Introduction
Skeletal muscle accounts for the majority of
insulin-stimulated glucose uptake and is thus a key organ for the
development of insulin resistance. Disordered lipid metabolism is
considered a major contributor to skeletal muscle insulin
resistance (1). Impaired insulin
signaling, as observed in subjects with obesity and type 2
diabetes, is strongly associated with an excess accumulation of
triacylglycerols (TAGs) within muscle fibers in skeletal muscle
(2–5). TAGs are mainly stored in neutral
lipid droplets (LDs), coated by lipid droplet-associated proteins
which are referred to as perilipins (6,7).
One of the 5 perilipins (PLINs), PLIN2, is a marker for LDs in
human skeletal muscle, and the levels of intramuscular PLIN2 and
triglycerides are closely correlated. Studies have reported
increased TAG accumulation and lipid droplet formation when PLIN2
is overexpressed in vitro (8–10).
Conversely, the knockdown of PLIN2 in macrophages has been shown to
decrease the size and number of cellular lipids and LDs (8). A previous study using a rat model of
diabetes demonstrated that increased levels of Plin2 in skeletal
muscle correlated with insulin resistance when the rats were fed a
high-fat diet (11). Plin2
antisense oligonucleotides have been shown to protect mice from
insulin resistance when fed high-fat diets (12). These studies indicate a connection
between PLIN2 dysregulation and the development of insulin
resistance; however, the molecular mechanisms underlying this
response are not yet fully understood.
Inflammation also contributes to the development of
insulin resistance (13–15). The nucleotide-binding domain,
leucine-rich repeat containing protein 3 (NLRP3) inflammasome is a
group of protein complexes that sense a diverse set of host-derived
stimuli, such as damage-associated molecular patterns (DAMPs) and
control the production of important pro-inflammatory cytokines,
such as interleukin (IL)-1β (16). The NRLP3 inflammasome enhances
insulin resistance by triggering inflammation in adipose tissue in
subjects with obesity (16,17).
In this study, we found that PLIN2 overexpression
led to the activation of the NLRP3 inflammasome in C2C12 cells,
providing evidence that the PLIN2 inhibition of insulin-induced
glucose uptake is linked to both defects in lipid metabolism and to
inflammation.
Materials and methods
Cell culture
C2C12 mouse myoblasts (American Type Culture
Collection, Manassas, VA, USA) were cultured in Dulbecco's modified
Eagle's medium (DMEM; Sigma Aldrich, St. Louis, MO, USA)
supplemented with 20% fetal bovine serum (FBS; Atlanta Biologicals,
Flowery Branch, GA, USA), penicillin (100 U/ml) and streptomycin
(100 μg/ml) (both from Sigma-Aldrich). All cells were
maintained in a 5% CO2 incubator at 37°C. After the
cryopreserved cells were thawed, they were subcultured for at least
2 passages, and experiments were performed at 24 h
post-seeding.
Preparation of PLIN2 expression
constructs
Human PLIN2 cDNA, including the open reading frame
and 3′ untranslated region (UTR), was amplified using
PfuTurbo DNA polymerase (Agilent Technologies, Santa Clara,
CA, USA) with primers that included the 5′ XhoI and
XbaI restriction sites. The polymerase chain reaction (PCR)
primers were as follows: forward,
5′-ctcgagaagaaaaatggcatccgttgcagttgatcca-3′ and reverse,
5′-tctagaaactggtctatcctgcagtgaattttattgaattc-3′. The thermal
cycling conditions were as follows: an initial denaturation step at
95°C for 2 min, followed by 35 cycles of amplification (95°C for 30
sec, 60°C for 30 sec, and 72°C for 2 min), and a final extension at
72°C for 10 min. Afterwards, an A nucleotide base overhang was
generated by the addition of Ex Taq DNA polymerase (Clontech,
Mountain View, CA, USA). The PCR product was inserted into the
pGEM-T Easy Vector (Promega, Madison,WI, USA). For the construction
of the mammalian expression vectors, TA clones were subcloned into
the XhoI and XbaI sites in the pCS2+
vector, which was kindly provided by Dr Louis Kunkel (Division of
Genetics and Genomics, Boston Children's Hospital and Harvard
Medical School, Boston, MA, USA).
