Introduction
Sepsis, also known as systemic inflammatory response
syndrome (SIRS), is caused by infection associated with organ
damage. Sepsis is a life-threatening condition characterized by
rapid progression and a high fatality rate. The mortality rate of
sepsis is complicated by heart failure and may reach 70–90%
(1). Cardiac dysfunction in
sepsis is a complex pathophysiological process and the mechanisms
underlying sepsis-induced myocardial dysfunction remain to be
elucidated. Pro-inflammatory cytokines (such as tumor necrosis
factor (TNF)-α and interleukin (IL)-1), endothelin-1, nitric oxide
and adhesion molecules act directly or indirectly to suppress
cardiac function in sepsis, which results in myocardial dysfunction
(2).
Apoptosis has been reported to play an important
role in sepsis-induced cardiac dysfunction (3). The pharmacological inhibition of
cardiac apoptosis prevents sepsis-induced myocardial dysfunction
(4). Although a number of
mediators and pathways have been associated with apoptosis which
occurs in septic myocardial dysfunction, the precise cause remains
elusive.
The ATP-sensitive K+ (KATP)
channels, first discovered in the heart more than 20 years ago,
represent a unique group of channels that are regulated
predominantly by the cellular metabolic state (5). The function of the cardiac
sarcolemmal KATP (sarcKATP) channel was
initially associated with the cardiac response to stress (5), and this was later confirmed by a
number of studies. The opening of sarcKATP channels
under conditions of metabolic stress affects excitability and other
membrane potential-related functions, such as Ca2+
loading, thus, helping to maintain cellular homeostasis during
cardiac challenge (5). Studies
suggest that the opening of KATP channels prevents
activation of the inflammatory process and the production of a
variety of pro-inflammatory factors in microglial cells (6), and blocks the apoptosis of
cardiocytes in ischemia-reperfusion (7). KATP channels also play an
essential role in the cardiovascular adaptive response during
stress (8). Furthermore, Buckley
et al proposed that the opening of KATP channels
may actually represent a protective mechanism against cellular
damage in endotoxemia (9).
Several researchers have reported that KATP channels
open in sepsis (10–12); however, whether or not they exert
a regulatory effect on the apoptosis of septic myocytes has yet to
be determined. It is well established that sepsis releases
lipopolysaccharide (LPS) into the circulation. LPS exerts a
deleterious effect on cardiac function and plays a significant role
in the development of acute and chronic heart failure (13). In the present study, we examined
the role of cardiac sarcKATP channels in the LPS-induced
apoptosis of cultured neonatal rat cardiomyocytes (NRCs).
Furthermore, we identified the downstream effects of cardiac
sarcKATP channel inhibition and activation by focusing
on the interaction between the sarcKATP channel and
mitochondrial calcium.
Materials and methods
Animals
The animal studies were conducted in accordance with
the guidelines of the Experimental Animal Center of Guangdong
Province (Guangzhou, China). This study was approved by the Ethics
Committee of Guangzhou University of Traditional Chinese Medicine
(Shenzhen, China). The rats were housed in a temperature- and
humidity-controlled room under a 12-h light/dark cycle prior to the
beginning of the experiments. No anesthetics were administered in
order to avoid interference with biochemical values.
Reagents
LPS from Escherichia coli serotype 055:B5 was
purchased from Sigma-Aldrich (St. Louis, MO, USA). A terminal
deoxynucleotidyl transferase-mediated dUTP nick end labeling
(TUNEL) kit was purchased from Roche (Mannheim, Germany). Assay
kits for the determination of caspase-3 activity were purchased
from Beyotime Institute of Biotechnology (Haimen, China).
Cultured NRCs
Primary cultures of NRCs were prepared from the
ventricles of 1–3-day-old Sprague-Dawley rats, as described
previously (14), with some
modifications. Briefly, the neonatal rats were decapitated, the
hearts were excised, and ventricular myocardium was sectioned into
1 mm3-thick slices and incubated with 0.25% trypsin (3–5
ml) in a shaker at 37°C for fractionated digestion. The tissue
pieces were allowed to settle, and all the supernatant was
collected, and centrifuged at 1,000 × g for 10 min. The supernatant
was discarded, and a single cell suspension was obtained with
Dulbecco's modified Eagle's medium (DMEM) containing 20% fetal
bovine serum (FBS). Ventricular myocytes were separated from the
faster-attaching nonmyocytes. The ventricular myocytes in the
supernatant were collected and plated on a 12-well culture plate.
The NRCs were used for experiments following a demonstration of
confluence and rhythmic contraction after 72 h. To explore the
roles of sarcKATP channel and mitochondrial calcium in
the LPS-induced apoptosis of myocytes, the following activators and
blockers were used: sarcKATP channel opener (P-1075, 100
µM), sarcKATP channel blocker (HMR-1098, 30
µM) and mitochondrial Ca2+ uniporter inhibitor
[ruthenium red (RR), 50 µM], respectively. They were applied
following stimulation with 25 µg/ml LPS or vehicle for 24 h.
