Instructions
Neuroblastoma, the most common extracranial solid
tumor in children, is a heterogeneous tumor that arises from the
neural crest (1). Patients with
neuroblastoma account for ~15% of childhood fatalities from cancer.
At the time of diagnosis, >70% of patients have metastatic
disease (2,3). The disease displays a remarkable
clinical diversity, ranging from spontaneous regression to fatal
progression and dissemination to privileged sites, such as
bone-marrow and liver (4,5). However, the molecular mechanisms
and/or intrinsic factors controlling neuroblastoma cancer
metastasis are not well understood.
The tumor microenvironment plays a crucial role in
orchestrating immune cell effectors/modulators, pro- and
anti-inflammatory cytokines, and chemokine production. The tumor
microenvironment does this by impacting, integrating and subverting
the immunity and inflammatory processes (6–9).
Numerous studies have demonstrated that the tumor microenvironment
not only responds to and supports carcinogenesis, but also actively
contributes to tumor initiation, progression and metastasis
(10). Mediators of inflammation
have long been known to increase metastatic dissemination (11,12). Furthermore, inflammatory cytokines
in the tumor microenvironment promote nuclear factor-κB (NF-κB)
signaling pathways activation, which may induce the expression of
several genes associated with malignant transformation (13).
Previously, it has become clear that NF-κB signaling
also has a critical role in cancer development and progression
(14). NF-κB actions as a dimer
composed of the RelA (p65) and NF-κB1 (p50) or NF-κB2 (p52)
subunits. In normal resting cells, NF-κB is sequestered in the
cytoplasm through binding to IκB. NF-κB activation involves its
release from its inhibitor, IκB, and its subsequent translocation
from the cytoplasm to the nucleus, where it binds to cognate
sequences in the promoter region of multiple genes. Regulating gene
expression by NF-κB is controlled mainly by the inhibitory IκB
proteins, which include IκB-α. Upon stimulation, IκB-α is rapidly
phosphorylated and degraded via the ubiquitin-proteasome pathway,
permitting activation and nuclear import of NF-κB (15). NF-κB was also shown to induce the
expression of CXCR4 (16).
Chemokines and their receptors were originally
described as essential mediators of leukocyte directional
migration, and have further emerged as crucial elements in all the
stages of tumor development (17–19). The binding of chemokines to their
cognate receptors elicits typical cellular responses, such as
directional migration. CXCR4 is the most frequently expressed
chemokine receptor on the tumor cells (20). The CXCR4 ligand is the small
chemokine SDF-1α. In addition to its critical role in tumor cell
growth, survival and angiogenesis in multiple cancers, the
CXCR4/SDF-1α pair has been shown to mediate homing and metastatic
secondary growth in SDF-1α-producing organs, such as liver and bone
marrow (21–23) and a study indicates that
neuroblastoma cells are equipped with a bone marrow homing system
that may mediate the establishment of bone marrow metastasis by
neuroblastoma cells (24).
To elaborate the role of the tumor microenvironment,
particularly the inflammatory cytokines in neuroblastoma cells
pathobiology, the NF-κB signaling activation was examined as a
potential mechanism by which cell metastasis is fostered. In the
present study, the expression of CXCR4 and NF-κB was examined in a
subset of neuroblastoma tumors and correlative analyses were
conducted with clinicopathological variables. Furthermore, the
inflammatory cytokine TNF-α acts as a regulator of functional CXCR4
expression on neuroblastoma cells in a NF-κB-dependent manner. The
inhibitor of NF-κB reduced CXCR4 expression on neuroblastoma cells
and resulted in decreased migriation towards SDF-1α in response to
TNF-α. Finally, the interaction of the tumor cells with macrophages
was shown to enhance CXCR4 expression in the neuroblastoma cells in
an NF-κB-dependent manner.
Materials and methods
Patients and tissue specimens
A total of 80 clinical neuroblastoma samples and 15
ganglioneuroma samples were collected from patients who had
undergone surgical resection at the Department of Pediatric
Surgery, The Affiliated Hospital of Qingdao University (Qingdao,
Shandong, China) between January 2006 and November 2013. The
patients who had undergone preoperative chemotherapy and
radiotherapy were not included. Hematoxylin and eosin-stained
slides from all the cases were reviewed to confirm the diagnoses.
All the neuroblastoma samples were classified according to the
International Neuroblastoma Staging System (INSS) classification
criteria (25). The 80 patients
included 48 males and 32 females aged from 1 month to 11 years
(median, 5 years). The clinicopathological characteristics of the
80 patients are summarized in Table
I. The patient consent was obtained, and the study was approved
by the Institutional Ethics Review Committee of the Affiliated
Hospital of Qingdao University.
 | Table INF-κB-p65 and CXCR4 expression in
neuroblastoma and their association with the clinicopathological
characteristics. |
Table I
NF-κB-p65 and CXCR4 expression in
neuroblastoma and their association with the clinicopathological
characteristics.
|
Characteristics | Total cases, n | NF-κB-p65
expression
| CXCR4 expression
|
|---|
| Low (n=32) | High (n=48) | P-value | Low (n=23) | High (n=57) | P-value |
|---|
| Age, years |
| <5 | 31 | 15 | 16 | 0.249 | 8 | 23 | 0.800 |
| ≥5 | 49 | 17 | 32 | | 15 | 34 | |
| Gender |
| Male | 48 | 17 | 31 | 0.356 | 11 | 37 | 0.209 |
| Female | 32 | 15 | 17 | | 12 | 20 | |
| Tumor size, cm |
| <3 | 41 | 23 | 28 | 0.244 | 10 | 31 | 0.461 |
| ≥3 | 39 | 9 | 20 | | 13 | 26 | |
| INSS stages |
| I, IIa, IVs | 21 | 13 | 8 | 0.021a | 12 | 9 | 0.0016a |
| IIb, III, IV | 59 | 19 | 40 | | 11 | 48 | |
| Metastasis |
| Absence | 18 | 12 | 6 | 0.013a | 11 | 7 | 0.002a |
| Presence | 62 | 20 | 42 | | 12 | 50 | |
Immunohistochemistry (IHC)
Immunohistochemistry for RELA (P65) and CXCR4 was
performed. All the specimens were fixed with 4% formaldehyde,
paraffin-embedded and cut into 4-μm serial sections.
Following deparaffinization and rehydration, tissue slides were
cooked to retrieve the antigen in a microwave oven with 10 mM
citrate buffer (pH 6) for 15 min. Endogenous peroxidase activity
was blocked with 3% hydrogen peroxide for 10 min at room
temperature and washed with phosphate-buffered saline (PBS).
Subsequently, the tissue slides were incubated with a purified
rabbit polyclonal antibody against RELA (P65) (1:800, Cell
Signaling Technology, USA) or CXCR4 (1:50, Abcam Cambridge, MA) at
4°C overnight. Following washing with PBS, the sections were
incubated with secondary antibodies (goat anti-rabbit polyclonal
antibody) for 30 min at 37°C. The sections were subsequently washed
three times with PBS and treated with diaminobenzidine. Finally,
the slides were counterstained with hematoxylin, dehydrated and
mounted. Negative controls were probed with PBS under the same
experimental conditions.
