Introduction
Neurodegenerative disorders are characterized by
loss or dysfunction of neurons in the central nervous system
(1,2). Oxidative stress and consequent
mitochondrial dysfunction contribute to the pathology of spinal
cord injury (3,4), stroke (5,6),
Parkinson's disease (7), and
Alzheimer's disease (8) via
production of reactive oxygen species (ROS), which include hydrogen
peroxide (H2O2), superoxide, singlet oxygen,
and the hydroxyl radical (9).
ROS play an important role in intracellular signal
transduction and gene expression in cell survival and organism
development (10). However,
excess accumulation of ROS can damage proteins, DNA and cell
membranes (11). ROS induce cell
death via the mitochondrial apoptosis pathway (12). Neurons are thought to be more
susceptible to ROS owing to increased oxidative metabolism and
fewer antioxidative enzymes (13,14). Suppression of ROS generation and
inhibition of apoptosis can therefore potentially prevent
neurodegeneration.
Cornus officinalis is among the most commonly
used Chinese medical herbs and has been used to treat kidney- and
brain-related diseases (15). Its
biological activities include antioxidant (16,17), anti-inflammatory (18) and antimicrobial (19) effects. Morroniside is the major
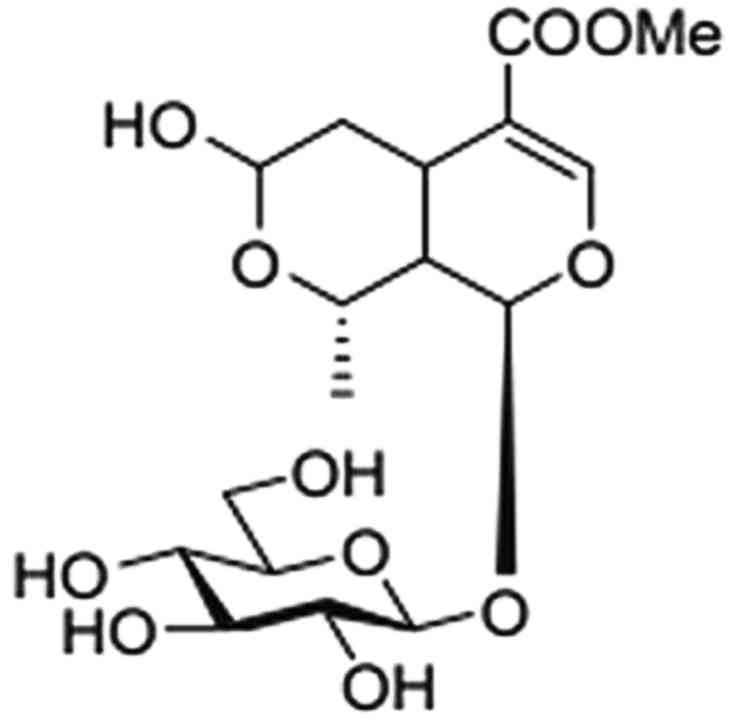
active ingredient of C. officinalis extract (20) (Fig.
1) and has been shown to reduce blood glucose, mitigate
cerebral ischemia-reperfusion injury, and modulate the immune
response (21). However, little
is known concerning the effects of morroniside on oxidative
stress-induced cell damage.
Materials and methods
Cell culture and treatment
SK-N-SH human neuroblastoma cells (Type Culture
Collection of the Chinese Academy of Sciences, Shanghai, China)
were cultured in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% (v/v) fetal bovine serum, 100 U/ml
penicillin/streptomycin (all from Invitrogen, Carlsbad, CA, USA) in
plastic 25-cm2 flasks at 37°C and under 5%
CO2, 95% air. Culture medium was changed every other day
and the cells were subcultured once attaining 70–80% confluency.
Morroniside (Phytomarker Ltd., Tianjin, China) was dissolved in
D-PBS (Invitrogen) and H2O2 (Sinopharm
Chemical Reagent Co. Ltd., Shanghai, China) was dissolved in
sterile distilled water. SK-N-SH cells were pretreated with
different concentrations of morroniside for 24 h, followed by
incubation with H2O2 (200–400 µM) for
24 h to induce injury.
Cell viability assay
The viability of the cells was assessed by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Briefly, SK-N-SH cells were plated into 96-well plates at
the density of 5×103 cells/well. Cells were preincubated
with morroniside (1, 10 and 100 µM) for 24 h before 200
µM H2O2 was administered. After a 24-h
incubation, 20 µl of a 5 mg/ml stock solution of MTT (Sigma,
St. Louis, MO, USA) was added to the culture medium and incubated
in the dark for 4 h at 37°C to allow for formazan formation. Then
MTT formazan crystals were dissolved in 100 µl dimethyl
sulfoxide (DMSO; Sigma) and spectrophotometrically determined at an
absorbance of 570 nm. The percentage of cell viability was measured
by normalization of all values to the control group (=100%).
Morphological observation
SK-N-SH cells were seeded into 6-well plates at the
density of 2×105 cells/well. When cell confluency
achieved 70–80%, the cells were pre-incubated with morroniside for
24 h before H2O2 (200 µM) was added to
induce cell damage for another 24 h. The growth and morphological
changes in each group were observed under an inverted phase
contrast microscope.
Cytotoxicity assay
Lactate dehydrogenase (LDH) assay was used to assay
H2O2-induced cytotoxicity. SK-N-SH cells were
seed into 96-well plates at a density of 5×103
cells/well. After cells were exposed to H2O2
(200 µM) for 24 h, a total of 120 µl of cell medium
was collected for LDH analysis according to the manufacturer's
protocol included in the cytotoxicity assay kit (Beyotime
Biotechnology, Nantong, China). The absorbance of each group was
read at 490 nm. Each group had five duplicate wells and the
experiments were repeated at least three times. Data are expressed
as the percentage of LDH release of the injury group
(H2O2-induced group).
Apoptosis analysis
In order to determine whether morroniside protects
against H2O2-induced apoptosis, Hoechst 33342
(Sigma), a fluorescent nuclear dye, was used to detect cell
apoptosis by observing the cell morphology. Briefly, SK-N-SH cells
were treated as described above and then washed with
phosphate-buffered saline (PBS) and incubated with Hoechst 33342
(10 µg/ml in PBS; Sigma) for 15 min at 37°C in the dark. The
cells were washed with PBS, fixed with cold 4% paraformaldehyde for
another 15 min in room temperature, washed with PBS again, and then
examined by fluroescence microscopy. The percentage of apoptotic
cells was calculated as the ratio of apoptotic cells to the total
cells counted. At least 500 cells were counted from more than 3
random microscopic fields.
Moreover, in order to confirm the anti-apoptotic
effect of morroniside, flow cytometry with Annexin V-FITC and
propidium iodide (PI) double staining (Beyotime Biotechnology) was
performed. After SK-N-SH cells were treated with
H2O2 (200 µM), 5×105 cells
were collected and counted for Annexin V-FITC and PI double
staining. Briefly, cells in different groups were washed three
times with cold PBS and stained with Annexin V-FITC for 15 min in
the dark. After cells were washed another three times, PI was added
and the fluorescence of each group was immediately analyzed by flow
cytometry.
Assessment of intracellular ROS
production
For visualization and analysis of intracellular ROS,
the oxidation sensitive probe DCFH-DA (Beyotime Biotechnology) was
used. After the treatment of H2O2, SK-N-SH
cells were exposed to 10 µM DCFH-DA for 20 min at 37°C in
the dark. The cells were washed with PBS for 3 times, and then DCF
fluorescence was observed using fluorescence microscopy and
quantified by fluorescence multi-well plate reader (BioTek,
Highland Park, VT, USA) with an excitation wavelength of 488 nm and
emission wavelength of 525 nm.