Cell transfection
To induce the overexpression of PLIN2 in the C2C12
cells, the cells (at 70% confluence) were transfected with the
pCS2+ vector expressing full-length human PLIN2
using Lipofectamine 2000 reagent (Life Technologies, Carlsbad, CA,
USA) according to the manufacturer's instructions. An empty
pCS2+ vector was transfected into the C2C12 cells as a
control. To reduce endogenous NLRP3 expression, the C2C12 cells
were transfected with 80 pmol of siRNA oligonucleotides (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) using Lipofectamine 2000
reagent. Non-targeted siRNA oligonucleotides (Santa Cruz
Biotechnology, Inc.) were used as controls. The C2C12 cells were
also co-transfected with the pCS2+ vector expressing
PLIN2 and siRNA oligonucleotides targeting NLRP3. At 48 h
post-transfection, the cells were harvested to extract the protein,
and 25 μg of protein were used for the detection of PLIN2 or
NLRP3 by western blot analysis.
Western blot analysis
The cells were lysed in lysis buffer (1% Triton
X-100, 150 mM NaCl, 20 mM Tris, pH 7.5) with protease inhibitor
cocktail and phenylmethylsulfonyl fluoride (PMSF) (both from Santa
Cruz Biotechnology, Inc.), and 25 μg of protein were
separated by electrophoresis. The resolved protein was transferred
onto polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford,
MA, USA). The membranes were blocked with 5% skim milk in
Tris-buffered saline (TBS) containing 0.1% Tween-20 (Thermo Fisher
Scientific, Waltham, MA, USA) for 1 h to reduce non-specific
binding and then incubated with antibodies to NLRP3 (sc-66846),
caspase-1 (sc-56036), IL-1β (sc-7884), β-actin (sc-47778; all from
Santa Cruz Biotechnologies, Inc.) and PLIN2 (ab52355; Abcam,
Cambridge, UK) at 4°C overnight. After washing 3 times with TBS
containing 0.1% Tween-20, the membranes were incubated with
horseradish peroxidase (HRP)-conjugated secondary antibody (ab6728,
ab97051; Abcam), and the signals were visualized using an enhanced
chemiluminescence (ECL) kit (Thermo Fisher Scientific) on a
ChemiDoc XRS+ imaging system (Bio-Rad, Hercules, CA,
USA).
Measurement of intracellular triglyceride
levels
At 48 h post-transfection with either
PLIN2-expressing vector or the empty vector, the C2C12 cells
were harvested in radioimmunoprecipitation (RIPA) lysis buffer
(Santa Cruz Biotechnologies, Inc.). The intracellular triglyceride
levels were measured in the cell lysates using a colorimetric
triglyceride quantification kit (ab65336; Abcam) according to the
manufacturer's instructions at a 570 nm wavelength on a microplate
reader (BioTek Instruments, Inc., Winooski, VT, USA). C2C12 cells
incubated with 400 μM palmitic acid (Sigma-Aldrich) for 24 h
were also subjected to this triglyceride assay as a positive
control.
Glucose uptake assay
At 48 h post-transfection with either
PLIN2-expressing vector or the empty vector, C2C12 cells
were washed twice with Krebs-Ringer-Phosphate-HEPES (KRPH) buffer
(20 mM HEPES, 5 mM KH2PO4, 1 mM
MgSO4, 1 mM CaCl2, 136 mM NaCl and 4.7 mM
KCl, pH 7.4) and starved of glucose by incubation with KRPH buffer
containing 0.2% bovine serum albumin (BSA) at 37°C for 40 min. The
cells were then stimulated with 100 nM insulin (Sigma-Aldrich) for
30 min in KRPH buffer supplemented with 0.2% BSA. Glucose transport
was determined by subsequent stimulation with
2-deoxy-D-glucose-6-phosphate (2DG6P) at a final concentration of
0.1 mM for 20 min, as previously described (18). The reaction was terminated by
washing the cells 4 times with ice-cold phosphate-buffered saline
(PBS). The cells were lysed in lysis buffer, and glucose uptake was
assessed using the Glucose Uptake Colorimetric assay kit (MAK083;
Sigma-Aldrich) in accordance with the manufacturer's instructions.
The absorbance was measured at a 412 nm wavelength on a microplate
reader (BioTek Instruments, Inc.). To confirm the effects of IL-1β
treatment on glucose uptake, the C2C12 cells were incubated with
murine IL-1β (PeproTech, Rocky Hill, NJ, USA) at 10, 25 and 50
ng/ml for 24 h. The cells were then subjected to the glucose uptake
assay described above.
Reverse transcription PCR (RT-PCR)
Total RNA was extracted from the C2C12 cells in each
experimental group using TRIzol reagent (Life Technologies). At 48
h post-transfection with PLIN2-overexpressing vector and a
combination of PLIN2- expressing vector with either
NLRP3 siRNA or non-targeted control siRNA, the C2C12 cells
were stimulated with 100 nM insulin for 30 min. This was followed
by RNA extraction to measure the expression levels of insulin
receptor substrate-1 (IRS-1). RNA was also extracted from the C2C12
cells treated with murine IL-1β at concentrations of 10, 25 and 50
ng/ml. First-strand cDNA was constructed with an oligo(dT) primer
using the Superscript III first-strand synthesis system (Life
Technologies) for RT-PCR. PCR was performed using Ex Taq DNA
polymerase as follows: an initial denaturation step at 95°C for 2
min, followed by 35 cycles of amplification (95°C for 30 sec, 56°C
for 30 sec, 72°C for 1 min) using the DNA Engine System (Bio-Rad).