The negative control included cells maintained in DMEM containing
10% FBS with or without inhibitors and not exposed to LPS
challenge.
Analysis of cardiomyocyte viability
The cytotoxic effects of LPS on cardiac myocytes
were measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) assay and the optimal exposure time and
dose of LPS was established. Exogenous MTT was reduced to insoluble
purple crystal sediment, which dissolves in dimethyl sulfoxide
(DMSO), within the cells by mitochondrial succinate dehydrogenase
in the viable cells, but not in the dead cells. The cells were
seeded in 96-well plates at a density of 5×104
cells/well. The cardiomyocytes were incubated with 20 µl MTT
solution (5 mg/ml; HyClone, Logan, UT, USA) for 4 h at 37°C. Next,
150 µl DMSO (HyClone) was added to each well to dissolve the
formazan crystals, and the plate was agitated for 10 min until all
the crystals were dissolved. The amount of MTT formazan was
quantified by determining the absorbance at 570 nm using a
microplate reader (ELX808; Biotek, Winooski, VT, USA). The
viability was calculated as follows: viability (%) = (A570,
sample-A570, blank)/(A570, control-A570, blank) ×100.
Assessment of apoptosis by TUNEL
assay
Apoptosis was analyzed by TUNEL assay (Roche) and
Hoechst 33258 staining (H1399; Invitrogen, Carlsbad, CA, USA)
according to the manufacturer's instructions. The TUNEL assay was
used in order to detect DNA strand breaks. Briefly, the NRCs were
grown on laminin-coated chamber slides and exposed to LPS challenge
as described above. Twenty-four hours later, the cells were fixed
in 4% paraformaldehyde (PFA) for 1 h, and the TUNEL assay was
performed, according to the manufacturer's instructions using a
commercial kit (Roche). Apoptotic nuclei stained with fluorescein
isothiocyanate (FITC; TUNEL assay) were visualized by confocal
microscopy (FV1000; Olympus, Tokyo, Japan). TO-PRO®-3
stain was used to stain all nuclei. TUNEL-positive cells were
counted and the number of apoptotic cells was expressed as a
percentage of the total number of cells.
Expression of recombinant adenovirus
vector containing subunits of KATP channel mutant
Kir6.2AAA in primary cultured NRCs
A recombinant adenovirus vector carrying
KATP channel mutant subunit Kir6.2AAA was constructed
and expressed in rat cardiomyocytes. Based upon primers for Kir6.2
sites, site-directed mutagenesis of Kir6.2 GFG amino acids into AAA
was performed by means of overlap polymerase chain reaction (PCR).
PCR products were cloned into a pShuttle vector for sequence
analysis. After PmeI linearization, it was transformed into
the adenovirus expression vector pAdEasy-1. The pAdEasy-1 vector
was then packaged into a liposome and transfected into primary
cultured rat cardiomyocytes. The green fluorescent protein
(GFP)-expressing vector (Invitrogen, Shanghai, China) was used as a
control. The expression of Kir6.2AAA was confirmed by RT-PCR and
western blot analysis. The effects on the LPS-induced apoptosis of
myocytes were observed in the presence or absence of LPS.
Caspase-3 activity
To measure caspase-3 enzymatic activity, the NRCs
were cultured for 48 h followed by treatment with P-1075 (100
µM), HMR-1098 (30 µM) and/or RR (50 µM) for 30
min following treatment with or without LPS (25 µg/ml). The
activity of caspase-3 was determined using a caspase assay kit
(Beyotime Institute of Biotechnology) based on the ability of
caspase-3 to change acetyl-Asp-Glu-Val-Asp p-nitroanilide
(Ac-DEVD-pNA) into a yellow formazan product p-nitroaniline (pNA).
Protein concentrations were determined using a Bradford protein
assay (Bio-Rad, Hercules, CA, USA).
Western blot analysis
Cultured myocytes were harvested and lysed for 30
min on ice in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1%
NP-40, 10% glycerol, 0.4 mM sodium orthovanadate, 10 mM sodium
pyrophosphate, 10 mM sodium fluoride, 0.5 mM dithiothreitol (DTT)
and 2 µl/ml protease inhibitor). Following 15 min of
centrifugation at 12,000 × g and 4°C, the supernatants were
obtained and used in subsequent experiments. Equal amounts of
protein from each sample were resolved on sodium dodecyl sulfate
(SDS)-polyacrylamide gel by electrophoresis, and transferred to
Immobilon polyvinylidene difluoride (PVDF) membranes (Millipore,
Billerica, MA, USA), blocked with 5% BSA in TBST (20 mM Tris-HCl,
137 mM NaCl, and 0.1% Tween-20, pH 7.5) at room temperature for 1
h. The membranes were incubated with primary antibodies overnight
at 4°C. Following incubation with horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:3,000; Forevergen,
Guangzhou, China), the immunoblots were exposed to enhanced
chemiluminescence (ECL) reagent (Forevergen). The bands were
quantified by optical density using glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) as a control. The primary antibodies were as
follows: anti-Bcl-2 (CST2870; 1:1,000; Cell Signaling Technology,
Inc., Danvers, MA, USA), anti-Bax (SC-493; 1:200; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), and anti-GAPDH (HC301;
1:5,000; TransGen Biotech, Beijing, China).
