Introduction
Atherosclerosis, which poses a serious threat to
human health, is an important public health concern. The
development of atherosclerosis involves a complex pathogenic
process which includes endothelial dysfunction, the entry and
retention of lipids, leukocyte adhesion, the migration and
proliferation of vascular smooth muscle cells (VSMCs) and the
increased synthesis of the extracellular matrix (1). Disorders in the functions of VSMCs
play a vital role in vascular proliferative diseases, including
atherosclerosis and restenosis following coronary angioplasty
(2). Atherogenic stimuli,
including cytokines, lipids, shear stress and reactive oxygen
species, provoke VSMCs migrating from the media to the intima, to
switch from a quiescent 'contractile' state to a 'synthetic'
phenotypic state and to enter the cell cycle. Cell cycle
progression is regulated by a range of regulatory proteins,
including cyclins, cyclin-dependent kinases (CDKs) and CDK
inhibitors (CDKIs). The cyclin D/CDK4/6 complex is active in the
early G1 phase of the cell cycle, regulating G1 phase progression,
whereas cyclin E/CDK2 or cyclin A/CDK2 are essential for the G1/S
phase transition. Cell cycle progression is blocked by CDKIs, such
as p27 and p21 (3).
Angiotensin II (Ang II), the main member of the
renin-angiotensin-aldosterone system (RAAS), is an important
regulator of the cardiovascular system (4). Ang II induces the growth and
migration of smooth muscle cells, which contributes to the
progression and development of cardiovascular disease through the
activation of the mitogen-activated protein kinase (MAPK) signaling
cascade, including extracellular signal-regulated kinase (ERK1/2),
JNK and p38 MAPK (5). MAPK
activation increases c-fos and c-jun gene expression
and promotes the translocation of activator protein-1 (AP-1) to the
nucleus, where it binds to the promoter of various target genes,
such as cyclin D1 (6).
Jumonji domain-containing 2A (JMJD2A), also known as
KDM4A, belongs to the Jumonji C (JmjC) domain-containing family of
JMJD2 proteins. It is a lysine trimethyl-specific histone
demethylase that catalyses the demethylation of trimethylated
histone H3 lysine 9 (H3K9me3) and H3K36 (H3K36me3) (7). Through JMJD2A activity, H3K9me3
demethylation contributes to an open chromatin state and is
connected with the transcriptional activation of promoter regions
(8).
In the present study, we investigated the role of
JMJD2A in the dysfunction of VSMCs following stimulation with Ang
II. In addition, we aimed to investigate whether IOX1, a known
JMJD2A inhibitor, can suppress the proliferation and migration of
VSMCs induced by Ang II, as well as the underlying mechanisms.
Materials and methods
JMJD2A inhibitor
The JMJD2A inhibitor, IOX1
[5-carboxy-8-hydroxyquinoline (5-carboxy-8-HQ)], was dissolved in
dimethyl sulfoxide (DMSO) (both from Sigma, St. Louis, MO, USA) to
yield a 250 µM stock solution and was subsequently diluted
in Dulbecco's modified Eagle's medium (DMEM; HyClone, Logan, UT,
USA) to the required concentration.
Primary culture of VSMCs
As previously described (9), VSMCs were isolated from the thoracic
aortas of male Sprague-Dawley rats (weighing, 150–180 g). All
animals used in our study were purchased from the Animal Center of
Renmin Hospital of Wuhan University. The experimental procedures
and animal care procedures were conducted in accordance with the
Institutional Animal Care and Use Committee of Wuhan University.
Briefly, rat thoracic aortas were harvested following euthanasia.
After scraping off the adventitia and endothelium, thoracic aortas
were cut into sections of 2×2 mm. The primary VSMCs were obtained
using the tissue explants adherent method and cultured in DMEM
(HyClone) supplemented with 10% fetal bovine serum (FBS; Gibco,
Grand Island, NY, USA) and 1% penicillin/streptomycin (PS; Beyotime
Institute of Biotechnology, Bejing, China) in an atmosphere of 5%
CO2, at 37°C. The purity of the VMSCs was assessed by
immunostaining with anti-smooth muscle α-actin antibody. VSMCs
between the third and fifth passages were used for the
experiments.
Cell proliferation assay
The cell proliferation assay was performed using a
Cell Counting Kit-8 (CCK8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). Briefly, approximately 8,000 VSMCs were seeded in
96-well plates in 200 µl of culture medium. Following
synchronization with DMEM containing 0.1% FBS for 24 h, the VSMCs
were pretreated with IOX1 at various concentrations (0, 50, 100 and
200 µM) for 2 h, and then stimulated with 1 µM Ang II
(Sigma) for 24 h. CCK8 solution (20 µl) was then added and
the OD value was measured at 450 nm using a microplate
spectrophotometer (Infinite M200; Tecan Group Ltd., Männedorf,
Switzerland).
