Introduction
Kidney transplantation is the optimal treatment for
patients with end-stage renal disease (ESRD) (1); however, a severe shortage of organs
is the permanent bottleneck which limits the availability of organ
transplantation as a treatment. To increase the donor pool, novel
and innovative means of increasing the number of suitable kidneys
for transplantation have been established, including expanded
criteria donation (ECD) and donation after cardiac death (DCD).
However, the organs procured from DCD donors are at greater risk of
serious ischemia-reperfusion injury (IRI). IRI is known to be a
risk factor for delayed graft function (DGF), rejection, renal
fibrosis and both poorer short- and long-term graft function as
well as reduced patient survival following transplantation
(2–5). Thus, methods to optimize the quality
of DCD kidneys and improve short-term and long-term outcomes are
urgently required. Hypothermic machine perfusion (HMP) and static
cold storage (CS) are methods that have been developed over the
past 30 years in order to maximize the benefit of donated kidneys
(6). In fact, HMP has been
reported to obtain better transplantation outcomes compared with CS
(7–10). Although this potential benefit of
HMP remains poorly understood, it may provide an opportunity to
improve organ quality in combination with pharmacological and gene
transfer therapies (11).
Inflammation is an invariable finding in acute and
chronic kidney injury (12) and
is a critical initiating and aggravating factor in kidney damage
(13). A pivotal mediator of this
inflammatory response is the transcription factor nuclear factor-κB
(NF-κB), which regulates the expression of adhesion molecules,
chemokines and other pro-inflammatory molecules in renal or
endothelial cells (ECs) (14–16). The best-characterized signaling
pathways that lead to NF-κB activation are those stimulated by
members of the interleukin-1 (IL-1)/Toll-like receptor family and
tumor necrosis factor-α (TNF-α) (17).
Traditionally, two forms of cell death have been
identified: apoptosis (programmed cell death) and necrosis
(18). However, another form of
cell death, known as necroptosis, has recently been identified
which has the characteristics of apoptosis and necrosis (19). Evidence suggests that apoptotic
pathways contribute to renal tubular injury (20,21). Receptor-interacting protein kinase
3 (RIPK3)-mediated necroptosis in donor kidneys may promote
inflammatory injury and has a major impact on renal IRI and graft
survival (22).
A20, also known as tumor necrosis factor,
alpha-induced protein 3 (TNFAIP3), was originally identified as a
TNF-inducible gene product of ECs (23). It has been demonstrated that A20
mRNA is markedly upregulated in the mouse kidney in response to
TNF-α treatment, and A20 knockout mice are hypersensitive to the
pro-inflammatory effects of TNF-α and lipopolysaccharide, which
results in premature death due to severe multi-organ inflammation
and cachexia (24,25). A20 is a potent anti-inflammatory
protein associated with multiple human autoimmune diseases and
human malignancies (26,27). A20 also enhances cell viability
and has been shown to protect ECs, proximal tubular epithelial
cells (TECs) and pancreatic islets from injury in response to
various stimuli including TNF-α and oxidative stress (28–30). A20 is a cytoplasmic zinc-finger
protein that has been characterized as a dual inhibitor of NF-κB
activation and cell death (31).
Furthermore, A20 inhibits RIPK3-dependent necroptosis thereby
attenuating inflammatory injury (32). Thus, measuring A20 at the mRNA and
protein levels has the potential to be diagnostic and prognostic of
transplantation outcomes and therefore may be important in
determining timely therapeutic interventions in order to prolong
graft survival (33). HMP is
known to be superior to CS as a method of donor organ storage
following cardiac death; therefore, in this study, we examined the
molecular mechanism underlying the protective effects of HMP on
donor organ storage.
Materials and methods
Animals
Eighteen healthy male rabbits (obtained from the
Animal Experiment Center of Wuhan University/Wan Qian Jia He
Experimental Animal Culture Center, Wuhan, China) weighing 3.1±0.2
kg (aged 12–13 weeks) were randomly allocated to three groups
(n=6/group).
All animal experiments were performed in accordance
with the Experimental Animal Regulations established by The
Ministry of Science and Technology of the People's Republic of
China and the Guidelines for the Care and Use of Laboratory Animals
published by the US National Institutes of Health. The study
received ethics approval from the Ethics Committee of Zhongnan
Hospital of Wuhan University. Prior to performing the experiments,
all the animals were subjected to an overnight fast with unlimited
access to water.
Establishment of the animal model
The rabbits were anesthetized and surgery was
performed at the appropriate room temperature. In the sham group,
the left kidney was subjected to warm ischemia for 25 min by
clamping the left renal pedicle which was followed by reperfusion
for 29 h. In the HMP group, the left renal pedicle was clamped for
25 min which was followed by recovered blood flow for 1 h. The
kidneys were then hypothermically (4–8°C) preserved in vivo
for 4 h in HCA-II solution (Shanghai Chang Zheng Hospital,
Shanghai, China) using HMP (n=6) or CS (n=6). In the HMP group, the
left kidneys were connected in vivo to the LifePort Kidney
Transporter (Organ Recovery System, Chicago, IL, USA). A mean
arterial pressure of 58±7.5 mmHg was maintained during the period
of perfusion. Following anastomosis of the vessels, a right
nephrectomy was performed and specimens were obtained 24 h later.
All procedures were identical in both groups, with the exception
that the kidneys were stored in polystyrene organ boxes (Zhejiang
Zhenhua Plastic Co., Ltd., Zhejiang, China) in the CS group.
Western blot analysis of A20, apoptosis
signal-regulating kinase 1 (ASK1), c-Jun N-terminal kinase (JNK),
phosphorylated (p-)JNK, pro-caspase-3, cleaved caspase-3, RIPK3,
mucosa-associated lymphoid tissue lymphoma translocation gene 1
(MALT1), NF-κB and IκBα expression
The kidney tissue was homogenized in RIPA buffer
containing a protease inhibitor and then centrifuged at 15,000 × g
for 20 min at 4°C in order to extract the total proteins. The
supernatants were collected and the total protein concentrations
were normalized using the BCA assay (Beyotime Institute of
Biotechnology, Shanghai, China). The proteins were separated by
10–12% sodium dodecyl sulfate (SDS)-polyacrylamide gel
electrophoresis and transferred to PVDF membranes (Millipore,
Billerica, MA, USA). The blots were incubated with the antibodies
specific for the following: A20 (RS-92803R), ASK1 (RS-90145R),
MALT1 (RS-96863R) and IκBα (RS-90167R) (Shanghai Ruiqi
Bio-Technology, Inc., Shanghai, China); caspase-3 (GB13009), and
JNK (GB17018)/p-JNK (GB13019-M) (Wuhan Goodbio Technology, Inc.,
Hubei, China); NF-κB (bs-0465R) and RIPK3 (bs-3551R) (Bioss Bio
Technology, Inc., Beijing, China). β-actin or GAPDH were used as
controls. Following incubation with anti-IgG, the proteins were
visualized using an ECL reagent followed by exposure to X-ray film.
Quantification of band density was determined using the Quantity
One software package (Bio-Rad Laboratories, Hercules, CA, USA).
Reverse transcription
quantitative-polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissue using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions. cDNA was synthesized using PrimeScript
RT reagent kit with cDNA eraser (Invitrogen) according to the
manufacturer's instructions. After PCR amplification, the products
of A20, XIAP, GADD45β, MnSOD and c-FLIP were separated by agarose
gel electrophoresis and visualized by ethidium bromide staining.
One-step real-time RT-PCR was performed using SYBR Premix Ex Taq™
(Takara, Hubei, China) in a real-time PCR machine (ABI 7900;
Applied Biosystems, Carlsbad, CA, USA) according to the
manufacturers' instructions. The primer pairs used are listed in
Table I. GAPDH and β-actin were
used as endogenous controls. The relative mRNA expression levels of
each target gene were normalized to those of the controls using the
2−ΔΔCT method.
 | Table ISequences of rabbit primers used for
comparative RT-qPCR. |
Table I
Sequences of rabbit primers used for
comparative RT-qPCR.