Determination of apoptosis by flow
cytometry
The apoptotic ratio was determined by flow cytometry
with Annexin V-FITC/PI staining, according to manufacturer's
instructions. Briefly, the NRCs were treated with P-1075 (100
µM), HMR-1098 (30 µM) and/or RR (50 µM) for 24
h in the presence or absence of LPS (25 µg/ml). Following
experimental treatment, the cells were collected, washed with
calcium-free phosphate-buffered saline (PBS) and resuspended in
binding buffer. The cells were treated with Annexin V-FITC and PI,
left in the dark at room temperature for 15 min, and analyzed using
a Beckton-Dickinson flow cytometer (Navios; Beckman Coulter, Brea,
CA, USA) [fluorescence-activated cell sorting (FACS) analysis].
Statistical analysis
Average values are presented in histograms and in
the text as the means ± SD. Data were analyzed using SPSS 11.5 for
Windows (SPSS, Inc., Chicago, IL, USA). The total number of cells
analyzed in each condition is given in the figure legends. The t
test was used to determine statistical significance. P-values in
the figures are represented by asterisks.
Results
NRC culture and LPS exposure
After continuous culture for 24 h, all adherent
cardiac myocytes were spindle-, diamond- or polygonal-shaped and
spontaneously beating at 60–100 beats/min. The cells migrated
across the surface and developed pseudopodia, forming irregular
star shapes and weaving into a network of mutual contacts (data not
shown). After 72 h, the cultured cells were arranged in radial
clusters (Fig. lA) beating
synchronously and autonomously at a rate of 100 beats/min. After
stable growth for 24–48 h, the cardiomyocytes were exposed to
different concentrations of LPS (0, 5, 25 or 50 µg/ml) for
various time periods (0, 4, 8 and 24 h) in order to determine the
optimal observation window. The viability of the cardiomyocytes was
determined using the MTT method, for the optimal dose and time
duration of LPS exposure. We found that LPS induced cardiomyocyte
death in a time- and concentration-dependent manner. Treating the
myocytes with 25 µg/ml LPS for 24 h resulted in a nearly 60%
survival rate (Fig. 1B), which
was an appropriate observation window for the follow-up
experiments.
Effects of sarcKATP channel
and mitochondrial calcium on the viability and LPS-induced
apoptosis of NRCs
To determine whether further activation or
inhibition of the sarcKATP channel affected the
viability and LPS-induced apoptosis of NRCs, the cells were exposed
to 25 µg/ml LPS for 24 h followed by treatment with 100
µM P-1075, 30 µM HMR-1098 and 50 µM RR.
Cellular viability is an important indicator of NRC
injury. Following LPS exposure, cellular viability was 62.6±4.0%,
and significantly reduced compared with the control group
(P<0.01). P-1075 markedly increased cellular viability to
71.3±7.1% (P<0.05, compared with the LPS group). HMR-1098
reduced cellular viability to 42.8±6.3% (P<0.01, compared with
the LPS group). These results indicated that the
sarcKATP channel significantly preserved cellular
viability (Fig. 1C).
Fig. 2A shows the
TUNEL and Hoechst staining images of the NRCs. As shown in the bar
graph in Fig. 2B, there was a
significant increase in apoptosis in the LPS group compared with
that in the control group (40.1±6.7% in the LPS group vs. 8.7±2.6%
in the control group, P<0.01). The percentage of apoptotic cells
was further increased in the presence of HMR-1098 (69.3±6.9% in the
LPS + HMR group, P<0.01 compared with the LPS group). The
percentage of apoptotic cells was decreased in the presence of
P-1075 (24.3±6.6% in the LPS + P group, P<0.01 compared with LPS
group). The application of RR resulted in the attenuation of
LPS-induced apoptosis in the presence of HMR-1098 (43.3±2.4% in the
LPS + RR group and 54.7±6.2% in the LPS + HMR + RR group,
respectively, P<0.01 vs. LPS + HMR group) and P-1075 (43.3±2.4%
in the LPS + RR group and 37.0±2.4%).