Cell migration assay
A Transwell chamber assay (Corning Inc., Corning,
NY, USA) was performed to evaluate the migration activity of the
VMSCs. Synchronized (105) VSMCs were seeded into the
upper chamber in 200 µl of serum-free medium in the presence
or absence of IOX1. Following inhibitor precondition for 2 h, 600
µl of DMEM supplemented with 1 µM Ang II were added
to the lower chamber followed by incubation for 8 h. Cells
remaining on the upper side of the membrane were gently wiped with
a cotton swab. The migrated cells were fixed with methanol and
stained with 0.5% crystal violet. The number of migrated cells was
counted in 5 random fields under a microscope (Olympus Corporation,
Tokyo, Japan) at a ×100 magnification.
Cell cycle analysis
Cell-cycle analysis was carried out by flow
cytometry. The VSMCs were cultured in 6-cm culture plates (Corning
Inc.) at a density of 5×105 cells/plate. At 50% cell
density, the VSMCs were synchronized for 24 h. Then VSMCs were then
pre-treated with IOX1 (200 µM), followed by treatment with
Ang II treatment for 24 h. The VMSCs were then trypsinized,
harvested and washed twice with cold phosphate-buffered saline
(PBS), and fixed with 70% cold ethanol and stored at 4°C overnight.
After staining with prop-idium iodide (PI solution: 50 mg/ml PI,
100 mg/ml RNase A) at 37°C for 30 min, the cell cycle distribution
was determined by flow cytometry (BD FACSCalibur; Becton, Dickinson
and Company, Franklin Lakes, NJ, USA). The percentage of cells in
the G0/G1, S and G2-M phase of the cell cycle was determined using
ModFitLT cell cycle analysis software (Verity Software House Inc.,
Topsham, ME, USA).
Western blot analysis
The synchronized VSMCs were pretreated with IOX1
(200 µM) for 2 h and then stimulated with Ang II for 0, 3,
6, 12 and 24 h and lysed in RIPA lysis buffer (Beyotime Institute
of Biotechnology) containing 1 mM PMSF and 10 mM phosphatase
inhibitor (Roche, Mannheim, Germany) on ice for 15 min, and
centrifuged for 5 min at 12,000 × g at 4°C. Total protein was
quantified using a BCA protein assay kit (Beyotime Institute of
Biotechnology) according to the manufacturer's instructions. Equal
amounts of protein (35 µg) were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto polyvinylidene difluoride (PVDF) membranes. The
PVDF membranes were blocked and incubated with antibodies against
JMJD2A (SAB3500095; Sigma), H3K9me3 (ab8898; Abcam, Cambridge, MA,
USA), cyclin D1 (2978T), cyclin E (4136S), p21 (2947T), p27 (3686T;
all from Cell Signaling Technology, Inc., Danvers, MA, USA) and
GAPDH (5632-1; Epitomics, Burlingame, CA, USA) overnight at 4°C.
The washed membranes were incubated with horseradish peroxidase
(HRP)-conjugated secondary antibodies (7074; Cell Signaling
Technology, Inc.) for 2 h at room temperature, and subsequently
detected using ECL reagent (Pierce, Rockford, IL, USA). The results
of the western blot analysis were quantified using Quantity One
software.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The mRNA expression levels of cyclin D1 and p21 were
measured by RT-qPCR. The synchronized VSMCs were pre-treated with
IOX1 (200 µM) for 2 h and then stimulated with Ang II for 12
h. Total RNA was extracted using the PicoPure RNA isolation kit
(Applied Biosystems Life Technologies, Carlsbad, CA, USA) according
to the manufacturer's instructions and reverse transcribed into
cDNA using a First-Strand Synthesis system (Invitrogen, Carlsbad,
CA, USA). This was performed using the SYBR system on the ABI Prism
7500 Sequence Detection System (Applied Biosystems Life
Technologies, Foster City, CA, USA). Each sample was run and
analyzed in triplicate. Data were normalized to GAPDH expression
and analyzed using the 2−ΔΔCt method. The primer
sequences used in the present study were as follows: cyclin D1
forward, 5′-TGTTCGTGGCCTCTAAGATGAAG-3′ and reverse,
5′-GGAAGTGTTCGATGAAATCGTG-3′; p21 forward,
5′-AGTATGCCGTCGTCTGTTCG-3′ and reverse, 5′-GAGTGCAAGACAGCGACAAG-3′;
and GAPDH forward, 5′-GACATGCCGCCTGGAGAAAC-3′ and reverse,
5′-AGCCCAGGATGCCCTTTAGT-3′.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assay was performed using a ChIP assay kit
(Pierce) according to the manfacturer's instructions. Briefly, the
synchronized VSMCs were pre-treated with IOX1 (200 µM) for 2
h and then stimulated with Ang II for 12 h and fixed with 1%
formaldehyde, washed and lysed. The cell lysates were sonicated to
create chromatin fragments of 400–500 bp in length, diluted in ChIP
dilution buffer and subjected to immunoprecipitation with
anti-H3K9me3 antibodies (ab8898; Abcam) overnight at 4°C. The
immune complexes were collected, washed and eluted with buffer.