Histological assessment
All the samples were evaluated in a blinded manner
by two independent pathologists without knowledge of any other
clinicopathological data. The score method to evaluate the
immunostaining results was performed by multiplying the stain
intensity by the stain area. The stain intensity score was as
follows: No staining (score 0), weak staining (score 1), moderate
staining (score 2) or strong staining (score 3). The percentage of
the extent of reactivity was scored as follows: <25% (score 1),
25–50% (score 2), 50–75% (score 3) or >75% (score 4) of tumor
cells. The total expression of NF-κB-p65 and CXCR4 was determined
as either negative or low expression (−), score ≤2; or positive or
high expression (+), score ≥3.
Cell culture and reagents
The human malignant neuroblastoma SH-SY5Y cell lines
and human monocytic cell line THP-1 were obtained from the Cell
Bank of Type Culture Collection of Chinese Academy of Science
(Shanghai, China). The cell lines were maintained in Dulbecco’s
Modified Eagle’s Medium (DMEM; Hyclone, Logan, USA) supplemented
with 10% fetal bovine serum (FBS; Hyclone), and the cells were
cultured at 37°C with 5% CO2 in humidified atmosphere.
Recombinant human TNF-α (Sigma-Aldrich, St. Louis, MO, USA) was
dissolved in 0.1% bovine serum albumin (BSA) in PBS, and stock
solution (10 μg/ml) was stored at −20°C. Recombinant human
SDF-1α (Life Technologies, Carlsbad, CA, USA) was prepared in 0.1%
BSA in PBS and stock solution (100 μg/ml) was stored at 4°C.
Phorbol myristate acetate (PMA), a THP-1 cells inducer
(Sigma-Aldrich), was prepared in PBS (320 nM) and maintained at 4°C
until required. Pyrrolidinedithiocarbamic acid ammonium salt
(PDTC), a NF-κB (p65) inhibitor (3) (Sigma-Aldrich) was prepared in PBS
(50 μM) and maintained at 4°C until required.
Co-culture of macrophage and SH-SY5Y cell
lines
THP-1 cells (1×106 per well) were seeded
into the upper insert of a 6-well transwell apparatus with 0.4
μm pore size (Corning Costar, Rochester, NY, USA) and
treated with 320 nM PMA for 24 h to generate macrophages. Following
a thorough wash to remove all PMA, PMA-treated THP-1 macrophages
were co-cultured with SH-SY5Y cells (in a 6-well plate;
1×106 cells per well) without direct contact. In the
co-culture system, SH-SY5Y cells were cultured with
THP-1-differentiated macrophages for 24 h and harvested for use in
subsequent experiments.
Inhibition of the NF-κB signaling pathway
in the co-culture system
A concentration of 50 μM PDTC was added to
the macrophage/cancer cell co-cultures. After incubation for 24 h,
the SH-SY5Y cells were harvested for RNA and protein extraction,
then reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) and western blots were performed for CXCR4 and NF-κB-p65
quantification. Co-culture of SH-SY5Y and TAM without PDTC was the
positive control and culture of SH-SY5Y alone without PDTC was the
negative control.
RT-qPCR
Total RNA was isolated from the cell lines by Trizol
(Takara, Dalian, China) according to the manufacturers’
instructions. cDNA was synthesized using the Takara Prime Script RT
reagent kit (Takara) in a total volume of 10 μl, containing
2 μl 5× Prime Script buffer, 0.5 μl Prime Script RT
Enzyme mix l, 0.5 μl Oligo dT Primer (50 μM), 0.5
μl Random 6 mers (100 μM) and 6.5 μl total
RNA. The conditions for RT were: 37°C for 15 min and 85°C for 5
sec. RT-qPCR was performed using the LightCycler system together
with the LightCycler DNA Master SYBR Green I kit (Takara). The
total volume was 20 μl, containing 10μl SYBR Premix
Ex Taq II (2×), 0.8 μl PCR Forward Primer (10 μM),
0.8 μl PCR Reverse Primer (10 μM), 2.0 μl
template (<100 ng) and 6.4 μl dH2O. The
conditions for PCR were: 1 cycle at 95°C for 30 sec, and
subsequently 40 cycles at 95°C for 5 sec and 20 sec at 60°C. The
reference gene, glyceraldehydes-3-phosphate dehydrogenase
(GAPDH) was used as an internal control. Gene expression was
quantified by the comparative CT method, normalizing CT values to
GAPDH and calculating the relative expression values. Primer
sequences were as follows: CXCR4, forward
5′-TGGCTGAAAAGGTGGTCTAG-3′, and reverse 5′-GATGCTGATCCCAATGTAGT-3′.
The amplification fragment was 333 basepairs (bp). GAPDH was
used as the internal control, and the primer sequence was forward,
5′-TCATGGGTGAACCATGAGAATG-3′, and reverse
5′-GGCATGGACTGTGGTCATGAG-3′. The amplification fragment was 146
bp.
Western blot analysis
The cells were washed twice with cold PBS (pH 7.0),
and lysed in radioimmunoprecipitation assay buffer [150 mM NaCl, 1%
Nonidet P-40, 1% deoxycholate, 0.1% SDS and 10 mM Tris-HCl (pH
8.0)] supplemented with protease inhibitors. The protein
concentration of each sample was assayed using the bicinchoninic
acid method kit (Pierce, Rockford, IL, USA). Equal amounts of
protein (50 μg) were subjected to SDS-PAGE on a 10% gel.
Subsequently, the protein was blotted onto a polyvinylidene
fluoride membrane. After blocking with 5% skimmed milk in 20 mM TBS
with 0.1% Tween for 1 h at room temperature with agitation, the
proteins were incubated with the indicated primary antibodies at
4°C overnight followed by incubation in mouse anti-rabbit secondary
antibody conjugated with horseradish peroxidase (1:6000; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) for 1 h. The proteins
were detected using the Pierce ECL Western Blotting substrate
(Santa Cruz Biotechnology, Inc.), with a 15 min exposure after
washing the membrane for imaging with the LAS-3000 image analyzer
(Life Science, Fujifilm Global, Shanghai, China). The primary
antibodies employed included anti-β-actin (1:2000; Santa Cruz
Biotechnology, Inc.), anti-NF-κB-p65 (1:1000; Abcam, Cambridge, MA,
USA), anti-CXCR4 (1:2000; Abcam), anti-p-IκB-α (1:1000; Santa Cruz
Biotechnology, Inc.) and anti-IκB-α (1:1000; Santa Cruz
Biotechnology, Inc.).
Transwell migration assay
The assays were performed using a transwell (Corning
Costar) containing a polycarbonate membrane filter (8-μm
pore size) for 24-well plates according to the manufacturer’s
instructions. SH-SY5Y cells (5×105) were pretreated for
24 h with TNF-α (20 ng/ml), the cells that were not exposed to
TNF-α were used as the controls. Subsequently, the pretreated cells
were harvested and seeded into the upper surface of the filter with
a volume of 200 μl DMEM containing 2% FBS in the presence or
absence of PDTC (50 μM) and placed into the lower wells
containing 500 μl complete medium with or without SDF-1α
(100 ng/ml) to induce cell migration. The migration transwell
chambers were incubated for 8 h at 37°C. Following incubation, the
transwell chambers were washed twice with PBS and the cells on the
bottom surface of the membrane were fixed in 95% ethanol for 10 min
at room temperature, stained with 0.1% crystal violet for 30 min
and washed three times with PBS. The number of migration cells in
ten randomly selected microscopic fields (magnification, ×200) per
membrane was counted.