Measurement of intracellular superoxide
anion production
Superoxide anion was detected with dihydroethidium
probes. Cells were treated with 2 µM dihydroethidium
(Beyotime Biotechnology) for 30 min at 37°C in dim light after
incubation with H2O2. Each well was washed
with cold PBS and then DMEM to remove the remaining probes. Then
cells were observed using fluorescence microscopy and the
fluorescence intensity was quantified by a fluorescence multi-well
plate reader.
Lipid peroxidation assay
Lipid peroxidation was monitored by measuring
malondiadehyde (MDA; Beyotime Biotechnology), a stable end product
of lipid peroxidation cascades using an MDA assay kit. SK-N-SH
cells were washed with ice-cold PBS and then harvested with RIPA
lysis buffer (Beyotime Biotechnology) after the treatment of
H2O2. Cell homogenates were centrifuged at
16,000 × g at 4°C for 10 min. The supernatant was used for MDA
assay and protein determination. The total protein concentrations
were measured using BCA Protein assay kit (Beyotime Biotechnology).
For MDA measurement, 100 µl samples were added into a 15-ml
tube followed by addition of 200 µl MDA working solution.
The mixture was heated at 100°C for 15 min, chilled to room
temperature, and centrifuged at 1,000 × g for 10 min. Supernatants
of 200 µl were transferred to 96-well plates, and the
absorbance of each group was read at 532 nm.
Determination of activity of superoxide
dismutase (SOD)
Cellular SOD levels were determined using a
Superoxide Dismutase assay kit (Jiancheng Bioengineering Institute,
Nanjing, China). SK-N-SH cells were pretreated with different
concentrations of morroniside for 24 h prior to exposure to
H2O2 (200 µM) for another 24 h. Cells
were washed twice with ice-cold PBS and harvested in RIPA lysis
buffer, and then total protein contents were determined with the
BCA protein assay kit (Beyotime Biotechnology). Samples were
collected and analyzed according to the manufacturer's
instructions. SOD levels were normalized to the protein
concentrations.
Monitoring mitochondrial membrane
potential (MMP)
SK-N-SH cells were treated with various dose of
morroniside in a 12-well plate for 24 h before
H2O2 (200 µM) for 24 h. A fluorescent
dye JC-1 (Beyotime Biotechnology) was added to achieve final
concentration of 10 µg/ml. Then cells were incubated for 30
min at 37°C in dim light. Cells were washed with PBS to remove the
excess dye, and then images were captured using fluorescence
microscopy within 30 min. For the JC-1 monomer, excitation
wavelength was 514 nm and maximum emission wavelength was 529 nm.
For the JC-1 polymer, the maximum excitation and emission
wavelength were 585 and 590 nm, respectively.
Measurement of caspase-3 activity
Activity of caspase-3 was measured with a commercial
caspase-3 activity assay kit (Beyotime Biotechnology), using
Ac-DEVD-pNA as the specific substrate. The cell pellets were lysed
on ice in lysis buffer for 15 min, and then the lysate was
centrifuged at 20,000 × g for 10 min at 4°C. Supernatant of 10
µl was collected and incubated with 10 µl Ac-DEVD-pNA
(2 mM) at 37°C for another 1–2 h in the dark. The absorbance values
were measured with a spectrofluorometer at 405 nm and were
corrected as protein content in the lysate. Protein concentration
was determined by Bradford protein assay kit (Beyotime
Biotechnology) according to the manufacturer's instructions.
Reverse transcription-polymerase chain
reaction (RT-PCR)
The mRNA expression levels of B cell lymphoma-2
(Bcl-2) and Bcl-2-associated X protein (Bax) were determined by
RT-PCR. Briefly, SK-N-SH cells were treated as mentioned above, and
the total RNA of each group was extracted using TRIzol agent
(Invitrogen) according to the manufacturer's instructions. Two
micrograms of total RNA was first reverse transcribed into cDNA,
and then routine PCR was performed as previously described
(22). Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was used as an internal control and products
were analyzed on 1% agarose gel. ImageJ software (NIH, Bethesda,
MD, USA) was used to quantify the optical density value of each
band. The sequences of specific primers (Sangon Biotech Inc.,
Shanghai, China) used for RT-PCR are listed in Table I.
 | Table ISequences of the primers and PCR
product sizes used in RT-PCR. |
Table I
Sequences of the primers and PCR
product sizes used in RT-PCR.
| Gene | Primer | Sequence | Size (bp) |
|---|
| GAPDH | Sense |
5′-AGAAGGCTGGGGCTCATTTG-3′ | 258 |
| Antisense |
5′-AGGGGCCATCCACAGTCTTC-3′ | |
| Bax | Sense |
5′-CCAAGGTGCCGGAACTGA-3′ | 57 |
| Antisense |
5′-CCCGGAGGAAGTCCAATGT-3′ | |
| Bcl-2 | Sense |
5′-CATGTGTGTGGAGAGCGTCAAC-3′ | 187 |
| Antisense |
5′-CTTCAGAGACAGCCAGGAGAAATC-3′ | |
Western blot analysis
Cells were collected at 24 h after exposure to
H2O2. In order to detect the expression of
Bcl-2 and Bax, SK-N-SH cells were washed with PBS and lysed using a
lysis buffer for 30 min on ice. Lysates were centrifuged at 16,000
× g for 10 min at 4°C, and the supernatant was saved for the total
protein determination. Protein concentrations were determined using
a commercially available BCA protein assay kit (Beyotime
Biotechnology). For western blot analysis, equivalent amounts of
protein of each sample (20 µg) were electrophoresed using
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE), and transferred to polyvinylidene difluoride membranes
(Millipore, Bedford, MA, USA). After the membranes were blocked
with 5% non-fat dry milk in Tris-buffered saline (TBS) for 1 h at
room temperature, primary antibodies (in TBST-5% BSA) against Bcl-2
(1:2,000; 12789-1-AP) and Bax (1:5,000; 60267-1-Ig) (both from
ProteinTech Group Inc, Chicago, IL, USA) were added and incubated
overnight at 4°C for determining signal transduction events. The
membranes were rinsed three times with TBST and incubated with an
appropriate HRP-conjugated secondary antibody (1:2,000; #04-15-06
and #04-18-06) (both from KPL, Gaithersburg, MD, USA) for 1 h at
37°C. An ECL kit (Millipore) was used to visualize membrane
immunoreactivity. Quantification was performed using a computerized
imaging program Quantity One (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All data are presented as mean ± standard deviation
of the mean (SD). Statistical analyses were performed by one-way
analysis of variance (ANOVA) with post hoc Tukey's t-test to
determine statistical significance. A value of P<0.05 was
considered to indicated statistically significant differences.
