Introduction
Prions are infectious agents without detectable
nucleic acids, which cause prion diseases or transmissible
spongiform encephalopathies (TSEs) in humans and various animal
species (1). The conversion from
a normal, endogenous membrane glycoprotein [cellular prion protein
(PrPC)] to a pathological, conformationally altered
isoform [scrapie prion protein (PrPSc)] is the crux of
prion biology. The most notable alteration in the secondary
structure during the conversion from PrPC to
PrPSc is a decrease in its α-helix content and a marked
increase in β-sheet content. Besides its infectivity in homologous,
and some heterologous species, PrPSc displays distinct
biochemical characteristics, including insoluble, easily formed
aggregates or fibrils, and partial resistance to proteolytic
digestion (2).
PrP is a conserved protein among mammals.
Full-length PrP is 253-254 amino acids (aa) long, with a signal
peptide (1-22 aa) in the N-terminus and a
glycosylphosphatidylinositol anchor (232-254 aa) in the C-terminus.
In the N-terminus, there is an octarepeat region that consists of
five octapeptides, which exert various biological activities,
including binding with copper, manganese and zinc metal ions,
promoting PrP internalization and interacting with other proteins,
such as glial fibrillary acidic protein and tubulin (3). Naturally occurring insertions or
deletions of octapeptide repeats in this region are associated with
the human prion disease, genetic or familial Creutzfeldt-Jacob
disease. In addition, insertions or deletions of octapeptide
repeats have been confirmed to induce similar pathogenicity in
rodent animals (4,5). PrP contains two N-linked
glycosylation sites at asparagine (Asn)181 and Asn197, which
contribute to the formation of three PrP isoforms: Di- mono- and
unglycosylated PrP, in host cells (6). N-linked glycan helps the folding
process of newly synthesized protein, whereas removal of PrP
glycosylation provokes cell apoptosis (7). Transgenic mice bearing a
glycosylation deficiency at either of the two glycan attachment
sites of PrPC exhibit increased sensitivity to bovine
spongiform encephalopathy and scrapie. In addition, a higher ratio
of unglycosylated PrP has been repeatedly detected in the brain
tissues of humans and animals with TSEs (8). These findings indicate the
importance of the maintenance of aa sequences and
post-translational modification of PrP protein.
In the C-terminal fragment of PrP, there are two
cysteines (Cys) at aa 179 and 214, which are critical for forming
intra- and/or intermolecular disulfide bridges (9). Formation of a disulfide bond serves
an important role in protein folding, stabilizing protein
conformational structure and biological function (10,11). In the present study, three
prokaryotic human PrP constructs: Wild-type PrP (PG5), mutant PrP
with insertion of seven extra octarepeats (PG12) and mutant PrP
with deletion of all five octarepeats (PG0), underwent redox in
vitro, in order to form a disulfide bond. Subsequently, the
biochemical features of these PrP proteins were evaluated.
Materials and methods
Plasmids
The generation of the following recombinant
prokaryotic protein-expressing plasmids: pQE30-huPrP23-231
containing five octapeptide repeats (wild-type PrP, PG5),
pQE30-huPG12 containing 12 octapeptide repeats (PG12) and
pQE30-huPG0 with deletion of all five octapeptide repeats (PG0) was
described previously (12).
Protein purification
The bacterially expressed recombinant polyhistidine
(His)-tagged PrPs were expressed and purified according to
previously described protocols (13). Briefly, the expressed PrPs were
recovered from urea-solubilized bacterial lysate following
sonication by immobilized nickel-based affinity chromatography. The
final protein product was >95% pure and was concentrated to 0.6
mg/ml with 10 mM NaAC. Proteins were aliquoted and stored at −80°C
for further analyses. Protein concentration of the dissolved
product was determined in triplicate using the bicinchoninic acid
(BCA) protein assay kit (Thermo Fisher Scientific, Inc., Waltham,
MA, USA) according to the manufacturer's protocol.
Redox of recombinant PrPs
Reduction of the purified PrPs was conducted based
on a previously described protocol, with modifications (14). The various PrPs (PG0, PG5 and
PG12) were initially incubated in redox buffer A (50 mM Tris-HCl,
100 mM DTT, 2.5 M GdnHCl, 3 M NaCl, pH 8.0) at 37°C for 16 h, in
order to produce thiol groups and free radicals. Subsequently, the
proteins were incubated with redox buffer B (150 mM
ICH2CONH2, 50 mM DTT, 500 mM Tris-HCl, pH
8.5) at 37°C for 6 h, in order to produce alkylation of thiol
groups to protect the free thiol groups. Proteins were then
carefully dialyzed in dialysis buffer A (10 mM NaAc, 50 mM NaCl, pH
4.0) and buffer B (10 mM NaAc, pH 5.0), respectively, to form
disulfide bonds via oxidation of thiol groups.
