/SD peaked at 24 h. Using the same model, we
found that CTRP3 protected MSCs from apoptosis in a
concentration-dependent manner. However, CTRP3 only inhibited the
early hypoxia/SD-induced apoptosis of MSCs, and had no effect on
late apoptosis or necrosis. CTRP3 had no apparent effect on MSC
proliferation, thus eliminating the potential interference between
the effects of proliferation and apoptosis.
In the present study, we examined the role of the
PI3K/Akt pathway in MSC survival and in modulating the
anti-apoptotic effects of CTRP3. PI3K/Akt is an important
pro-survival pathway, and the phosphorylation of Akt plays a major
role in anti-apoptotic functions in various types of cells
(26–29). CTRP3 has been shown to protect
cardiomyocytes from apoptosis through the PI3K/Akt pathway
(17), and this pathway has been
shown to exert a significant protective effect against MSC
apoptosis (21–23). Our results revealed that CTRP-3
stimulated Akt phosphorylation and prevented MSCs from
hypoxia/SD-induced apoptosis, while inhibition experiments with the
PI3K inhibitor, LY294002, indicated that CTRP3 protected MSCs from
apoptosis via the PI3K/Akt pathway.
A previous study demonstrated that hypoxia/SD
induces apoptosis in MSCs through the mitochondrial apoptotic
pathway (19). The present study
demonstrated that this process can be inhibited by CTRP3, which
inhibited hypoxia/SD-induced mitochondrial-dependent apoptosis by
increasing the Bcl-2/Bax ratio and the mitochondrial membrane
potential, and by inhibiting the release of cytochrome c and
the activation of caspase-3; these effects were mediated by the
PI3K/Akt pathway. The above results were confirmed by the fact that
the effects of CTRP3 on the mitochondrial apoptotic pathway were
blocked by the PI3K inhibitor, LY294002. The finding that CTRP3
affected early apoptosis, but had no effect on late apoptosis or
necrosis, is in accordance with a previous study that demonstrated
the involvement of the mitochondrial apoptotic pathway in the early
apoptotic process (30).
In conclusion, the results of the present study
suggest that CTRP3 promotes MSC survival under conditions mimicking
the ischemic myocardium. CTRP3 protects MSCs from
hypoxia/SD-induced mitochondrial apoptosis through the PI3K/Akt
signaling pathway. These findings highlight a potential novel
therapeutic strategy for protecting MSCs from apoptosis, and form
the basis for the future clinical exploitation of CTRP3 and MSCs in
cardiac regeneration therapies.
Acknowledgements
We thank Dr Wei Liu for her expert assistance with
the experimental design and excellent technical assistance, and Dr
Bo Sun for her assistance with the FACS analysis. Dr Wei Liu and Dr
Bo Sun are members of the Key Laboratory of Myocardial Ischemia
Mechanism and Treatment (Harbin Medical University), Ministry of
Education. This study was supported by grants from the Key
Laboratory of Myocardial Ischemia, Harbin Medical University,
Ministry of Education (no. KF201315).
References
|
1
|
Lopez AD, Mathers CD, Ezzati M, Jamison DT
and Murray CJ: Global and regional burden of disease and risk
factors, 2001: systematic analysis of population health data.
Lancet. 367:1747–1757. 2006. View Article : Google Scholar
|
|
2
|
Orlic D, Kajstura J, Chimenti S, et al:
Bone marrow cells regenerate infarcted myocardium. Nature.
410:701–705. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stamm C, Westphal B, Kleine HD, et al:
Autologous bone-marrow stem-cell transplantation for myocardial
regeneration. Lancet. 361:45–46. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wollert KC, Meyer GP, Lotz J, et al:
Intracoronary autologous bone-marrow cell transfer after myocardial
infarction: the BOOST randomised controlled clinical trial. Lancet.
364:141–148. 2004. View Article : Google Scholar
|
|
5
|
Zhang M, Methot D, Poppa V, Fujio Y, Walsh
K and Murry CE: Cardiomyocyte grafting for cardiac repair: graft
cell death and anti-death strategies. J Mol Cell Cardiol.
33:907–921. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geng YJ: Molecular mechanisms for
cardiovascular stem cell apoptosis and growth in the hearts with
atherosclerotic coronary disease and ischemic heart failure. Ann NY
Acad Sci. 1010:687–697. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chao W, Shen Y, Li L and Rosenzweig A:
Importance of FADD signaling in serum deprivation- and
hypoxia-induced cardiomyocyte apoptosis. J Biol Chem.
277:31639–31645. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bonavita F, Stefanelli C, Giordano E, et
al: H9c2 cardiac myoblasts undergo apoptosis in a model of ischemia
consisting of serum deprivation and hypoxia: inhibition by PMA.