Results
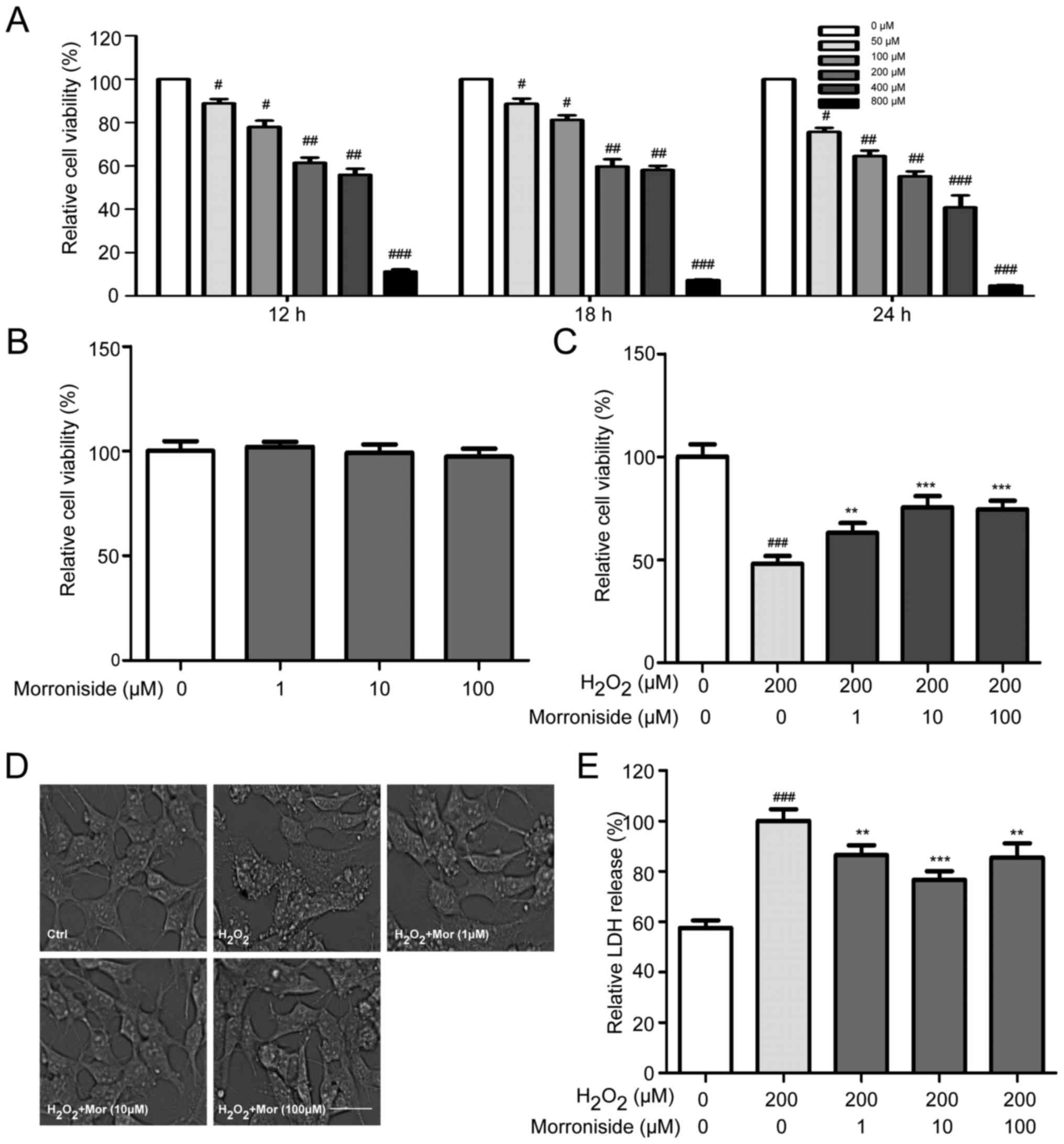
Morroniside attenuates
H2O2-induced cell death
H2O2-induced cell injury was
evaluated using the MTT assay. H2O2 reduced
cell viability in a dose- and time-dependent manner, with a
survival rate of 50% after 24 h in the presence of 200 µM
H2O2 relative to the untreated controls
(Fig. 2A). Cells were treated
with various concentrations of morroniside for 24 h to determine
whether the H2O2-induced decrease in
viability would be mitigated. Morroniside had no effect on the
survival of SK-N-SH cells that were not treated with
H2O2 (Fig.
2B). However, pretreatment with morroniside attenuated the
H2O2-induced decrease in cell survival in a
dose-dependent manner, with the greatest effect observed at 10
µM (Fig. 2C).
Morroniside reverses
H2O2-induced morphological changes
SK-N-SH cells treated for 24 h with 200 µM
H2O2 showed morphological changes including
loss of cell projections; a round, swollen, or shrunken cell body;
and aggregation. These changes were blocked by pretreatment with
morroniside (Fig. 2D).
Morroniside inhibits
H2O2-induced LDH release
The LDH release assay showed that
H2O2 increased LDH activity in the
supernatant of the SK-N-SH cell cultures relative to the control
group. This effect was suppressed upon treatment with 1, 10 and 100
µM morroniside (P<0.05) (Fig. 2E).
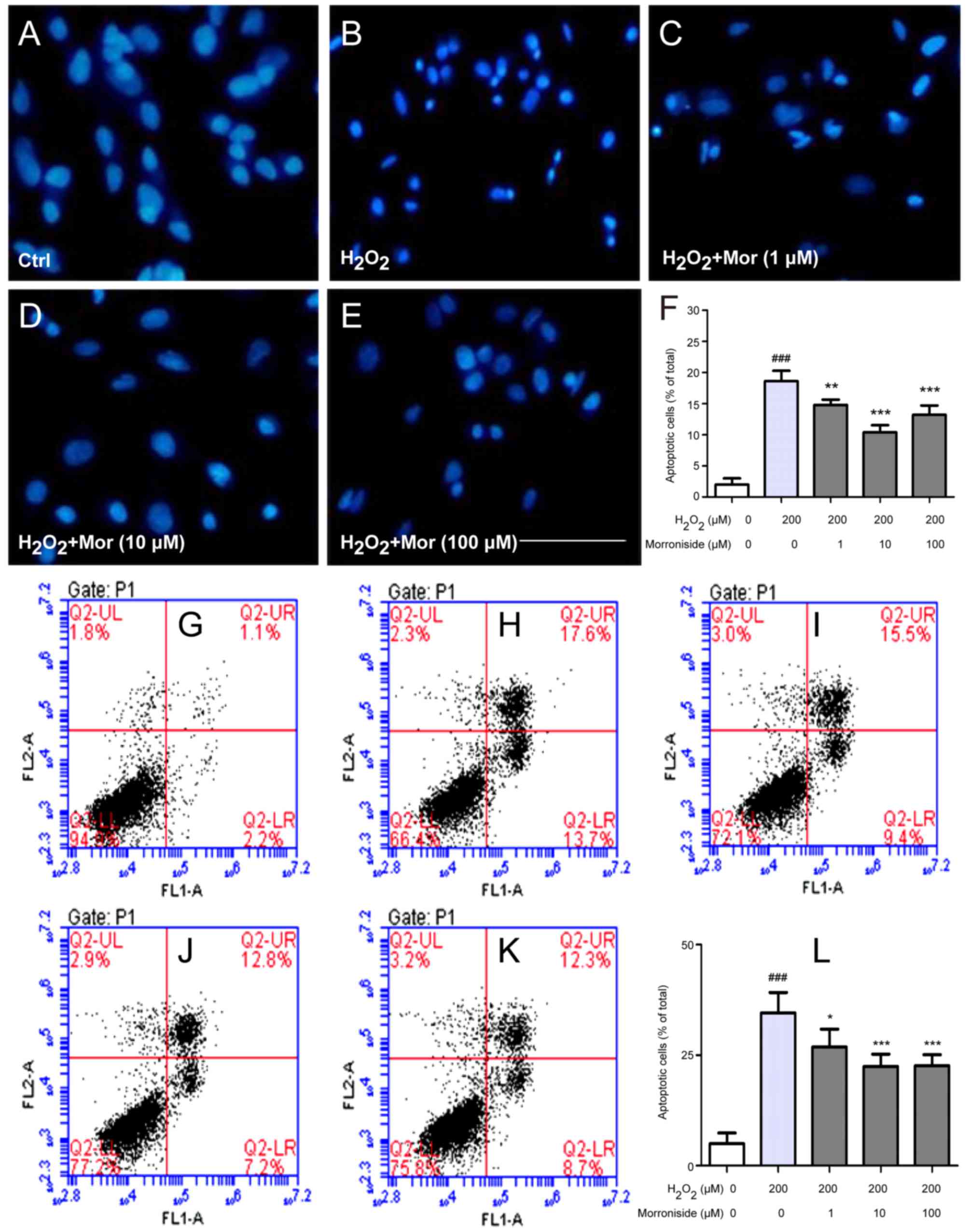
Morroniside protects SK-N-SH cells from
H2O2-induced apoptosis
We investigated whether the
H2O2-induced decrease in SK-N-SH cell
survival was due to apoptosis by Hoechst 33342 and Annexin V/PI
staining. Control cells stained with Hoechst 33342 had large,
oval-shaped nuclei (Fig. 3A).