Sedimentation experiments
Samples were centrifuged for 30 min at 20,000 × g at
4°C. The supernatants and pellets were separately collected and the
protein pellets were resuspended in a volume equal to the volume of
the supernatants. The fractions were separated by 12% SDS-PAGE and
were then analyzed by Commassie blue staining.
Circular dichroism (CD) analysis
Various samples of redox recombinant PrPs were
dissolved in 1mM MES solution (pH 5.5) at 10μM final
concentration. Three samples from each recombinant PrP solution
were independently evaluated by CD analysis. CD spectra were
recorded in a 0.1 cm path length quartz cell at room temperature
under constant nitrogen flush using a Jasco J720 spectropolarimeter
(JASCO, Easton, MD, USA). Subsequently, three spectra were
accumulated and the appropriate blanks were subtracted. The values
were expressed as molar ellipticity (θ). Estimates of percentage
secondary structure were obtained using K2D analysis programs.
Thioflavin T (ThT) fluorescence
PrPs that did or did not undergo redox were
incubated in assembly buffer containing 50 mM Tris-Cl (pH 7.4), 150
mM KCl and 10 mM ATP at 37°C for various durations. For ThT assays,
10 µl (0.6 µg) of each preparation was mixed with ThT
solution (180 µl) containing 50 mM glycine-OH (pH 8.5) and 5
µM ThT at room temperature for 1 min (1 mM stock solution in
water; Sigma T3516; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
The fluorescence of each sample was measured using a
spectropolarimeter (F-4500; Hitachi, Ltd., Tokyo, Japan) at 485 nm
using an excitation wavelength of 440 nm.
Proteinase K (PK) resistance assay
Recombinant PrPs that did or did not undergo redox
were assessed for PK sensitivity following treatment with 6.25,
12.5 and 25 µg/ml PK (Roche Diagnostics Gmbh, Mannheim,
Germany) at 37°C for 20 min or 1 h. The reaction was terminated by
boiling for 5 min with SDS-PAGE sample loading buffer.
Western blot analysis
Protein samples were separated by 12% SDS-PAGE and
were electrotransferred onto nitrocellulose (NC) membranes. All
protein extracts were quantified using BCA reagent (Merck,
Kenilworth, NJ, USA) prior to resolution with SDS-PAGE and
electrotransfer to NC membranes (Whatman; GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA). Membranes were blocked with 5%
(w/v) bovine serum albumin in 1X Tris-buffered saline containing
0.1% Tween-20 (TBST) at room temperature for 2 h and were then
probed with 1:5,000-diluted PrP specific monoclonal antibody 3F4
(MAB1562; EMD Millipore, Billerica, MA, USA) at 4°C over-night.
After washing with TBST, membranes were subsequently incubated with
1:5,000-diluted goat anti-mouse secondary antibodies (cat. no.
