Introduction
X-linked hypophosphatemic rickets (XLHR; OMIM307800)
is a disorder associated with renal phosphate wasting, which
results in hypophosphatemia, growth retardation, abnormal bone
mineralization, rickets and osteomalacia (1,2).
XLHR accounts for >80% of cases of familial hypophosphatemic
rickets and occurs in ~1 per 20,000 live births (3–5).
Due to loss of the physiological response to low levels of blood
phosphate, patients with XLHR are reported to have normal or low
serum levels of 1, 25-dihydroxyvitamin D3 (1,25(OH)2D), a normal
serum level of parathyroid hormone, increased activity of serum
alkaline phosphatases, and resistance to high doses of vitamin D
(6,7).
XLHR is caused by variants of the
phosphate-regulating neutral endopeptidase homolog, X-linked
(PHEX) gene (NC_000023.11; OMIM:300550), which is located at
Xp22.11 and contains 22 exons (8,9).
According to the Human Gene Mutation Database (HGMD professional
2016.4 release), a total of 510 different PHEX gene variants
have been identified, among which point variants, including
missense/nonsense variants and splicing variants, are the most
common and account for 45.09% (230 in 510) of all cases.
The PHEX gene encodes a protein, namely a
phosphate-regulating neutral endopeptidase, belonging to the type
II integral membrane zinc-dependent endopeptidase family. It
consists of an extracellular C-terminal region, a transmembrane
domain, and an N-terminal cytoplasmic tail (10). This protein is predominantly
expressed in mineralized tissues, including the calvaria, long bone
and teeth, and regulates the synthesis of fibroblast growth factor
23 through indistinct mechanisms (12–15). The aberrance of this protein leads
to a reduction in renal phosphate reabsorption, partly via a
decline in renal proximal tubular cell type II sodium-phosphate
co-transporters, and results in abnormal bone mineralization by
affecting fibroblast growth factor receptor signaling in osteocytes
(15,16).
The present study reported on two variants of the
PHEX gene in two Chinese families affected by XLHR. The
primary aim was to provide a genetic diagnosis for the affected
family members and to analyze the underlying genotype-phenotype
correlations. Based on the results, it is likely to be possible to
provide a prenatal diagnosis and medical suggestions to families
carrying affected fetuses.
Materials and methods
Subjects and ethics statement
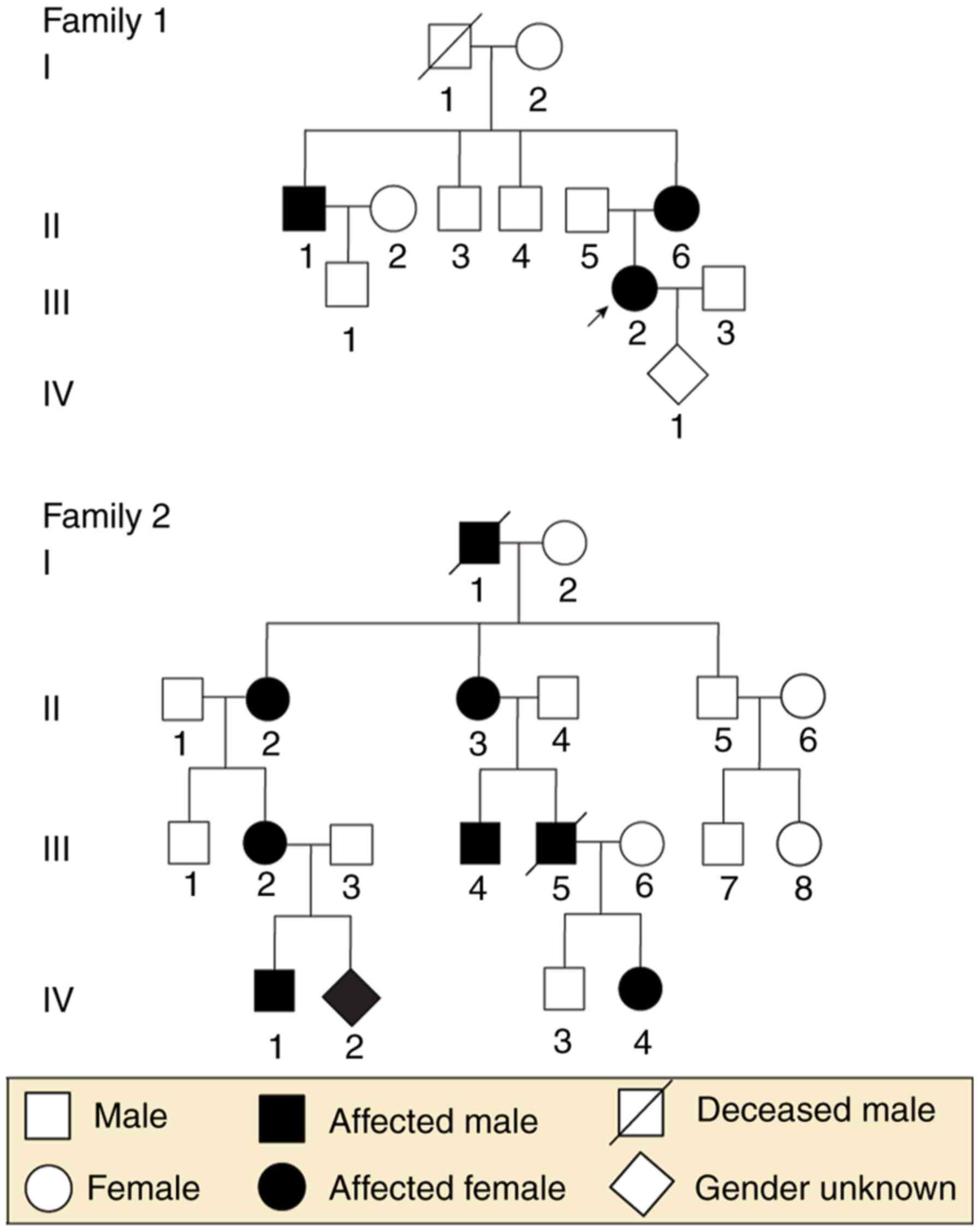
Two unrelated families, including a total of 23
individuals (nine individuals and one fetus in Family 1; 12
individuals and one fetus in Family 2) from the Department of
Obstetrics and Gynecology at West China Second University Hospital,
Sichuan University (Sichuan, China), were enrolled in the present
study. To be specific, the nine members in Family 1 included two
affected females and one affected male, and the 12 members of
Family 2 included four affected females and two affected males. The
present study was performed in the Prenatal Diagnosis Center of
Sichuan Province (Sichuan, China) and was approved by the ethics
committee of West China Second University Hospital, Sichuan
University. The two families provided written informed consent
prior to collection of blood samples, pedigree structures and
clinical data. Fetal DNA was acquired from backup amniotic fluid
samples, which were used for fetal chromosome analysis at the
Prenatal Diagnosis Center of Sichuan Province. The family trees are
shown in Fig. 1.