Statistical analysis
Statistical analysis was performed using the SPSS
software 17.0. The data are expressed as the mean ± standard
deviation. The Student’s t-test and one-way analysis of
variance test were used to compare data between the different
groups. The association between NF-κB-p65, CXCR4 expression and
clinicopathological parameters was analyzed using the χ2
test or the Fisher’s exact test. The possible association of
NF-κB-p65 and CXCR4 immunoreactivity was assessed using the
Fisher’s exact test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression of NF-kB-p65 and CXCR4 in
neuroblastoma compared to ganglioneuroma tissues
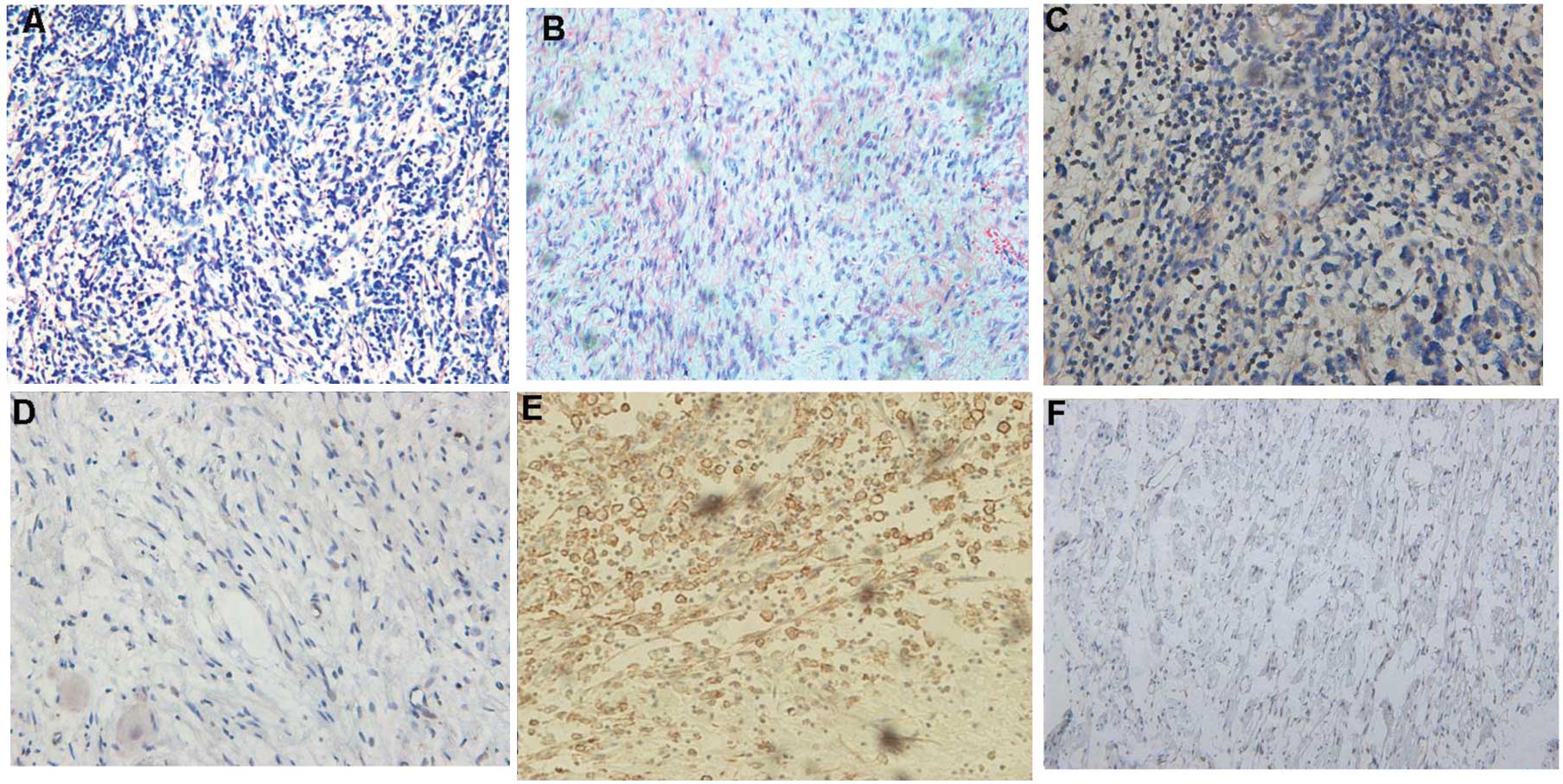
NF-kB-p65 protein showed cytoplasm and nucleus
staining, while CXCR4 was observed predominantly at the cell
membrane (Fig. 1). The expression
rate of NF-kB-p65 positive in the neuroblastoma group was 73.6%
(59/80), which was significantly higher than the rate of 20.0%
(3/15) in the ganglioneuroma group, and the difference between the
two groups was statistically significant (P=0.0001). The expression
rate of CXCR4 positive was 70.0% (56/80) in the neuroblastoma
group, which was significantly higher than the rate of 25.0% (3/12)
in the ganglioneuroma group, and the difference between the two
groups was statistically significant (P=0.0008) (Fig. 1, Table II).
| Table IIExpression of NF-κB-p65 and CXCR4 in
neuroblastoma compared to ganglioneuroma tissues. |
Table II
Expression of NF-κB-p65 and CXCR4 in
neuroblastoma compared to ganglioneuroma tissues.