32723; Thermo Fisher Scientific, Inc.) and reactive signals were
visualized using an enhanced chemiluminescence kit (PE, Waltham,
MA, USA). Images were captured using a ChemiDoc™ XRS+ Imager
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Densitometric
analysis of western blot analyses was conducted using ImageJ
software version 1.44 (National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
All of the experiments were performed at least three
times, with consistent results. Statistical analysis was performed
using GraphPad Prism6 Software (GraphPad Software, Inc., La Jolla,
CA, USA). All data values are presented as the means ± standard
deviation. P-values for differences between two groups were
determined by two-tailed t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
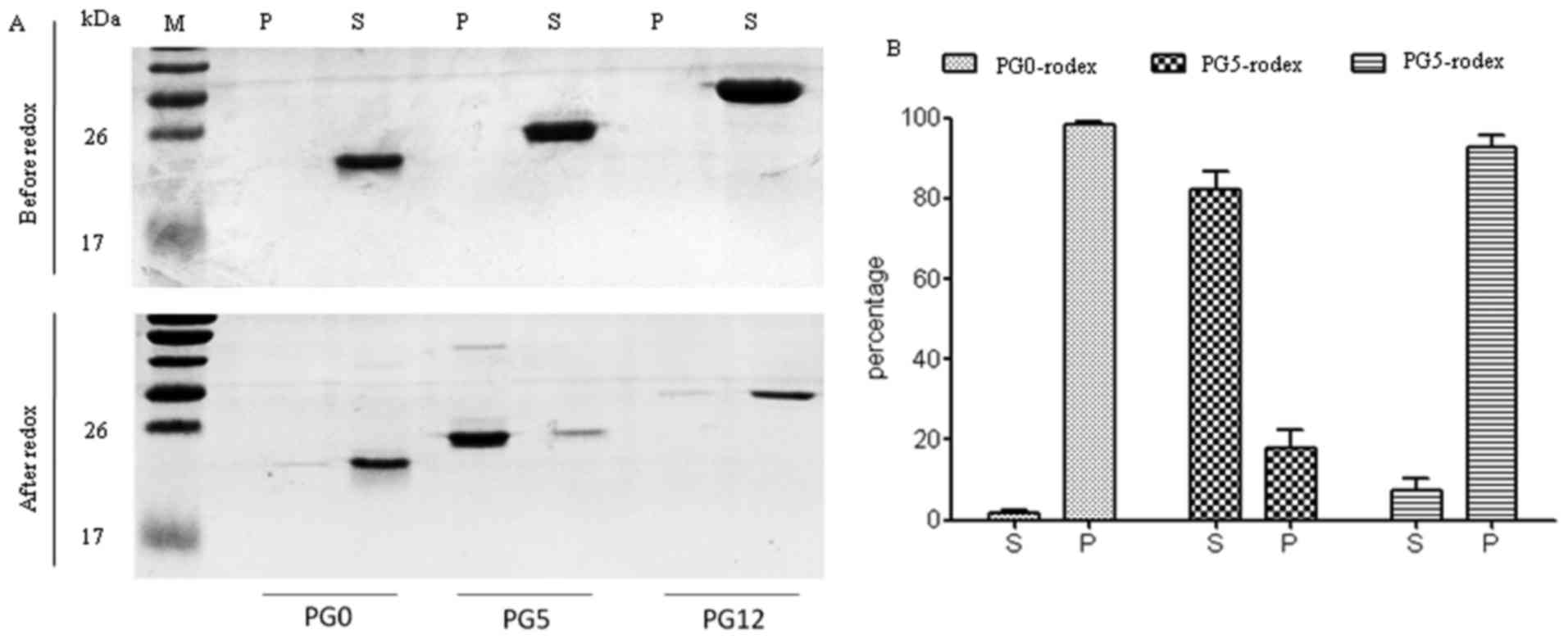
Effects of redox on the sedimentation
features of three recombinant human PrPs
The expression of recombinant human His-tagged PrPs
(PG0, PG5 and PG12) was induced in Escherichia coli strain
M15. SDS-PAGE of the purified products revealed that PG0 was
20-kDa, PG5 was 25-kDa and PG12 was 30-kDa; these proteins were
specifically recognized by the PrP 3F4 monoclonal antibody by
western blotting (data not shown). To determine the possible
effects of redox on the sedimentation characteristics of these
three PrP constructs, PG0, PG5 and PG12 preparations were
centrifuged before and after redox. SDS-PAGE revealed that almost
all PrPs were present in the supernatant fractions of all three PrP
constructs prior to redox (Fig.
1A, upper panel). Notably, following redox, the majority of PG5
transferred to the pellet fraction, whereas PG0 and PG12 mutants
remained in the supernatant fraction (Fig. 1A, lower panel). Densitometric
analysis of the gray values of PrP protein bands demonstrated that
~80% PG5 was present in the pellet following redox (Fig. 1B).