Protein-DNA cross-links were reversed overnight at 65°C and the DNA
was extracted. ChIP-enriched DNA samples were analyzed by RT-qPCR
using primers specific for cyclin D1 or p21. Data were analyzed
using the 2-ΔΔCt method and normalized with input
samples. The results are expressed as a percentage (%) of the
control values. The primer sequences were follows: cyclin D1
forward, CTCTGCCCGGCTTTGATCTCT-3′ and reverse,
5′-AGGCTGCAGGACTTTGCAACT-3′; and p21 forward,
5′-GTTCAGCCCTGGAACCGAAG-3′ and reverse,
5′-GTACCAAACACCCTTCACCTGGTAC-3′.
Statistical analysis
All results were analyzed using either a Student's
t-test for between-group comparisons or the one-way ANOVA for
multiple comparisons using SPSS 13 software. Data are presented as
the means ± SEM of 3 or 6 experiments, as indicated in the figure
legends. A value of p<0.05 was considered to indicate a
statistically significant difference.
Results
JMJD2A and H3K9me3 protein expression
levels in Ang II-stimulated VSMCs
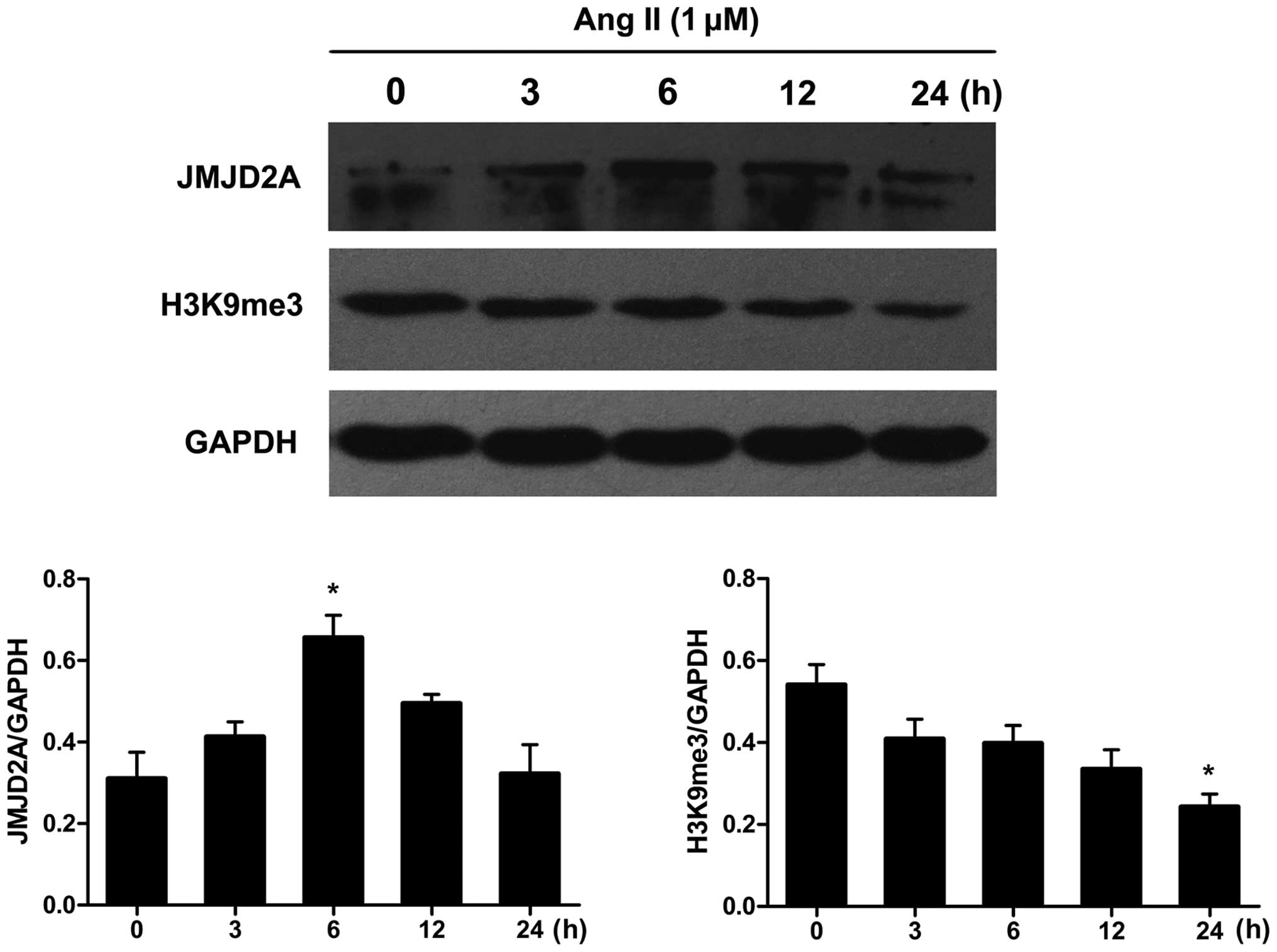
Subconfluent, quiescent VSMCs were exposed to Ang II
for 0, 3, 6, 12 and 24 h. Western blot analysis was performed to
assess the total protein levels of JMJD2A and its primary target,
H3K9me3, in Ang II-stimulated VSMCs. The time course of JMJD2A and
H3K9me3 protein expression is shown in Fig. 1. Ang II induced a slight increase
in the JMJD2A protein levels at an early time point (3 h). The
levels peaked at around 6 h and were maintained at these levels
until the 12-h time point. Conversely, the protein expression of
H3K9me3 decreased in a time-dependent manner in the Ang
II-stimulated VSMCs.