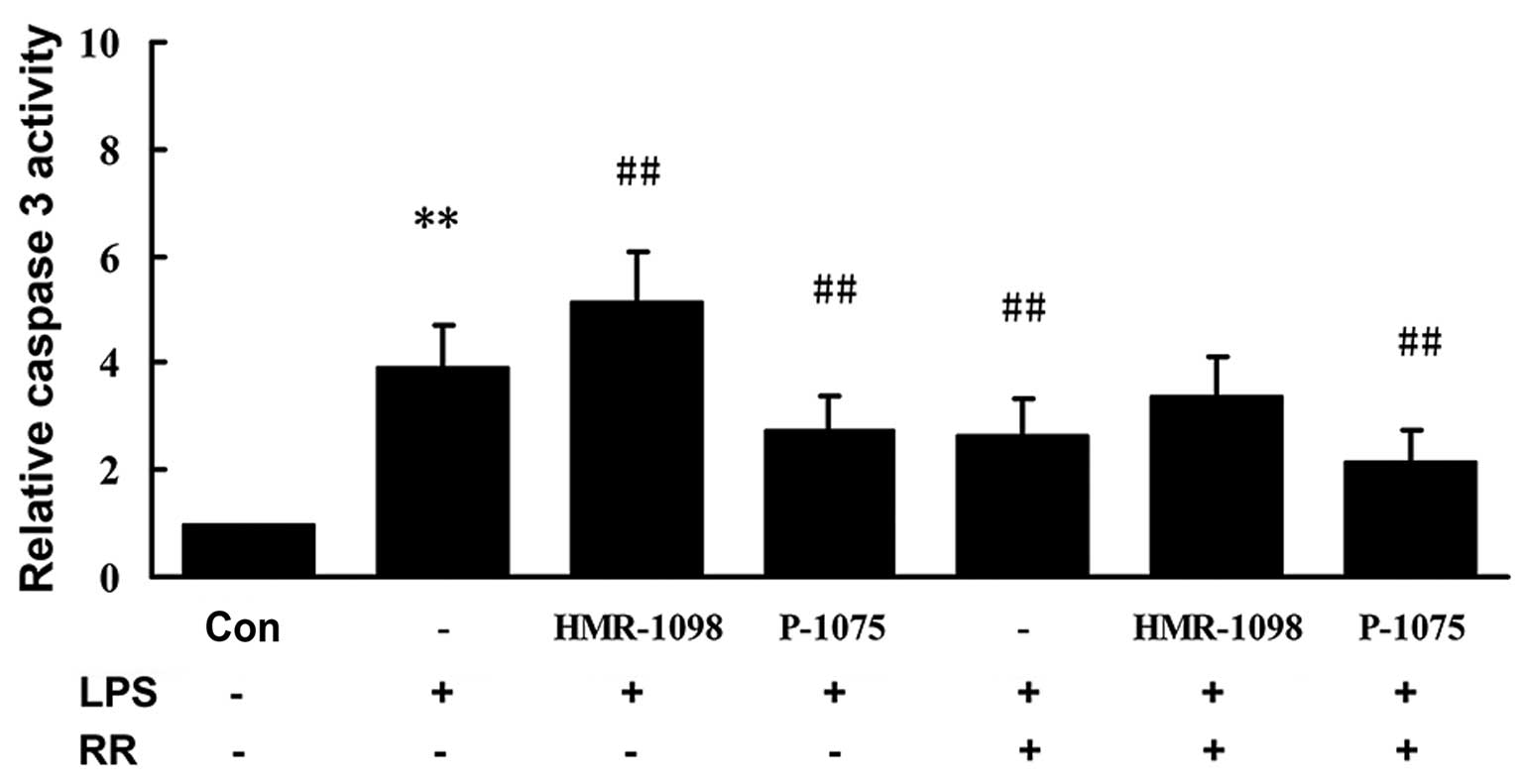
Effects of sarcKATP channel
and mitochondrial calcium on caspase-3 activity in NRCs
LPS-induced apoptosis of cardiomyocytes is
multifactorial. We examined the activity of caspase-3 in the NRCs.
As shown in Fig. 3, caspase-3
activity was markedly higher 24 h after LPS stimulation in the LPS
group compared with that in the control group (P<0.01).
Caspase-3 activity was suppressed markedly in the LPS + P-1075
group compared with the LPS group (P<0.01). Caspase-3 activity
was significantly increased in the LPS + HMR-1098 group (P<0.01,
compared with LPS group), which was partly attenuated in the
presence of 50 µM RR (P<0.01, compared with the LPS
group).