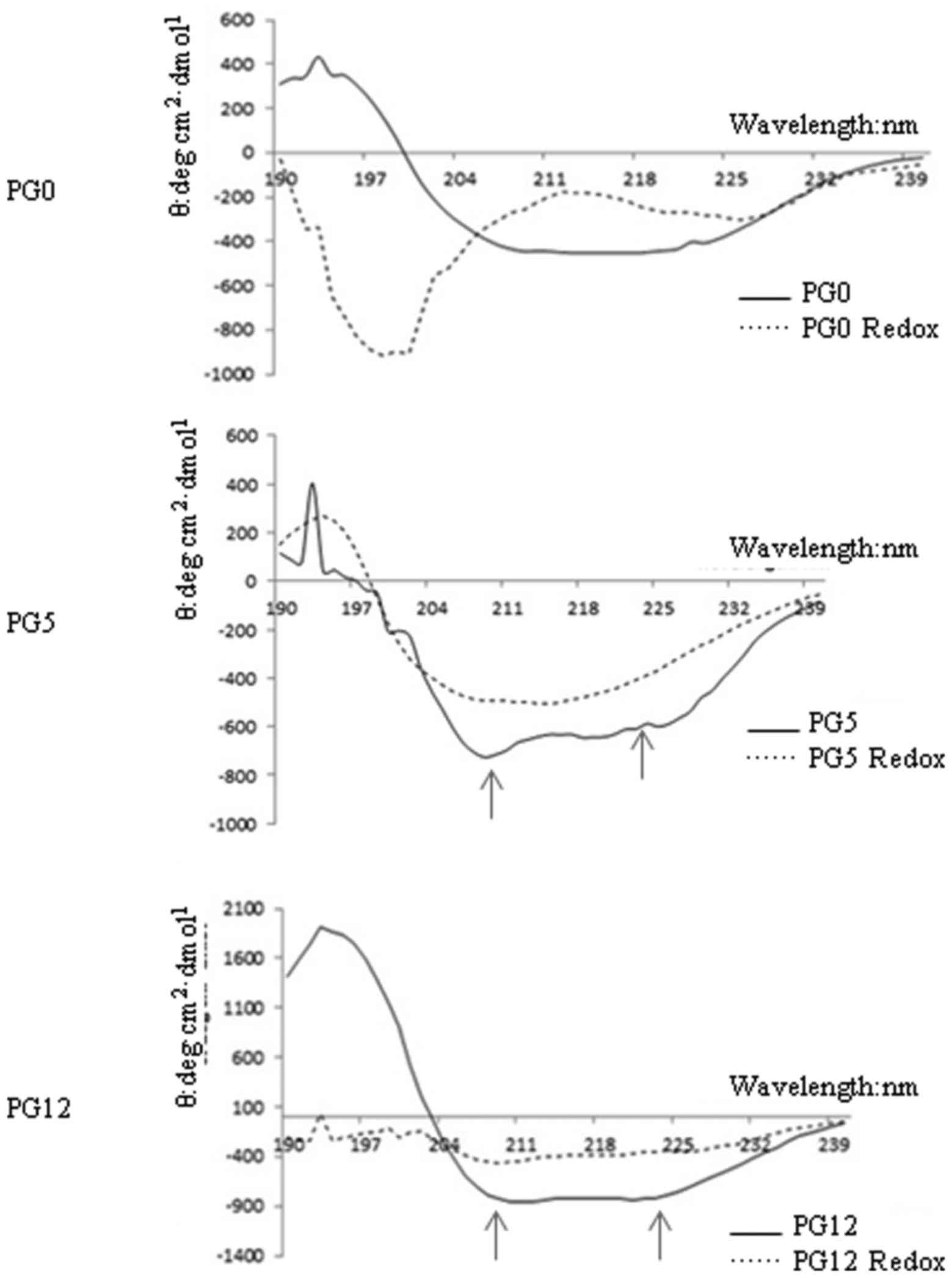
Effects of redox on the secondary
structure of the three recombinant human PrPs
To determine the effects of redox on PrP structure,
the secondary structures of PrP proteins were examined before and
after redox by far-ultraviolet (UV) CD spectra. As shown in
Fig. 2, redox-untreated PG5 and
PG12 displayed two maximum UV absorbance peaks (at ~210 and 222 nm)
representing α-helix predominance, whereas redox-untreated PG0
exhibited a UV absorbance peak at ~216 nm, indicating β-sheet
enrichment. Following redox, the two UV absorbance peaks in PG5 and
PG12 disappeared. The contents of the main structures of the three
PrP constructs prior to and following redox were evaluated and
summarized in Table I. As
expected, the α-helix content of the three PrP constructs was
markedly reduced by redox, particularly in PG5 and PG0. In
addition, the β-sheet content was markedly increased in
redox-treated PG5, whereas random-coil content was predominant in
redox-treated PG0. In redox-treated PG12, there was a marked
increase in random-coil content; however, the increase in β-sheet
content was limited (Table
I).
 | Table Iα-helix and β-sheet contents of three
human prion protein constructs prior to and after redox. |
Table I
α-helix and β-sheet contents of three
human prion protein constructs prior to and after redox.