| Variables | Total case, n | NF-κB-p65
expression
| CXCR4 expression
|
|---|
| Low, n | High, n | P-value | Low, n | High, n | P-value |
|---|
| Neuroblastoma | 80 | 21 | 59 | 0.0001 | 24 | 56 | 0.0008 |
| Ganglioneuroma | 15 | 13 | 2 | 12 | 3 | | |
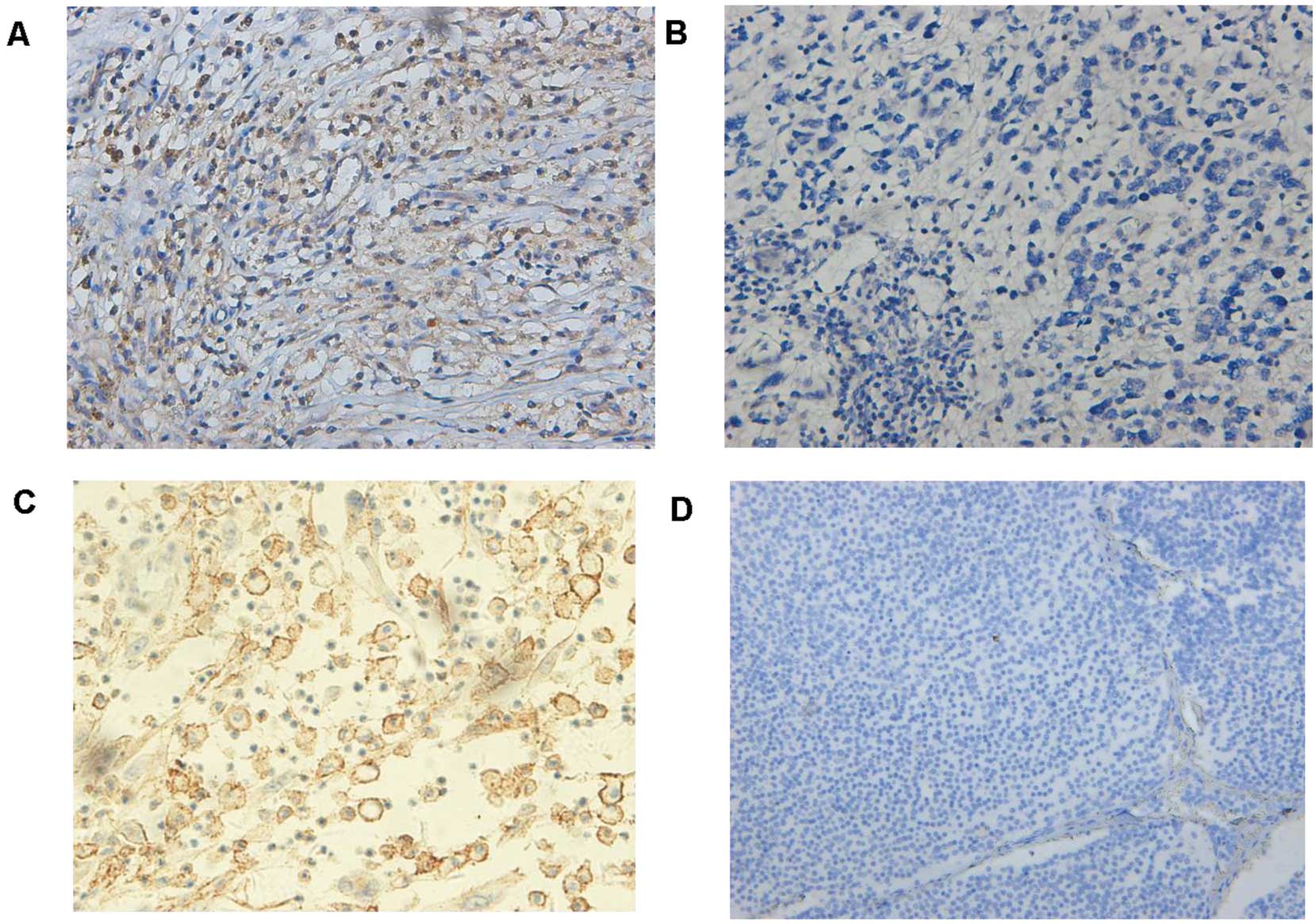
Expression of NF-kB-p65 and CXCR4 in
neuroblastoma and their association with clinicopathological
characteristics
The level of NF-kB-p65 and CXCR4 expression was
divided into the high and low groups according to the cut-off value
stated in the aforementioned methods. There were significantly
positive correlations between NF-kB-p65, CXCR4 expression and INSS
stage (P=0.021) or metastasis (P=0.013). However, no statistical
differences were found between clinicopathological factors (age,
gender and tumor size) and NF-kB-p65, CXCR4 expression (Table I). Notably, there was a positive
correlation between the expression of NF-kB-p65 and CXCR4 in the 80
neuroblastoma samples (P=0.0052, Fisher’s exact test) (Table III, Fig. 2).
| Table IIICorrelation of NF-κB-p65 and CXCR4
expression in neuroblastoma patient samples (P=0.0052). |
Table III
Correlation of NF-κB-p65 and CXCR4
expression in neuroblastoma patient samples (P=0.0052).
| NF-κB-p65 | CXCR4 positive,
n | CXCR4 negative,
n | Total, n |
|---|
| Positive | 21 | 18 | 39 |
| Negative | 9 | 32 | 41 |
| Total | 30 | 50 | 80 |
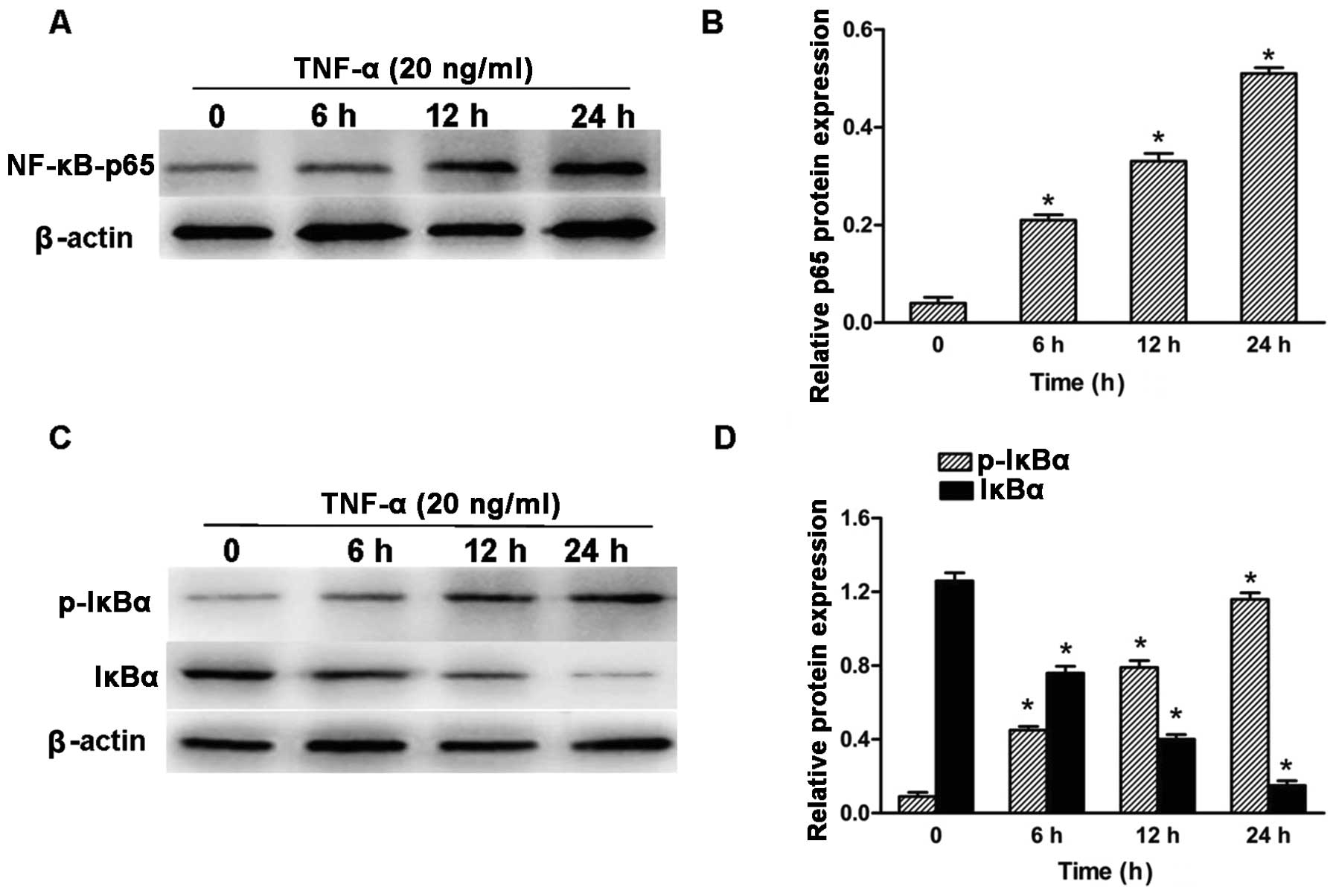
NF-kB contributes to TNF-α-mediated CXCR4
upregulation in neuroblastoma cells
As the levels of CXCR4 appeared to correlate with
NF-kB-p65 expression in neuroblastoma tumor samples, the role of
NF-κB-p65 on CXCR4 expression was investigated in vitro. In
response to an appropriate signal, the cytoplasmic inhibitor IκB-α
is phosphorylated on serine and degraded, thus dissociating from
the NF-κB (p65-p50) heterodimer. As a result, NF-κB heterodimer