| Gene | Forward | Reverse |
|---|
| A20 |
5′-AGACCGAGGAAGATTTGAAGAC-3′ |
5′-CGTTAATCAGATGCGTCGTG-3′ |
| ASK1 |
5′-GTTCGCCTTGGACAGTATCAT-3′ |
5′-CTCGTGGTCATCTTCTACATCC-3′ |
| IκBα |
5′-CCATCAACTACAACGGCCACA-3′ |
5′-ACTTCAACAAGAGCGACACCAG-3′ |
| XIAP |
5′-GAAGCCCAATGAAGACCCT-3′ |
5′-CTCCCTGAAACTGAATCCC-3′ |
| GADD45β |
5′-TCTTGGGTGATCGAGGACTGGC-3′ |
5′-CGCCTCCTTCTTCTGTCTTTGCT-3′ |
| MnSOD |
5′-CTTTGGGTCCTTTGACAAGTT-3′ |
5′-AAGTGTCCCTGCTCCTTATTG-3′ |
| c-FLIP |
5′-CCCAGCACCGAGACTATGA-3′ |
5′-GCTTTGGCTTCCCTATGAG-3′ |
| RIPK3 |
5′-GACCTCAAACCCTCCAACATC-3′ |
5′-CTAGACACTGCCTCTGCCAACT-3′ |
| TNF-α |
5′-GCCGTCTCCTACCCGAACAAG-3′ |
5′-CACAGGGCAATGATCCCAAAG-3′ |
| β-actin |
5′-TGGCTCTAACAGTCCGCCTAG-3′ |
5′-AGTGCGACGTGGACATCCG-3′ |
| HMGB1 |
5′-ATCCTGGCCTGTCCATTGGTG-3′ |
5′-TTTCAGCCTTGACGACTCCCT-3′ |
| MALT1 |
5′-GCGATGCCTATGTCACCGATTT-3′ |
5′-ACGTTCACCTCCTGCTTCTCCT-3′ |
| GAPDH |
5′-GCTGAACGGGAAACTCACTG-3′ |
5′-CGAAGGTAGAGGAGTGGGTG-3′ |
TdT-mediated biotin-16-dUTP nick-end
labeling (TUNEL) assay
Apoptotic cell death was evaluated using the
One-Step TUNEL Apoptosis assay kit (Beyotime Institute of
Biotechnology). Briefly, the apoptotic cells were identified by the
addition of digoxigenin-deoxynucleoside triphosphate (dNTP)
fragments to the 3′-OH DNA termini by TdT followed by labeling with
peroxidase- or Rhodamine-linked anti-digoxigenin antibodies and
visualization with either diaminobenzidine (DAB; Beyotime Institute
of Biotechnology) followed by light microscopy or fluorescence
microscopy (Olympus Corporation, Tokyo, Japan). As a positive
control, sections were incubated with DNase I for 10 min at room
temperature (25°C) prior to the fluorescent labeling procedure. The
cells labeled with green fluorescence were described as apoptotic
cells. The cells were labeled with 4′,6-diamidino-2-phenylindole
(DAPI; Beyotime Institute of Biotechnology) for nuclear
staining.
Immunohistochemical analysis of A20 and
RIPK3 expression
The expression of A20 and RIPK3 in paraffin-embedded
tissue sections was analyzed by immunohistochemistry (IHC).
Following deparaffinization and antigen retrieval in 10 mM sodium
citrate buffer (pH 6.0) using the pressure cooker method at full
power for 4 min, the tissue sections were exposed to 3%
H2O2 for 10 min. The tissue sections were
then blocked for 30 min at room temperature with 5% bovine serum
albumin (BSA). The sections were incubated with anti-A20 (1:40) and
anti-RIPK3 (1:200) antibodies overnight at 4°C in a humid chamber
and then incubated with the horseradish peroxidase (HRP)-conjugated
secondary detection antibody for 30 min at 37°C. The sections were
then incubated with DAB chromogen, counterstained with hematoxylin
(Beyotime Institute of Biotechnology) and finally dehydrated,
cleared and mounted with neutral gum. The sections were washed with
several changes of Tris-buffered saline (TBS)-0.3% Tween buffer
between each step.
Enzyme-linked immunosorbent assay
(ELISA)
TNF-α levels were measured using a specific ELISA
kit (Elabscience Biotechnology Co., Ltd., Hubei, China) according
to the manufacturer's instructions. The homogenates were first
centrifuged at 10,000 × g for 25 min in order to remove solid
tissue debris and the supernatant was collected and assayed.
Measurement of reactive oxygen species
(ROS) and malonaldehyde (MDA) levels
The levels of ROS and MDA are established markers
used to determine the extent of tissue oxidative damage and cell
viability. The kidney tissues were thawed following homogenization
according to the manufacturer's instructions. (Jiancheng Technology
Co., Ltd., Nanjing, China). The supernatant was assayed using the
Multiskan MK3 (Thermo Fisher Scientific, Waltham, MA, USA).
Statistical analysis
The results are presented as the means ± standard
deviation (SD). Statistical analysis was performed by one-way
analysis of variance (ANOVA) after proving the assumption of
normality (Shapiro-Wilk testing) and then followed by LSD multiple
comparison tests when F was significant. The software SPSS 17.0 for
Windows (SPSS, Inc., Chicago, IL, USA) was used. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of A20, but not of other NF-κB
target genes significantly increases in the HMP group compared with
that in the CS group
Fig. 1A shows the
results of the analysis of A20 and MALT1 expression obtained by
western blot analysis and RT-qPCR in the different treatment
groups. HMP treatment significantly increased A20 expression
compared with that in the CS group (P<0.01). The expression of
MALT1 (in the kidney), which cleaves A20 (34), in the HMP group was lower than
that in the CS group (P<0.01). Fig. 1B and C show the results of RT-qPCR
of A20, X-linked inhibitor of apoptosis protein (XIAP), manganese
superoxide dismutase (MnSOD), growth arrest and DNA
damage-inducible 45β (GADD45β) and cellular FLICE-inhibitory
proteins (c-FLIP) expression in the different treatment groups. It
has been proposed that a subset of NF-κB target genes, including
XIAP, MnSOD, GADD45β, and c-FLIP, are capable of antagonizing JNK
signaling (35–37). In the present study, the
expression of XIAP and GADD45β were marginally or poorly induced
whereas MnSOD levels were slightly lower in both the HMP and CS
groups compared with those in the sham group. Although the
expression of c-FLIP was increased in both the HMP and CS groups
compared with that in the sham group, there was no significant
difference among the three groups. Thus, these findings clearly
indicate that A20 rather than the JNK inhibitors, plays a
predominant role in the suppression of JNK signaling in
apoptosis.
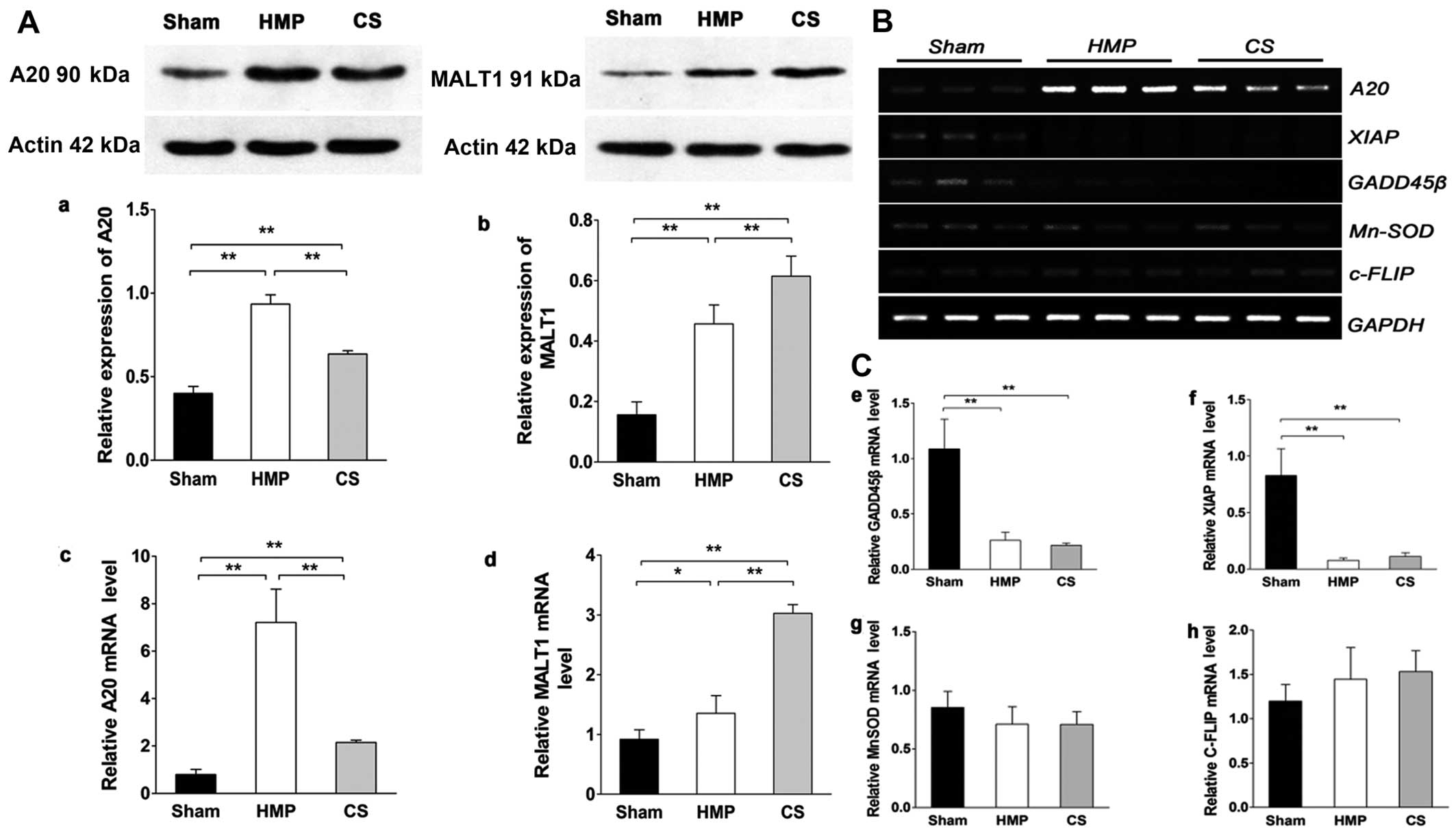 | Figure 1(A) A20 and mucosa-associated
lymphoid tissue lymphoma translocation gene 1 (MALT1) expression in
the three groups were analyzed by western blot analysis and
RT-qPCR. Representative blots are shown. Western blot analysis of
(a) A20 and (b) MALT1 protein. β-actin was used as the control.