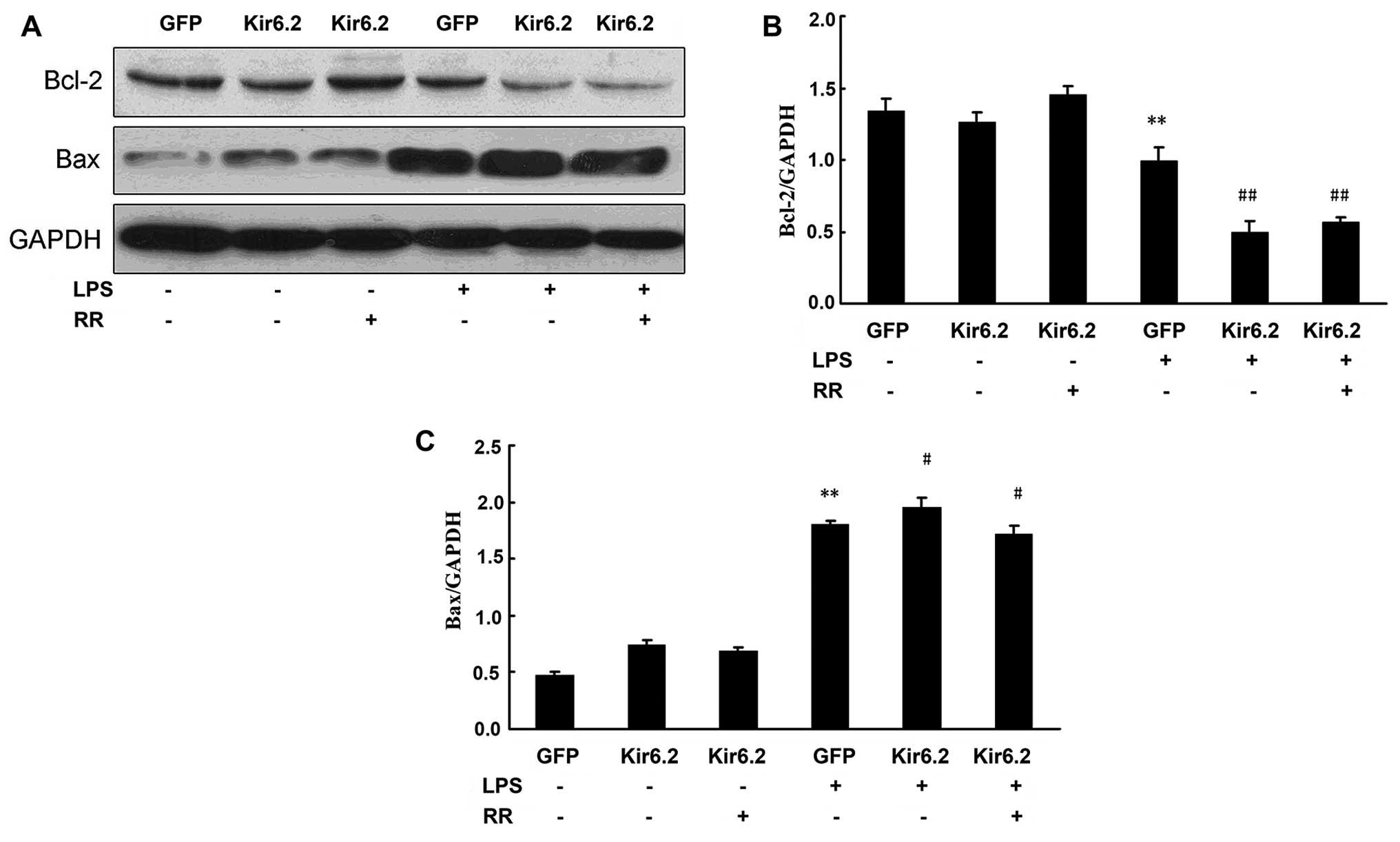
Effects of sarcKATP channel
and mitochondrial calcium on the expression of the
apoptosis-related proteins Bcl-2 and Bax
In order to determine whether the
sarcKATP channel modulates some of the anti- and
pro-apoptotic regulators, the protein levels of Bcl-2 and Bax were
assessed in the present study by western blot analysis (Fig. 4A). As also shown in Fig. 4B and C, LPS stimulus downregulated
Bcl-2 expression and upregulated Bax expression, and P-1075
increased the Bcl-2 protein level and decreased the Bax protein
level in the LPS-exposed NRCs. By contrast, HMR-1098 decreased the
Bcl-2 protein level and increased the Bax protein level in the
LPS-exposed NRCs. The results suggested that sarcKATP
prevented the apoptosis of NRCs by increasing Bcl-2 expression and
inhibiting Bax expression (Fig.
4)
Effect of Kir6.2AAA overexpression on
LPS-induced apoptosis
The recombinant adenovirus carrying the gene
fragment Kir6.2AAA and GFP was constructed successfully, with a
viral titer of 2.64×1011 VP/ml. Following infection with
recombinant adenovirus Kir6.2AAA, the rat myocytes expressed GFP
and emitted green fluorescence (Fig.
5A). The expression of Kir6.2AAA was significantly upregulated
in recombinant adenovirus vector Ad-Kir6.2AAA-infected myocytes and
this was confirmed by RT-PCR (data not shown). The increased
expression of Kir6.2AAA was confirmed in rat myocytes by western
blot analysis, which suggested that the recombinant adenovirus
carrying the gene fragment Kir6.2AAA and GFP was constructed
successfully and expressed correctly in the rat myocytes (data not
shown).