H2O2 (200 µM) treatment increased the
fraction of cells with condensed or fragmented nuclei (18.60±1.67
vs. 2.00±1.00%; P<0.01) (Fig. 3B
and F). These defects were rescued by pretreatment with
morroniside (1, 10 and 100 µM), which reduced the fraction
of apoptotic cells (14.80±0.84, 10.40±1.14 and 13.20±1.48%,
respectively vs. 18.60±1.67%; P<0.01) (Fig. 3C–F). Consistent with these
findings, there were few cells positive for Annexin V/PI staining
detected in the control group (Fig.
3G). H2O2 (200 µM) treatment
increased the percentage of Annexin V+/PI+
cells (34.58±4.59 vs. 4.96±2.44%; P<0.001) (Fig. 3H and L), which was reduced by
morroniside (1, 10 and 100 µM) treatment (26.86±4.05,
22.42±2.81 and 22.62±2.48%, respectively vs. 34.58±4.59%;
P<0.05) (Fig. 3I–L).
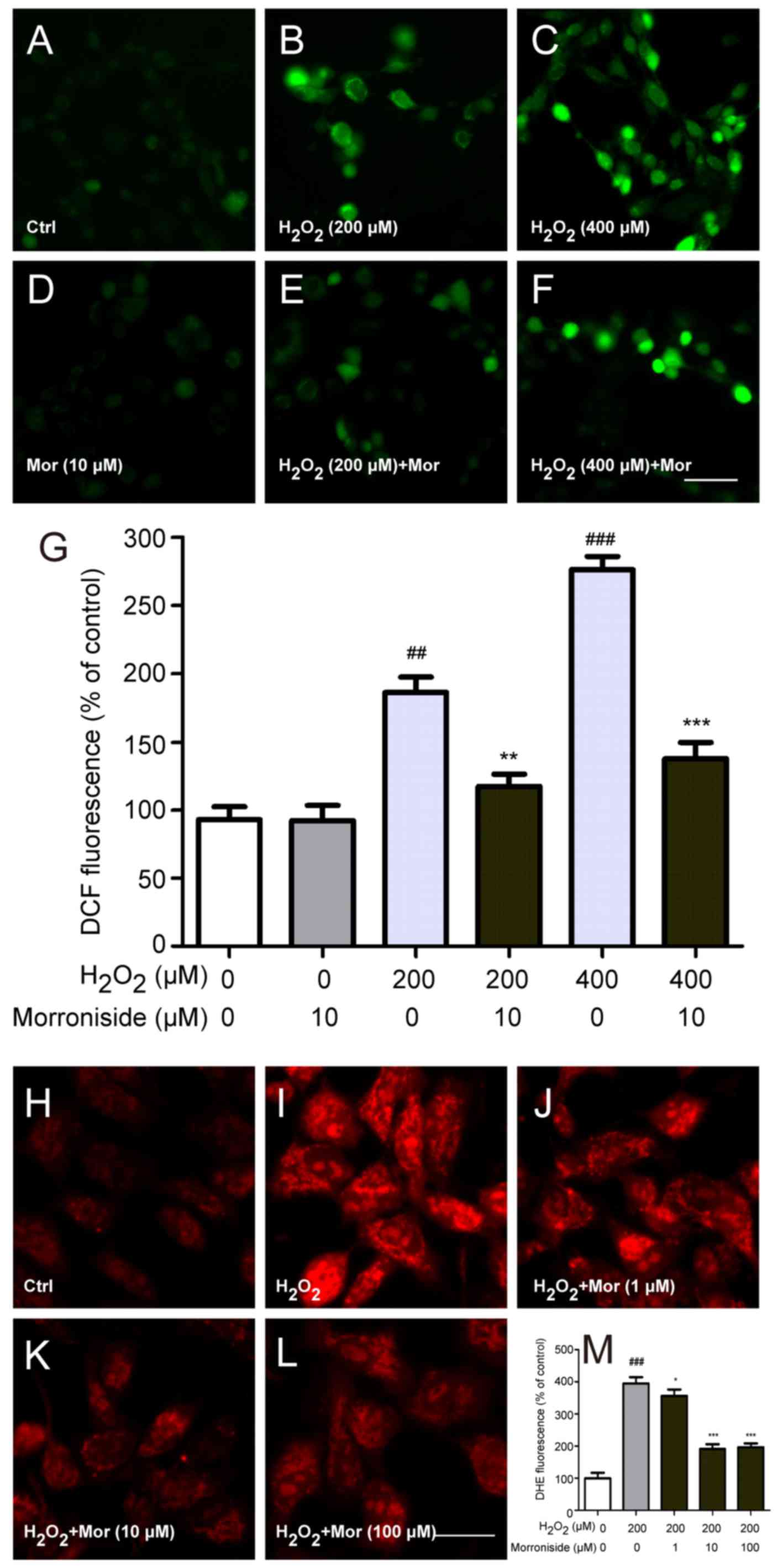
Morroniside inhibits
H2O2-induced elevation in intracellular ROS
level
Intracellular ROS levels were detected with
2,7′-dichlorofluorescein (DCF) diacetate. SK-N-SH cells exposed to
200 and 400 µM H2O2 for 24 h showed
dose-dependent increases in intracellular DCF fluorescence
(P<0.01) (Fig. 4A–C and G),
indicating that ROS levels were elevated. However, 10 µM
morroniside pretreatment abrogated these increases in fluorescence
intensity compared to the H2O2-treated groups
(P<0.01) (Fig. 4E–G),
suggesting a protective effect against
H2O2-induced free radical formation.
Superoxide anion is a type of ROS that is associated
with oxidative stress. To determine whether it is involved in
H2O2-induced cytotoxicity, we measured
intracellular superoxide anion levels with the dihydroethidium
probe (Fig. 4H–M). The
fluorescence intensity of SK-N-SH cells increased markedly after
incubation for 24 h with H2O2 (P<0.001)
(Fig. 4I and M). However,
pretreatment with 1, 10 and 100 µM morroniside significantly
reduced intracellular superoxide anion levels.
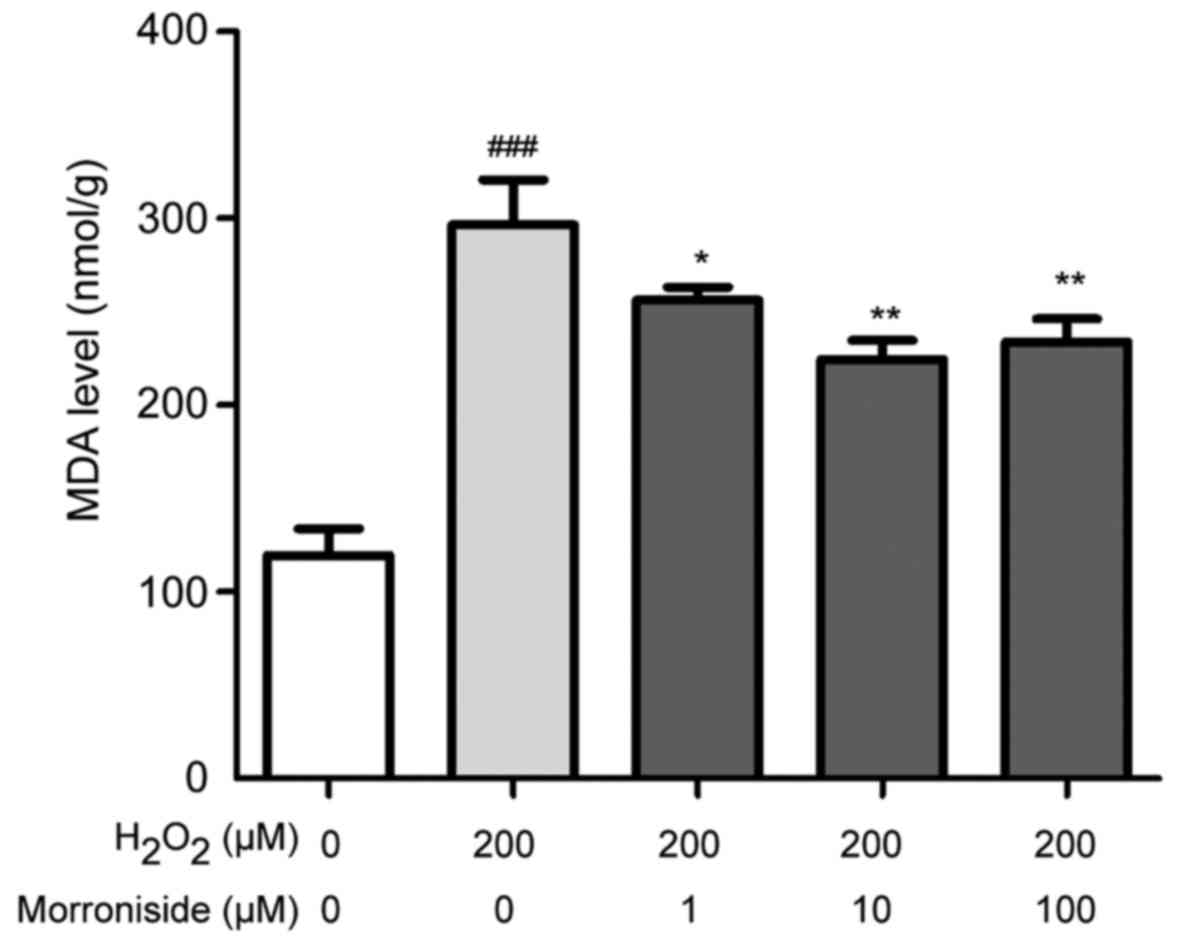
Morroniside suppresses
H2O2-induced lipid peroxidation
Lipid peroxidation levels were monitored by
detecting the stable end products of lipid peroxidation using an
MDA assay kit. Under normal conditions, cellular MDA content was
~119.47±14.14 nmol/g protein (Fig.