RT-qPCR analysis of (c) A20 and (d) MALT1 mRNA levels. Graphs
represent the statistical analysis of relative A20 mRNA levels
after normalization against β-actin. In the hypothermic machine
perfusion (HMP) group, A20 expression was increased compared with
that in the cold storage (CS) and sham groups (both
**P<0.01). By contrast, MALT1 expression was reduced
compared with that in the CS group (**P<0.01).
Results represent the means ± SD of three experiments,
*P<0.05. (B and C) In the HMP group, the expression
of A20, but not of other nuclear factor-κB (NF-κB) target genes was
significantly increased compared with the levels in the CS group.
Levels of particular transcripts were quantified by real-time PCR
using gene-specific primers. The amount of each target transcript
was normalized against the levels of GAPDH transcript. (B) The mRNA
expression of A20, XIAP, GADD45β, MnSOD and c-FLIP was examined in
renal tissue. After PCR amplification, the products were separated
by agarose gel electrophoresis and visualized by ethidium bromide
staining. (C-e-g) Compared with the sham group, X-linked inhibitor
of apoptosis protein (XIAP) and growth arrest and DNA
damage-inducible 45β (GADD45β) were marginally or poorly induced
(all **P<0.01), whereas manganese superoxide
dismutase (MnSOD) levels were slightly lower in both the HMP and CS
groups compared with those in the sham group (both P>0.05). (h)
Both the HMP and CS groups showed increased expression of cellular
FLICE-inhibitory proteins (c-FLIP) compared with that in the sham
group, although there were no significant differences among the
three groups. Data represent the means ± SD of three
experiments. |
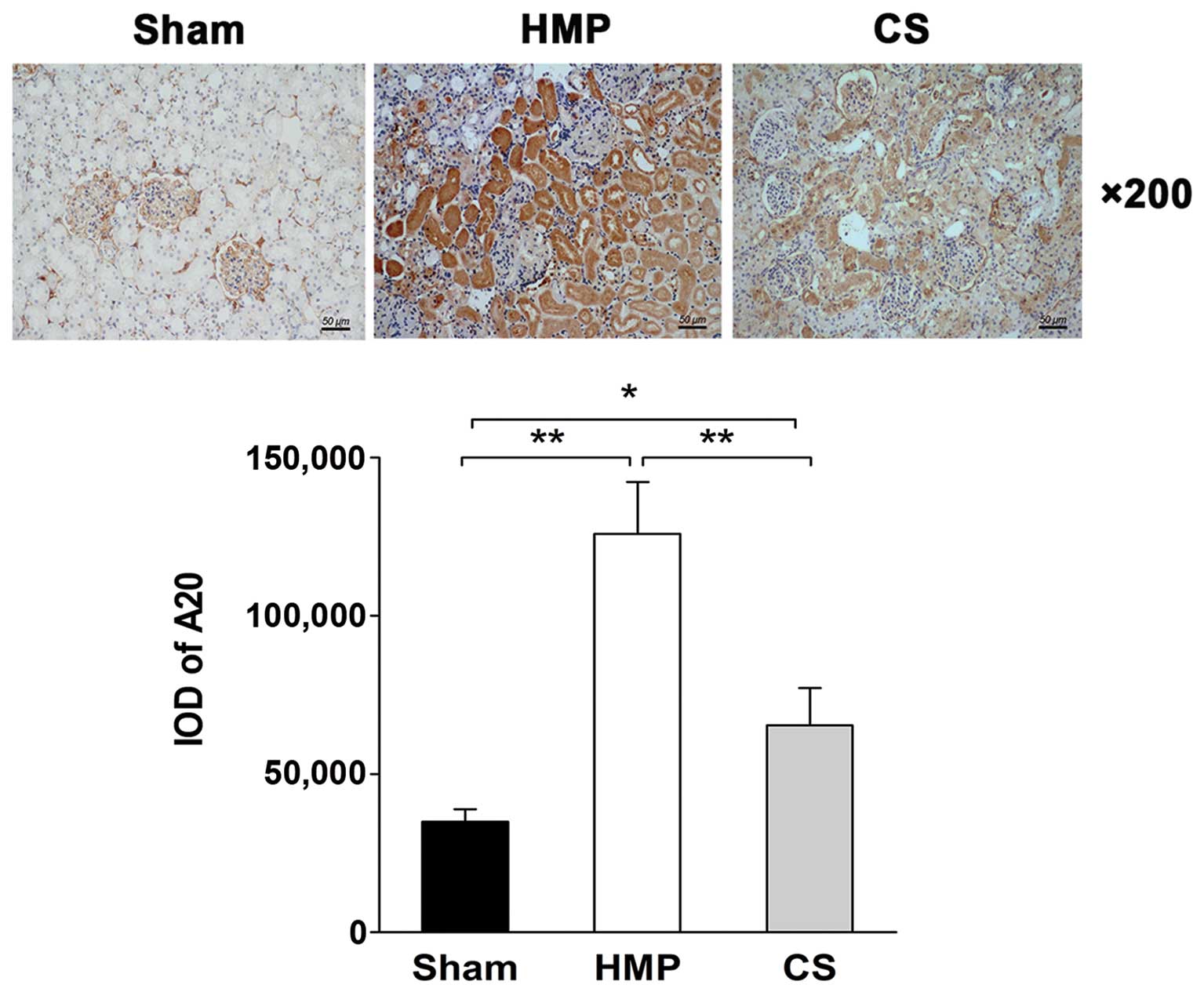
Immunohistochemical analysis reveals
higher A20 expression in the HMP group compared with that in the CS
group
The immunohistochemical analysis of the location and
levels of A20 expression in the kidneys in the HMP and CS groups is
shown in Fig. 2. A20 was
expressed predominantly in the TECs, with significantly more
abundant expression observed in the HMP group compared with that in
the CS group (P<0.01).
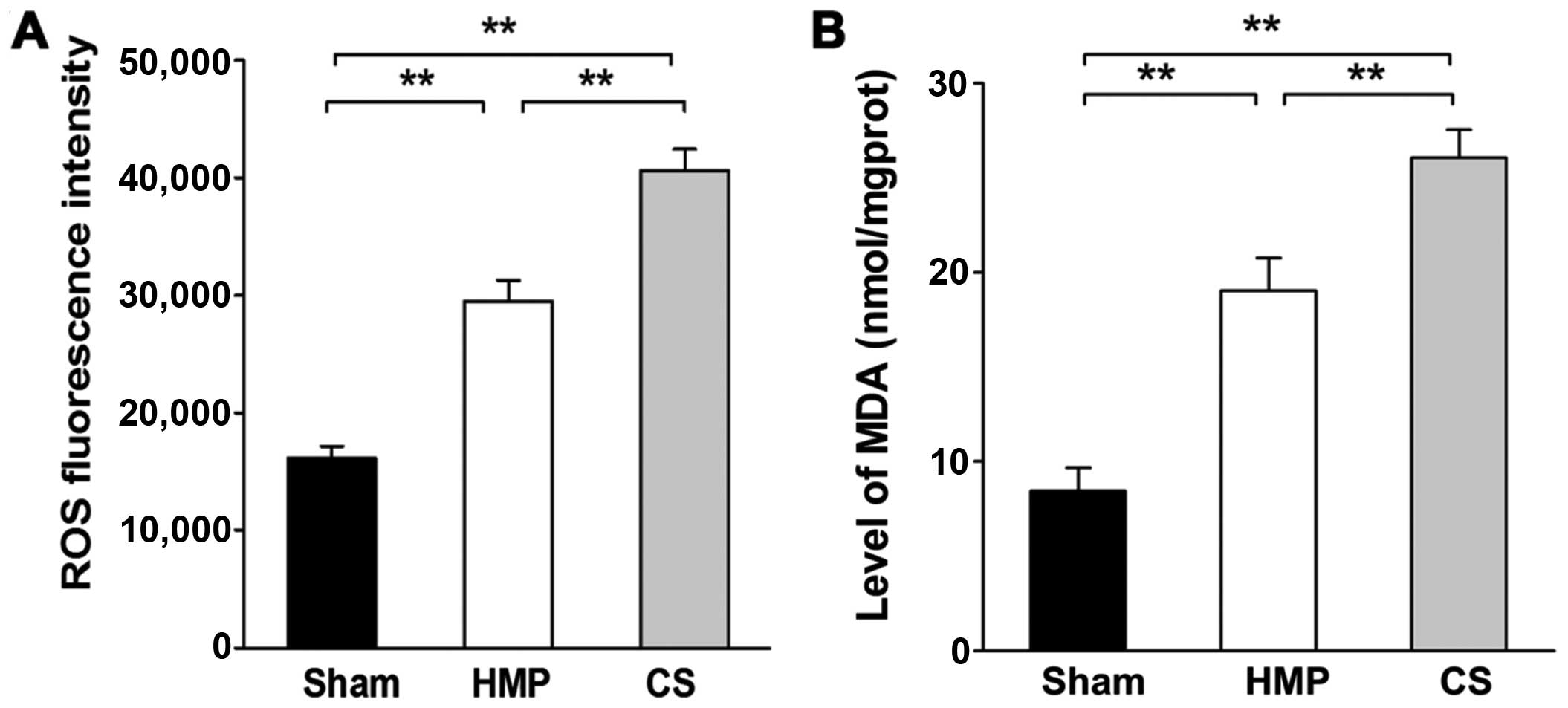
Extent of kidney injury significantly
decreases in the HMP group compared with that in the CS group
Renal cellular injury was detected by the
measurement of ROS levels and MDA, which are important indicators
of oxidative damage. Significantly higher levels of ROS and MDA
were detected in the CS group compared with those in the HMP group
(both P<0.01) (Fig. 3).