IRS-I (209 bp) was amplified using the following primers:
forward, 5′-CGATGGCTTCTCAGACGTG-3′ and reverse,
5′-CAGCCCGCTTGTTGATGTTG-3′. The internal control gene, GAPDH
(395 bp), was amplified using the following primers: forward,
5′-GTC TTC TCC ACC ATG GAG AAG GCT-3′ and reverse, 5′-CAT GCC AGT
GAG CTT CCC GTT CA-3′.
Statistical analysis
Values are expressed as the means ± standard error
of the mean (SEM). Statistical significance was analyzed using the
Student's t-test with GraphPad Prism 6 software (GraphPad Software
Inc., San Diego, CA, USA). For all analyses, a value of P<0.05
was considered to indicate a statistically significant
difference.
Results
PLIN2 overexpression leads to the
cellular accumulation of triglycerides
To determine the effects of PLIN2 on the
accumulation of intracellular triglycerides, we overexpressed
full-length human PLIN2 in the C2C12 cells using 2 methods:
transient transfection and treatment with palmitic acid. Palmitic
acid is a saturated fatty acid that activates peroxisome
proliferator-activated receptor γ (PPAR-γ), which in turn acts as a
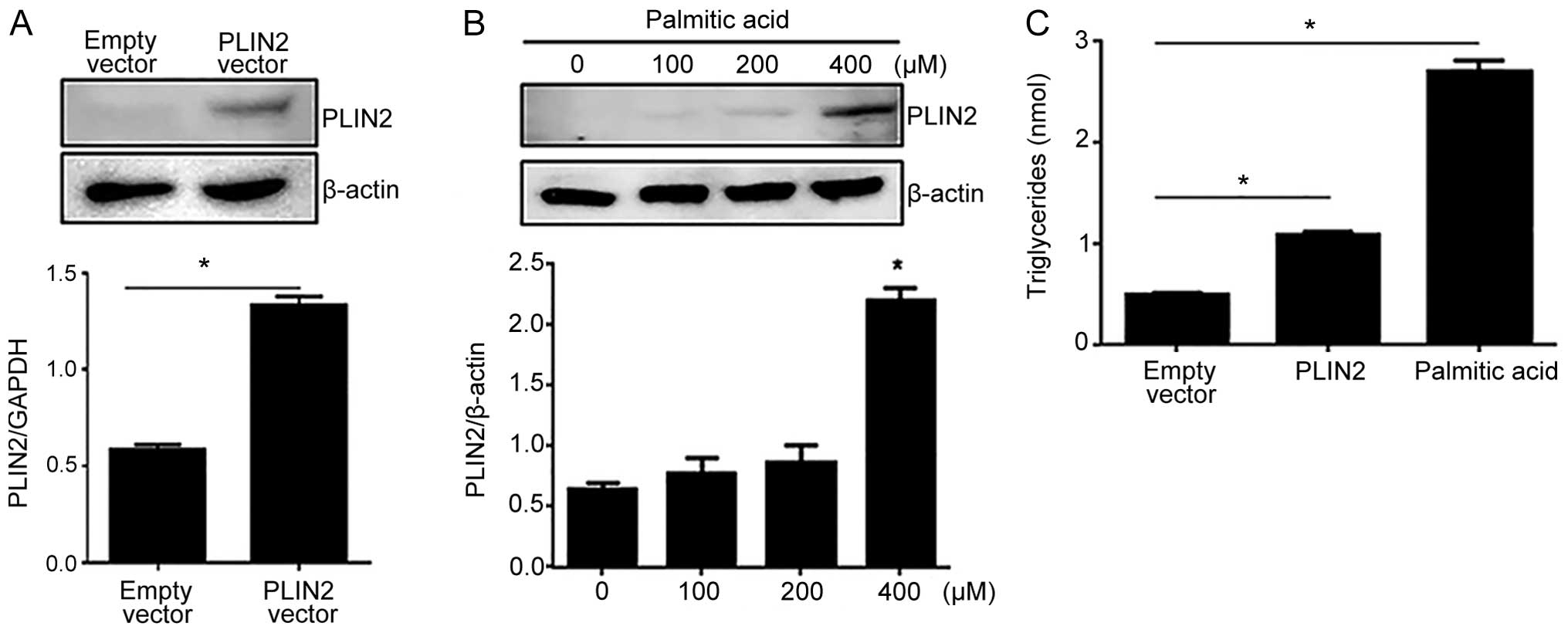
transcriptional activator of PLIN2 (19–21). Western blot analysis demonstrated
a significant induction of PLIN2 expression following transfection
with the PLIN2-overexpressing vector (Fig. 1A). Various concentrations (0, 100,
200 and 400 μM) of palmitic acid were used to mimic PLIN2
induction. Western blot analysis indicated that treatment of the
C2C12 cells with 400 μM of palmitic acid resulted in
significantly higher PLIN2 levels compared to treatment with 0, 100
or 200 μM of palmitic acid (Fig. 1B). We then measured the
intracellular triglyceride levels in the C2C12 cells in which the
overexpression of PLIN2 was induced using either method:
transient transfection with the vector expressing PLIN2 or
treatment with 400 μM palmitic acid, the latter serving as a
positive control. Both treatments yielded intracellular
triglyceride levels that were significantly higher than those in
the cells transfected with the empty vector; however, the cells
treated with palmitic acid displayed a more dramatic elevation in
intracellular triglyceride levels than the cells transfected with
the PLIN2-overexpressing vector (Fig. 1C).