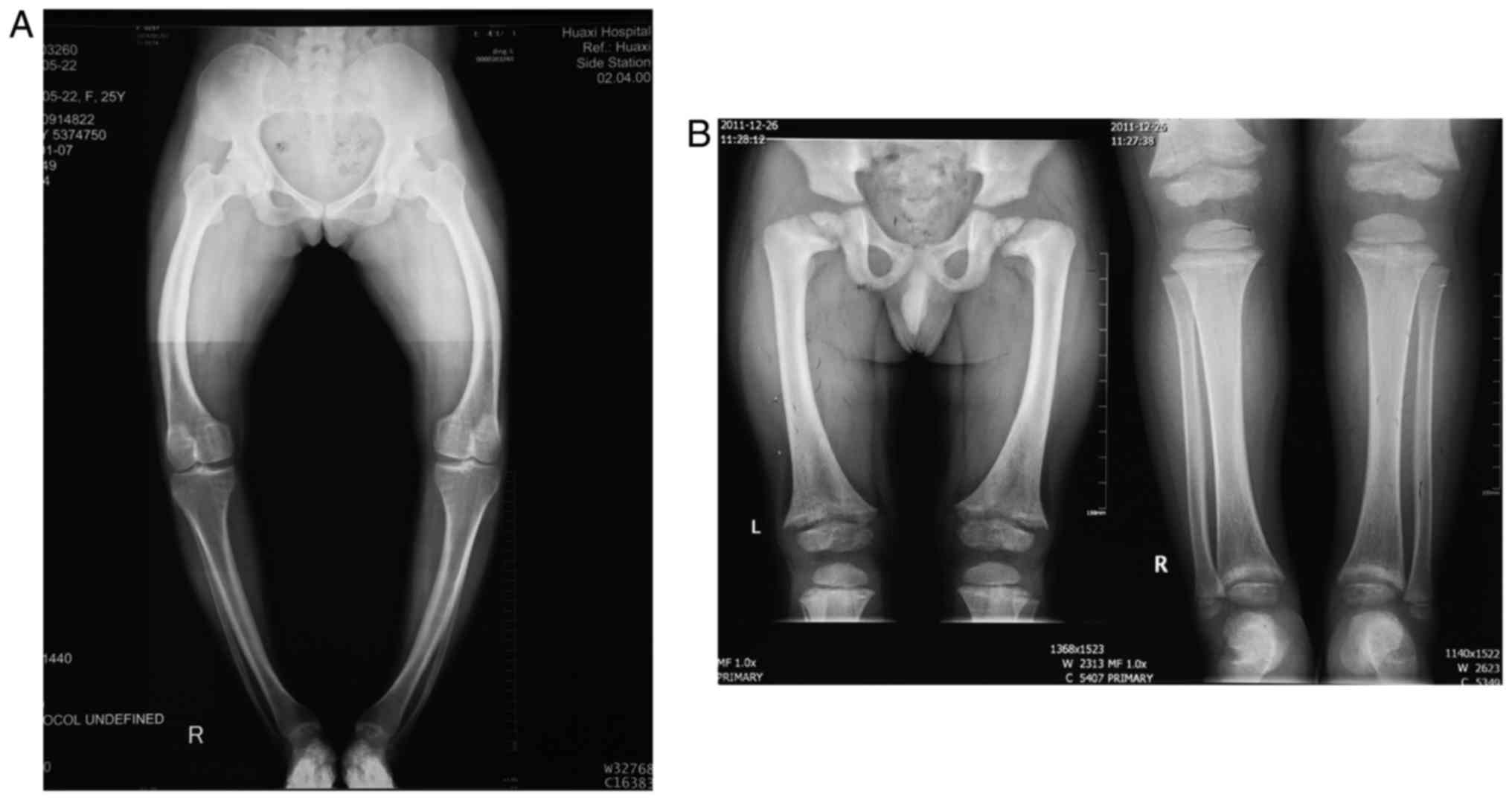
The proband from Family 1 was a 28-year-old pregnant
woman with a family history of rickets, who was short in stature
(146 cm) and presented with lower-extremity bowing. The T-score of
her left foot was-2.0, demonstrating decreased bone mineral density
(normal: T-score>-1; decreased: -1> T-score>-2.5;
osteoporosis: T-score<-2.5). The X-ray of her lower extremities
indicated knock knees, and a bilateral bowed femur, tibia and
fibula (Fig. 2A). The patient had
a low serum phosphate level (0.64 mmol/l), normal serum calcium
level (2.05 mmol/l) and normal serum alkaline phosphatase (328.8
U/l). Despite the lack of laboratory tests and X-ray images of the
proband's brother and sister, both were short in stature and had
lower-extremity bowing, indicating the probability that they had
XLHR.
In Family 2, the proband was a 9-year-old boy, who
also had signs and a family history of rickets. He was reported to
be 110 cm and 35 kg, complained of spontaneous dental abscesses and
had only three saprodontias in his mouth. An X-ray of his limbs
(aged 4 years old) showed decreased bone mineral density,
enlargement of the long bones, rough metaphysis with a cup-shaped
change, uneven density of his bilateral distal femoral ossification
centers, a narrowed collodiaphyseal angle and knock knees,
suggesting active rickets (Fig.
2B). The boy had a low serum phosphate level (1.20 mmol/l),
normal serum calcium level (2.20 mmol/l) and elevated serum
alkaline phosphatase (412.2 U/l). He was receiving oral phosphate,
calcitriol and an active form of vitamin D. His affected mother and
three other family members exhibited the same distinctive clinical
manifestations of a short stature with lower-extremity bowing.
DNA and RNA preparation
For the adults and children, genomic DNA was
extracted from peripheral whole blood samples, and fetal DNA was
isolated from amniotic fluid using the Gentra Puregene Blood kit
(Qiagen GmbH, Hilden, Germany) according to the manufacturer's
protocol. RNA was carefully obtained from the peripheral blood
mononuclear cells using TRIzol reagent according to the
manufacturer's protocol; these cells were isolated from the
peripheral whole blood samples using Ficoll-Hypaque density
gradient centrifugation at 800 × g for 20 min. DNA was stored at
−20°C prior to use, and RNA immediately underwent reverse
transcription polymerase chain reaction (RT-PCR) analysis following
isolation.