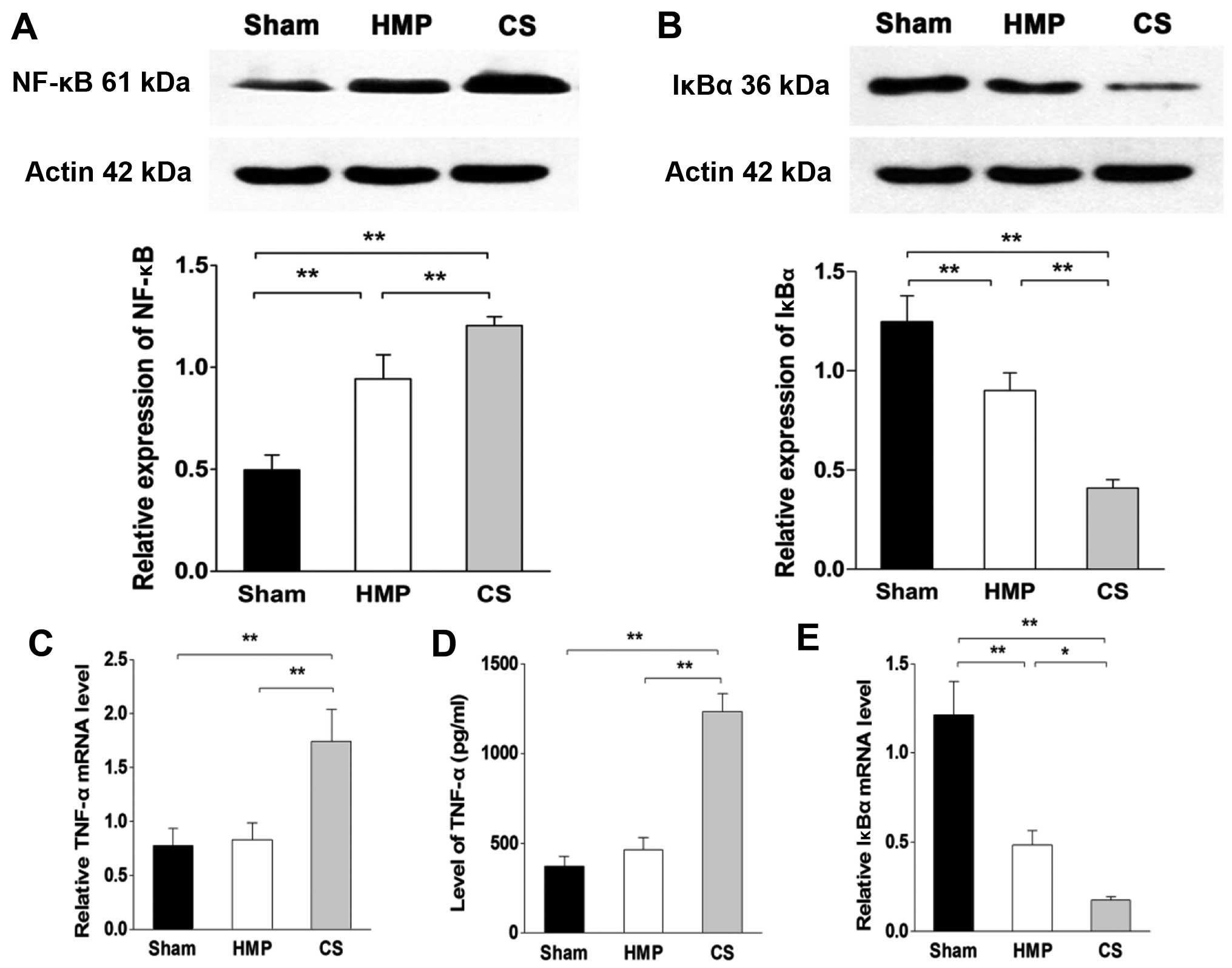
Inflammation of the kidneys decreased in
the HMP group compared with that in the CS group
In the basal state, NF-κB is sequestered in the
cytoplasm by inhibitory IκB proteins. When IκB kinase β (IKKβ) is
activated by pro-inflammatory signaling, it triggers the
degradation of IκBα, thereby promoting the nuclear translocation
and activation of NF-κB. To verify the degree of inflammation in
the kidney, we analyzed the levels of TNF-α by ELISA and RT-qPCR.
Furthermore, we evaluated the expression levels of NF-κB and IκBα
using western blot analysis and RT-qPCR. The HMP group showed
significantly decreased levels of TNF-α and NF-κB compared with
those in the CS group (both P<0.01), whereas there was no
significant difference in the expression of TNF-α between the HMP
and sham group (P>0.05). By contrast, the expression of IκBα in
the HMP group was significantly higher than that in the CS group
(protein level, P<0.01; mRNA level, P<0.05) (Fig. 4).
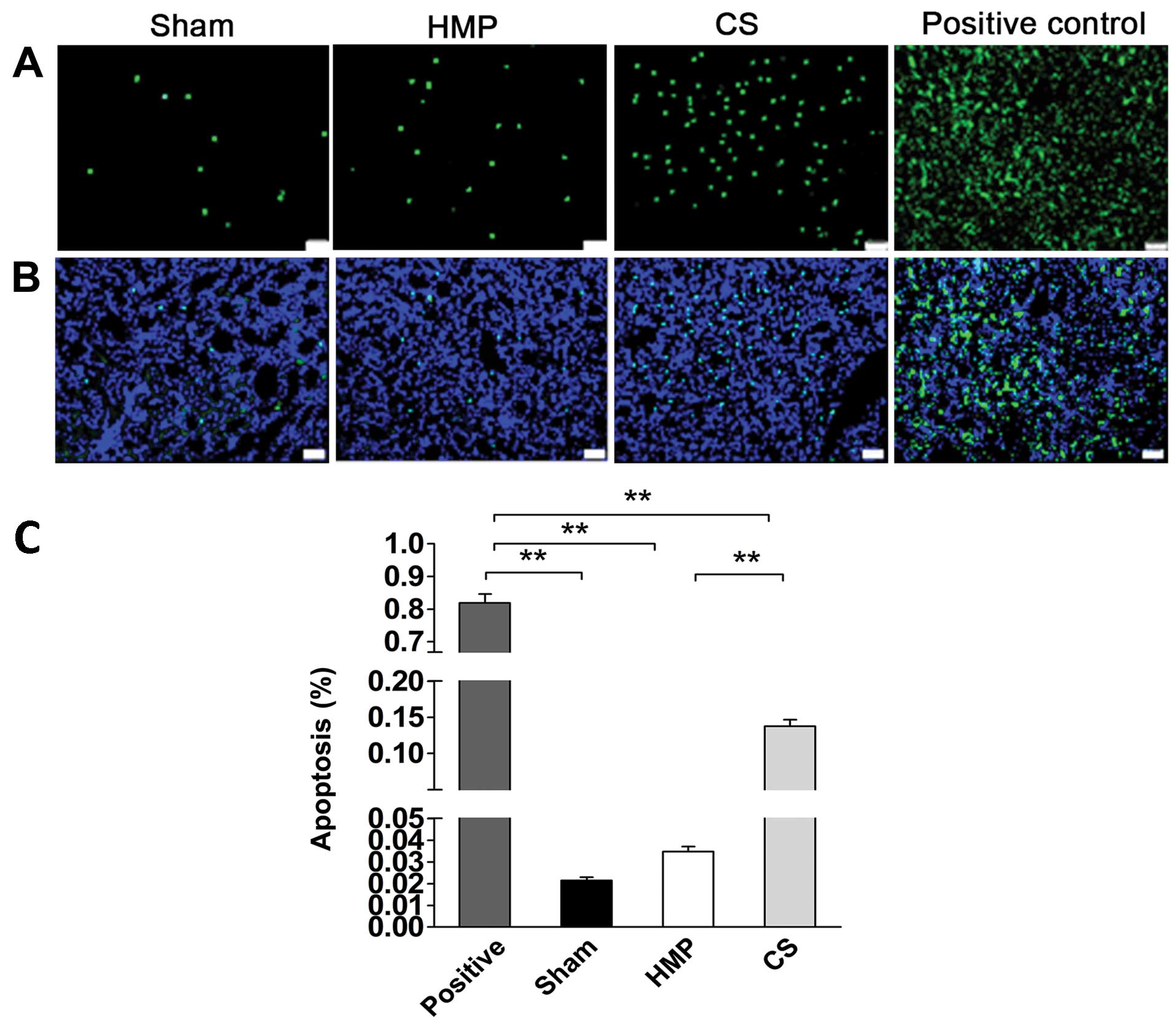
Apoptosis of renal cells significantly
decreases in the HMP group compared with that in the CS group
We detected apoptosis of renal cells using TUNEL
assay. The rate of apoptosis was approximately 4-fold lower in the
HMP group compared with that in the CS group (P<0.01) (Fig. 5), HMP 3.48±0.5%, and CS
13.77±2.0%.