Adenovirus infection has little effect on cellular
viability. Following LPS exposure, cellular viability of the GFP +
LPS group was significantly reduced (72.1±3.6%) compared with the
GFP group (P<0.01). Overexpression of Kir6.2AAA (Kir6.2 group)
markedly reduced the cellular viability to 43.4±2.8% (P<0.01,
compared with GFP + LPS group). RR treatment increased the cellular
viability to 62.0±6.9% (P<0.05, compared with GFP + LPS group)
(Fig. 5B).
Apoptosis was measured by flow cytometry (Fig. 7A). Adenovirus infection had little
effect on myocardial apoptosis: apoptotic rate of the Ad-GFP group
was 3.4±0.7%; apoptotic rate of the Ad-Kir6.2AAA group was
3.7±0.7%; apoptotic rate of the Ad-Kir6.2AAA + RR group was
4.1±0.6%. The apoptotic rate of the myocytes significantly
increased following LPS exposure: apoptotic rate of the Ad-GFP +
LPS group was 14.9±2.8%; apoptotic rate of the Ad-Kir6.2AAA + LPS
was 36.0±3.8%; apoptotic rate of the Ad-Kir6.2AAA + RR + LPS group
was 19.4±3.7% (Fig. 7B). The
results suggested that in the model of LPS-induced apoptosis in
NRCs, overexpression of Kir6.2AAA further promoted the apoptosis of
myocytes, and RR treatment protected the myocytes from LPS-induced
apoptosis.
The effect of Kir6.2AAA overexpression on Bcl-2 and
Bax protein levels was detected by western blot analysis (Fig. 6A). Our results showed that
adenovirus infection has no effect on the protein expression of
Bcl-2 and Bax. LPS downregulated Bcl-2 expression and upregulated
Bax expression in the GFP + LPS group. Overexpression of Kir6.2AAA
(Kir6.2 group) markedly decreased the Bcl-2 protein level and
increased the Bax protein level in the LPS-exposed NRCs (Fig. 6B and C). RR treatment increased
the Bcl-2 protein level and decreased the Bax protein level in the
LPS-exposed NRCs (Fig. 6).
Discussion
In the present study, we examined the role of the
sarcKATP channel in the LPS-induced apoptosis of NRCs.
Our findings suggest that the sarcKATP channel exerts a
cardioprotective effect against apoptosis induced by LPS and is
mediated by mitochondrial Ca2+ in rat
cardiomyocytes.
The protective role of the sarcKATP
channels has been proposed since the time of their discovery due to
their ability to sense intracellular metabolic conditions (15). In 1983, Noma (5) identified a KATP channel
in membrane patches prepared from isolated guinea pig ventricular
myocytes. Subsequently, KATP channels were discovered in
other tissues, including the brain, smooth muscle, skeletal muscle,
intestine, kidney and pancreas. The channel coupled myocardial
energy metabolism to membrane electrochemistry, which highlighted
its role in all types of ion channels. The metabolic gating of the
sarcKATP channel is attributed to its molecular
composition. Structurally, KATP channels are composed of
two distinct proteins, an inwardly rectifying potassium channel
(Kir) pore subunit and the sulfonylurea receptor (SUR), which may
have a regulatory role in modulating the sensitivity of the channel
to ATP, other nucleotides, and pharmacological agonists or
antagonists (16). The cardiac
sarcolemmal KATP channel is composed of an octomeric
complex of two types of subunits, the Kir6.2 and the SUR2A
subunit.
The cardiac sarcKATP channel is the
sensory receptor of cellular energy metabolism and highly sensitive
to fluctuations in its microenvironment. It is blocked by ATP in
the cell, normally closed, and opened under various pathological or
stress conditions. ATP production is reduced in sepsis, myocardial
ischemia and hypoxia as well as other stress conditions, which
results in the channel opening followed by cell membrane
hyperpolarization, closing of voltage-dependent Ca2+
channels and reduced internal calcium current. Simultaneously, the
opening of sarcKATP channels causes the outflow of
K+ and accelerated repolarization, which results in
shortening the duration of the cellular action potential and
reduced inflow of Ca2+. In addition, due to the increase
in ion outflow and rapid recovery of resting membrane potential,
Ca2+ is easily released into the cytoplasm by
Na+/Ca2+ exchange under a lower resting
membrane potential. These mechanisms alleviate the extent of
cellular damage from cytosolic Ca2+ overload. On the
other hand, intracellular myocardial [Ca2+]i levels are
reduced, leading to depression of the myocardical contraction,
thereby reducing the cellular ATP consumption (5). Thus, sarcKATP channels
mediate endogenous protective effects in myocytes by sensing and
regulating the cellular levels of energy metabolism through
electrical feedback, which helps the myocytes survive metabolic
crisis. Genetic deletion or alterations to the Kir6.2 subunit of
the sarcKATP channel results in a loss of both ischemic
preconditioning and adaptation to stress (16–19) In addition, a reduction in the
number of functional sarcKATP channels eliminates the
transient action potentials during metabolic inhibition, leading to
reduced metabolic stress tolerance, and increased Ca2+
loading during metabolic inhibition (20).