Variant screening of the PHEX gene
To determine the PHEX gene variants, all 22
exons of the PHEX gene in the proband were initially
amplified, followed by sequencing of the amplification products.
Once a variant of the PHEX gene was identified in proband,
DNA samples of the remaining healthy and affected family members
were screened for this variant. Finally, the fetal PHEX gene
was assessed only when the variant site was verified in all
affected family members.
Information on the PHEX gene was obtained
from the online NCBI database (https://www.ncbi.nlm.nih.gov/pubmed; RefSeq
NM_000444.5). In total, 22 pairs of primers were designed for PCR
amplification of all 22 exon regions and intron/exon boundaries of
the PHEX gene using an online tool (http://www.yeastgenome.org/cgi-bin/web-primer).
The sequences of the primers are listed in Table I. The PCR conditions were as
follows: 95°C for 3 min, 95°C for 30 sec, 72°C for 1 min (34
cycles) and 72°C for 5 min. The PCR products were submitted to
Sanger sequencing on an automated ABI PRISM 3130 sequencer (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
were then prepared with the BigDye Terminator v3.1 Cycle Sequencing
kit (Applied Biosystems; Thermo Fisher Scientific, Inc.).
 | Table IPrimers used to amplify 22 exons and
cDNA of the phosphate-regulating neutral endopeptidase homolog,
X-linked gene. |
Table I
Primers used to amplify 22 exons and
cDNA of the phosphate-regulating neutral endopeptidase homolog,
X-linked gene.
| Exon | Forward primer
(5′–3′) | Reverse primer
(5′–3′) | Annealing
temperature (°C) | Product size
(bp) |
|---|
| DNA | | | | |
| 1 |
TTCCTGACGGCAGTTTCTTA |
AGGCAAACAGCCCTATACCT | 55.0 | 360 |
| 2 |
TGGGTTTTGGAATACCGTGT |
GGAATTCATAGCCAGCGGA | 57.0 | 530 |
| 3 |
CGGCCTATCCATTCACTTTGA |
TTCTGCAAACTTTCCTTTCAA | 57.2 | 513 |
| 4 |
TCTGGAGGTTGGAATTGTGA |
TGGCTTCTGGATTCTCTACCA | 55.5 | 486 |
| 5 |
CCACCCCACCTCTTTTACCTA |
ATGGCACCCCAAAAGGCTAAT | 58.9 | 436 |
| 6 |
TGGGATGCAGACGATTTCAT |
TCCTGGGTATCTCTTCTGACG | 56.0 | 504 |
| 7 |
TTGCATAATGCCTGCACCAT |
ATGGGCAATGACACAAAACG | 58.2 | 409 |
| 8 |
CACCAAAGCCTTGAAAAACTT |
CTGAGCCAATGCCAACAATT | 56.0 | 477 |
| 9 |
TCTGTTTTGTTCTCTCTCCCC |
TACACCAGACAGTGCTTTTGG | 55.0 | 343 |
| 10 |
ATGGAGCTTTGCCAACTGTT |
GAAAACAATGTGTGGCCTCA | 55.5 | 454 |
| 11 |
TGACCTCAGGTGATCTGCTCA |
AAGGCTGACATTAGCCTGTTG | 56.5 | 496 |
| 12 |
GAGTCATTTCTCATGCAAGCC |
CCTCCTGGTGAAACAAAAATC | 55.0 | 366 |
| 13 |
CCTTCACAGTGGCTTGCATAT |
TGATCTGCCTGGCATATTCA | 56.0 | 377 |
| 14 |
TGTGACTGATGCAGCTTCTCT |
AGAAATGGGGGACCTGTAATT | 55.5 | 423 |
| 15 |
AATCTCTCCCCTACCTAACCC |
CCCTGAGAAGACCCTGAAAAT | 55.0 | 394 |
| 16 |
ATCTCTTAGAGGGCTCCCAGT |
TAAGATGGCTTTCCTGTCCA | 55.0 | 397 |
| 17 |
AGCAGTTTATCTTGGCTTTCC |
AAAGTCTACCCCGATCACCAA | 56.4 | 377 |
| 18 |
TGTTCCCTGCTGTTATGACTG |
TGATTCAGCAGGTATGGGGTA | 56.4 | 405 |
| 19 |
TTGCTGAGGATAGTTTGCCA |
TGACTTCACACCCCAAAAAG | 55.0 | 452 |
| 20 |
GCTAGGAATGGAAAGAACAGC |
TTTCTTTGATCAAGGGAGCA | 55.0 | 451 |
| 21 |
CCTGGGCACATATACGATTCT |
TGGGATTTTTTTCAGATCACC | 56.0 | 462 |
| 22 |
TGGCAGATAGTAATATGGGCA |
ATGAAGGCTCAGTGCAGCCTC | 58.9 | 555 |
| cDNA | | | | |
| 1 |
ATGGAAGCAGAAACAGGGAG |
TCAAGGTAGTCTTCCCTCACG | 55.5 | 710.0 |
| 2 |
GCCAAAATCCTTTATTCATCC |
TCTTTAAAGTACTGCGGGACG | 56.0 | 644.0 |
| 3 |
TGGGCTACATCAAGAAGGTCA |
TGGAAATCGGATCTGGTTGGT | 58.2 | 710.0 |
| 4 |
AGAAGCCGACTACTTTGGCAA |
ATAAACCAGCGTCCCAGCTA | 56.9 | 698.0 |
All sequencing results were aligned with the
referenced PHEX gene sequences from the NCBI databse using
the online Pairwise Sequence Alignment tool (www.ebi.ac.uk/Tools/psa/emboss_water/nucleotide.html).
To predict the outcomes of variants, the SIB Bioinformatics
Resource Portal database (http://www.expasy.org/), the Human Gene Variant
Database (http://www.hgmd.cf.ac.uk) and the
PolyPhred database (http://droog.gs.washington.edu/polyphred/) were used.
Additionally, to search for the suspected aberrant splicing
variants, Alamut® Alamut Interactive Biosoftware
(version 2.9.0; Rouen, France) was used, which contains the four
algorithms SpliceSiteFinder, MaxEntScan, NNSplice and GVGD.
RT-PCR analysis
The aberrantly spliced transcripts were verified by
sequencing the entire cDNA of the PHEX gene. An initial cDNA
strand was obtained from RT-PCR of total RNA using the RT reagent
kit with gDNA Eraser (Takara Biotechnology Co., Kyoto, Japan) under
the condition: 37°C for 15 min and 85°C for 5 sec. A total of 2
μl cDNA product was then used as a template to amplify the
four overlapping fragments with the four pairs of primers. Primer
sequences are listed in Table I.