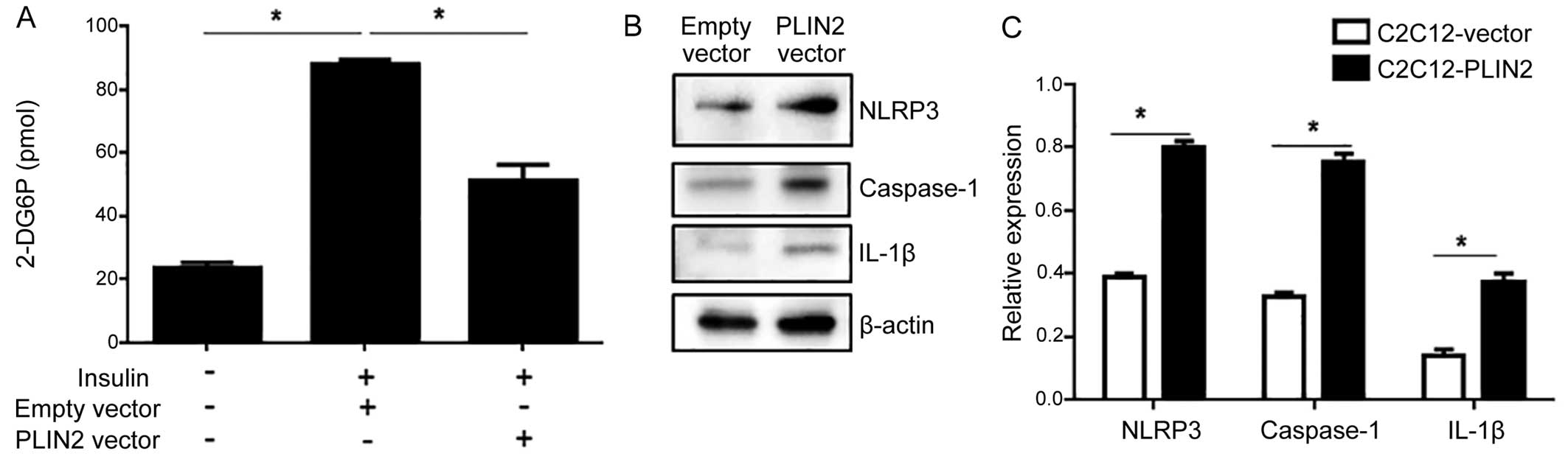
PLIN2 overexpression impairs
insulin-stimulated glucose uptake
Since increased levels of intracellular
triglycerides are associated with an impaired glucose uptake
(22), we hypothesized that
glucose uptake would be reduced in C2C12 cells that overexpress
PLIN2. Following transfection with the PLIN2-expressing
vector, the C2C12 cells were subjected to an insulin-stimulated
glucose uptake assay. C2C12 cells transfected with the empty vector
responded with a significant escalation of glucose uptake in
response to insulin stimulation, whereas the C2C12 cells that
overexpressed PLIN2 exhibited an impaired glucose uptake in
response to insulin stimulation (Fig.
2A).