The role of sarcKATP channels in cardiac
sepsis remains poorly understood. Many scholars have confirmed that
the channel was activated or upregulated in response to LPS
challenge. Since the sarcKATP channel plays an important
role in maintaining cellular homeostasis, it mediates several
cellular pathological and physiological activities, such as
apoptosis. In the present study, P-1075, a selective
sarcKATP opener, reduced LPS-induced apoptosis. By
contrast, HMR-1098, a selective sarcKATP blocker, or the
overexpression of Kir6.2AAA increased LPS-induced apoptosis in the
cultured NRCs, as measured by flow cytometry and the TUNEL assay,
which was consistent with the results of a previous study (10). LPS induced the apoptosis of
cardiomyocytes. The sarcKATP channel prevented the
apoptosis of cardiomyocytes induced by oxidative stress (21) and the apoptosis of the bladder
smooth muscle induced by acute urinary retention (22). The channel also exerts a
protective effect against ischemia-reperfusion-induced apoptosis in
testicular damage (23).
As the cellular power houses, mitochondria are not
only associated closely with the sarcKATP channel, but
play a central role in apoptosis. Since mitochondria generate the
majority of cellular ATP and its metabolite ADP in the cell, which
are the main regulating factors of sarcKATP channels,
mitochondria invariably affect the activity of sarcKATP
channels. On the other hand, mitochondria also regulate ionic
steady state in the cell. Under different stress conditions,
mitochondria act as a buffer and reduce intracellular
Ca2+ overload. However, uncontrolled Ca2+
overload initiates cell death including apoptosis (24). Mitochondrial Ca2+
overload triggers the opening of the mitochondrial permeability
transport channel (mPTP), and the mitochondrial permeability
transition (MPT), followed by phagocytosis by lysosomes. When a
large number of mitochondria reach MPT, cytochrome c and
apoptosis-inducing factor enter the cytoplasm, to activate cellular
apoptosis, and even necrosis (25). In sepsis, mitochondrial function
is impaired in cardiomyocytes and ATP production is reduced, which
decreases sarcoplasm/endoplasmic reticulum (S/ER) calcium ATPase
(SERCA) activity, leading to reduced levels of Ca2+ in
the S/ER, increased levels of cytoplasmic Ca2+,
resulting in mitochondrial Ca2+ accumulation (26). Opening sarcKATP
channels reduces intracellular Ca2+overload by cell
membrane hyperpolarization, shortening the action potential
duration. Stimulating Na+-Ca2+ exchange also
affects the mitochondrial Ca2+ load. In the present
study, the mitochondrial calcium transport inhibitor RR partially
attenuated the pro-apoptotic effect of HMR-1098. The results
suggested that the mitochondrial pathway is involved in the
sarcKATP-channel-mediated apoptosis of NRCs induced by
LPS.
The present study provides new evidence correlating
sarcKATP channel activity with downstream mitochondrial
function. By elucidating the mechanism responsible for the
sarcKATP channel-mediated regulation of apoptosis, we
have provided a new perspective for the possible pathogenesis of
sepsis being associated with cardiac dysfunction, which should
facilitate the development of novel targeted therapies and
strategies.
Acknowledgments
This study was supported by the grants from the
National Natural Science Foundation of China (no. 81101450)
References
|
1
|
Merx MW and Weber C: Sepsis and the heart.
Circulation. 116:793–802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zanotti-Cavazzoni SL and Hollenberg SM:
Cardiac dysfunction in severe sepsis and septic shock. Curr Opin
Crit Care. 15:392–397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lancel S, Joulin O, Favory R, Goossens JF,
Kluza J, Chopin C, Formstecher P, Marchetti P and Neviere R:
Ventricular myocyte caspases are directly responsible for
endotoxin-induced cardiac dysfunction. Circulation. 111:2596–2604.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suzuki J, Bayna E, Dalle Molle E and Lew
WY: Nicotine inhibits cardiac apoptosis induced by
lipopolysaccharide in rats. J Am Coll Cardiol. 41:482–488. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Noma A: ATP-regulated K+
channels in cardiac muscle. Nature. 305:147–148. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou F, Yao HH, Wu JY, Ding JH, Sun T and
Hu G: Opening of microglial K(ATP) channels inhibits
rotenone-induced neuroinflammation. J Cell Mol Med. 12:1559–1570.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu H, Zhang HY, Zhu X, Shao Z and Yao Z:
Preconditioning blocks cardiocyte apoptosis: role of
K(ATP) channels and PKC-epsilon. Am J Physiol Heart Circ
Physiol. 282:H1380–H1386. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kane GC, Liu XK, Yamada S, Olson TM and
Terzic A: Cardiac KATP channels in health and disease. J
Mol Cell Cardiol. 38:937–943. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Buckley JF, Singer M and Clapp LH: Role of
KATP channels in sepsis. Cardiovasc Res. 72:220–230.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang ZW, Chen JK, Ni M, Zhao T, Deng YP,
Tao X, Jiang GJ and Shen FM: Role of Kir6.2 subunits of
ATP-sensitive potassium channels in endotoxemia-induced cardiac
dysfunction. Cardiovasc Diabetol. 12:752013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kuo JH, Chen SJ, Shih CC, Lue WM and Wu
CC: Abnormal activation of potassium channels in aortic smooth
muscle of rats with peritonitis-induced septic shock. Shock.