IOX1 suppresses the proliferation and
migration of VMSCs induced by Ang II
Since the expression of JMJD2A was upregulated in
the Ang II-stimulated VSMCs, we intended to block JMJD2A with the
specific inhibitor, IOX1, and to examine the effect of JMJD2A
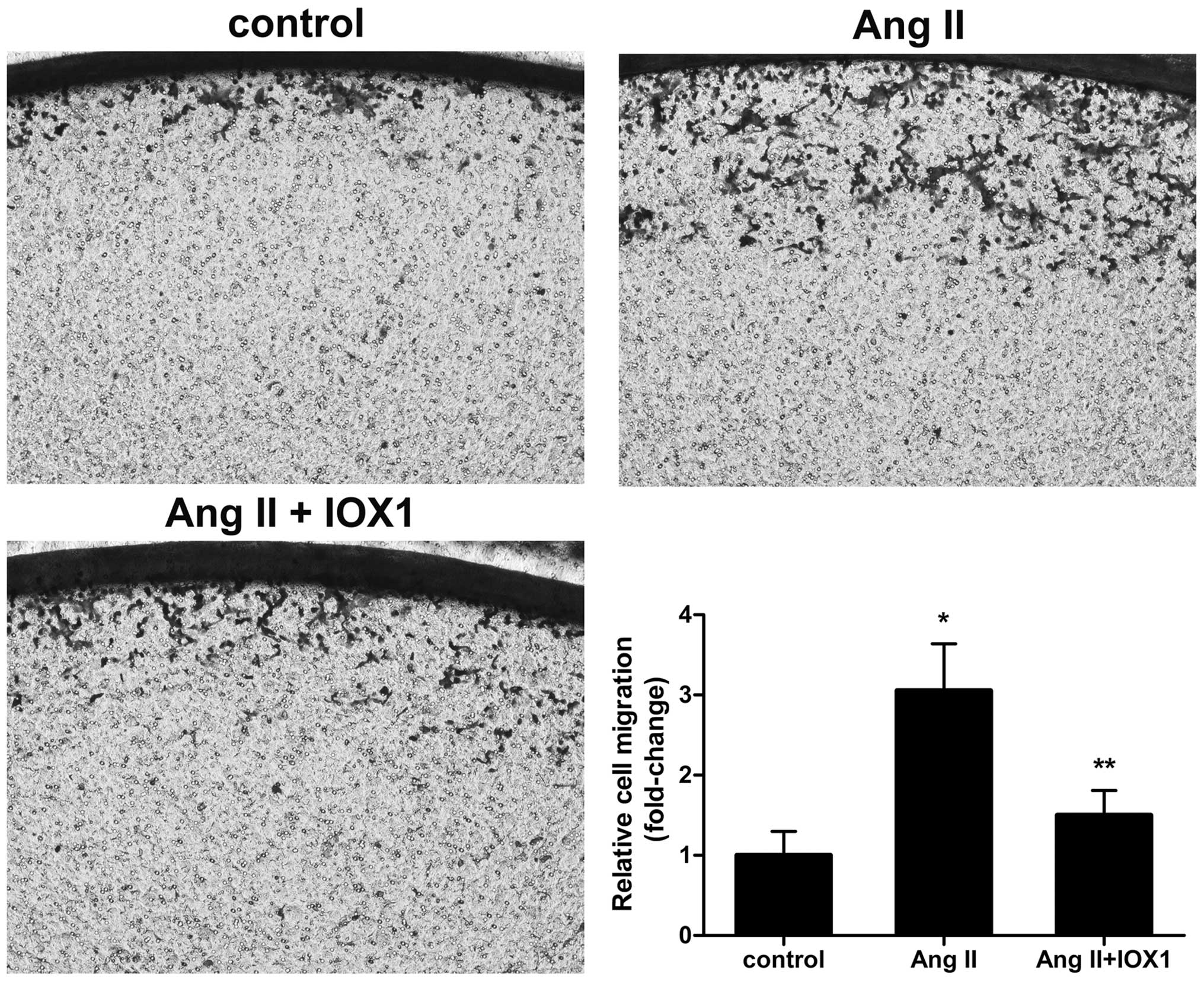
inhibition on Ang II-stimulated VSMCs. A significant increse in
proliferation was observed in the subconfluent, quiescent VSMCs
following stimulation with Ang II in the absence of serum (Fig. 2). The VSMCs pre-treated with IOX1
(0, 50, 100 and 200 µM) exhibited a decrease in
proliferation in a dose-dependent manner. The most prominent
suppressive effect on VSMC proliferation was observed following
treatment with IOX1 at 200 µM (decreased in proliferation of
44.73%). We then wished to examine whether IOX1 suppresses the
migration of Ang II-stimulated VSMCs. Pretreated VSMCs pre-treated
with 200 µM IOX1 were stimulated with Ang II. The enhanced
migration of the Ang II-stimulated VSMCs was indeed suppressed by
pretreatment with IOX1 (Fig. 3).