PLIN2 impairs insulin-stimulated glucose
uptake activity through the activation of the NLRP3
inflammasome
We then investigated the potential involvement of
the NLRP3 inflammasome in PLIN2-induced insulin resistance. This
pathway has been implicated in recognizing certain non-microbial
'danger signals' that lead to caspase-1 activation and the
subsequent production of IL-1β. As excessive fat is also known to
activate the NLRP3 inflammasome (17), we hypothesized that this
inflammasome would be activated by accumulating levels of
triglycerides in C2C12 cells that overexpress PLIN2. PLIN2
overexpression led to an increased protein expression of NLRP3,
caspase-1 and IL-1β, suggesting the activation of the NLRP3
inflammasome (Fig. 2B and C). To
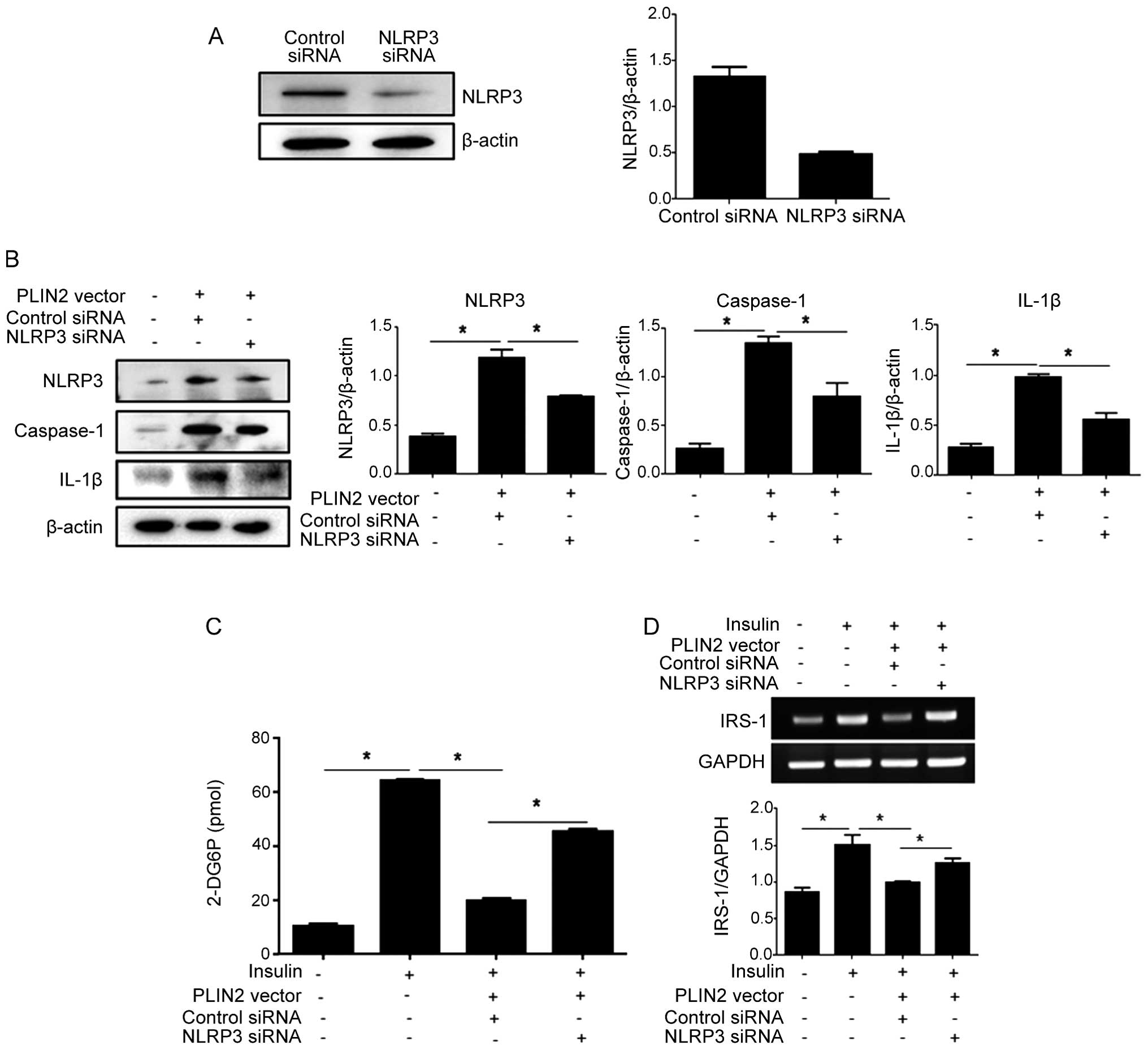
confirm that PLIN2 affects cellular glucose uptake through the
activation of the NLRP3 inflammasome, we performed NLRP3
knockdown experiments by transfection with siRNA. siRNA targeting
NLRP3 effectively reduced the endogenous expression of NLRP3
protein (Fig. 3A). Subsequently,
the C2C12 cells were co-transfected with PLIN2-expressing
vector and NLRP3 siRNA, followed by a glucose uptake assay.