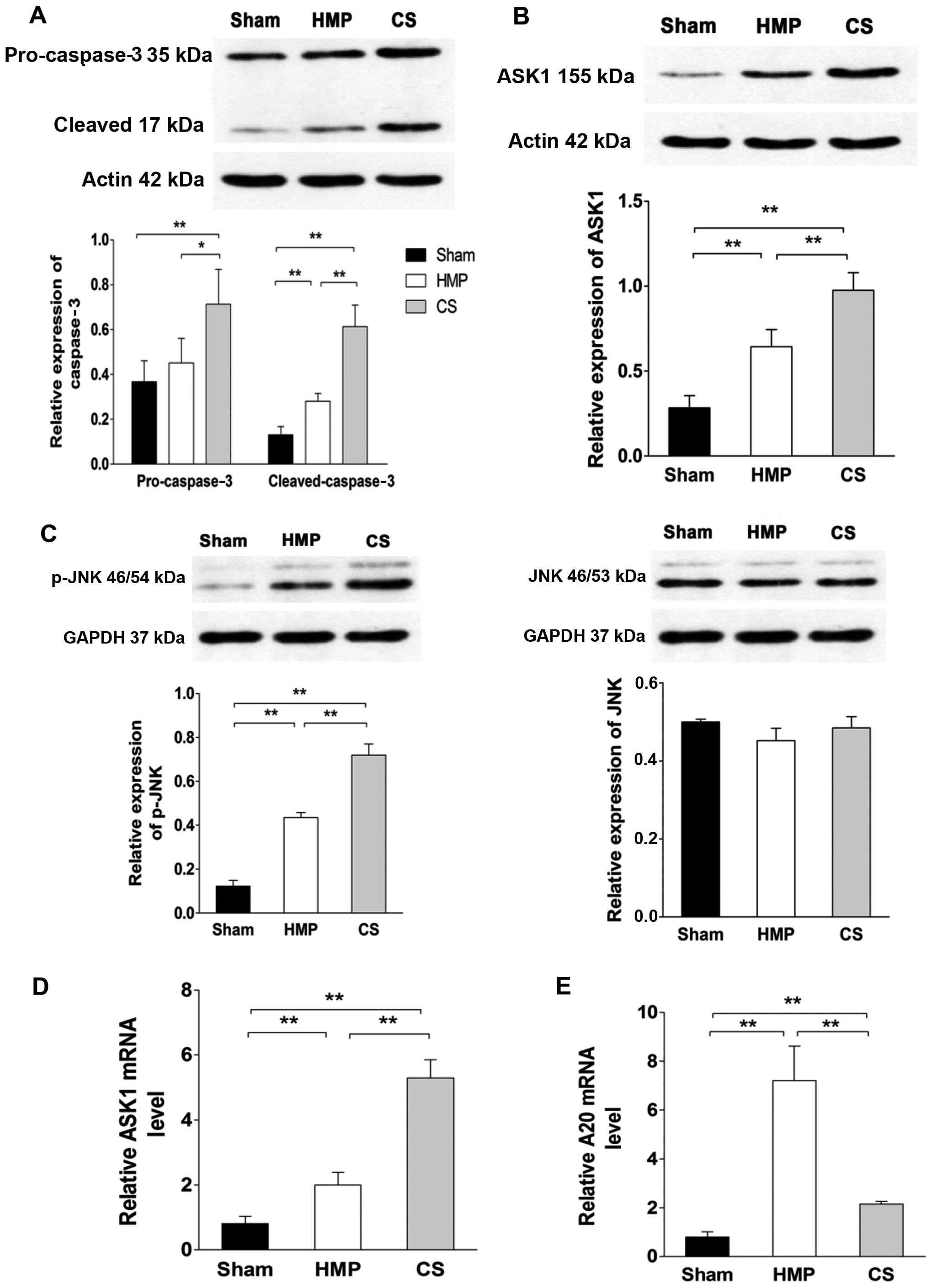
Expression of ASK1, p-JNK and cleaved
caspase-3 significantly decreases in the HMP group compared with
that in the CS group
To evaluate whether A20 decreased apoptosis through
the ASK1-JNK pathway, the expression of ASK1, JNK and its activated
form p-JNK were evaluated by western blot analysis and RT-qPCR
(Fig. 6). HMP treatment reduced
the activation of ASK1, JNK, specifically reducing the expression
of p-JNK compared with that in the CS group (all P<0.01).
Compared with the CS group, pro-caspase-3 expression was decreased
in the HMP group (P<0.05); furthermore, the expression of its
activated form was more significantly decreased in the HMP group
(P<0.01). Compared with the sham group, both the HMP and CS
groups showed increased expression of p-JNK and cleaved caspase-3
(all P<0.01) (Fig. 6).
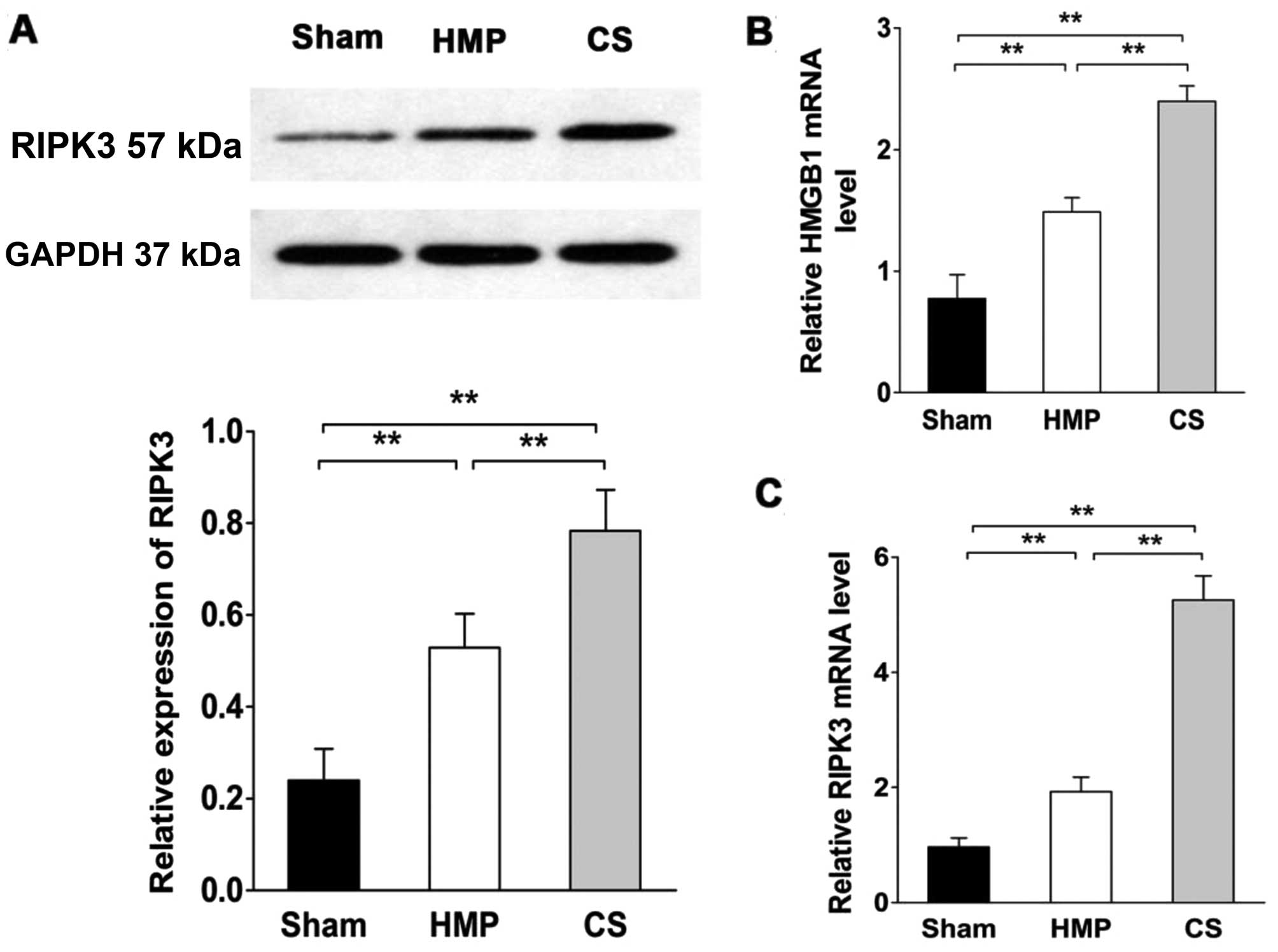
RIPK3 expression significantly decreases
compared with that in the CS group
RIPK3 expression was evaluated by western blot
analysis and RT-qPCR (Fig. 7).
RIPK3 expression was significantly lower in the HMP group compared
with that in the CS group (P<0.01). Compared with the sham
group, both the HMP and CS groups showed increased expression of
RIPK3 (both P<0.01). We also measured high mobility group box 1
(HMGB1) release in order to confirm the necrosis of renal cells
(38). HMGB1 expression was
significantly lower in the HMP group compared with that in the CS
group (P<0.01). Both the HMP and CS groups showed increased
expression of HMGB1 compared with that in the sham group (both
P<0.01).