These data indicated that IOX1 inhibited the proliferation and
migration of VSMCs stimulated with Ang II.
IOX1 blocks the cell cycle progression of
VSMCs stimulated with Ang II
We then examined the effects of IOX1 on the cell
cycle of VSMCs. Following absolute synchronization, the VSMCs were
stimulated with Ang II for 24 h in the presence or absence of IOX1.
Cell cycle analysis using flow cytometry revealed that there was a
marked increase in the percentage of Ang II-stimulated cells in the
S phase compared with the quiescent (unstimulated) cells. This
increase in the S phase cell population was decreased by IOX1
treatment (Fig. 4). The
percentage of cells in the G0/G1 phase were increased in the cells
pre-treated with IOX1 (Fig. 4),
indicating that IOX1 significantly inhibited the proliferation of
the VSMCs by slowing down the progression of the cell cycle from
the G0/G1 to the S phase.
Effect of IOX1 on the expression of cell
cycle-related genes
Considering that IOX1 suppressed the proliferation
of VSMCs and blocked cell cycle progression, particularly at the
G1/S phase transition, we measured the expression levels of various
cell cycle-related proteins in the Ang II-stimulated VSMCs in the
presence or absence of IOX1 (50, 100 and 200 µM).
Stimulation with Ang II increased the expression of cyclin D1 and
cyclin E (Fig. 5). This increase
in cyclin D1 expression was attenuated by IOX1 in a
concentration-dependent manner. However, IOX1 had a marginal effect
on cyclin E expression. In addition, we measured the protein
expression levels of the CDKIs, p21 and p27. The levels of p27 were
not influenced by Ang II, whereas the p21 levels were decreased in
the Ang II-stimulated VSMCs. When the cells were pre-treated with
to IOX1, p21 expression was upregulated in the Ang II-stimulated
VSMCs. To determine whether the changes in the protein expression
levels of cyclin D1 and p21 are regulated by IOX1 at the
transcriptional level, RT-qPCR was performed to measure their mRNA
expression levels. The mRNA expression levels were similar to the
protein levels. IOX1 decreased cyclin D1 mRNA expression and
upregulated p21 mRNA expression (Fig.
6). These data suggested that IOX1 inhibited the Ang II-induced
VSMC proliferation through selective regulation of the cell
cycle-related proteins, cyclin D1 and p21.
IOX1 mediates cyclin D1 and p21
expression by regaining H3K9me3 at the promoters of these
genes
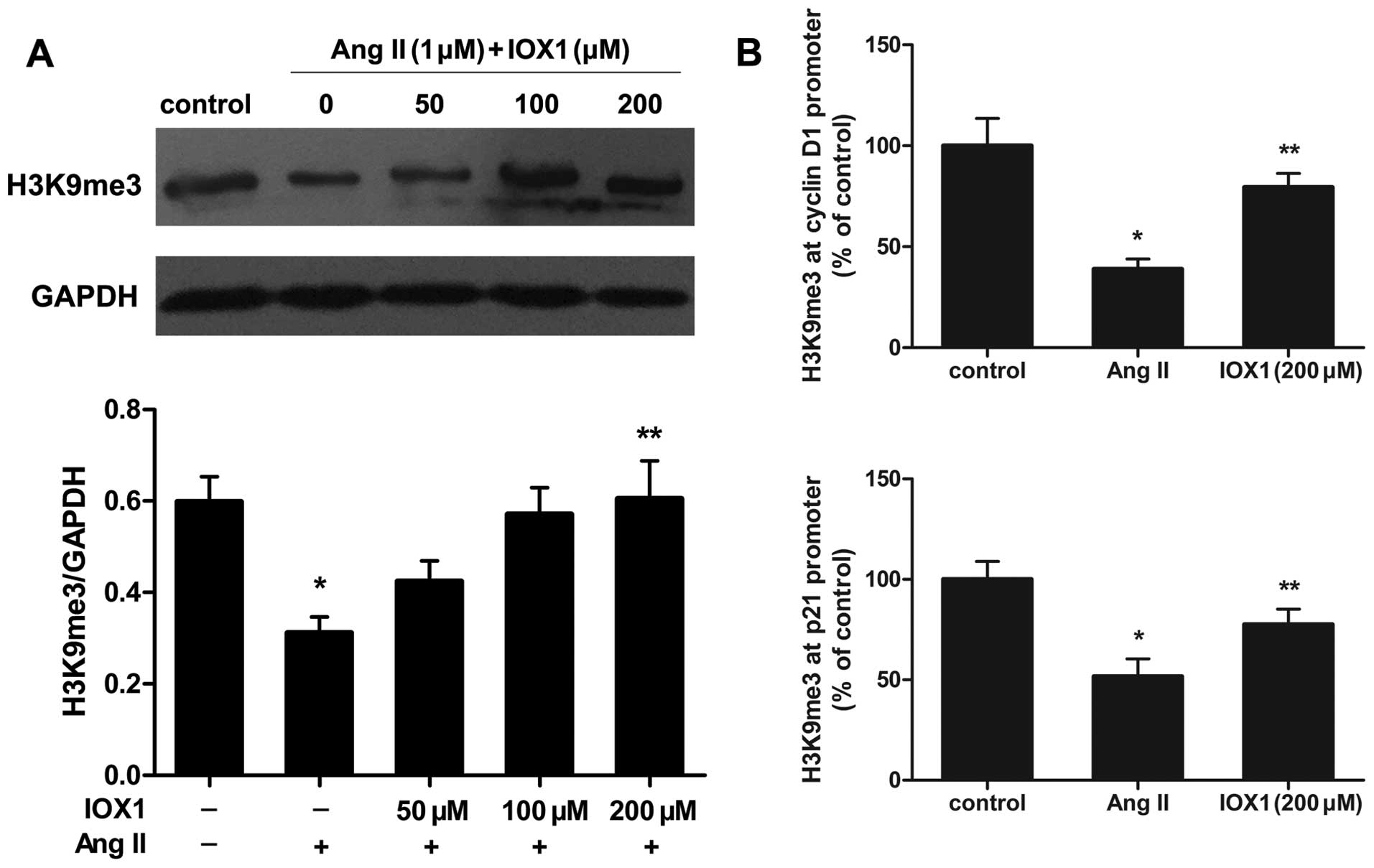
To determine whether IOX1 epigenetically mediates
the expression of cyclin D1 and p21 in Ang II-stimulated VSMCs, we
first examined the global changes in H3K9me3, the main substrate of
JMJD2A. IOX1 enhanced the total protein levels of H3K9me3 which
were reduced in the Ang II-stimulated VSMCs (Fig. 7A). To investigate the mechanisms
through which IOX1 regulates cyclin D1 and p21 expression, ChIP