5). In cells treated with 200 µM
H2O2 for 24 h, the level was increased to
296.28±24.25 nmol/g protein (P<0.001). However, pretreatment
with 1, 10 and 100 µM morroniside reduced MDA levels to
256.26±6.70, 223.63±12.47 and 224.35±10.18 nmol/g protein,
respectively (P<0.05).
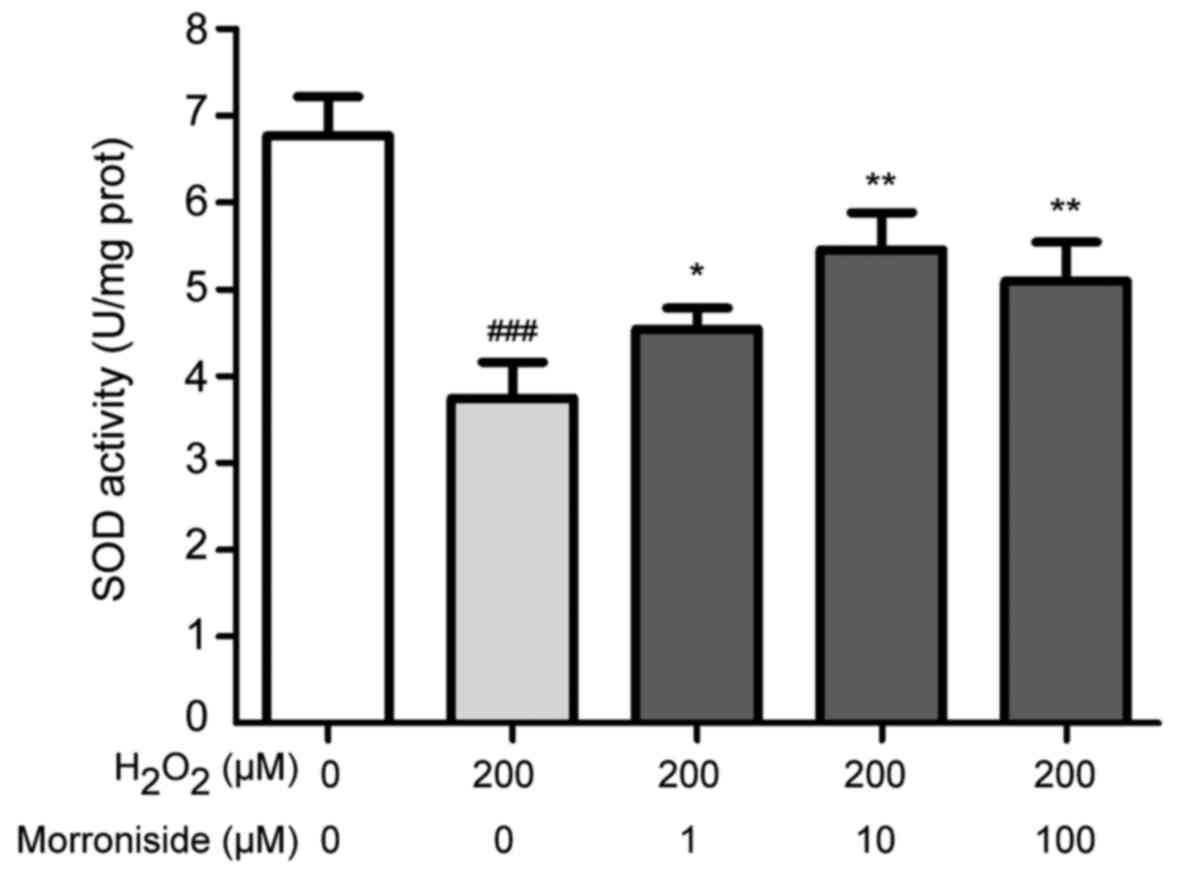
Morroniside inhibits
H2O2-induced SOD depletion
To investigate the antioxidant properties of
morroniside, we measured intracellular levels of SOD. Treatment
with 200 µM H2O2 for 24 h reduced
intracellular SOD levels from 6.77±0.45 to 3.74±0.41 U/mg
(P<0.001). This was reversed by morroniside pretreatment at
concentrations ranging from 1 to 100 µM; for example, a
concentration of 10 µM enhanced SOD level by over 45%
relative to the H2O2-treated group (Fig. 6).