| Structure | PG0 | PG0 redox | PG5 | PG5 redox | PG12 | PG12 redox |
|---|
| α-helix | 17.1 | 3.7 | 24.9 | 1.9 | 55.4 | 22.0 |
| β-sheet | 53.9 | 22.7 | 36.2 | 52.6 | 33.8 | 38.2 |
| Random-coil | 29.0 | 73.6 | 38.9 | 45.5 | 10.9 | 39.9 |
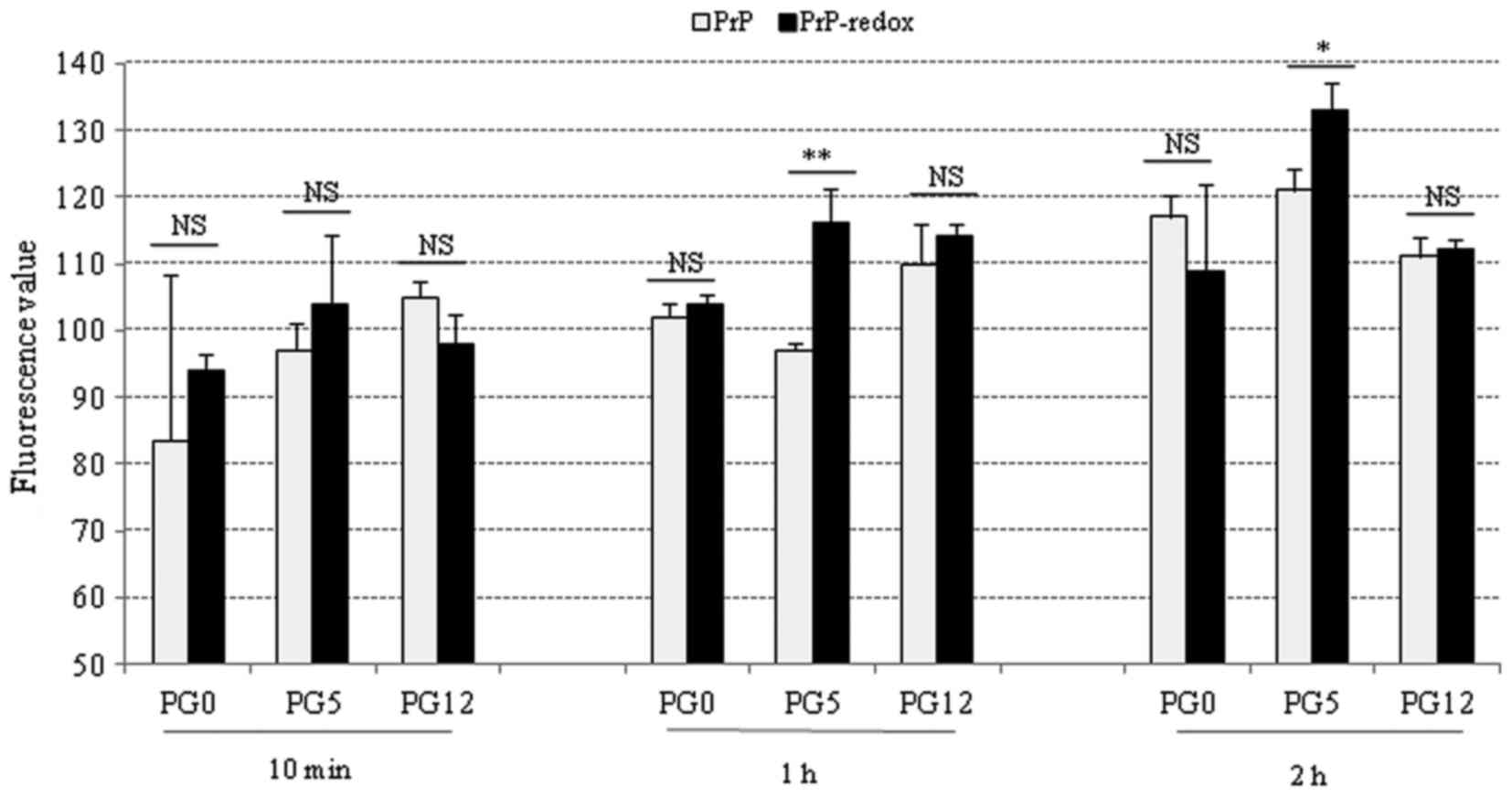
Effects of redox on the fibril formation
of the three recombinant human PrPs
To determine the potential effects of redox on PrP
fibril formation, various PrP proteins were subjected to ThT assays
prior to and following redox. Following incubation in assembling
buffer for 10 min, or 1 and 2 h, the fluorescence value of each
sample was measured. Generally, the fluorescence values of all
tested PrP samples were increased along with prolonging the
incubation times. In the 10-min preparations, redox-treated PG0 and
PG5 exhibited slightly higher fluorescence values compared with in
the untreated PrPs; however, none of these results were significant
(Fig. 3, left panel).