32:74–79. 2009. View Article : Google Scholar
|
|
12
|
Shi W, Cui N, Wu Z, Yang Y, Zhang S, Gai
H, Zhu D and Jiang C: Lipopolysaccharides up-regulate Kir6.1/SUR2B
channel expression and enhance vascular KATP channel
activity via NF-kappaB-dependent signaling. J Biol Chem.
285:3021–3029. 2010. View Article : Google Scholar
|
|
13
|
Charalambous BM, Stephens RC, Feavers IM
and Montgomery HE: Role of bacterial endotoxin in chronic heart
failure: the gut of the matter. Shock. 28:15–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen M, Wu RX, Zhao L, Li J, Guo HT, Fan
R, Cui Y, Wang YM, Yue SQ and Pei JM: Resveratrol attenuates
ischemia/reperfusion injury in neonatal cardiomyocytes and its
underlying mechanism. PLoS One. 7:e512232012. View Article : Google Scholar
|
|
15
|
Nichols CG: KATP channels as
molecular sensors of cellular metabolism. Nature. 440:470–476.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sellitto AD, Al-Dadah AS, Schuessler RB,
Nichols CG and Lawton JS: An open sarcolemmal adenosine
triphosphate-sensitive potassium channel is necessary for
detrimental myocyte swelling secondary to stress. Circulation.
124(Suppl 11): S70–S74. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suzuki M, Sasaki N, Miki T, Sakamoto N,
Ohmoto-Sekine Y, Tamagawa M, Seino S, Marbán E and Nakaya H: Role
of sarcolemmal K(ATP) channels in cardioprotection against
ischemia/reperfusion injury in mice. J Clin Invest. 109:509–516.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zingman LV, Hodgson DM, Bast PH, Kane GC,
Perez-Terzic C, Gumina RJ, Pucar D, Bienengraeber M, Dzeja PP, Miki
T, et al: Kir6.2 is required for adaptation to stress. Proc Natl
Acad Sci USA. 99:13278–13283. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gumina RJ, Pucar D, Bast P, Hodgson DM,
Kurtz CE, Dzeja PP, Miki T, Seino S and Terzic A: Knockout of
Kir6.2 negates ischemic preconditioning-induced protection of
myocardial energetics. Am J Physiol Heart Circ Physiol.
284:H2106–H2113. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rainbow RD, Lodwick D, Hudman D, Davies
NW, Norman RI and Standen NB: SUR2A C-terminal fragments reduce
KATP currents and ischaemic tolerance of rat cardiac
myocytes. J Physiol. 557:785–794. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marinovic J, Ljubkovic M, Stadnicka A,
Bosnjak ZJ and Bienengraeber M: Role of sarcolemmal ATP-sensitive
potassium channel in oxidative stress-induced apoptosis:
mitochondrial connection. Am J Physiol Heart Circ Physiol.
294:H1317–H1325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ohmasa F, Saito M, Oiwa H, Tsounapi P,
Shomori K, Kitatani K, Dimitriadis F, Kinoshita Y and Satoh K:
Pharmacological preconditioning of ATP-sensitive potassium channel
openers on acute urinary retention-induced bladder dysfunction in
the rat. BJU Int. 110:E245–E252. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsounapi P, Saito M, Dimitriadis F,
Kitatani K, Kinoshita Y, Shomori K, Takenaka A and Satoh K: The
role of K ATP channels on ischemia-reperfusion injury in the rat
testis. Life Sci. 90:649–656. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dorn GW II: Apoptotic and non-apoptotic
programmed cardiomyocyte death in ventricular remodelling.
Cardiovasc Res. 81:465–473. 2009. View Article : Google Scholar :
|
|
25
|
Nishida K, Yamaguchi O and Otsu K:
Crosstalk between autophagy and apoptosis in heart disease. Circ
Res. 103:343–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hassoun SM, Marechal X, Montaigne D,
Bouazza Y, Decoster B, Lancel S and Neviere R: Prevention of
endotoxin-induced sarcoplasmic reticulum calcium leak improves
mitochondrial and myocardial dysfunction. Crit Care Med.
36:2590–2596. 2008. View Article : Google Scholar : PubMed/NCBI
|