translocates from the cytosol to the nucleus and induces the
expression of target genes containing NF-κB response elements
(24). Previous studies (26,27) have shown that TNF-α treatment of
various cell types stimulated NF-κB activation. To determine
whether TNF-α activated NF-κB signaling in human neuroblastoma
SH-SY5Y cells, SH-SY5Y neuroblastoma cells were cultured in the
presence of 20 ng/ml TNF-α for the indicated time and subsequently
the total cell lysates were collected and subjected to western blot
analysis for NF-κB (p65), phosphorylated-IκB-α and IκB-α. Following
exposure to TNF-α, SH-SY5Y cell lines showed nuclear translocation
of NF-κB (p65) in the indicated time (Fig. 3A and B). Exposure to TNF-α
increased IκB-α phosphorylation levels in the neuroblastoma SH-SY5Y
cell lines, which was accompanied by a marked decrease in IκB-α
protein expression (Fig. 3C and
D).
To further explore the role of TNF-α in the
upregulation of CXCR4 expression, SH-SY5Y neuroblastoma cells were
treated with TNF-α for various time and concentrations. RT-qPCR and
western blotting detection revealed that the expression of CXCR4
was upregulated significantly in a time- and dose-dependent manner
(Fig. 4A–D). The CXCR4 promoter
region has been shown to contain NF-κB response elements (16), thus, whether the NF-κB pathway
played a role in the induction of CXCR4 in response to TNF-α was
investigated. SH-SY5Y cells were pre-treated with PDTC, which is a
potent antioxidant inhibitor of NF-κB (28). The cells were subsequently treated
with 20 ng/ml TNF-α for 24 h. RT-qPCR and western blot analysis
showed that PDTC clearly inhibited TNF-α-induced CXCR4 expression
in SH-SY5Y cells (Fig. 4E and
F).
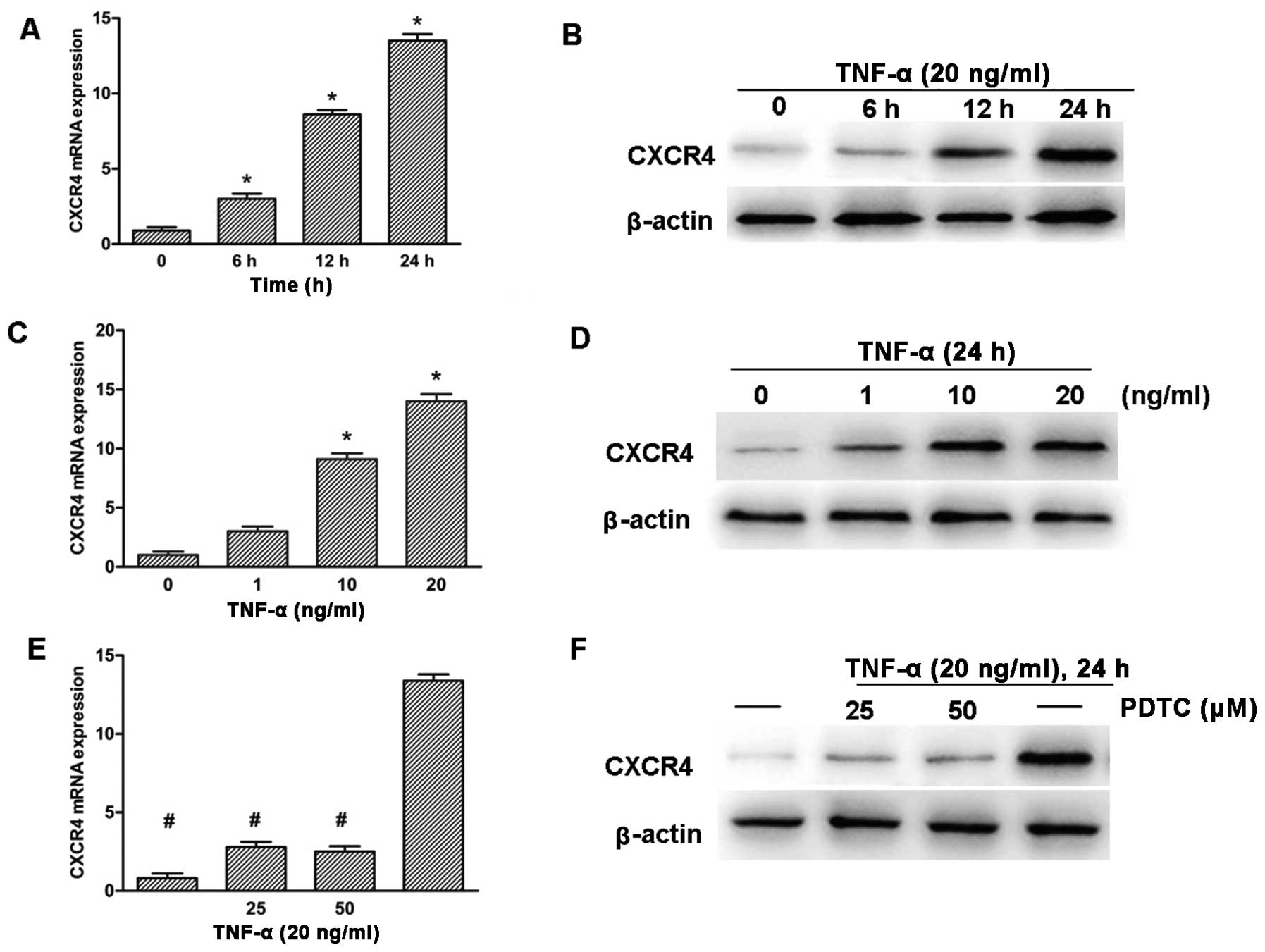 | Figure 4TNF-α promotes CXCR4 expression
through NF-κB pathway activition in SH-SY5Y cells. (A and B) Cells
were treated with TNF-α (20 ng/ml) for various time, as indicated.