Redox-treated PG5 exhibited a significantly higher fluorescence
value compared with in untreated PG5 at 1 and 2 h, whereas no
significance was detected in PG0 and PG12 PrPs between the
redox-treated and untreated groups at 1 and 2 h(Fig. 3, middle and right panels). These
results indicated that redox may increase fibril formation of
wild-type PrP, but may not affect the fibril formation of mutated
PrPs with insertion or deletion of octarepeats.
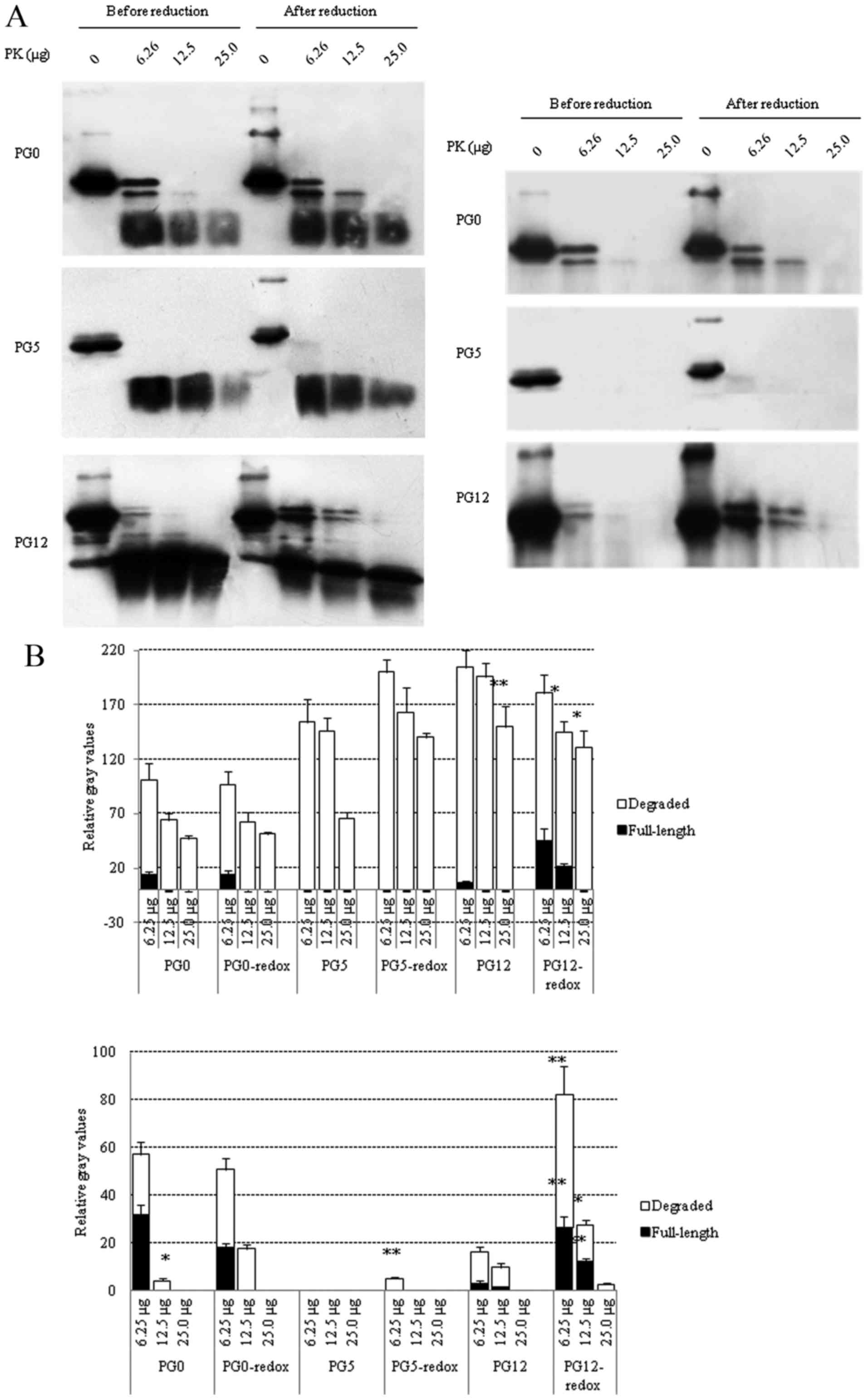
Effects of redox on PK resistance of the
three recombinant human PrPs
To evaluate the effects of redox on PK resistance,
three PrP constructs were digested with various doses of PK before
and after redox, and the reactions were terminated at 20 min or 1 h
post-digestion. All samples were subjected to PrP-specific western
blot analyses and the gray values were normalized to those of the
PrP without PK (0 µg). In the preparations digested for 20
min (Fig. 4A, left panel),
residual full-length PrP signals were observed in redox-treated and
untreated PG0 at the lowest PK dosage (6.25 µg),
redox-treated PG12 (6.25 and 12.5 µg PK) and redox-untreated
PG12 (6.25 µg PK), but not in PG5 reactions. Analysis of the
degraded PrP in the blots revealed that redox-treated PG5 contained
more residual signals than untreated PG5. In the preparations
digested for 1 h (Fig. 4B), the
PrP signal almost disappeared in PG5 reactions, with the exception
of an extremely weak residual signal in redox-treated PG5 following
treatment with the lowest dose of PK (6.25 µg). Residual
full-length PrP was still detectable in PG0 reactions (6.25
µg PK) with or without redox; however, the degraded PrP
signals were reduced in untreated PG0 following treatment with 12.5
µg PK compared with in the redox-treated PG0. Notably, more
full-length PrP signals were observed in redox-treated PG12
compared with in untreated PG12 following treatment with 6.25
µg PK, and PrP signals were detectable in redox-treated PG12
only following treatment with 12.5 µg PK. These data
indicated that redox can increase PK resistance of wild-type and
mutated recombinant human PrPs. The PrP mutants with insertion or
deletion of octarepeats possessed stronger PK-resistant
activities.
Discussion
The formation of disulfide bridges is one of
numerous types of post-translational protein modification. Proteins
in which two cysteines have formed a disulfide bond are oxidized,
whereas proteins without disulfide bonds are reduced, or are known
to be in the thiol state (15). A
previous study verified that the conformational structures,
chemical characteristics and biological functions of various
proteins may be markedly altered during the transformation between