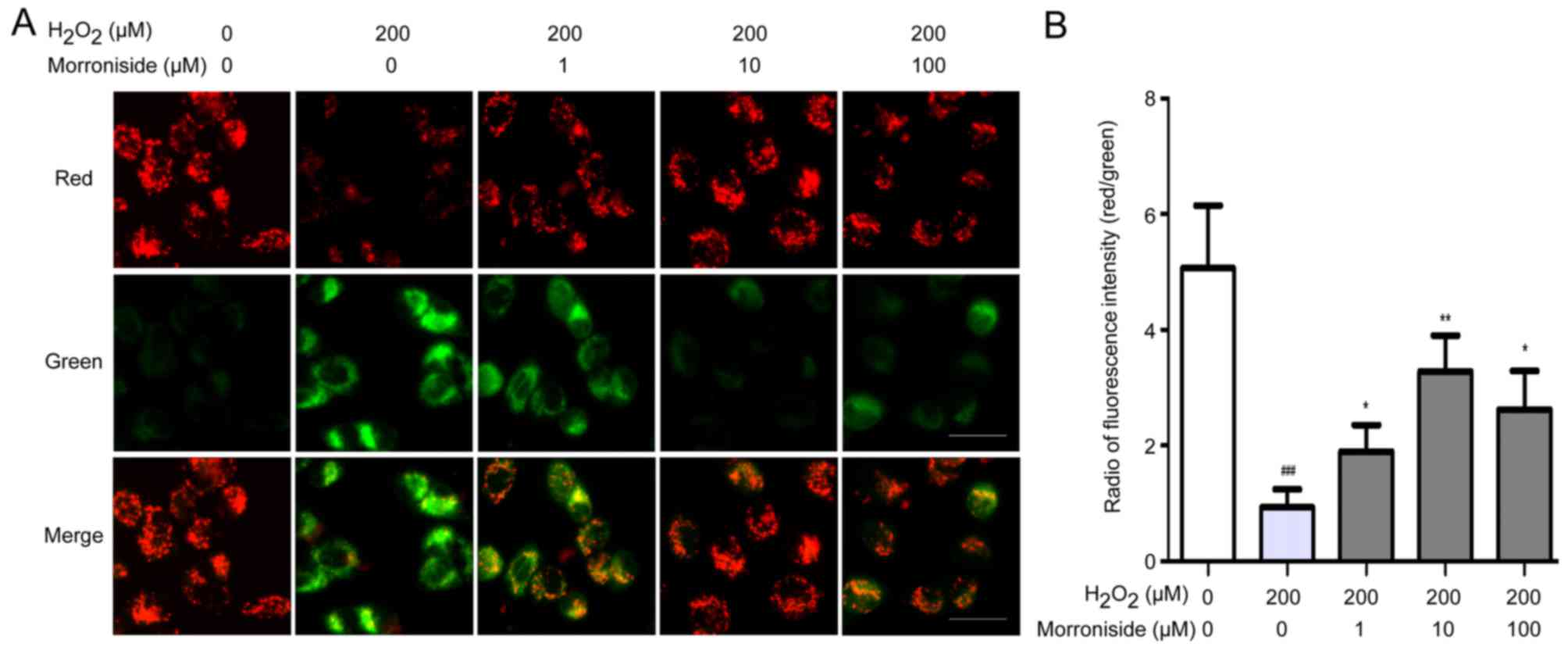
Morroniside suppresses
H2O2-induced decreases in MMP
We investigated whether morroniside could suppress
the H2O2-induced decrease in MMP using JC-1
dye. SK-N-SH cells treated with 200 µM
H2O2 for 24 h showed an obvious decrease in
MMP, as evidenced by a decrease in the ratio of red to green
fluorescence (Fig. 7). This
effect was mitigated by treatment with morroniside, most obviously
at a concentration of 10 µM.
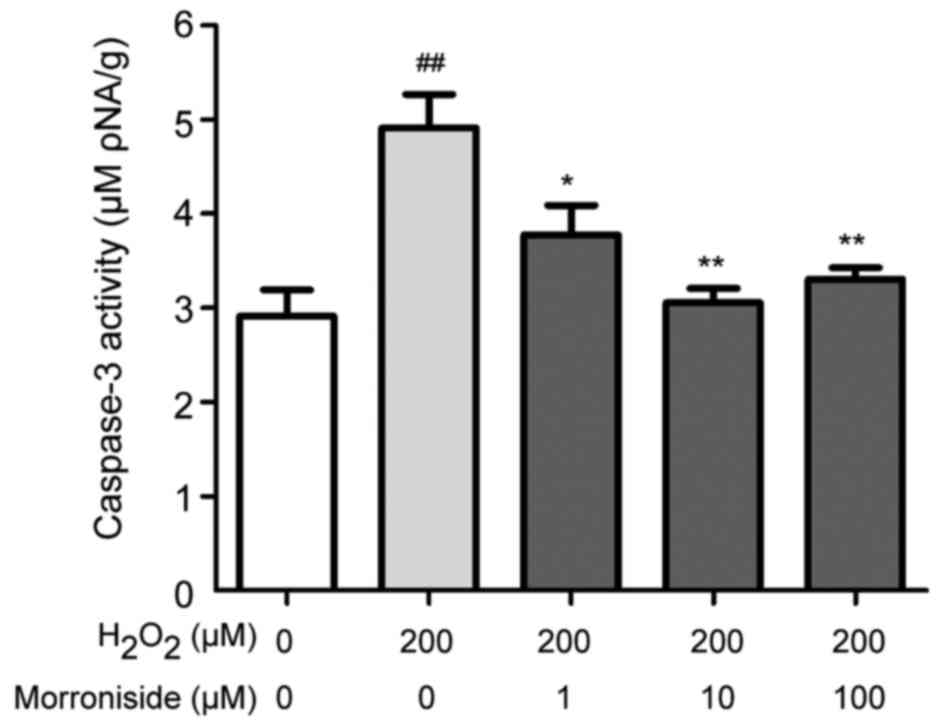
Morroniside inhibits
H2O2-induced caspase-3 level
Caspase-3 is a key protein in the regulation of
apoptosis. SK-N-SH cells treated with 200 µM
H2O2 for 24 h showed a nearly 2-fold increase
in the caspase-3 level relative to the control cells (P<0.01)
(Fig. 8). Pretreatment with
morroniside (1, 10 and 100 µM) suppressed the
H2O2-induced upregulation of caspase-3
(3.77±0.31, 3.05±0.16 and 3.31±0.12 µM pNA/g, respectively
vs. 4.91±0.36 µM pNA/g; P<0.05).
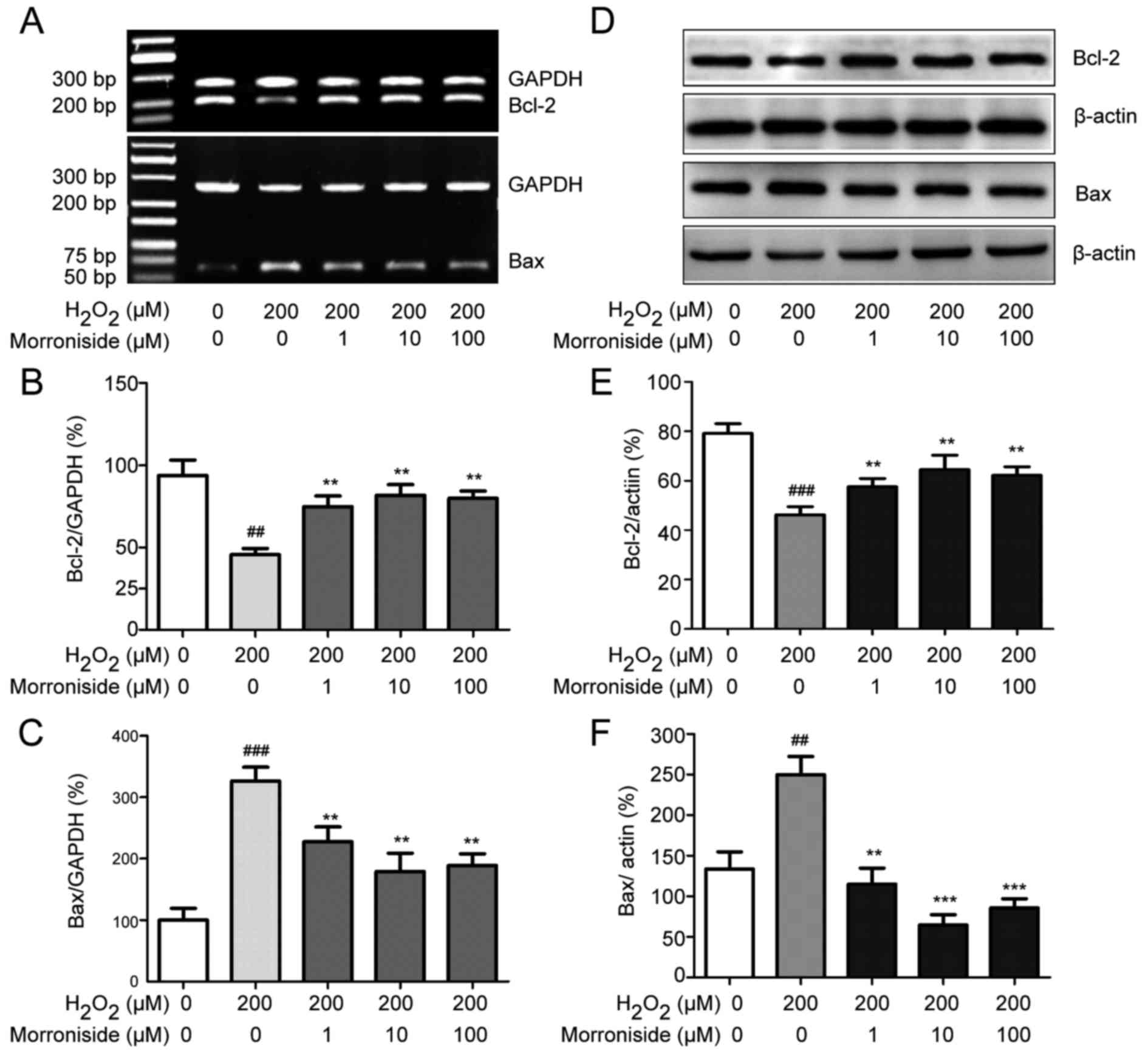
Morroniside modulates expression of
apoptosis-related proteins
The expression levels of the apoptosis-related genes
Bcl-2 and Bax were altered in the SK-N-SH cells by
H2O2 administration; the Bcl-2 mRNA level was
downregulated while that of Bax was upregulated (Fig. 9A–C). These changes were abolished
by pretreatment with 1–100 µM morroniside. Similarly,
western blot analysis revealed that H2O2 (200
µM) decreased the Bcl-2 level (P<0.001) and increased
that of Bax (P<0.01) after 24 h (Fig. 9D–F). However, pretreatment with 1,
10 and 100 µM morroniside restored Bcl-2 and Bax expression
levels compared to these levels in the
H2O2-induced group.