(C and D) Cells were treated with TNF-α for various concentrations,
as indicated, for 24 h. (E and F) Cells were treated with NF-κB
inhibitor PDTC (25 and 50 μM) for 2 h and were subsequently
treated with TNF-α (20 ng/ml) for 24 h. CXCR4 expression was
analyzed by RT-qPCR (A, C and E) and western blot analysis (B, D
and F). GAPDH and β-actin were used as the loading control.
(*P<0.05 vs. untreated group, #P<0.05
vs. TNF-α-treated group). TNF-α, tumor necrosis factor-α; CXCR4,
CXC chemokine receptor-4; NF-κB, nuclear factor-κB; PDTC,
pyrrolidinedithiocarbamic acid ammonium salt; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
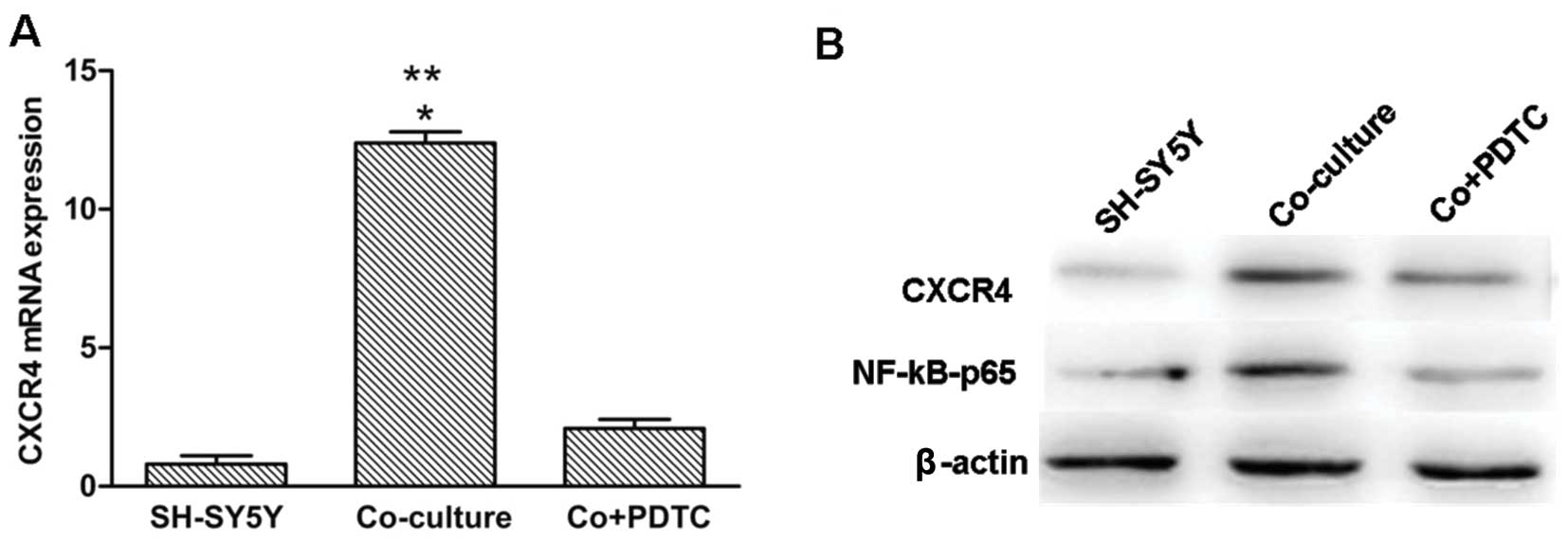
Upregulation of CXCR4 in SH-SY5Y cells is
mediated by the NF-κB signaling pathway in the co-culture
system
In the present study, PDTC, a specific inhibitor of
the NF-κB pathway was added to the macrophages/SH-SY5Y co-culture
system for 24 h at a concentration of 50 μM. This treatment
resulted in a significant reduction in CXCR4 mRNA and
protein levels in the co-cultured SH-SY5Y cells when compared to
the SH-SY5Y cells, either in the positive or negative control.
NF-κB P65 protein was significantly decreased in the SH-SY5Y cells
in the presence of PDTC (Fig. 5A and
B).
NF-κB mediates the migration towards
SDF-1α in neuroblastoma cells
To evaluate the expression of NF-kB in regulating
the migration of neuroblastoma cells towards SDF-1α, the transwell
migration assay was performed. As shown in Fig. 6, TNF-α pre-treated cells showed a
significant increase in migration towards SDF-1α as compared to
cells exposed to SDF-1α alone or TNF-α pre-treated without SDF-1α
in the lower well (Fig. 6B–D,
P<0.05). Following knockdown of NF-κB expression with PDTC, the
migration of the TNF-α pre-treated cells towards SDF-1α was
significantly decreased (Fig. 6D and
F, P<0.05).
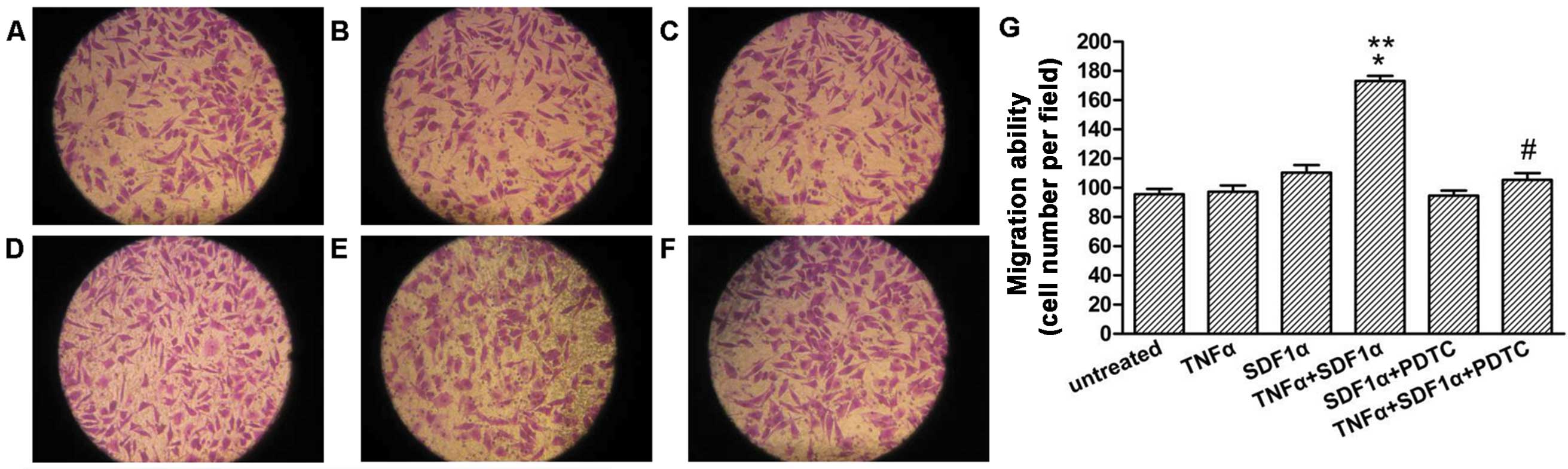 | Figure 6NF-κB mediates migration towards
SDF-1α in the SH-SY5Y cells. SH-SY5Y cells pretreated or untreated
with TNF-α (20 ng/ml) for 24 h were seeded in the migration
chambers in the presence or absence of PDTC (50 μM) and
placed into wells in the presence or absence of SDF-1α (100 ng/ml).