these two states (16). The
present study hypothesized that following redox, the three
recombinant human PrPs would exhibit reduced α-helix content and
increased PK resistance. The results of the present study
demonstrated that redox produced distinct effects on the different
PrP constructs. These findings indicated that the number of
octarepeats in PrP affects its biochemical features.
Under the experimental conditions, wild-type PrP was
revealed to be more sensitive to redox compared with the two PrP
mutants. In addition to the reduction in α-helix content and the
increase in PK resistance, oxidized wild-type PrP contained
increased β-sheet content, and easily formed aggregates and
fibrils. The biochemical phenotype of normal PrP can be influenced
by numerous small molecules, including metal ions (17). Exposure of recombinant human PrP
to Mn2+ increases β-sheet content and enhances the
formation of aggregates in vitro (18). Furthermore, saturation of
recombinant mouse PrP with Cu2+ has been reported to
efficiently enhance conversion to PK-resistant PrP in protein
misfolding cyclic amplification (19). Notably, in a previous study, the
biochemical features of oxidized PG5, formed via redox, share
similarity with Cu2+- and Mn2+-treated PrP,
even with pathological PrPSc (20). Therefore, it may be hypothesized
that, as the propagating substrate for prions, increased oxidation
of PrPC may benefit the conversion from PrPC
to PrPSc. Further evaluation of the redox state in the
microenvironment of the central nervous system during prion
infection may help to improve understanding of prion biology.
Although the crystal structure of pathological
PrPSc remains to be elucidated, it is well-known that
the PrPSc molecule is rich in β-sheet. Alterations to
the conformational structures, e.g., a reduction in β-sheet content
and dissociation of prion rods, may reduce, and even remove, prion
infectivity (21). It has been
suggested that changes to covalent bonds occur during conversion to
PrPSc, besides conformational changes (22). PrP has the ability to
self-aggregate and form oligomers, which can be observed in
vitro and in vivo (23). Increased amounts of the oligomeric
form of PrP can be achieved via redox on the thiol group within the
PrP peptide, thus highlighting the importance of disulfide bond
formation in this activity (24).
Furthermore, the reduction and alkylation of PrP in vitro
has been proposed to inhibit conversion to PrPSc. Using
the alkylating antitumor drug, mechlorethamine, prion replication
in vitro is efficiently inhibited (25), which may indicate that formation
of the PrP oligomer, and even PrPSc, depends on
formation of an intermolecular disulfide bond.