assays were performed us ing anti-H3K9me3 to examine the effects of
IOX1 on the cyclin D1 and p21 promoters in the Ang II-stimulated
VSMCs. The results revealed that the levels of H3K9me3 in the
cyclin D1 and p21 promoter regions were decreased in the Ang
II-stimulated cells compared to to the untreated control cells.
Pre-treatment with 200 µM of IOX1 increased the levels of
H3K9me3 in the promoters of these genes (Fig. 7B).
Discussion
The abnormal growth of VSMCs has been shown to
contribute to the development of occlusive vascular diseases, such
as atherosclerosis and restenosis following coronary angioplasty
(2). It is important to elucidate
the underlying mechanisms responsible for the dysfunction of VSMCs
in a pathogenic environment. In this study, we found that the
levels of JMJD2A, a newly recognized histone demethylase, were
increased in the Ang II-stimulated VSMCs, and this was accompanied
by suppressed global levels of H3K9me3. Thus, we utilized IOX1, a
selective inhibitor of the Jumonji protein subtypes, to examine the
effects of the inhibition of JMJD2A on VSMC proliferation and
migration. We used Ang II as a proliferative agent which has been
demonstrated to be a critical mediator of cardiovascular disease
progression (4). IOX1 suppressed
the proliferation and migration of VSMCs induced by Ang II,
exerting anti-proliferative and anti-migratory effects by
regulating the expression of cell cycle-related proteins, cyclin D1
and p21. expression. ChIP assays demonstrated that IOX1 altered the
expression of the cell cycle proteins by modifying the level of
H3K9me3 at the promoters of these genes.
JMJD2A, also known as KDM4A, belongs to the JmjC
domain-containing family of JMJD2 proteins. It is a lysine
trimethyl-specific histone demethylase that catalyses the
demethylation of H3K9me3 and H3K36me3 (7). It has been suggested that there is
an alteration in the JMJD2A expression pattern in several types of
cancer (10–15). JMJD2A is involved in the
epigenetic regulation of tumor-related gene expression which
depends on the demethylation of H3K9me3 or the interaction with the
tumor suppressor, retinoblastoma protein (Rb), histone deacetylases
(HDACs), and the nuclear receptor co-repressor (N-CoR) (16,17). Previous studies have identified
some H3K9me3-related histone methyltransferases and demeth-ylases,
such as Suv39h1, G9a and JMJD1A, which participate in the
epigenetic regulation of dysfunctional VSMCs in atherosclerosis
(18–20). To the best of our knowledge, there
is no study available to date on the connection between
atherosclerosis and JMJD2A. In the present study, we demonstrated
that Ang II induced total JMJD2A expression, and this was
accompanied by a suppressed global expression of H3K9me3. Based on
the above results, we postulated that JMJD2A and its substrate,
H3K9me3, participate in the dysfunction of Ang II-stimulated
VSMCs.