FEBS Lett. 536:85–91. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kishore U, Gaboriaud C, Waters P, et al:
C1q and tumor necrosis factor superfamily: modularity and
versatility. Trends Immunol. 25:551–561. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wong GW, Wang J, Hug C, Tsao TS and Lodish
HF: A family of Acrp30/adiponectin structural and functional
paralogs. Proc Natl Acad Sci USA. 101:10302–10307. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schaffler A, Ehling A, Neumann E, et al:
Genomic organization, promoter, amino acid sequence, chromosomal
localization, and expression of the human gene for CORS-26
(collagenous repeat-containing sequence of 26-kDa protein). Biochim
Biophys Acta. 1630:123–129. 2003. View Article : Google Scholar
|
|
12
|
Weigert J, Neumeier M, Schaffler A, et al:
The adiponectin paralog CORS-26 has anti-inflammatory properties
and is produced by human monocytic cells. FEBS Lett. 579:5565–5570.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Compton SA and Cheatham B: CTRP-3:
blocking a toll booth to obesity-related inflammation.
Endocrinology. 151:5095–5097. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kopp A, Bala M, Buechler C, et al:
C1q/TNF-related protein-3 represents a novel and endogenous
lipopolysaccharide antagonist of the adipose tissue. Endocrinology.
151:5267–5278. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kopp A, Bala M, Weigert J, et al: Effects
of the new adiponectin paralogous protein CTRP-3 and of LPS on
cytokine release from monocytes of patients with type 2 diabetes
mellitus. Cytokine. 49:51–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hofmann C, Chen N, Obermeier F, et al:
C1q/TNF-related protein-3 (CTRP-3) is secreted by visceral adipose
tissue and exerts antiinflammatory and antifibrotic effects in
primary human colonic fibroblasts. Inflamm Bowel Dis. 17:2462–2471.
2011. View Article : Google Scholar
|
|
17
|
Yi W, Sun Y, Yuan Y, et al: C1q/tumor
necrosis factor-related protein-3, a newly identified adipokine, is
a novel antiapoptotic, proangiogenic, and cardioprotective molecule
in the ischemic mouse heart. Circulation. 125:3159–3169. 2012.
View Article : Google Scholar
|
|
18
|
Pittenger MF, Mackay AM, Beck SC, et al:
Multilineage potential of adult human mesenchymal stem cells.
Science. 284:143–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu W, Chen J, Cong X, Hu S and Chen X:
Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem
cells. Stem Cells. 24:416–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen J, Baydoun AR, Xu R, et al:
Lysophosphatidic acid protects mesenchymal stem cells against
hypoxia and serum deprivation-induced apoptosis. Stem Cells.
26:135–145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mangi AA, Noiseux N, Kong D, et al:
Mesenchymal stem cells modified with Akt prevent remodeling and
restore performance of infarcted hearts. Nat Med. 9:1195–1201.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hahn JY, Cho HJ, Kang HJ, et al:
Pre-treatment of mesenchymal stem cells with a combination of
growth factors enhances gap junction formation, cytoprotective
effect on cardiomyocytes, and therapeutic efficacy for myocardial
infarction. J Am Coll Cardiol. 51:933–943. 2008. View Article : Google Scholar
|
|
23
|
Lu G, Haider HK, Jiang S and Ashraf M:
Sca-1+ stem cell survival and engraftment in the
infarcted heart: dual role for preconditioning-induced connexin-43.
Circulation. 119:2587–2596. 2009.
|
|
24
|
Orlic D, Kajstura J, Chimenti S, et al:
Mobilized bone marrow cells repair the infarcted heart, improving
function and survival. Proc Natl Acad Sci USA. 98:10344–10349.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li Q, Wang L, Tan W, et al: Identification
of C1qTNF-related protein 4 as a potential cytokine that stimulates
the STAT3 and NF-κB pathways and promotes cell survival in human
cancer cells. Cancer Lett. 308:203–214. 2011.PubMed/NCBI
|
|
26
|
Zhu P, Tan MJ, Huang RL, et al:
Angiopoietin-like 4 protein elevates the prosurvival intracellular
O2(−):H2O2 ratio and confers
anoikis resistance to tumors. Cancer Cell. 19:401–415. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Albrecht-Schgoer K, Schgoer W, Holfeld J,
et al: The angiogenic factor secretoneurin induces coronary
angiogenesis in a model of myocardial infarction by stimulation of
vascular endothelial growth factor signaling in endothelial cells.
Circulation. 126:2491–2501. 2012. View Article : Google Scholar
|
|
28
|
Kanwar JR, Kamalapuram SK and Kanwar RK:
Survivin signaling in clinical oncology: a multifaceted dragon. Med
Res Rev. 33:765–789. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shortt J, Martin BP, Newbold A, et al:
Combined inhibition of PI3K-related DNA damage response kinases and
mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood.
121:2964–2974. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu R, Chen J, Cong X, Hu S and Chen X:
Lovastatin protects mesenchymal stem cells against hypoxia- and
serum deprivation-induced apoptosis by activation of PI3K/Akt and
ERK1/2. J Cell Biochem. 103:256–269. 2008. View Article : Google Scholar
|