The PCR conditions were as follows: 95°C for 3 min, 95°C for 30
sec, 72°C for 1 min (34 cycles) and 72°C for 5 min. The final PCR
products were sequenced and aligned with the coding region of the
referenced PHEX gene from the NCBI database.
RT-quantitative PCR analysis
To examine the fold changes in the gene expression
of PHEX affected by the suspicious abnormal splice donor
site in Family 2, semi-quantitative analysis of the gene expression
of PHEX in the proband and that of his affected mother was
performed through qPCR analysis using the cDNA, which was derived
from the peripheral blood total RNA. The samples were prepared with
10 μl of SYBR-Green Master mix (Thermo Fisher Scientific,
Inc.) and 0.25 μl of primers (forward
5′-TTCGCTTGTGATGGCTGGAT-3′ and reverse 5′-CTGTATGGCTTCGGTGTCCC-3′)
in a final reaction volume of 20 μl. The samples from one
normal male and one normal female were used as controls, and the
GAPDH gene was used as an internal reference gene. The RT-qPCR
analysis was performed under the following conditions: 95°C for 5
min, followed by 40 cycles of 95°C for 15 sec and 60°C for 30 sec,
with a final extension at 72°C for 5 min. The 2−ΔΔCq
method was used to assess the relative changes in the gene
expression of PHEX according to the standard procedure
(17).
Results
Variant screening
The PHEX gene is the only gene in which
pathogenic variants known to cause XLHR have been reported.
Therefore, the present study primarily focused on examining the
PHEX gene in the two families. By directly sequencing all 22
exons and the intron/exon boundaries of the PHEX gene of the
proband, two separate point variants were identified.
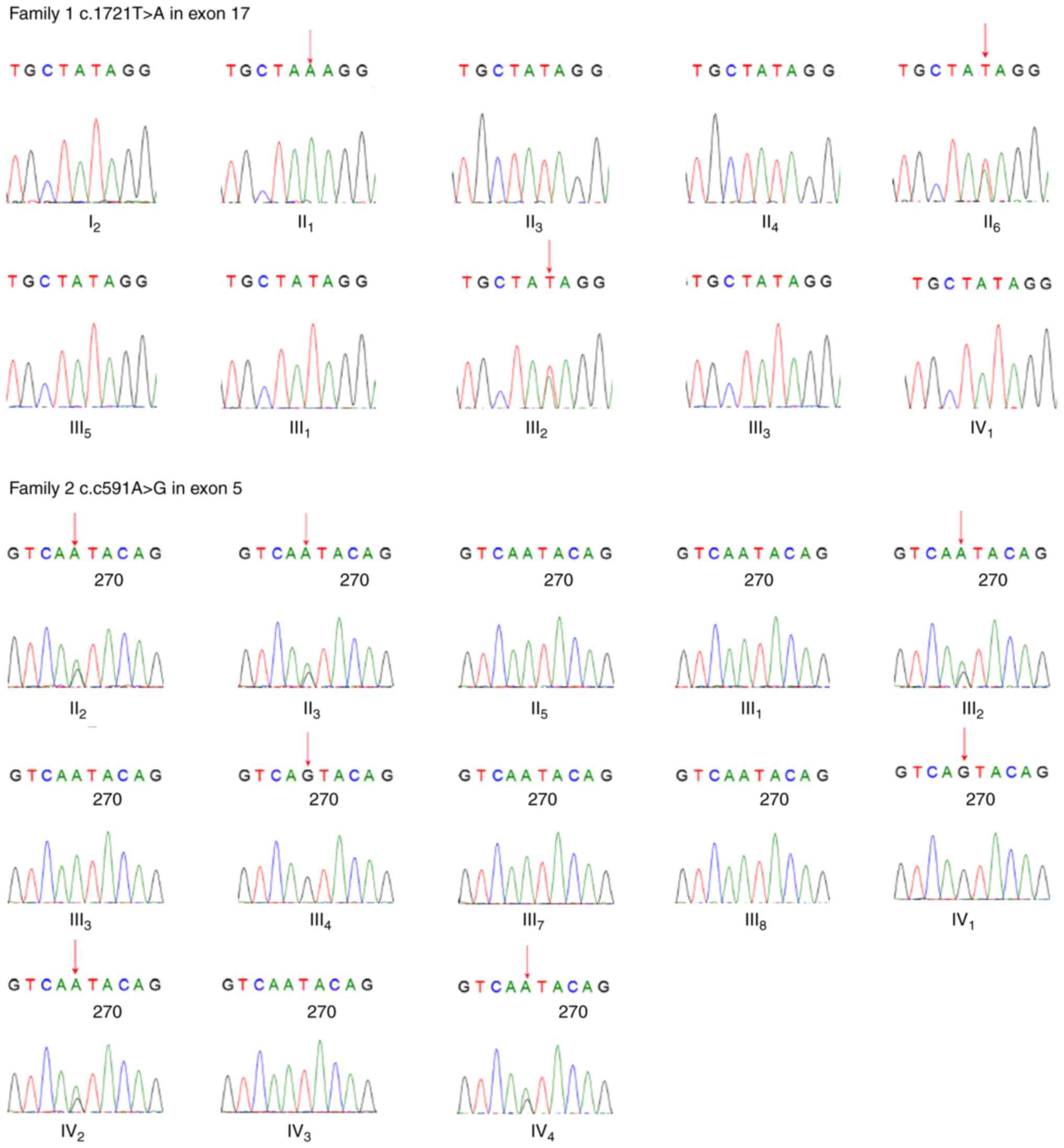
For the proband in Family 1, a novel missense
heterozygous variant c.1721T>A in exon 17 was found, which
resulted in a change of the 574th amino acid from isoleucine to
lysine, p.574Ile>Lys. The sequencing results of exon 17 in the
remaining affected family members verified the variant in this
pedigree: A single peak (A) in the affected male (II1)
and heterozygous peaks (A/T) in the affected female
(II6) as males receive only one copy of the X
chromosome. This PHEX gene variant was not found in healthy
family members. Therefore, it was observed that individuals showing
clinical symptoms of XLHR had this variant, whereas the healthy
individuals did not. However, the proband's grandmother
(I2) reported no complaints of any signs of clinical
rickets, and gene analysis showed no variant in the PHEX
gene. Subsequently, the present analyzed 15 short-sequence tandem
repeat regions on chromosomes 21, 18,13, X and Y of the grandmother
(I2) and her four children (II1,
II3, II4 and II6) via RT-PCR
analysis, and found the healthy grandmother (I2) was
their biological mother, suggesting the possibility of the gonadal
mosaicism in the grandmother.