The cells were allowed to migrate for 24 h. Representative figures
indicate the average number of migrated cells per field. (A)
Untreated control. (B) TNF-α pretreated group. (C) Untreated
control, SDF-1α-exposed group. (D) TNF-α pretreated, SDF-1α-exposed
group. (E) PDTC pretreated group, SDF-1α-exposed group. (F) TNF-α,
PDTC pretreated, SDF-1α-exposed group. (G) Migration ability for
the different treatments. Results are representative of five
independent experiments. *P<0.05 vs. TNF-α pretreated
group; **P<0.05 vs. untreated, SDF-1α exposed group;
#P<0.05 vs. TNF-α pretreated, SDF-1α exposed group.
NF-κB, nuclear factor-κB; TNF-α, tumor necrosis factor-α; SDF-1α,
stromal cell-derived factor-1α; PDTC, pyrrolidinedithiocarbamic
acid ammonium salt. |
Discussion
Neuroblastoma is the most common malignant tumour in
infancy, its high degree of malignancy and early metastasis are
critical factors that affect the cure rate of neuroblastoma
patients. However, the molecular and cellular mechanisms regulating
neuroblastoma metastatic spread remain largely elusive.
Evidence indicating that inflammatory mediators
affect genetic stability and cause persistent epigenetic
alterations indicates that inflammatory components of the tumor
microenvironment impact on the fundamental mechanisms responsible
for the generation of metastatic variants. Inflammatory cytokines
produced by the tumor or inflammatory cells in the tumor
microenvironment promote tumor progression through the induction of
genes dependent on NF-κB signaling pathway (29–31).
The role of the NF-κB signaling system in connecting
inflammation and cancer is currently well accepted (32); furthermore, NF-κB is increasingly
recognized as a crucial element in numerous steps of cancer
initiation and progression. Elevated NF-κB activity is observed in
various types of cancer, including neuroblastoma (33). The activation of NF-κB induces the
expression of various molecules, including cyclooxygenase-2, matrix
metallopeptidase-9 and adhesion molecules, such as intracellular
adhesion molecule 1, vascular cell adhesion molecule 1 and
endothelial-leukocyte adhesion molecule 1, all of which have been
linked with cancer cell invasion and metastasis (34). The inhibition of NF-κB activity is
believed to suppress neuroblastoma cell migration and invasion.
TNF-α has been shown to induce NF-κB activation (35). The NF-κB complex is normally
confined to the cytosol through its interaction with the IκB
protein; upon stimulation, IκB is degraded and NF-κB is activated.
In the present study, IκB-α phosphorylation and NF-κB nuclear
translocation were observed in TNF-α-stimulated SH-SY5Y cells.
These results are in agreement with a previous study, which
revealed that TNF-α operates via activation of NF-κB pathways
(36).
The CXCR4 chemokine receptor, which has been closely
linked with cancer cell growth, invasion, angiogenesis and
metastasis, has been found to be overexpressed in various types of
tumor, including breast cancer, ovarian cancer, glioma, pancreatic
cancer, prostate cancer, acute myeloid leukemia as compared to
normal cells, which show little or no CXCR4 expression (37–39). A previous study indicated that a
CXCR4/SDF-1α axis may be involved in attracting neuroblastoma cells
to bone marrow, which was one of the favorable sites of metastasis
formation by neuroblastoma (24);
however, the mechanism responsible for its upregulation has not
been completely elucidated. Although what leads to the
overexpression of CXCR4 in cancer cells remains unclear, studies
point to genetic and microenvironmental factors (35). Hypoxia in the tumor
microenvironment (40) and NF-κB
(16) have been indicated in
CXCR4 overexpression. In the present study, there was a
significant positive correlation between the expression status of
NF-κB-p65 and that of CXCR4 in neuroblastoma tissues. TNF-α was
also shown to induce CXCR4 expression in neuroblastoma cells in a
time- and dose-dependent manner. In addition, blocking the NF-κB
pathway with PDTC suppressed TNF-α-induced CXCR4 expression. There
was another clear upregulation of CXCR4 expression in SH-SY5Y cells
following co-culture with macrophages, an alternative source of
TNF-α in the neuroblastoma microenvironment. Notably, this
upregulation was inhibited by the NF-κB inhibitor, PDTC.
Overexpression of CXCR4, whose involvement in
various human tumors is well known, was frequently observed in
neuroblastoma tissues to increase neuroblastoma metastasis. In the
present study, there was a marked increase in migration towards
SDF-1α in TNF-α pre-treated SH-SY5Y cells and the treatment with a
NF-κB inhibitor, PDTC, resulted in a significant suppression of
SH-SY5Y cell migration towards SDF-1α.
Taken together, these findings led to the conclusion
that TNF-α partially functions through the NF-κB signaling pathway
to upregulate CXCR4 expression to foster neuroblastoma
metastasis. Inflammatory factors in the tumor microenvironment
activated NF-κB; constitutive NF-κB activation further upregulates
major inflammatory factors, such as TNF-α, interleukin (IL)-6, IL-1
and IL-8, which are potent activators for NF-κB. Thus, it is
believed that NF-κB and inflammation constitute a positive feedback
loop to promote tumor cell survival and progression (41). However, the possibility of other
transcription factors, in addition to NF-κB, contributing to the
TNF-α-mediated upregulation of CXCR4 should be considered.
For instance, hepatocyte growth factor and hypoxia inducible
factor-1 are able to activate the transcription of CXCR4
(42).
Of note in the present study, the
immunohistochemical analysis revealed significantly higher
expression of NF-κB and CXCR4 in neuroblastoma tissues when
compared to ganglioneuroma tissues, which further supports the
increasing data that NF-κB and CXCR4 are abnormally expressed in
neuroblastoma cells. Furthermore, there were significant
correlations between the high level of NF-κB-p65, CXCR4 expression,
clinical metastasis and INSS stages, which indicated the utility of
NF-κB and CXCR4 as predictive biomarkers and therapeutic targets in
neuroblastoma.
In conclusion, the inflammatory factor, TNF-α,
promoted human SH-SY5Y cell metastasis through activation of NF-κB
and upregulation of CXCR4 expression. Inhibition of the
TNF-α-activated NF-κB pathway suppressed cell migration in the
SH-SY5Y cells.
The present data suggests that the TNF-α-activated
NF-κB/CXCR4/SDF-1α pathway may be a potential regulator of
neuroblastoma cell metastasis. Targeting inflammatory cytokines or
NF-κB signaling pathways, and ultimately CXCR4, may be a
therapeutic strategy in neuroblastoma.