Discussion
Oxidative stress caused by ROS is implicated in the
pathogenesis of most chronic diseases (23,24). ROS accumulation results in
cellular damage (5). Loss of
neurons due to oxidative stress plays a key role in the development
of cerebrovascular and/or neurodegenerative diseases (1,8,25).
Neurons are thought to be more susceptible to oxidative damage than
other cell types due to their high oxygen consumption and low
antioxidant capabilities (26).
In the present study, we found that H2O2, a
freely diffusible ROS, induced apoptosis in the human neuroblastoma
SK-N-SH cells.
Usually, antioxidants, especially those derived
from natural sources, have many therapeutic applications (27,28). Morroniside is a constituent of
C. officinalis, a traditional Chinese medicine (29), and has demonstrated protective
effects against cerebral ischemic damage (15), indicating a potential role of
morroniside on restoring injured neurons. However, little is known
concerning the underlying mechanisms. In this study, we
investigated the neuroprotective role of morroniside in SK-N-SH
cells treated with H2O2.
Exposure of cells to exogenous
H2O2 can induce ROS overproduction (30), leading to peroxidation of membrane
lipids and disruption of cellular integrity (31). The present study showed that
pretreatment with morroniside reversed
H2O2-induced apoptosis by blocking ROS
production.
SOD is a component of the antioxidant machinery in
neurons (32). In most
situations, ROS produced through metabolic processes are quenched
by intracellular defense systems, including SOD (33). Previous studies have shown that
depletion of cellular glutathione leads to accumulation of ROS and
mitochondrial dysfunction (34).
We found here that morroniside treatment increased SOD activity in
the SK-N-SH cells, which prevented ROS accumulation. Thus, we
propose that the cytoprotective effects of morroniside are
associated with its antioxidant capacity, for which, antioxidants
such as glutathione or SOD are indispensable.
Overproduction of ROS disrupts the intracellular
redox equilibrium, resulting in apoptosis (35). ROS target mitochondrial membrane
proteins, leading to activation of the mitochondrial permeability
transition pore, which decreases the MMP (36) and releases cytochrome c
into the cytosol, where it binds to apoptotic protease activating
factor 1 and forms the apoptosome, which activates pro-apoptotic
caspases (37). Indeed, we
observed that H2O2 treatment reduced the MMP
and activated the pro-apoptotic protein caspase-3. These effects
were blocked by pretreatment with morroniside, suggesting that
morroniside exerts protective effects by blocking the mitochondrial
apoptotic pathway.
H2O2 has been reported to
modulate the levels of Bcl-2 and Bax, two genes associated with
mitochondrial apoptosis, in various cell types including cortical
neurons (30), endothelial cells
(38), and HT22 cells (39), specifically by suppressing Bcl-2
and stimulating Bax expression (30). This was observed in our study at
both the mRNA and protein levels following administration of
H2O2 for 24 h. Moreover, morroniside
treatment reversed the changes in Bcl-2 and Bax expression induced
by H2O2. The pro-apoptotic protein Bax is
activated during apoptosis, and its relocation from the cytoplasm
to the mitochondrial outer membrane leads to formation of the
mitochondrial permeability transition pore and downstream events.
Bax accumulated in the mitochondrial outer membrane binds to the
anti-apoptotic protein Bcl-2 and maintains it in an inactive state
(37). Bcl-2 also acts as an
antioxidant by suppressing ROS levels and the mitochondrial
apoptosis pathway (40). Taken
together, these findings suggest that morroniside blocks
mitochondrial apoptosis via modulation of Bcl-2 and Bax
expression.
In conclusion, in the present study, morroniside
was found to protect against H2O2-induced
oxidative damage in SK-N-SH neuronal cells. This effect was
mediated by inhibition of ROS-induced oxidative stress as well as
suppressing and stimulating Bax and Bcl-2 expression, respectively,
leading to inhibition of the mitochondrial apoptotic pathway. Thus,
morroniside can potentially be used for the prevention and
treatment of neurodegenerative diseases.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (no. 81471277) and the Program
of Anhui Province for Academic and Technical Leader in
University.
References
|
1
|
Destée A, Mutez E, Kreisler A,
Vanbesien-Maillot C and Chartier-Harlin MC: Parkinson disease:
Genetics and neuronal death. Rev Neurol (Paris). 165:F80–F85.
2009.In French.
|
|
2
|
Zeineddine R and Yerbury JJ: The role of
macropinocytosis in the propagation of protein aggregation
associated with neurodegenerative diseases. Front Physiol.
6:2772015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jia Z, Zhu H, Li J, Wang X, Misra H and Li
Y: Oxidative stress in spinal cord injury and antioxidant-based
intervention. Spinal Cord. 50:264–274. 2012. View Article : Google Scholar
|
|
4
|
Bains M and Hall ED: Antioxidant therapies
in traumatic brain and spinal cord injury. Biochim Biophys Acta.
1822:675–684. 2012. View Article : Google Scholar
|
|
5
|
Rodrigo R, Fernández-Gajardo R, Gutiérrez
R, Matamala JM, Carrasco R, Miranda-Merchak A and Feuerhake W:
Oxidative stress and pathophysiology of ischemic stroke: Novel
therapeutic opportunities. CNS Neurol Disord Drug Targets.
12:698–714. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shirley R, Ord EN and Work LM: Oxidative
stress and the use of antioxidants in stroke. Antioxidants Basel.
3:472–501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dias V, Junn E and Mouradian MM: The role
of oxidative stress in Parkinson's disease. J Parkinsons Dis.
3:461–491. 2013.PubMed/NCBI
|
|
8
|
Lovell MA and Markesbery WR: Oxidative DNA
damage in mild cognitive impairment and late-stage Alzheimer's
disease. Nucleic Acids Res. 35:7497–7504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khojah HM, Ahmed S, Abdel-Rahman MS and
Hamza AB: Reactive oxygen and nitrogen species in patients with
rheumatoid arthritis as potential biomarkers for disease activity
and the role of antioxidants. Free Radic Biol Med. 97:285–291.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dröge W: Free radicals in the
physiological control of cell function. Physiol Rev. 82:47–95.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Turrens JF: Mitochondrial formation of
reactive oxygen species. J Physiol. 552:335–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee HG and Yang JH: PKC-δ mediates
TCDD-induced apoptosis of chondrocyte in ROS-dependent manner.