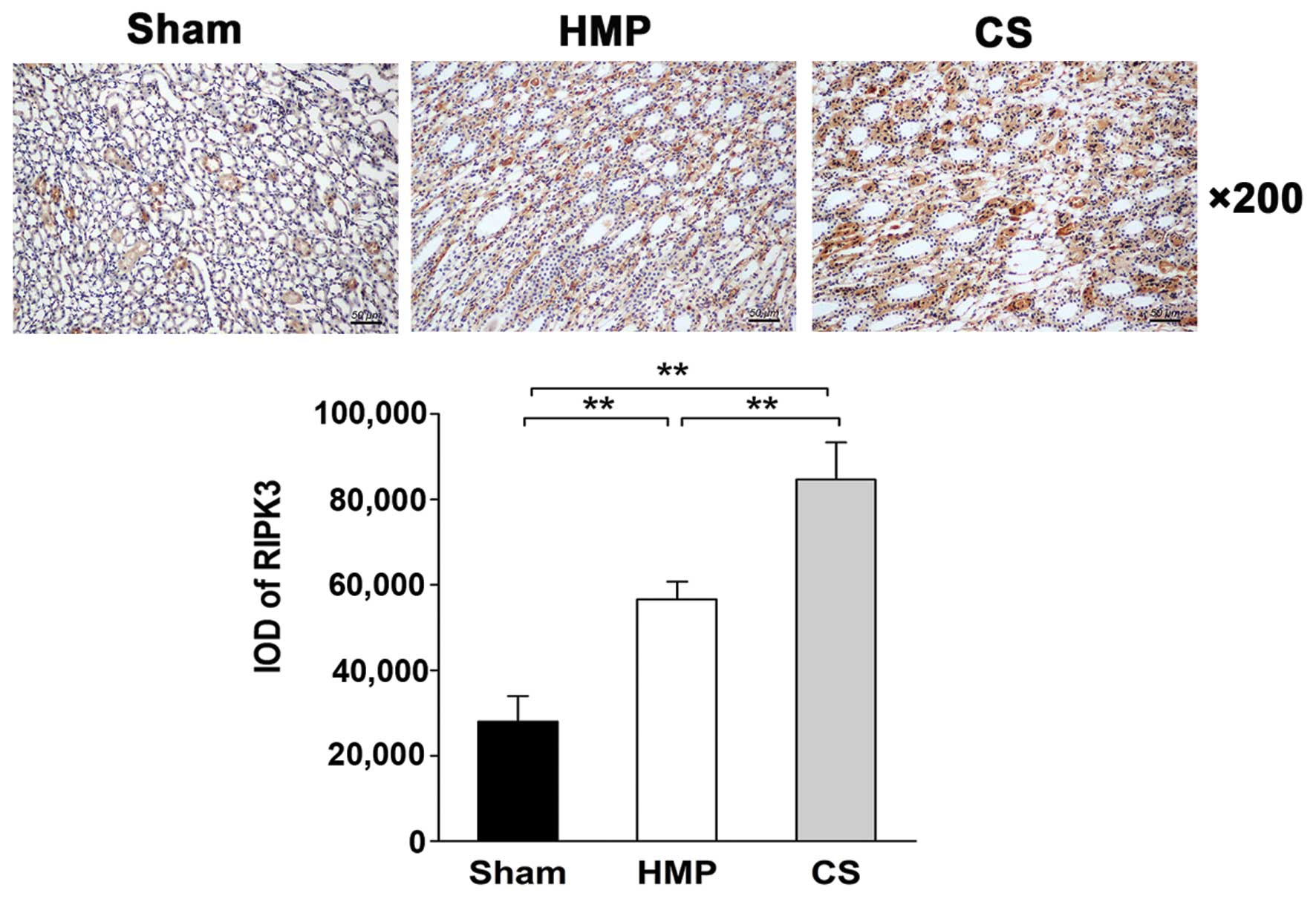
Immunohistochemical analysis of
paraffin-embedded sections reveals low RIPK3 expression in the HMP
group
The expression and location of RIPK3 in the kidney
was analyzed by IHC. Only low levels of RIPK3 protein was expressed
in the HMP group, whereas significantly higher levels were detected
in the CS group (P<0.01). RIPK3 protein was expressed mainly in
the TECs (Fig. 8).
Discussion
The renal transplant waiting list continues to grow
each year, as does the demand for donor organs. To expand the
existing pool of organs, the use of kidneys obtained from donors
after cardiac death is increasing in most countries. However, there
is ongoing controversy regarding the quality of some DCD and ECD
grafts compared with standard criteria organs. Such allografts are
prone to higher rates of DGF, primary non-function and decreased
long-term graft survival (5,39,40). The findings of the multicenter
Eurotransplant trial published in 2009 demonstrated that HMP
reduced the incidence of DGF (26.5% with CS to 20.8% with HMP) in
kidneys obtained from the most common types of deceased donors
(8). The three-year follow-up
data from this trial also demonstrated improved graft survival with
HMP (9). HMP is known to be more
appropriate for donor organ storage after cardiac death than CS;
however, clarification of the underlying mechanism is urgently
required in order to further improve donor organ quality through
HMP.
Kidney transplantation is invariably associated with
organ damage, including IRI. Following an ischemic episode, the
apoptosis and necrosis of TECs occurs initially, and the following
EC injury further exacerbates ongoing ischemia of the tubular
epithelium as well as the inflammatory response (41,42). TECs are known to be the most
susceptible cell type to inflammatory injury, which results in
tubular necrosis (43). A
previous study has shown the benefit of blocking necroptosis in
renal IRI (44). In addition,
apoptosis is one of the principal causes of cell death in isolated
kidneys and cultured renal tubular cells following IRI (45). Therefore, controlling inflammation
and cell death, particularly of TECs, is an attractive therapeutic
strategy to reverse acute kidney injury, halt chronic disease and
protect renal allografts.
The anti-inflammatory function of A20 is well
documented in A20 knockout mice, which are cachectic at birth and
die within 3 weeks of birth as a result of uncontrolled
inflammation (24). Moreover,
TNF-α markedly increases A20 mRNA expression in the kidney
(24) and da Silva et al
demonstrated that inflammation induces the NF-κB-dependent protein
A20 in human renal proximal TECs (RPTECs) (46). These findings are consistent with
our results demonstrating that A20 is expressed in rabbit TECs.
Hypoxia stimulates NF-κB signaling in epithelial and macrophage
cell lines by preventing the repression of IKKβ by oxygen-dependent
prolyl hydroxylases (47). In
addition, NF-κB may be activated during ischemia/reperfusion (I/R)
by changes in the cellular redox potential, which occurs in
response to hypoxia (during ischemia) followed by re-oxygenation
(during reperfusion) (48). NF-κB
transcription factors act as central regulators of inflammation
(16). NF-κB is usually
sequestered in the cytoplasm in association with inhibitor of
NF-κBα, IκBα. Pro-inflammatory signaling leads to IκBα degradation,
thereby promoting the nuclear translocation and activation of
NF-κB. It is well established that A20, as one of the NF-κB target
genes, is involved in a negative feedback loop to block NF-κB
activation (24,31). It has been demonstrated that A20
protects vascular ECs, hepatocytes and pancreatic β cells from
inflammation (49–51). This is consistent with our results
showing the potent anti-inflammatory effect of A20 in TECs. In the
HMP group, A20 reduced the expression of NF-κB and TNF-α compared
with that in the CS group.
A20 was initially characterized as an inhibitor of
TNF-induced apoptosis (52). As
an anti-apoptotic protein, A20 has been demonstrated to protect
breast cancer MCF-7 cells, fibrosarcoma WEHI164 cells, embryonic
fibroblast NIH 3T3 cells, ECs and L929 cells from TNF-mediated
apoptosis or necrosis (28,52–54). A20 acts early in the TNF-α-induced
signaling cascade by blocking both TNF-α-induced rapid activation
of JNK and processing of the receptor-associated caspase-8
(55). Daniel et al also
showed that A20 targets the TNF-α-induced apoptotic pathway by
inhibiting the proteolytic cleavage of caspases-8 and -2 as well as
caspases-3 and -6 (28). Caspase
inhibition using shRNA in order to silence caspase-8 or transgenic
overexpression of the endogenous caspase-8 inhibitor c-FLIP has
been shown to protect renal TECs against TNF-α-induced apoptosis
in vitro and ischemic kidney injury in vivo (56,57). As an alternative mechanism
underlying the anti-apoptotic effects of A20, Won et al
proposed that A20 binds to ASK1, an important mitogen-activated
protein kinase kinase (MAPKK) kinase in the JNK signaling cascade,
and mediates ASK1 degradation, leading to the suppression of JNK
activation and eventually the inhibition of apoptosis (58). Moreover, A20 may inhibit apoptosis
through the suppression of pro-inflammatory cytokines (59). The mitochondria, an organelle
found in most cells, are key sites for integrating the death
receptor and mitochondrial apoptotic pathway during exogenous
stress (60). The caspase family
plays an important role in mediating apoptosis. Among them,
caspase-3 acts as a key execution molecule that functions in many
ways in the transduction of apoptotic signals (61). In the present study, we found that
A20 expression in the HMP group was significantly increased
compared with that in the CS group. Furthermore, the results of
western blot analysis or RT-qPCR revealed that the expression of
ASK1, p-JNK and cleaved caspase-3 was significantly reduced
compared with that in the CS group. Moreover, TUNEL assay analysis
of apoptosis in our study showed that the rate of apoptosis was
approximately 4-fold lower in the HMP group compared with that in
the CS group. These results are consistent with those of a previous
study showing 5-fold lower levels of myocyte apoptosis in the HMP
group compared with those in the CS group in an ex vivo rat
heart transplantation model (62). In the present study, we
demonstrated that A20 is predominantly expressed in the TECs, with
significantly more abundant expression in the HMP group compared
with that in the CS group. This suggests that HMP decreased the
rate of renal cell apoptosis and the subsequent apoptotic cell
death pathway during IRI by suppressing the ASK1-JNK signaling
cascade.