In Family 2, the sequencing results of the
PHEX gene of the proband showed a point variant c.591A>G
in exon 5, which caused no change in the amino acid sequence
(p.197, glutamic acid). This variant was also observed in the other
affected family members, including four females (II2,
II3, III2 and IV4), one male
(III4) and the fetus (IV2). The affected
females showed heterozygous peaks (A/G) at this site and the
affected male showed only one peak (G), whereas the healthy females
showed homozygous peaks (A) and the healthy male showed one peak
(A). This variant was only detected in the affected male and female
family members, and the inheritance patterns of the disease
appearing in this pedigree strictly followed X-linked dominant
inheritance. It was hypothesized that this variant likely
originated in the proband's great grandfather, who had similar
rickets symptoms according to his children's recall, although it is
difficult to make definitive conclusions as the man had passed
away. All sequencing results are shown in Fig. 3.
Pathogenicity analysis of variants
In Family 1, the c.1721T>A variant resulted in
the 574th amino acid of the protein (phosphate-regulating neutral
endopeptidase) changing from isoleucine to lysine (p.Ile574Lys).
This non-synonymous substitution is located at peptidase M13, the
C-terminal domain of the protein, which is close to the mental
binding (p.580, histidine) and active site (p.581, glutamic acid).
According to the Align GVDV prediction (18,19), this variant scored 10.12 for
Grantham variation and 93.77 for Grantham deviation, and was
classified as C55, suggesting likely pathogenicity. Additionally,
the SIFT prediction software sorted this variant as deleterious. In
conclusion, the variant c.1721T>A of the PHEX gene was
identified to cause XLHR in this pedigree, and it was possible that
the variant was inherited from the grandmother, who may have had
gonadal mosaicism of the PHEX gene.
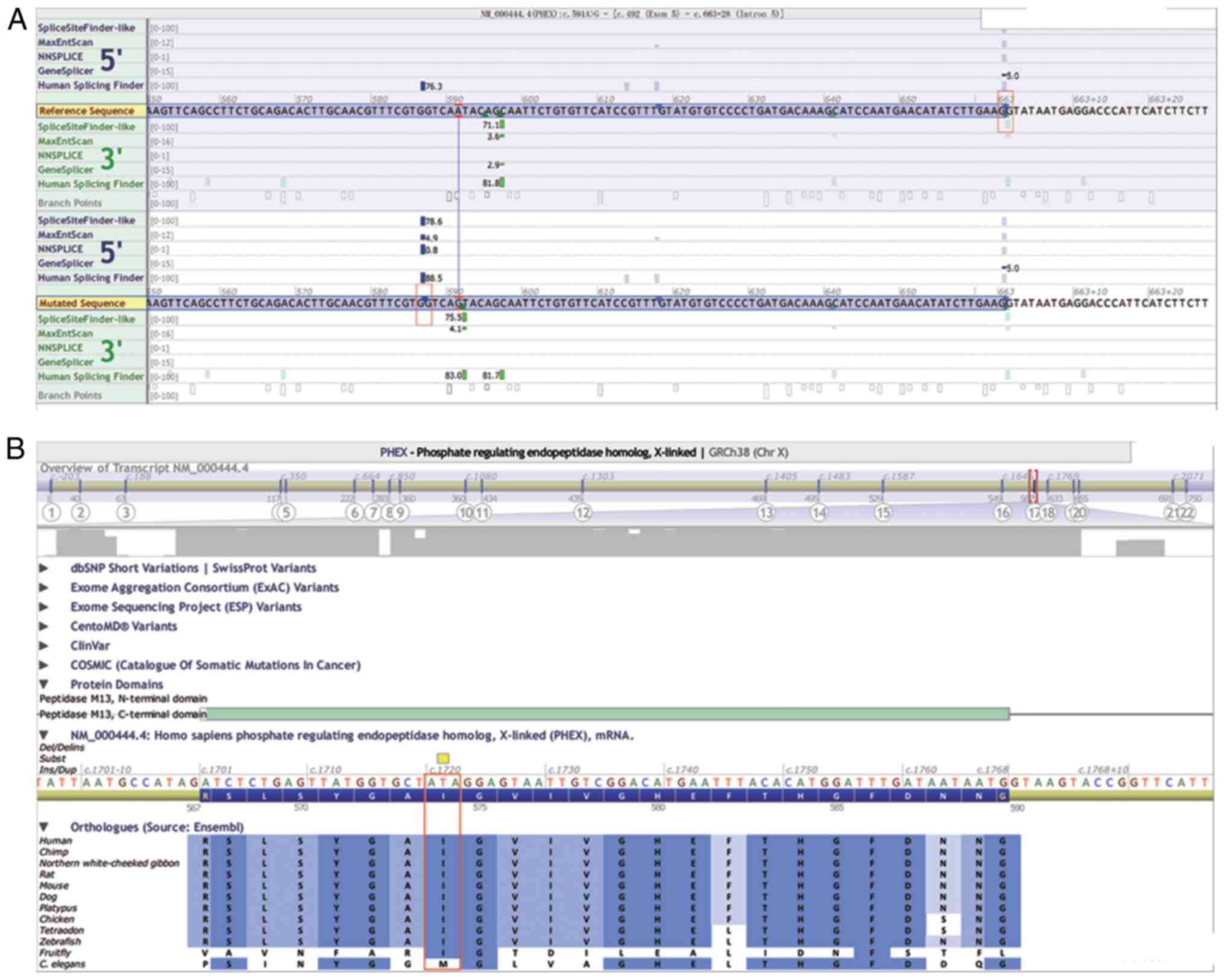
Theoretically, the c.591A>G variant in Family 2,
which also encodes for the 197th glutamine residue, did not lead to
a change in its amino acid sequence. However, based on the
prediction of Alamut Interactive Biosoftware, this synonymous
variant resulted in an abnormal splice donor site between c.586 and
c.587 in the middle of exon 5, with the remainder of the sequences
in the 3′ site of exon 5 being cut off during mRNA splicing
activity (Fig. 4A). The Ile in
this variant site is highly conserved among 12 different species,
suggesting its potential importance to protein function (Fig. 4B).