Acknowledgments
The present study was supported by grants from the
Natural Science Foundation of China (NSFC 81272986 to Professor
Qian Dong).
References
|
1
|
Maris JM and Matthay KK: Molecular biology
of neuroblastoma. J Clin Oncol. 17:2264–2279. 1999.PubMed/NCBI
|
|
2
|
Aronson MR, Smoker WR and Oetting GM:
Hemorrhagic intracranial parenchymal metastases from primary
retroperitoneal neuroblastoma. Pediatr Radiol. 25:284–285. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ara T and DeClerck YA: Mechanisms of
invasion and metastasis in human neuroblastoma. Cancer Metastasis
Rev. 25:645–657. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ciccarone V, Spengler BA, Meyers MB,
Biedler JL and Ross RA: Phenotypic diversification in human
neuroblastoma cells: expression of distinct neural crest lineages.
Cancer Res. 49:219–225. 1989.PubMed/NCBI
|
|
5
|
Brodeur GM: Neuroblastoma: biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mantovani A, Marchesi F, Portal C,
Allavena P and Sica A: Linking inflammation reactions to cancer:
novel targets for therapeutic strategies. Adv Exp Med Biol.
610:112–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mantovani A, Garlanda C and Allavena P:
Molecular pathways and targets in cancer-related inflammation. Ann
Med. 42:161–170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu M and Polyak K: Microenvironmental
regulation of cancer development. Curr Opin Genet Dev. 18:27–34.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Balkwill F: Tumour necrosis factor and
cancer. Nat Rev Cancer. 9:361–371. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Giavazzi R, Garofalo A, Bani MR, et al:
Interleukin 1-induced augmentation of experimental metastases from
a human melanoma in nude mice. Cancer Res. 50:4771–4775.
1990.PubMed/NCBI
|
|
13
|
Ditsworth D and Zong WX: NF-kappaB: key
mediator of inflammation-associated cancer. Cancer Biol Ther.
3:1214–1216. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pahl HL: Activators and target genes of
Rel/NF-kappaB transcription factors. Oncogene. 18:6853–6866. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Helbig G, Christopherson KW 2nd,
Bhat-Nakshatri P, et al: NF-kappaB promotes breast cancer cell
migration and metastasis by inducing the expression of the
chemokine receptor CXCR4. J Biol Chem. 278:21631–21638. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baggiolini M, Dewald B and Moser B: Human
chemokines: an update. Annu Rev Immunol. 15:675–705. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rot A and von Andrian UH: Chemokines in
innate and adaptive host defense: basic chemokinese grammar for
immune cells. Annu Rev Immunol. 22:891–928. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vicari AP and Caux C: Chemokines in
cancer. Cytokine Growth Factor Rev. 13:143–154. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Balkwill F: Cancer and the chemokine
network. Nat Rev Cancer. 4:540–550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun X, Cheng G, Hao M, et al:
CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer
Metastasis Rev. 29:709–722. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Muller A, Homey B, Soto H, et al:
Involvement of chemokine receptors in breast cancer metastasis.
Nature. 410:50–56. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang J, Sun Y, Song W, Nor JE, Wang CY and
Taichman RS: Diverse signaling pathways through the SDF-1/CXCR4
chemokine axis in prostate cancer cell lines leads to altered
patterns of cytokine secretion and angiogenesis. Cell Signal.
17:1578–1592. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Geminder H, Sagi-Assif O, Goldberg L, et
al: A possible role for CXCR4 and its ligand, the CXC chemokine
stromal cell-derived factor-1, in the development of bone marrow
metastases in neuroblastoma. J Immunol. 167:4747–4757. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tabyaoui I, Tahiri-Jouti N, Serhier Z, et
al: Immunohistochemical expression of CD44s in human neuroblastic
tumors: Moroccan experience and highlights on current data. Diagn
Pathol. 8(39): 2013
|
|
26
|
Lu Y, Jeong YT, Li X, et al: Emodin
isolated from Polygoni cuspidati radix inhibits TNF-α and IL-6
release by blockading NF-κB and MAP kinase pathways in mast cells
stimulated with PMA plus A23187. Biomol Ther (Seoul). 21:435–441.
2013. View Article : Google Scholar
|
|
27
|
Hong GE, Kim JA, Nagappan A, et al:
Flavonoids identified from Korean Scutellaria baicalensis Georgi
inhibit inflammatory signaling by suppressing activation of NF-κB
and MAPK in RAW 264.7 cells. Evid Based Complement Alternat Med.
2013(912031): 2013
|
|
28
|
Dai L, Gu L, Ding C, Qiu L and Di W: TWEAK
promotes ovarian cancer cell metastasis via NF-kappaB pathway
activation and VEGF expression. Cancer Lett. 283:159–167. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aggarwal BB: Nuclear factor-kappaB: the
enemy within. Cancer Cell. 6:203–208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shishodia S and Aggarwal BB: Nuclear
factor-kappaB activation mediates cellular transformation,
proliferation, invasion angiogenesis and metastasis of cancer.
Cancer Treat Res. 119:139–173. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahn KS and Aggarwal BB: Transcription
factor NF-kappaB: a sensor for smoke and stress signals. Ann NY
Acad Sci. 1056:218–233. 2005. View Article : Google Scholar
|
|
32
|
Karin M: NF-kappaB as a critical link
between inflammation and cancer. Cold Spring Harb Perspect Biol.
1:a0001412009. View Article : Google Scholar
|
|
33
|
Okera M, Bae K, Bernstein E, et al:
Evaluation of nuclear factor kappaB and chemokine receptor CXCR4
co-expression in patients with prostate cancer in the Radiation
Therapy Oncology Group (RTOG) 8610. BJU Int. 108:E51–E58. 2011.
View Article : Google Scholar
|
|
34
|
Sethi G and Tergaonkar V: Potential
pharmacological control of the NF-kappaB pathway. Trends Pharmacol
Sci. 30:313–321. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kulbe H, Hagemann T, Szlosarek PW,
Balkwill FR and Wilson JL: The inflammatory cytokine tumor necrosis
factor-alpha regulates chemokine receptor expression on ovarian
cancer cells. Cancer Res. 65:10355–10362. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu Y and Zhou BP:
TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and
invasion. Br J Cancer. 102:639–644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Proudfoot AE: Chemokine receptors:
multifaceted therapeutic targets. Nat Rev Immunol. 2:106–115. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fernandis AZ, Prasad A, Band H, Klosel R
and Ganju RK: Regulation of CXCR4-mediated chemotaxis and
chemoinvasion of breast cancer cells. Oncogene. 23:157–167. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Murphy PM: Chemokines and the molecular
basis of cancer metastasis. N Engl J Med. 345:833–835. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schioppa T, Uranchimeg B, Saccani A, et
al: Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp
Med. 198:1391–1402. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Karin M: NF-kappaB and cancer: mechanisms
and targets. Mol Carcinog. 45:355–361. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Esencay M, Newcomb EW and Zagzag D: HGF
upregulates CXCR4 expression in gliomas via NF-kappaB: implications
for glioma cell migration. J Neurooncol. 99:33–40. 2010. View Article : Google Scholar : PubMed/NCBI
|