Generally, necroptosis is defined as cell death
mediated through a pathway that depends on the RIPK1-RIPK3 complex
that may be inhibited by necrostatin-1 (Nec-1) (63). Necroptosis is induced by a class
of death receptors that includes tumor necrosis factor receptor
(TNFR)1, TNFR2 and Fas. Among them, the TNF-α/TNFR-induced pathway
is the most widely studied. Necroptotic death typically triggers
inflammation in vivo due to the release of intracellular
molecules from dying cells (19).
Necrosis results in the loss of membrane integrity and the release
of HMGB1 and other damage-associated molecular patterns (DAMPs)
that promote inflammatory responses (64,65). The contribution of RIPK1-dependent
necroptosis to kidney failure has also been observed in models of
I/R and it may rescued by Nec-1 inhibitor (44). A recently study showed that
RIPK1/3 is a regulator of TNF-α-mediated necroptosis in renal TECs
and RIPK3-deficient allografts had improved renal function and
longer rejection-free survival compared with kidneys from wild-type
mice (22). Recently Onizawa
et al showed that A20 used its deubiquitinating motif to
restrict RIPK3 ubiquitination and the formation of necroptotic
RIPK1-RIPK3 complexes and also restricted RIPK3-dependent
necroptosis in multiple cell types, although not in renal cells
(32). In the present study,
RIPK3 protein was expressed mainly in the TECs and in the HMP
group, RIPK3 expression was significantly lower than that in the CS
group. In addition the HMP group exhibited reduced HMGB1 expression
compared with that in the CS group. These results indicate that,
compared with CS, HMP induces greater A20 expression during IRI,
hence restricting RIPK3-dependent necroptosis and inflammation in
TECs.
In the present study, we have demonstrated that A20
may be induced at low temperatures, and that HMP significantly
increases A20 expression compared with CS. Thus, we concluded that
HMP significantly inhibits inflammation and decreases the apoptosis
and necroptosis of renal cells during IRI by inducing the
expression of A20. With regard to the mechanism, we suggest that
the higher levels of A20 expression in the HMP group are due to the
lower levels of MALT1, which cleaves A20 (34), compared with those in the CS
group; however the detailed mechanism remains to be fully
elucidated and further studies are required to better delineate the
molecular targets of A20 in organs stored using the HMP
technique.
Acknowledgments
The present study was funded by the National Natural
Science Foundation of China (grant no. 81570079).
References
|
1
|
Wolfe RA, Ashby VB, Milford EL, Ojo AO,
Ettenger RE, Agodoa LY, Held PJ and Port FK: Comparison of
mortality in all patients on dialysis, patients on dialysis
awaiting transplantation, and recipients of a first cadaveric
transplant. N Engl J Med. 341:1725–1730. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nicholson ML, Metcalfe MS, White SA,
Waller JR, Doughman TM, Horsburgh T, Feehally J, Carr SJ and Veitch
PS: A comparison of the results of renal transplantation from
non-heart-beating, conventional cadaveric, and living donors.
Kidney Int. 58:2585–2591. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perico N, Cattaneo D, Sayegh MH and
Remuzzi G: Delayed graft function in kidney transplantation.
Lancet. 364:1814–1827. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Curci C, Castellano G, Stasi A, Divella C,
Loverre A, Gigante M, Simone S, Cariello M, Montinaro V, Lucarelli
G, et al: Endothelial-to-mesenchymal transition and renal fibrosis
in ischaemia/reperfusion injury are mediated by complement
anaphylatoxins and Akt pathway. Nephrol Dial Transplant.
29:799–808. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gueler F, Gwinner W, Schwarz A and Haller
H: Long-term effects of acute ischemia and reperfusion injury.
Kidney Int. 66:523–527. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kosieradzki M and Rowiński W:
Ischemia/reperfusion injury in kidney transplantation: mechanisms
and prevention. Transplant Proc. 40:3279–3288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
St Peter SD, Imber CJ and Friend PJ: Liver
and kidney preservation by perfusion. Lancet. 359:604–613. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moers C, Smits JM, Maathuis MH, Treckmann
J, van Gelder F, Napieralski BP, van Kasterop-Kutz M, van der Heide
JJ, Squifflet JP, van Heurn E, et al: Machine perfusion or cold
storage in deceased-donor kidney transplantation. N Engl J Med.
360:7–19. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moers C, Pirenne J, Paul A and Ploeg RJ;
Machine Preservation Trial Study Group: Machine perfusion or cold
storage in deceased-donor kidney transplantation. N Engl J Med.
366:770–771. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Y, Fu Z, Zhong Z, Wang R, Hu L,
Xiong Y, Wang Y and Ye Q: Hypothermic machine perfusion decreases
renal cell apoptosis during ischemia/reperfusion injury via the
Ezrin/AKT pathway. Artif Organs. 40:129–135. 2016. View Article : Google Scholar
|
|
11
|
Schold JD, Kaplan B, Howard RJ, Reed AI,
Foley DP and Meier-Kriesche HU: Are we frozen in time? Analysis of
the utilization and efficacy of pulsatile perfusion in renal
transplantation. Am J Transplant. 5:1681–1688. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eddy AA: Progression in chronic kidney
disease. Adv Chronic Kidney Dis. 12:353–365. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akcay A, Nguyen Q and Edelstein CL:
Mediators of inflammation in acute kidney injury. Mediators
Inflamm. 2009:1370722009. View Article : Google Scholar
|
|
14
|
Kono H, Nakagawa K, Morita S, Shinoda K,
Mizuno R, Kikuchi E, Miyajima A, Umezawa K and Oya M: Effect of a
novel nuclear factor-κB activation inhibitor on renal
ischemia-reperfusion injury. Transplantation. 96:863–870. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Latanich CA and Toledo-Pereyra LH:
Searching for NF-kappaB-based treatments of ischemia reperfusion
injury. J Invest Surg. 22:301–315. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cao CC, Ding XQ, Ou ZL, Liu CF, Li P, Wang
L and Zhu CF: In vivo transfection of NF-kappaB decoy
oligodeoxynucleotides attenuate renal ischemia/reperfusion injury
in rats. Kidney Int. 65:834–845. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hatada EN, Krappmann D and Scheidereit C:
NF-kappaB and the innate immune response. Curr Opin Immunol.
12:52–58. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Daemen MA, van't Veer C, Denecker G,
Heemskerk VH, Wolfs TG, Clauss M, Vandenabeele P and Buurman WA:
Inhibition of apoptosis induced by ischemia-reperfusion prevents
inflammation. J Clin Invest. 104:541–549. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Linkermann A and Green DR: Necroptosis. N
Engl J Med. 370:455–465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kaushal GP, Basnakian AG and Shah SV:
Apoptotic pathways in ischemic acute renal failure. Kidney Int.
66:500–506. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kaushal GP, Kaushal V, Hong X and Shah SV:
Role and regulation of activation of caspases in cisplatin-induced
injury to renal tubular epithelial cells. Kidney Int. 60:1726–1736.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lau A, Wang S, Jiang J, Haig A, Pavlosky
A, Linkermann A, Zhang ZX and Jevnikar AM: RIPK3-mediated
necroptosis promotes donor kidney inflammatory injury and reduces
allograft survival. Am J Transplant. 13:2805–2818. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Opipari AW Jr, Boguski MS and Dixit VM:
The A20 cDNA induced by tumor necrosis factor alpha encodes a novel
type of zinc finger protein. J Biol Chem. 265:14705–14708.
1990.PubMed/NCBI
|
|
24
|
Lee EG, Boone DL, Chai S, Libby SL, Chien
M, Lodolce JP and Ma A: Failure to regulate TNF-induced NF-kappaB
and cell death responses in A20-deficient mice. Science.
289:2350–2354. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Boone DL, Turer EE, Lee EG, Ahmad RC,
Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, et
al: The ubiquitin-modifying enzyme A20 is required for termination
of Toll-like receptor responses. Nat Immunol. 5:1052–1060. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Catrysse L, Vereecke L, Beyaert R and van
Loo G: A20 in inflammation and autoimmunity. Trends Immunol.