NLRP3 knockdown resulted in decreased levels of NLRP3,
caspase-1 and IL-1β in the cells overexpressing PLIN2 (Fig. 3B), and increased
insulin-stimulated glucose uptake (Fig. 3C). The non-targeted control siRNA
did not increase glucose uptake in the cells that overexpressed
PLIN2.
Suppression of IRS-1 by IL-1β mediates
the effects of the NLRP3 inflammasome on insulin-induced glucose
uptake
IRS-1, one of the major substrates of the insulin
receptor kinase, is essential for the activatation of PI3K in
response to insulin, which leads to the phosphorylation of protein
kinase B (also known as Akt) and subsequent glucose uptake
(23). The downregulation of
IRS-1 seems to be the major mechanism involved in the alteration of
insulin signaling and glucose transport (24). Thus, we examined the effects of
PLIN2 and NLRP3 manipulations on the mRNA expression
levels of IRS-1 in the C2C12 cells. PLIN2 overexpression
(with transfection with control siRNA) led to a decrease in IRS-1
expression. However, PLIN2 over-expression accompanied by
NLRP3 knockdown via siRNA transfection was associated with
an increased IRS-1 expression in response to insulin
stimulation (Fig. 3D). IL-1β, the
final step in the NLRP3 inflammasome, has been demonstrated to
reduce insulin-induced glucose uptake in adipocytes by affecting
IRS-1 expression (18,25). To determine whether IL-1β
treatment affects insulin-induced glucose uptake in myoblasts, we
treated the C2C12 cells with IL-1β at concentrations of 0, 10, 25
and 50 ng/ml for 24 h prior to measuring insulin-induced glucose
uptake. IL-1β was found to inhibit glucose transport upon insulin
stimulation in a dose-dependent manner (Fig. 4A). We also found that IL-1β
decreased endogenous IRS-1 expression in the C2C12 cells (Fig. 4B).
Discussion
Skeletal muscle is a major organ which is involved
in insulin-stimulated glucose uptake and thus in the development of
insulin resistance. Previous studies have demonstrated that excess
lipid accumulation contributes to the pathogenesis of insulin
resistance, thus suggesting potential therapeutic interventions
(2,26,27). Aside from the lipid overload
hypothesis, inflammation has been considered to be a pivotal
contributor to insulin resistance. Pro-inflammatory cytokines, such
as tumor necrosis factor-α (TNF-α), IL-6 and monocyte
chemoattractant protein-1 (MCP-1) have been identified as promoters
of insulin resistance; however, these findings were mainly related
to adipose tissue (13,28).
In this study, we attempted to identify an
association between lipid accumulation and the inflammatory pathway
that leads to insulin resistance in C2C12 cells. We found that the
knockdown of NLRP3 attenuated PLIN2-induced insulin
resistance in C2C12 cells. PLIN2 is a lipid droplet-associated
protein that is abundantly expressed in skeletal muscle. It is well
known that the expression of PLIN2 correlates with the accumulation
of intramuscular lipids (8–10).
We observed in this study that C2C12 cells which overexpressed
PLIN2 contained significantly greater amounts of triglycerides.
Furthermore, we also noted that PLIN2 overexpression led to
the upregulation of IL-1β through NLRP3-caspase-1 upregulation.
The NLRP3 inflammasome consists of a group of
protein complexes that are composed of the Nod-like receptor
protein NLRP3 (also termed cryopyrin or NALP3), adapter proteins, a
caspase recruitment domain (Cardinal) and caspase-1. The NLRP3
inflammasome has been implicated in the production of mature IL-1β
and IL-18 in response to a variety of signals (16,17). One of the signals, DAMPs, is
host-derived and is released as a result of perturbations of tissue
homeostasis caused by microbial or non-microbial insults, conveying
a general sense of tissue under stress (16,17). Although the normal activation of
the NLRP3 inflammasome contributes to host defense, excessive
activation results in inflammatory diseases mediated by the
pro-inflammatory cytokine, IL-1β (16,17). It has been previously reported
that the NLRP3 inflammasome instigates obesity-induced
autoinflammation and insulin resistance (29). Considering that muscle is a
critical organ involved in insulin sensitivity, we can speculate
that the NLRP3 inflammasome links excess fat and insulin resistance
in myoblasts. In the present study, we demonstrated that C2C12
cells overexpressing PLIN2 exhibited increased levels of NLRP3,
caspase-1 and IL-1β. As PLIN2 impaired glucose uptake activity,
this indicates a potential correlation between NLRP3 and glucose
uptake in C2C12 cells in response to insulin. The knockdown of
NLRP3 by RNA interference significantly inhibited insulin
resistance which is typically induced in PLIN2-overexpressing C2C12
cells, suggesting that the NLRP3 inflammasome mediates
PLIN2-induced insulin resistance. Decreased levels of IRS-1 have
been reported to alter insulin signaling in adipocytes and myocytes
(24,25). In this study, we observed that
IRS-1 expression was decreased in C2C12 cells that overexpressed
PLIN2 in the context of insulin exposure, whereas NLRP3
knockdown increased the expression of IRS-1. IL-1β, the final
product of the NLRP3 inflammasome, has been reported to reduce
insulin-induced glucose uptake in adipocytes, mainly by inhibiting
IRS-1 expression (18). In the
present study, we observed the inhibitory effect of IL-1β on the
expression of IRS-1 in C2C12 cells, and on this basis we suggest
that inflammatory cytokines directly target muscle cells, as well
as adipocytes in the context of insulin signaling disturbances.