The cell cycle is a complex process that involves
multiple regulatory proteins, including cyclins, CDKs and CDKIs. In
line with previous studies, in the present study, we demonstrated
that, following stimulation with Ang II, the expression of cyclin
D1 and cyclin E increased markedly (21,22). The Ang II-induced cyclin D1
expression was significantly inhibited by IOX1, whereas the
expression of cyclin E was not affected. In addition, treatment of
the VSMCs with IOX1 increased p21 expression that had been
decreased by stimulation with Ang II. These results suggested that
IOX1 blocks cell cycle progression by regulating cyclin D1 and p21
expression. The abnormal expression of cyclin D1 is a hallmark of
proliferative diseases, including cancer and atherosclerosis
(23,24). Cyclin D1 is regulated
transcriptionally and post-transcriptionally (25). In this study, we confirmed that
IOX1 inhibited the expression of cyclin D1 by increase H3K9me3
expression in the promoter region, reaffirming that H3K9me3 is
related to heterochromatin formation and transcriptional repression
(26). It has been demonstrated
that the removal of H3K9me3 by JMJD2A is required for the
recruitment of AP-1 to chromatin, thereby promoting the positive
feedback loop that induces the activation of AP-1 (12). Previous studies have reported that
AP-1 transcription factors regulate cyclin D1 gene expression
(27). IOX1 may suppress cyclin
D1 expression by inhibiting the activation of AP-1. Cyclin D1 is a
target gene of estrogen receptor α (ERα) (28). Berry et al (13) demonstrated that the downregulation
of JMJD2A reduced the transcription of a seminal ERα target gene,
cyclin D1, which was consistent with our results. Our results,
however, seemed to be self-contradictory as IOX1 increased p21
expression. Our observation was in agreement with that of the study
by Kim et al, demonstrating that JMJD2A depletion led to
increased levels of H3K9me3 at the p21 promoter and to increased
levels of p21 (15).
IOX1, a cell-permeable derivative of
8-hydroxyquinoline, is the most potent broad-spectrum inhibitor of
the 2-oxoglutarate (2OG)-dependent JmjC family of histone lysine
demethyl-ases (29). It exerts
its inhibitory effect on JMJDs by chelating with the Fe(II) ion to
form a bidentate structure positioned at the active site (30). It was demonstrated to inhibit
JMJD2A activity in a cell (29).
To the best of our knowledge, there is no study available to date
on the role of IOX1 in VSMCs. We proved that IOX1 inhibited VSMC
proliferation and migration induced by Ang II. IOX1 may also
inhibit other JmjC-domain containing histone lysine demethylases.
Thus, there may be other potential mechanisms involved and further
investigations are warranted.
In conclusion, in this study, we investigated the
inhibitory effect of IOX1 on the proliferation and migration of
VMSCs induced by Ang II. We found that this effect was mediated
through the regulation of the expression of the cell cycle-related
proteins, cyclin D1 and p21. This regulatory effect was exerted by
restoring H3K9me3 expression at the promoter regions of these
genes. IOX1 may be thuse prove to be a potential therapeutic agent
in the treatment of atherosclerosis.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of the People's Republic of China grants
(nos. 81170195 and 81200156) and the Fundamental Research Funds for
the Central Universities (no. 20120141120079). We would like to
thank Professor Yanhong Tang, Professor Xi Wang, Dr Teng Wang and
Dr Ping Hu from the Renmin Hospital of Wuhan University for their
excellent technical assistance.
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Doran AC, Meller N and McNamara CA: Role
of smooth muscle cells in the initiation and early progression of
atherosclerosis. Arterioscler Thromb Vasc Biol. 28:812–819. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
King RW, Jackson PK and Kirschner MW:
Mitosis in transition. Cell. 79:563–571. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weiss D, Sorescu D and Taylor WR:
Angiotensin II and atherosclerosis. Am J Cardiol. 87:25C–32C. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mehta PK and Griendling KK: Angiotensin II
cell signaling: physiological and pathological effects in the
cardiovascular system. Am J Physiol Cell Physiol. 292:C82–C97.
2007. View Article : Google Scholar
|
|
6
|
Klein EA and Assoian RK: Transcriptional
regulation of the cyclin D1 gene at a glance. J Cell Sci.
121:3853–3857. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Z, Zang J, Kappler J, Hong X,
Crawford F, Wang Q, Lan F, Jiang C, Whetstine J, Dai S, et al:
Structural basis of the recognition of a methylated histone tail by
JMJD2A. Proc Natl Acad Sci USA. 104:10818–10823. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Whetstine JR, Nottke A, Lan F, Huarte M,
Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M and Shi
Y: Reversal of histone lysine trimethylation by the JMJD2 family of
histone demethylases. Cell. 125:467–481. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang J, Chen J, Yang J, Xu C, Ding J,
Yang J, Guo Q, Hu Q and Jiang H: Sodium ferulate inhibits
neointimal hyperplasia in rat balloon injury model. PLoS One.
9:e875612014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shin S and Janknecht R: Activation of
androgen receptor by histone demethylases JMJD2A and JMJD2D.