Chemosphere. 81:1039–1044. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hamann K, Durkes A, Ouyang H, Uchida K,
Pond A and Shi R: Critical role of acrolein in secondary injury
following ex vivo spinal cord trauma. J Neurochem. 107:712–721.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hamann K and Shi R: Acrolein scavenging: A
potential novel mechanism of attenuating oxidative stress following
spinal cord injury. J Neurochem. 111:1348–1356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li X, Huo C, Wang Q, Zhang X, Sheng X and
Zhang L: Identification of new metabolites of morroniside produced
by rat intestinal bacteria and HPLC-PDA analysis of metabolites in
vivo. J Pharm Biomed Anal. 45:268–274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park CH, Noh JS, Kim JH, Tanaka T, Zhao Q,
Matsumoto K, Shibahara N and Yokozawa T: Evaluation of morroniside,
iri glycoside from Corni Fructus, on diabetes-induced alterations
such as oxidative stress, inflammation, and apoptosis in the liver
of type 2 diabetic db/db mice. Biol Pharm Bull. 34:1559–1565. 2011.
View Article : Google Scholar
|
|
17
|
Ai HX, Wang W, Sun FL, Huang WT, An Y and
Li L: Morroniside inhibits H2O2 cells.
Zhongguo Zhong Yao Za Zhi -induced apoptosis in cultured nerve.
33:2109–2112. 2008.In Chinese.
|
|
18
|
Jang SE, Jeong JJ, Hyam SR, Han MJ and Kim
DH: Ursolic acid isolated from the seed of Cornus officinalis
ameliorates colitis in mice by inhibiting the binding of
lipopolysaccharide to Toll-like receptor 4 on macrophages. J Agric
Food Chem. 62:9711–9721. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu HH, Hur JM, Seo SJ, Moon HD, Kim HJ,
Park RK and You YO: Protective effect of ursolic acid from Cornus
officinalis on the hydrogen peroxide-induced damage of HEI-OC1
auditory cells. Am J Chin Med. 37:735–746. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim SH, Lee MK, Ahn M-J, Lee KY, Kim YC
and Sung SH: Efficient method for extraction and simultaneous
determination of active constituents in Cornus officinalis by
reflux extraction and high performance liquid chromatography with
diode array detection. J Liq Chromatogr Relat Technol. 32:822–832.
2009. View Article : Google Scholar
|
|
21
|
Li CY, Li L, Li YH, Ai HX and Zhang L:
Effects of extract from Cornus officinalis on nitric oxide and
NF-kappaB in cortex of cerebral infarction rat model. Zhongguo
Zhong Yao Za Zhi. 30:1667–1670. 2005.In Chinese.
|
|
22
|
Hu JG, Fu SL, Zhang KH, Li Y, Yin L, Lu PH
and Xu XM: Differential gene expression in neural stem cells and
oligodendrocyte precursor cells: a cDNA microarray analysis. J
Neurosci Res. 78:637–646. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rani V, Deep G, Singh RK, Palle K and
Yadav UC and Yadav UC: Oxidative stress and metabolic disorders:
Pathogenesis and therapeutic strategies. Life Sci. 148:183–193.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miranda-Díaz AG, Pazarín-Villaseñor L,
Yanowsky-Escatel FG and Andrade-Sierra J: Oxidative stress in
fiabetic nephropathy with early chronic kidney disease. J Diabetes
Res. 2016:70472382016. View Article : Google Scholar
|
|
25
|
Forte M, Conti V, Damato A, Ambrosio M,
Puca AA, Sciarretta S, Frati G, Vecchione C and Carrizzo A:
Targeting nitric oxide with natural derived compounds as a
therapeutic strategy in vascular diseases. Oxid Med Cell Longev.
2016:73641382016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karpinska A and Gromadzka G: Oxidative
stress and natural antioxidant mechanisms: the role in
neurodegeneration. From molecular mechanisms to therapeutic
strategies. Postepy Higieny i Medycyny Doswiadczalnej (Online).
67:43–53. 2013. View Article : Google Scholar
|
|
27
|
Turkez H, Sozio P, Geyikoglu F, Tatar A,
Hacimuftuoglu A and Di Stefano A: Neuroprotective effects of
farnesene against hydrogen peroxide-induced neurotoxicity in vitro.
Cell Mol Neurobiol. 34:101–111. 2014. View Article : Google Scholar
|
|
28
|
Tian LL, Wang XJ, Sun YN, Li CR, Xing YL,
Zhao HB, Duan M, Zhou Z and Wang SQ: Salvianolic acid B, an
antioxidant from Salvia miltiorrhiza, prevents 6-hydroxydopamine
induced apoptosis in SH-SY5Y cells. Int J Biochem Cell Biol.
40:409–422. 2008. View Article : Google Scholar
|
|
29
|
Ding X, Wang MY, Yu ZL, Hu W and Cai BC:
Studies on separation, appraisal and the biological activity of
5-HMF in Cornus officinalis. Zhongguo Zhong Yao Za Zhi. 33:392–396.
4842008.In Chinese.
|
|
30
|
Jiang X, Nie B, Fu S, Hu J, Yin L, Lin L,
Wang X, Lu P and Xu XM: EGb 761 protects hydrogen peroxide-induced
death of spinal cord neurons through inhibition of intracellular
ROS production and modulation of apoptotic regulating genes. J Mol
Neurosci. 38:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu H, Shen J, Liu H, Shi Y, Li L and Wei
M: Morroniside and loganin extracted from Cornus officinalis have
protective effects on rat mesangial cell proliferation exposed to
advanced glycation end products by preventing oxidative stress. Can
J Physiol Pharmacol. 84:1267–1273. 2006. View Article : Google Scholar
|
|
32
|
Hilton JB, White AR and Crouch PJ:
Endogenous Cu in the central nervous system fails to satiate the
elevated requirement for Cu in a mutant SOD1 mouse model of ALS.
Metallomics. 8:1002–1011. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hwang KA, Hwang YJ and Song J: Antioxidant
activities and oxidative stress inhibitory effects of ethanol
extracts from Cornus officinalis on raw 264.7 cells. BMC Complement
Altern Med. 16:1962016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bharat S, Cochran BC, Hsu M, Liu J, Ames
BN and Andersen JK: Pretreatment with R-lipoic acid alleviates the
effects of GSH depletion in PC12 cells: Implications for
Parkinson's disease therapy. Neurotoxicology. 23:479–486. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Deng G, Su JH, Ivins KJ, Van Houten B and
Cotman CW: Bcl-2 facilitates recovery from DNA damage after
oxidative stress. Exp Neurol. 159:309–318. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nguyen TT, Stevens MV, Kohr M, Steenbergen
C, Sack MN and Murphy E: Cysteine 203 of cyclophilin D is critical
for cyclophilin D activation of the mitochondrial permeability
transition pore. J Biol Chem. 286:40184–40192. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Del Gaizo Moore V and Letai A: BH 3
profiling - measuring integrated function of the mitochondrial
apoptotic pathway to predict cell fate decisions. Cancer Lett.
332:202–205. 2013. View Article : Google Scholar
|
|
38
|
Li DW, Li JH, Wang YD and Li GR:
Atorvastatin protects endothelial colony forming cells against
H2O2 induced oxidative damage by regulating
the expression of Annexin A2. Mol Med Rep. 12:7941–7948.
2015.PubMed/NCBI
|
|
39
|
Xu H, Luo P, Zhao Y, Zhao M, Yang Y, Chen
T, Huo K, Han H and Fei Z: Iduna protects HT22 cells from hydrogen
peroxide-induced oxidative stress through interfering
poly(ADP-ribose) polymerase-1-induced cell death (parthanatos).
Cell Signal. 25:1018–1026. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Neuzil J, Wang XF, Dong LF, Low P and
Ralph SJ: Molecular mechanism of 'mitocan'-induced apoptosis in
cancer cells epitomizes the multiple roles of reactive oxygen
species and Bcl-2 family proteins. FEBS Lett. 580:5125–5129. 2006.
View Article : Google Scholar : PubMed/NCBI
|