In conclusion, the present study provides a fresh
view of the role of LDs and associated proteins in cellular
metabolism by demonstrating the following novel insights. First,
insulin-mediated cellular glucose uptake inversely correlated with
PLIN2 expression in myoblasts. Second, components of the NLRP3
inflammasome were significantly upregulated in
PLIN2-overexpressing myoblast cells, while the
downregulation of NLRP3 inhibited PLIN2-induced insulin
resistance. Taken together, these data suggest that PLIN2
influences cellular glucose uptake and transport by interacting
with the NLRP3 inflammasome. Since insulin resistance can be
characterized by inefficient glucose uptake in muscle and fat
cells, our findings provide a molecular mechanism that links
cellular lipid contents and the inflammatory response underlying
the pathogenesis of insulin resistance. Future studies using
animals fed a high-fat diet may help determine whether the
suppression of the activation of the NLRP3 inflammasome would
protect the animals against type 2 diabetes mellitus.
Acknowledgments
The authors thank the Department of Pediatrics at
the University of Florida College of Medicine for supporting this
study.
References
|
1
|
Rachek LI: Free fatty acids and skeletal
muscle insulin resistance. Prog Mol Biol Transl Sci. 121:267–292.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moors CC, van der Zijl NJ, Diamant M,
Blaak EE and Goossens GH: Impaired insulin sensitivity is
accompanied by disturbances in skeletal muscle fatty acid handling
in subjects with impaired glucose metabolism. Int J Obes.
36:709–717. 2012. View Article : Google Scholar
|
|
3
|
Sinha R, Dufour S, Petersen KF, LeBon V,
Enoksson S, Ma YZ, Savoye M, Rothman DL, Shulman GI and Caprio S:
Assessment of skeletal muscle triglyceride content by (1)H nuclear
magnetic resonance spectroscopy in lean and obese adolescents:
relationships to insulin sensitivity, total body fat, and central
adiposity. Diabetes. 51:1022–1027. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kaneko S, Iida RH, Suga T, Fukui T, Morito
M and Yamane A: Changes in triacylglycerol-accumulated fiber type,
fiber type composition, and biogenesis in the mitochondria of the
soleus muscle in obese rats. Anat Rec (Hoboken). 294:1904–1912.
2011. View
Article : Google Scholar
|
|
5
|
Gray RE, Tanner CJ, Pories WJ, MacDonald
KG and Houmard JA: Effect of weight loss on muscle lipid content in
morbidly obese subjects. Am J Physiol Endocrinol Metab.
284:E726–E732. 2003. View Article : Google Scholar
|
|
6
|
Ducharme NA and Bickel PE: Lipid droplets
in lipogenesis and lipolysis. Endocrinology. 149:942–949. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martin S and Parton RG: Lipid droplets: A
unified view of dynamic organelle. Nat Rev Mol Cell Biol.
7:373–378. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Larigauderie G, Cuaz-Pérolin C, Younes AB,
Furman C, Lasselin C, Copin C, Jaye M, Fruchart JC and Rouis M:
Adipophilin increases triglyceride storage in human macrophages by
stimulation of biosynthesis and inhibition of beta-oxidation. FEBS
J. 273:3498–3510. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fukushima M, Enjoji M, Kohjima M, Sugimoto
R, Ohta S, Kotoh K, Kuniyoshi M, Kobayashi K, Imamura M, Inoguchi
T, et al: Adipose differentiation related protein induces lipid
accumulation and lipid droplet formation in hepatic stellate cells.