Verification of abnormal splice donor
site
To determine the effect of the suspected splicing of
variant c.591A>G in PHEX gene exon 5 in mRNA, RT-PCR was
performed using peripheral blood total RNA for all affected family
members from Family 2. The cDNA of the PHEX gene was
amplified using the general PCR method, followed by direct
sequencing. The sequencing results revealed a 77-bp deletion
between c.586 and c.662 of PHEX cDNA in the patients; it
occurred in the terminal region (3′ site) of exon 5, indicating an
abnormal splicing donor site existing in the middle of exon 5
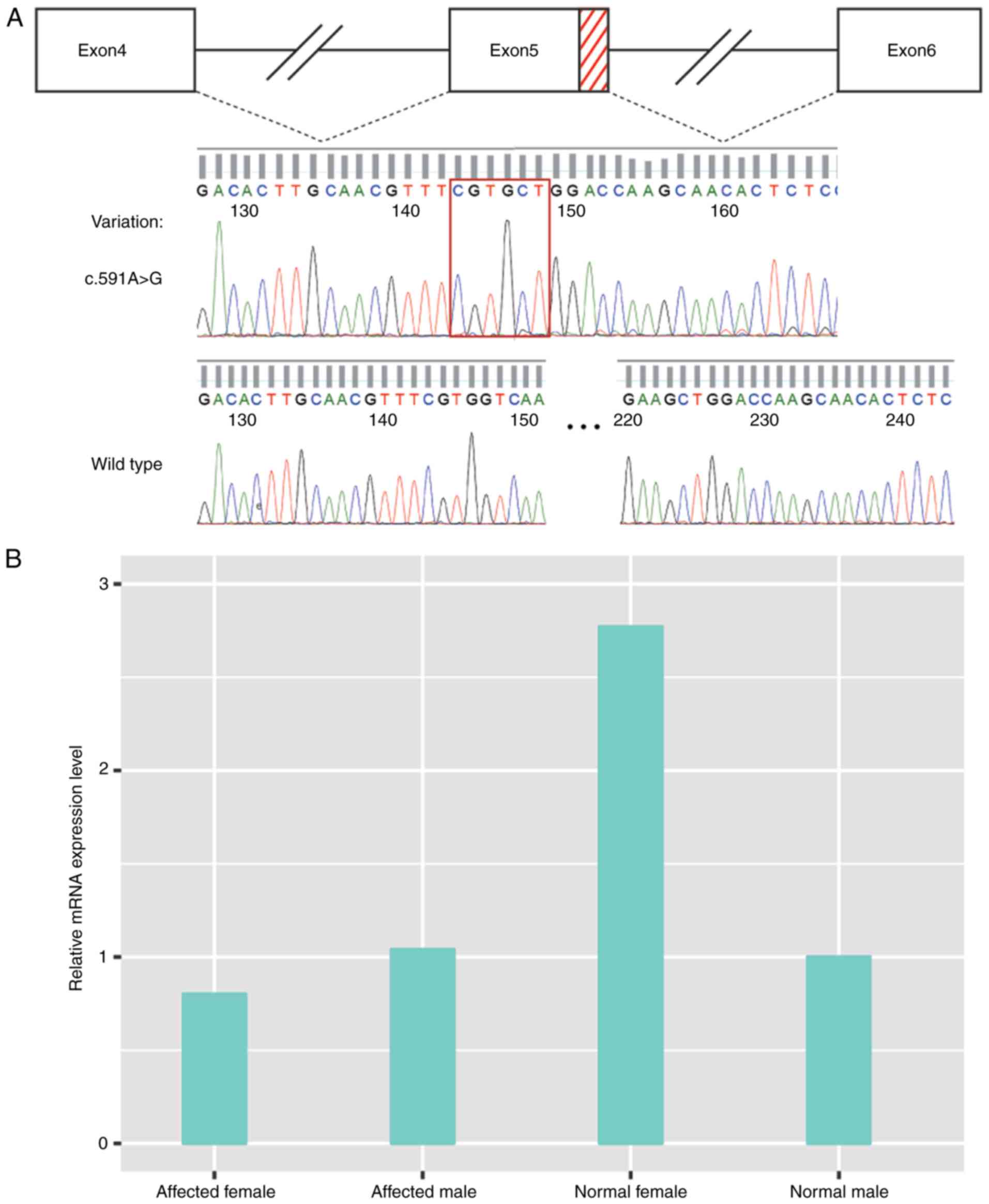
(Fig. 5A).
Analysis of relative gene expression of
PHEX
To determine the change in the gene expression of
PHEX in patients with the c.591A>G splicing variant in
Family 2, the present study performed semi-quantitative assessment
of mRNA derived from the PHEX gene of the proband and his
mother through RT-qPCR. The PHEX gene was located on
chromosome X, which causes the differences in expression levels in
females and males; therefore, a normal female and normal male were
used as controls when assessing the respective gene expression in
the affected male (proband) and affected female (his mother). As
shown in Fig. 5, the average
2−∆∆Cq of the proband/normal male and proband/normal
female was 1.05 and 0.44, respectively, and that of the normal
male/normal female was 0.36, suggesting almost the same PHEX
gene expression level in the proband as in normal male. However,
the average 2−∆∆Cq of the proband's mother/normal female
was 0.43 and that of the proband's mother/normal male was 0.78,
whereas the value for the normal female/normal male was 2.77; this
indicated that the gene expression of PHEX was downregulated
in the proband's mother, compared with that in the normal female.
The value was even lower, compared with that of a normal male, who
has only one copy of chromosome X. The results of the relative
analysis of the gene expression of PHEX are shown in
Fig. 5B.
Prenatal diagnosis
Two fetuses, whose mothers were genetically
diagnosed with XLHR, received a prenatal diagnosis of the specific
PHEX gene variant in their own family. Their DNA was
acquired from the backup amniotic fluid and analyzed using direct
sequencing. For the fetus (IV1) from Family 1, this
sequencing map showed homozygous peaks (T) in the familial variant
site (c.1721T>A), indicating that the fetus was unaffected,
which was then confirmed by physical and laboratorial examinations
post-delivery. For the fetus (IV2) from Family 2,
heterozygous peaks (A/G) were observed in its familial variant site
(c.591A>G), indicating that it was a female fetus and likely to
be affected with XLHR.
Discussion
The present study reported on two unrelated families
with XLHR, and identified one point variant of the PHEX gene
in each family. Several other types of hypophosphatemic rickets
have also been reported, including the autosomal dominant type
(OMIM 193100) caused by the abnormal fibroblast growth factor 23
gene (20), and autosomal
recessive HR (OMIM 241520) caused by either the ectonucleotide
pyrophosphatase/phosphodiesterase-1 gene or dentin matrix protein 1
gene (21–24). However, based on the clinical
symptoms, biochemical findings and modes of inheritance appearing
in the family, the present study focused on the PHEX gene,
which is the only causative gene for XLHR (25). In Family 1, a novel variant
NM_000444.5: c.1721T>A involved an the amino acid substitution
of p.Ile574Lys was identified. Due to their composition, polarity
and volume, the moderate physicochemical difference between Ile and
Lys is a Grantham distance 102 (26), which exceeded the average Grantham
distance (92.06) for disease-associated variants (27). Considering the highly conserved
amino acid Ile in this variation site among 12 species, although no
current data is available concerning variant c.1721T>A, it is
likely to be pathogenic. The healthy I2 member of this
family had four children; two of whom were affected, including one
male (II1) and one female (II6). According to
the results of the PCR analysis (data not shown), I2 was
the biological mother of the two affected individuals. Similarly, a
father with a somatic mosaic for the PHEX variant has been
reported, leaving one of his daughters affected (28); indicating the possibility of the
gonadal mosaic for her PHEX gene variant.