35:22–31. 2014. View Article : Google Scholar
|
|
27
|
Ma A and Malynn BA: A20: Linking a complex
regulator of ubiquitylation to immunity and human disease. Nat Rev
Immunol. 12:774–785. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Daniel S, Arvelo MB, Patel VI, Longo CR,
Shrikhande G, Shukri T, Mahiou J, Sun DW, Mottley C, Grey ST and
Ferran C: A20 protects endothelial cells from TNF-, Fas-, and
NK-mediated cell death by inhibiting caspase 8 activation. Blood.
104:2376–2384. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kunter U, Daniel S, Arvelo MB, Choi J,
Shukri T, Patel VI, Longo CR, Scali ST, Shrikhande G, Rocha E, et
al: Combined expression of A1 and A20 achieves optimal protection
of renal proximal tubular epithelial cells. Kidney Int.
68:1520–1532. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Grey ST, Longo C, Shukri T, Patel VI,
Csizmadia E, Daniel S, Arvelo MB, Tchipashvili V and Ferran C:
Genetic engineering of a suboptimal islet graft with A20 preserves
beta cell mass and function. J Immunol. 170:6250–6256. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Coornaert B, Carpentier I and Beyaert R:
A20: Central gatekeeper in inflammation and immunity. J Biol Chem.
284:8217–8221. 2009. View Article : Google Scholar :
|
|
32
|
Onizawa M, Oshima S, Schulze-Topphoff U,
Oses-Prieto JA, Lu T, Tavares R, Prodhomme T, Duong B, Whang MI,
Advincula R, et al: The ubiquitin-modifying enzyme A20 restricts
ubiquitination of the kinase RIPK3 and protects cells from
necroptosis. Nat Immunol. 16:618–627. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bodonyi-Kovacs G, Strom TB and Putheti P:
A20 - a biomarker of allograft outcome: a showcase in kidney
transplantation. Adv Exp Med Biol. 809:103–116. 2014. View Article : Google Scholar
|
|
34
|
Coornaert B, Baens M, Heyninck K, Bekaert
T, Haegman M, Staal J, Sun L, Chen ZJ, Marynen P and Beyaert R: T
cell antigen receptor stimulation induces MALT1
paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat
Immunol. 9:263–271. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
De Smaele E, Zazzeroni F, Papa S, Nguyen
DU, Jin R, Jones J, Cong R and Franzoso G: Induction of gadd45beta
by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature.
414:308–313. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang G, Minemoto Y, Dibling B, Purcell NH,
Li Z, Karin M and Lin A: Inhibition of JNK activation through
NF-kappaB target genes. Nature. 414:313–317. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chang L, Kamata H, Solinas G, Luo JL,
Maeda S, Venuprasad K, Liu YC and Karin M: The E3 ubiquitin ligase
itch couples JNK activation to TNFalpha-induced cell death by
inducing c-FLIP(L) turnover. Cell. 124:601–613. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vanden Berghe T, Grootjans S, Goossens V,
Dondelinger Y, Krysko DV, Takahashi N and Vandenabeele P:
Determination of apoptotic and necrotic cell death in vitro and in
vivo. Methods. 61:117–129. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Barba J, Zudaire JJ, Robles JE, Rosell D,
Berian JM and Pascual I: Complications of kidney transplantation
with grafts from expanded criteria donors. World J Urol.
31:893–900. 2013. View Article : Google Scholar
|
|
40
|
McLaren AJ, Jassem W, Gray DW, Fuggle SV,
Welsh KI and Morris PJ: Delayed graft function: risk factors and
the relative effects of early function and acute rejection on
long-term survival in cadaveric renal transplantation. Clin
Transplant. 13:266–272. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Padanilam BJ: Cell death induced by acute
renal injury: a perspective on the contributions of apoptosis and
necrosis. Am J Physiol Renal Physiol. 284:F608–F627. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bonegio R and Lieberthal W: Role of
apoptosis in the pathogenesis of acute renal failure. Curr Opin
Nephrol Hypertens. 11:301–308. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bonventre JV and Zuk A: Ischemic acute
renal failure: an inflammatory disease? Kidney Int. 66:480–485.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Linkermann A, Bräsen JH, Himmerkus N, Liu
S, Huber TB, Kunzendorf U and Krautwald S: Rip1
(receptor-interacting protein kinase 1) mediates necroptosis and
contributes to renal ischemia/reperfusion injury. Kidney Int.
81:751–761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Toronyi E: Role of apoptosis in the kidney
after reperfusion. Orv Hetil. 149:305–315. 2008.In Hungarian.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
da Silva CG, Maccariello ER, Wilson SW,
Putheti P, Daniel S, Damrauer SM, Peterson CR, Siracuse JJ,
Kaczmarek E and Ferran C: Hepatocyte growth factor preferentially
activates the anti-inflammatory arm of NF-κB signaling to induce
A20 and protect renal proximal tubular epithelial cells from
inflammation. J Cell Physiol. 227:1382–1390. 2012. View Article : Google Scholar :
|
|
47
|
Rius J, Guma M, Schachtrup C, Akassoglou
K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG and Karin M:
NF-kappaB links innate immunity to the hypoxic response through
transcriptional regulation of HIF-1alpha. Nature. 453:807–811.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li C and Jackson RM: Reactive species
mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol
Cell Physiol. 282:C227–C241. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Grey ST, Arvelo MB, Hasenkamp W, Bach FH
and Ferran C: A20 inhibits cytokine-induced apoptosis and nuclear
factor kappaB-dependent gene activation in islets. J Exp Med.
190:1135–1146. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Arvelo MB, Cooper JT, Longo C, Daniel S,
Grey ST, Mahiou J, Czismadia E, Abu-Jawdeh G and Ferran C: A20
protects mice from D-galactosamine/lipopolysaccharide acute toxic
lethal hepatitis. Hepatology. 35:535–543. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lutz J, Luong A, Strobl M, Deng M, Huang
H, Anton M, Zakkar M, Enesa K, Chaudhury H, Haskard DO, et al: The
A20 gene protects kidneys from ischaemia/reperfusion injury by
suppressing pro-inflammatory activation. J Mol Med Berl.
86:1329–1339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Opipari AW Jr, Hu HM, Yabkowitz R and
Dixit VM: The A20 zinc finger protein protects cells from tumor
necrosis factor cytotoxicity. J Biol Chem. 267:12424–12427.
1992.PubMed/NCBI
|
|
53
|
Hess S, Gottfried E, Smola H, Grunwald U,
Schuchmann M and Engelmann H: CD40 induces resistance to
TNF-mediated apoptosis in a fibroblast cell line. Eur J Immunol.
28:3594–3604. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Heyninck K, Denecker G, De Valck D, Fiers
W and Beyaert R: Inhibition of tumor necrosis factor-induced
necrotic cell death by the zinc finger protein A20. Anticancer Res.
19:2863–2868. 1999.
|
|
55
|
Lademann U, Kallunki T and Jäättelä M: A20
zinc finger protein inhibits TNF-induced apoptosis and stress
response early in the signaling cascades and independently of
binding to TRAF2 or 14-3-3 proteins. Cell Death Differ. 8:265–272.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Du C, Guan Q, Yin Z, Zhong R and Jevnikar
AM: IL-2-mediated apoptosis of kidney tubular epithelial cells is
regulated by the caspase-8 inhibitor c-FLIP. Kidney Int.
67:1397–1409. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Du C, Wang S, Diao H, Guan Q, Zhong R and
Jevnikar AM: Increasing resistance of tubular epithelial cells to
apoptosis by shRNA therapy ameliorates renal ischemia-reperfusion
injury. Am J Transplant. 6:2256–2267. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Won M, Park KA, Byun HS, Sohn KC, Kim YR,
Jeon J, Hong JH, Park J, Seok JH, Kim JM, et al: Novel
anti-apoptotic mechanism of A20 through targeting ASK1 to suppress
TNF-induced JNK activation. Cell Death Differ. 17:1830–1841. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nagamachi A, Nakata Y, Ueda T, Yamasaki N,
Ebihara Y, Tsuji K, Honda Z, Takubo K, Suda T, Oda H, et al:
Acquired deficiency of A20 results in rapid apoptosis, systemic
inflammation, and abnormal hematopoietic stem cell function. PLoS
One. 9:e874252014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zamzami N and Kroemer G: The mitochondrion
in apoptosis: how Pandora's box opens. Nat Rev Mol Cell Biol.
2:67–71. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Cho BB and Toledo-Pereyra LH:
Caspase-independent programmed cell death following ischemic
stroke. J Invest Surg. 21:141–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Peltz M, He TT, Adams GA IV, Koshy S,
Burgess SC, Chao RY, Meyer DM and Jessen ME: Perfusion preservation
maintains myocardial ATP levels and reduces apoptosis in an ex vivo
rat heart transplantation model. Surgery. 138:795–805. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Galluzzi L, Vitale I, Abrams JM, Alnemri
ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry
WS, Fulda S, et al: Molecular definitions of cell death
subroutines: Recommendations of the Nomenclature Committee on Cell
Death 2012. Cell Death Differ. 19:107–120. 2012. View Article : Google Scholar :
|
|
64
|
Chan FK: Fueling the flames: mammalian
programmed necrosis in inflammatory diseases. Cold Spring Harb
Perspect Biol. 4:a0088052012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kaczmarek A, Vandenabeele P and Krysko DV:
Necroptosis: the release of damage-associated molecular patterns
and its physiological relevance. Immunity. 38:209–223. 2013.
View Article : Google Scholar : PubMed/NCBI
|