In Vitro Cell Dev Biol Anim. 41:321–324. 2005. View Article : Google Scholar
|
|
10
|
Listenberger LL, Ostermeyer-Fay AG,
Goldberg EB, Brown WJ and Brown DA: Adipocyte
differentiation-related protein reduces the lipid droplet
association of adipose triglyceride lipase and slows
triacylglycerol turnover. J Lipid Res. 48:2751–2761. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Minnaard R, Schrauwen P, Schaart G,
Jorgensen JA, Lenaers E, Mensink M and Hesselink MK: Adipocyte
differentiation-related protein and OXPAT in rat and human skeletal
muscle: involvement in lipid accumulation and type 2 diabetes
mellitus. J Clin Endocrinol Metab. 94:4077–4085. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Varela GM, Antwi DA, Dhir R, Yin X,
Singhal NS, Graham MJ, Crooke RM and Ahima RS: Inhibition of ADRP
prevents diet-induced insulin resistance. Am J Physiol Gastrointest
Liver Physiol. 295:G621–G628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maury E and Brichard SM: Adipokine
dysregulation, adipose tissue inflammation and metabolic syndrome.
Mol Cell Endocrinol. 314:1–16. 2010. View Article : Google Scholar
|
|
14
|
Sell H and Eckel J: Monocyte chemotactic
protein-1 and its role in insulin resistance. Curr Opin Lipidol.
18:258–262. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tack CJ, Stienstra R, Joosten LA and Netea
MG: Inflammation links excess fat to insulin resistance: the role
of the interleukin-1 family. Immunol Rev. 249:239–252. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Strowig T, Henao-Mejia J, Elinav E and
Flavell R: Inflammasomes in health and disease. Nature.
481:278–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Benetti E, Chiazza F, Patel NS and Collino
M: The NLRP3 Inflammasome as a novel player of the intercellular
crosstalk in metabolic disorders. Mediators Inflamm.
2013:6786272013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jager J, Grémeaux T, Cormont M, Le
Marchand-Brustel Y and Tanti JF: Interleukin-1beta-induced insulin
resistance in adipocytes through down-regulation of insulin
receptor substrate-1 expression. Endocrinology. 148:241–251. 2007.
View Article : Google Scholar :
|
|
19
|
Liu X, Xue R, Ji L, Zhang X, Wu J, Gu J,
Zhou M and Chen S: Activation of farnesoid X receptor (FXR)
protects against fructose-induced liver steatosis via inflammatory
inhibition and ADRP reduction. Biochem Biophys Res Commun.
450:117–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dalen KT, Schoonjans K, Ulven SM,
Weedon-Fekjaer MS, Bentzen TG, Koutnikova H, Auwerx J and Nebb HI:
Adipose tissue expression of the lipid droplet-associating proteins
S3-12 and perilipin is controlled by peroxisome
proliferator-activated receptor-gamma. Diabetes. 53:1243–1252.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Petrighi Polidori G, Lomax MA and Docherty
K: Palmitate enhances the differentiation of mouse embryonic stem
cells towards white adipocyte lineages. Mol Cell Endocrinol.
361:40–50. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bartels ED, Ploug T, Størling J,
Mandrup-Poulsen T and Nielsen LB: Skeletal muscle apolipoprotein B
expression reduces muscular triglyceride accumulation. Scand J Clin
Lab Invest. 74:351–357. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Taniguchi CM, Emanuelli B and Kahn CR:
Critical nodes in signalling pathways: Insights into insulin
action. Nat Rev Mol Cell Biol. 7:85–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang WM, Jeong HJ, Park SY and Lee W:
Induction of miR-29a by saturated fatty acids impairs insulin
signaling and glucose uptake through translational repression of
IRS-1 in myocytes. FEBS Lett. 588:2170–2176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao D, Madi M, Ding C, Fok M, Steele T,
Ford C, Hunter L and Bing C: Interleukin-1β mediates
macrophage-induced impairment of insulin signaling in human primary
adipocytes. Am J Physiol Endocrinol Metab. 307:E289–E304. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abdul-Ghani MA and DeFronzo RA:
Pathogenesis of insulin resistance in skeletal muscle. J Biomed
Biotechnol. 2010:4762792010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kraegen EW and Cooney GJ: Free fatty acids
and skeletal muscle insulin resistance. Curr Opin Lipidol.
19:235–241. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rotter V, Nagaev I and Smith U:
Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1
adipocytes and is, like IL-8 and tumor necrosis factor-alpha,
overexpressed in human fat cells from insulin-resistant subjects. J
Biol Chem. 278:45777–45784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vandanmagsar B, Youm YH, Ravussin A,
Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM and Dixit
VD: The NLRP3 inflammasome instigates obesity-induced inflammation
and insulin resistance. Nat Med. 17:179–188. 2011. View Article : Google Scholar : PubMed/NCBI
|