Biochem Biophys Res Commun. 359:742–746. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kogure M, Takawa M, Cho HS, Toyokawa G,
Hayashi K, Tsunoda T, Kobayashi T, Daigo Y, Sugiyama M, Atomi Y, et
al: Deregulation of the histone demethylase JMJD2A is involved in
human carcinogenesis through regulation of the G(1)/S transition.
Cancer Lett. 336:76–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ding X, Pan H, Li J, Zhong Q, Chen X, Dry
SM and Wang CY: Epigenetic activation of AP1 promotes squamous cell
carcinoma metastasis. Sci Signal. 6:ra282013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Berry WL, Shin S, Lightfoot SA and
Janknecht R: Oncogenic features of the JMJD2A histone demethylase
in breast cancer. Int J Oncol. 41:1701–1706. 2012.PubMed/NCBI
|
|
14
|
Kauffman EC, Robinson BD, Downes MJ,
Powell LG, Lee MM, Scherr DS, Gudas LJ and Mongan NP: Role of
androgen receptor and associated lysine-demethylase coregulators,
LSD1 and JMJD2A, in localized and advanced human bladder cancer.
Mol Carcinog. 50:931–944. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim TD, Shin S, Berry WL, Oh S and
Janknecht R: The JMJD2A demethylase regulates apoptosis and
proliferation in colon cancer cells. J Cell Biochem. 113:1368–1376.
2012. View Article : Google Scholar
|
|
16
|
Gray SG, Iglesias AH, Lizcano F,
Villanueva R, Camelo S, Jingu H, Teh BT, Koibuchi N, Chin WW,
Kokkotou E and Dangond F: Functional characterization of JMJD2A, a
histone deacetylase- and retinoblastoma-binding protein. J Biol
Chem. 280:28507–28518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang D, Yoon HG and Wong J: JMJD2A is a
novel N-CoR-interacting protein and is involved in repression of
the human transcription factor achaete scute-like homologue 2
(ASCL2/Hash2). Mol Cell Biol. 25:6404–6414. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Villeneuve LM, Reddy MA, Lanting LL, Wang
M, Meng L and Natarajan R: Epigenetic histone H3 lysine 9
methylation in metabolic memory and inflammatory phenotype of
vascular smooth muscle cells in diabetes. Proc Natl Acad Sci USA.
105:9047–9052. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weng X, Cheng X, Wu X, Xu H, Fang M and Xu
Y: Sin3B mediates collagen type I gene repression by interferon
gamma in vascular smooth muscle cells. Biochem Biophys Res Commun.
447:263–270. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lockman K, Taylor JM and Mack CP: The
histone demethylase, Jmjd1a, interacts with the myocardin factors
to regulate SMC differentiation marker gene expression. Circ Res.
101:e115–e123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen D, Liu J, Rui B, Gao M, Zhao N, Sun
S, Bi A, Yang T, Guo Y, Yin Z and Luo L: GSTpi protects against
angiotensin II-induced proliferation and migration of vascular
smooth muscle cells by preventing signal transducer and activator
of transcription 3 activation. Biochim Biophys Acta. 1843:454–463.
2014. View Article : Google Scholar
|
|
22
|
Kim JE and Choi HC: Losartan inhibits
vascular smooth muscle cell proliferation through activation of
AMP-activated protein kinase. Korean J Physiol Pharmacol.
14:299–304. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Molenaar JJ, Ebus ME, Koster J, van Sluis
P, van Noesel CJ, Versteeg R and Caron HN: Cyclin D1 and CDK4
activity contribute to the undifferentiated phenotype in
neuroblastoma. Cancer Res. 68:2599–2609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsaousi A, Williams H, Lyon CA, Taylor V,
Swain A, Johnson JL and George SJ: Wnt4/β-catenin signaling induces
VSMC proliferation and is associated with intimal thickening. Circ
Res. 108:427–436. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Musgrove EA: Cyclins: roles in mitogenic
signaling and oncogenic transformation. Growth Factors. 24:13–19.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lachner M, O'Carroll D, Rea S, Mechtler K
and Jenuwein T: Methylation of histone H3 lysine 9 creates a
binding site for HP1 proteins. Nature. 410:116–120. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen Q, Uray IP, Li Y, Krisko TI, Strecker
TE, Kim HT and Brown PH: The AP-1 transcription factor regulates
breast cancer cell growth via cyclins and E2F factors. Oncogene.
27:366–377. 2008. View Article : Google Scholar
|
|
28
|
Shaulian E and Karin M: AP-1 as a
regulator of cell life and death. Nat Cell Biol. 4:E131–E136. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schiller R, Scozzafava G, Tumber A,
Wickens JR, Bush JT, Rai G, Lejeune C, Choi H, Yeh TL, Chan MC, et
al: A cell-permeable ester derivative of the JmjC histone
demethylase inhibitor IOX1. ChemMedChem. 9:566–571. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
King ON, Li XS, Sakurai M, Kawamura A,
Rose NR, Ng SS, Quinn AM, Rai G, Mott BT, Beswick P, et al:
Quantitative high-throughput screening identifies
8-hydroxyquinolines as cell-active histone demethylase inhibitors.
PLoS One. 5:e155352010. View Article : Google Scholar : PubMed/NCBI
|