The present study identified a splice-site variant
(NM_000444.5: c.591A>G) in Family 2 with eight affected
individuals of both genders. Through direct sequencing of cDNA, an
abnormal splicing donor site was identified, which was 5 bp before
the variant site; therefore, the aberrant mRNA failed to be
translated into an intact protein due to a deletion and an advanced
termination codon (TGA) at the 211th codon, which may impair the
physiological function of the protein. Therefore, the variant
c.591A>G is likely to be responsible for this disease in all
members of Family 2. In the present study, it was found that the
gene expression level of PHEX in the proband was similar to
that of the normal male from the same family, almost half of that
of the normal female. However, the expression level in the
proband's mother, an affected female, was lower than that of the
normal female, and even lower than that of the normal male. A
similar expression level of PHEX was observed in another
female patient (II2) from the same family. However, the
general gene expression level of PHEX in female patients was
not available due to the lack of RNA samples of female patients
from the same family. The difference in gender may have led to the
differences in expression levels of PHEX in the proband and
his mother, which resulted from different splicing patterns; an
alternative explanation is that it is associated with individual
differences, which may explain the phenotypic diversity among
patients (29,30). Further experiments concerning gene
expression regulation may shed light on these results; however, it
is difficult to draw clear conclusions from the present study
alone.
At present, 75 splicing variants of the PHEX
gene have been identified, accounting for 14.70% of the total
variants (HGMD professional 2016.4 release), however, none of these
occur in exons. Each mRNA is derived from pre-mRNA through a
precise splicing reaction, which is one of the pivotal components
of gene expression, involving a multi-component machine known as
the spliceosome (30,31). The 5′ splice site (5′ss), the 3′
splice site (3′ss), and the branch point sequence constitute the
three core splicing signals, which are essential to the splicing
reaction (32,33). It is known that >80% of all
3′ss variants occur in the invariant AG sites of 3′ss ends
(34). Therefore, the pre-mRNA of
the PHEX gene in patients contained a c.586gtggtcagta
sequence in the middle of exon 5, resembling a 3′ss motif, which
can be recognized by the spliceosome, permitting aberrant splicing
in exon 5.
XLHR is a lifelong metabolic disease, and patients
begin to suffer from renal phosphate wasting at birth. To correct
rickets/osteomalacia, radiographic abnormalities and skeletal
deformities, treatments for children are required at the onset of
diagnosis, continuing until long bone growth is complete (35). For affected parents with a family
history of XLHR, prenatal diagnosis for pathogenic variants of the
PHEX gene is important; doing so allows pediatricians to
obtain the individual's genetic information, which is valuable for
providing an accurate diagnosis and devising a treatment plan. In
the present study, two pregnant women (18 weeks gestation) with
XLHR were included to obtain a prenatal diagnosis for their babies.
The DNA of each fetus was obtained from backup amniotic fluid and
the specific PHEX gene variants were assessed. The woman
from Family 2, who delivered an affected boy, conceived a baby with
the same PHEX gene variant as her own; the woman from Family
1 carried a baby without the PHEX gene variant. The two
babies had normal karyotypes (data not shown). Unlike the affected
older brother, who received treatments until 4 years of age
following diagnosis, the affected baby in Family 2 obtained
sufficient medical care according to physical examinations and
laboratory tests based on early genetic tests. One aim of prenatal
diagnosis is to assist parents in adequately preparing for the
affected baby, including financial support and psychological
preparation; parents can turn to pediatricians for early treatments
as soon as possible, which can be beneficial in terms of reducing
disorders, including growth retardation or rickets.
The baby from Family 1 was confirmed to be a healthy
girl in the follow-up visit 1 month following birth; however, the
fetus from Family 2 had not yet been born, therefore, continued
follow-up was planned for the baby from Family 2 following
delivery. The follow-up visits for the babies planned to last for 3
years.
In conclusion, the present study reported on two
unrelated pedigrees with XLHR and performed genetic analysis of the
PHEX gene in the two families. Through direct sequencing of
the PHEX gene, one missense variant (NM_000444.5:
c.1721T>A) was identified in exon 17 in Family 1, and a splicing
variant (NM_000444.5: c.591A>G) was identified in exon 5 in
Family 2. The results of RT-PCR analysis verified the aberrant
splicing site caused by variant c.591A>G, which resulted in a 77
bp loss in the 3′ site region of exon 5 on splicing. In the female
patient, the c.591A>G variant led to downregulation of the gene
expression of PHEX >2-fold, compared with that in the
healthy female. However, this fold change was not observed in the
male patient, who exhibited a similar gene expression level of
PHEX to that of the normal male. Based on the evidence
concerning the genotype-phenotype correlations between the specific
variant and XLHR in each family, it was possible to provide a
prenatal diagnosis for the fetuses and provide medical suggestions
to the affected families.
Abbreviations:
|
XLHR
|
X-linked hypophosphatemic rickets
|
Acknowledgments
This study was supported by a research grant from
the National Natural Science Foundation of China (grant no.
81270060).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Pavone V, Testa G, Gioitta Iachino S,
Evola FR, Avondo S and Sessa G: Hypophosphatemic rickets: Etiology,
clinical features and treatment. Eur J Orthop Surg Traumatol.
25:221–226. 2015. View Article : Google Scholar
|
|
2
|
Cho HY, Lee BH, Kang JH, Ha IS, Cheong HI
and Choi Y: A clinical and molecular genetic study of
hypophosphatemic rickets in children. Pediatr Res. 58:329–333.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chandran M, Chng CL, Zhao Y, Bee YM, Phua
LY and Clarke BL: Novel PHEX gene mutation associated with X linked
hypophosphatemic rickets. Nephron Physiol. 116:17–218. 2010.
View Article : Google Scholar
|
|
4
|
Tenenhouse HS: X-linked hypophosphataemia:
A homologous disorder in humans and mice. Nephrol Dial Transplant.
14:333–341. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Beck-Nielsen SS, Brock-Jacobsen B, Gram J,
Brixen K and Jensen TK: Incidence and prevalence of nutritional and
hereditary rickets in southern Denmark. Eur J Endocrinol.
160:491–497. 2009. View Article : Google Scholar
|
|
6
|
Albright F, Butler AM and Bloomberg E:
Rickets resistant to vitamin d therapy. Am J Dis Child. 54:529–547.
1937.
|
|
7
|
Stickler GB and Morgenstern BZ:
Hypophosphataemic rickets: Final height and clinical symptoms in
adults. Lancet. 2:902–905. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
A gene (PEX) with homologies to
endopeptidases is mutated in patients with X-linked
hypophosphatemic rickets. The HYP Consortium. Nat Genet.
11:130–136. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Filisetti D, Ostermann G, von Bredow M,
Strom T, Filler G, Ehrich J, Pannetier S, Garnier JM, Rowe P,
Francis F, et al: Non-random distribution of mutations in the PHEX
gene, and under-detected missense mutations at non-conserved
residues. Eur J Hum Genet. 7:615–619. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo R and Quarles LD: Cloning and
sequencing of human PEX from a bone cDNA library: Evidence for its
developmental stage-specific regulation in osteoblasts. J Bone
Miner Res. 12:1009–1017. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lipman ML, Panda D, Bennett HP, Henderson
JE, Shane E, Shen Y, Goltzman D and Karaplis AC: Cloning of human
PEX cDNA. Expression, subcellular localization, and endopeptidase
activity. J Biol Chem. 273:13729–13737. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Du L, Desbarats M, Viel J, Glorieux FH,
Cawthorn C and Ecarot B: cDNA cloning of the murine Pex gene
implicated in X-linked hypophosphatemia and evidence for expression
in bone. Genomics. 36:22–28. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu S, Tang W, Fang J, Ren J, Li H, Xiao Z
and Quarles LD: Novel regulators of Fgf23 expression and
mineralization in Hyp bone. Mol Endocrinol. 23:1505–1518. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Onishi T, Umemura S, Shintani S and
Ooshima T: Phex mutation causes overexpression of FGF23 in teeth.
Arch Oral Biol. 53:99–104. 2008. View Article : Google Scholar
|
|
15
|
Farrow EG and White KE: Recent advances in
renal phosphate handling. Nat Rev Nephrol. 6:207–217. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martin A, Liu S, David V, Li H, Karydis A,
Feng JQ and Quarles LD: Bone proteins PHEX and DMP1 regulate
fibro-blastic growth factor Fgf23 expression in osteocytes through
a common pathway involving FGF receptor (FGFR) signaling. FASEB J.
25:2551–2562. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Mathe E, Olivier M, Kato S, Ishioka C,
Hainaut P and Tavtigian SV: Computational approaches for predicting
the biological effect of p53 missense mutations: A comparison of
three sequence analysis based methods. Nucleic Acids Res.
34:1317–1325. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tavtigian SV, Deffenbaugh AM, Yin L,
Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A and Thomas
A: Comprehensive statistical study of 452 BRCA1 missense
substitutions with classification of eight recurrent substitutions
as neutral. J Med Genet. 43:295–305. 2006. View Article : Google Scholar
|
|
20
|
ADHR Consortium: Autosomal dominant
hypophosphataemic rickets is associated with mutations in FGF23.
Nat Genet. 26:345–348. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lorenz-Depiereux B, Schnabel D, Tiosano D,
Häusler G and Strom TM: Loss-of-function ENPP1 mutations cause both
generalized arterial calcification of infancy and
autosomal-recessive hypophosphatemic rickets. Am J Hum Genet.
86:267–272. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan
B, Yu X, Rauch F, Davis SI, Zhang S, et al: Loss of DMP1 causes
rickets and osteomalacia and identifies a role for osteocytes in
mineral metabolism. Nat Genet. 38:1310–1315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lorenz-Depiereux B, Bastepe M, Benet-Pagès
A, Amyere M, Wagenstaller J, Müller-Barth U, Badenhoop K, Kaiser
SM, Rittmaster RS, Shlossberg AH, et al: DMP1 mutations in
autosomal recessive hypophosphatemia implicate a bone matrix
protein in the regulation of phosphate homeostasis. Nat Genet.
38:1248–1250. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Beck-Nielsen SS, Brixen K, Gram J and
Brusgaard K: Mutational analysis of PHEX, FGF23, DMP1, SLC34A3 and
CLCN5 in patients with hypophosphatemic rickets. J Hum Genet.
57:453–458. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Econs MJ and Francis F: Positional cloning
of the PEX gene: New insights into the pathophysiology of X-linked
hypophosphatemic rickets. Am J Physiol. 273:F489–F498.
1997.PubMed/NCBI
|
|
26
|
Grantham R: Amino acid difference formula
to help explain protein evolution. Science. 185:862–864. 1974.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Subramanian S and Kumar S: Evolutionary
anatomies of positions and types of disease-associated and neutral
amino acid mutations in the human genome. BMC Genomics. 7:3062006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goji K, Ozaki K, Sadewa AH, Nishio H and
Matsuo M: Somatic and germline mosaicism for a mutation of the PHEX
gene can lead to genetic transmission of X-linked hypophosphatemic
rickets that mimics an autosomal dominant trait. J Clin Endocrinol
Metab. 91:365–370. 2006. View Article : Google Scholar
|
|
29
|
Wang ET, Sandberg R, Luo S, Khrebtukova I,
Zhang L, Mayr C, Kingsmore SF, Schroth GP and Burge CB: Alternative
isoform regulation in human tissue transcriptomes. Nature.
456:470–476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ward AJ and Cooper TA: The pathobiology of
splicing. J Pathol. 220:152–163. 2010.
|
|
31
|
Wahl MC, Will CL and Lührmann R: The
spliceosome: Design principles of a dynamic RNP machine. Cell.
136:701–718. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Z and Burge CB: Splicing regulation:
From a parts list of regulatory elements to an integrated splicing
code. RNA. 14:802–813. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yeo G and Burge CB: Maximum entropy
modeling of short sequence motifs with applications to RNA splicing
signals. J Comput Biol. 11:377–394. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Krawczak M, Reiss J and Cooper DN: The
mutational spectrum of single base-pair substitutions in mRNA
splice junctions of human genes: Causes and consequences. Hum
Genet. 90:41–54. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carpenter TO, Imel EA, Holm IA, Jan de
Beur SM and Insogna KL: A clinician's guide to X-linked
hypophosphatemia. J Bone Miner Res. 26:1381–1388. 2011. View Article : Google Scholar : PubMed/NCBI
|