Maintenance of the correct number of octarepeats in
PrP is critical for protein functions. Insertion or deletion of
octarepeats in the PrP gene is directly associated with inherited
human prion diseases. Our previous study confirmed that the
molecular interaction of PrP with tubulin depends on octarepeats
(26). In addition, although the
binding activity of PrP to tubulin is closely associated with the
number of octarepeats, the regulation on microtubule polymerization
is also associated with the number of octarepeats; mutants which
exhibited strong inhibition on microtubule polymerization (27). The neuroprotective effects of PrP
are also associated with the number of octarepeats, both insertions
and deletions of octarepeats exert marked cytotoxic effects
(28). In the present study,
compared with wild-type PG5, PG0 and PG12 exhibited very limited
alterations in sedimentation and fibril formation, and increased
random-coil content following redox. However, whether the
octarepeat region affects the formation of a disulfide bond during
redox under these experimental conditions remains unclear. The
distinct outcomes detected among the various PrP constructs after
redox emphasized the importance of the correct number of
octarepeats within PrP with regards to its biological features.
Under the experimental conditions of the present
study, PG0 and PG12 maintained solubility after redox.
Coincidentally, the formation of fibrils in oxidized PG0 and PG12
was not increased; the exact reason for this is currently unknown.
CD spectra detected marked increases in the random-coil content of
redox-treated PG0 and PG12. Furthermore, β-sheet content was
reduced in redox-treated PG0 and exhibited little change in
redox-treated PG12. Those secondary structural alterations in PG0
and PG12 following redox may be associated with the detected
biochemical phenotypes.
In accordance with previous data (29), the PK resistance of PG0 and PG12
was much stronger than PG5 in the present study. After redox, the
PK resistance of oxidized PG5 and PG12 was markedly increased,
whereas that of oxidized PG0 was only slightly increased. PK
resistance of PrPSc is believed to be associated with an
increase in β-sheet content; therefore, the redox-induced increase
in β-sheet content and decrease in α-helix content in oxidized PG5
may be associated with its increased PK resistance. Conversely, the
increases in PK resistance of PG12 and PG0 seem to not be directly
associated with an increase in β-sheet content, since redox did not
induce a significant increase in β-sheet in PG12, and β-sheet
content was reduced in PG0 following redox. These findings
indicated that besides detectable alterations in the secondary
structure of PrP because of formation of disulfide bond, other
unknown conformational changes associated with an alteration in the
number of octarepeats may be involved in the appearance of PK
resistance in PrP mutants. This partly indicates the treatment
direction of genetic CJD.
Acknowledgments
The present study was supported by the Chinese
National Natural Science Foundation Grants (grant nos. 81630062 and
81572048), the China Mega-Project for Infectious Disease (grant
nos. 2011ZX10004-101 and 2012ZX10004215) and the SKLID Development
Grant (grant nos. 2015SKLID503 and 2016SKLID603).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Colby DW and Prusiner SB: Prions. Cold
Spring Harb Perspect Biol. 3:a0068332011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prusiner SB, Scott MR, DeArmond SJ and
Cohen FE: Prion protein biology. Cell. 93:337–348. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stellato F, Minicozzi V, Millhauser GL,
Pascucci M, Proux O, Rossi GC, Spevacek A and Morante S:
Copper-zinc cross-modulation in prion protein binding. Eur Biophys
J. 43:631–642. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paucar M, Xiang F, Moore R, Walker R,
Winnberg E and Svenningsson P: Genotype-phenotype analysis in
inherited prion disease with eight octapeptide repeat insertional
mutation. Prion. 7:501–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Beck JA, Mead S, Campbell TA, Dickinson A,
Wientjens DP, Croes EA, Van Duijn CM and Collinge J:
Two-octapeptide repeat deletion of prion protein associated with
rapidly progressive dementia. Neurology. 57:354–356. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wiseman FK, Cancellotti E, Piccardo P,
Iremonger K, Boyle A, Brown D, Ironside JW, Manson JC and Diack AB:
The glycosylation status of PrPC is a key factor in
determining transmissible spongiform encephalopathy transmission
between species. J Virol. 89:4738–4747. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang Y, Chen L, Pan HZ, Kou Y and Xu CM:
Glycosylation modification of human prion protein provokes
apoptosis in HeLa cells in vitro. BMB Rep. 42:331–337. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuczius T and Kelsch R: Effects of metal
binding on solubility and resistance of physiological prions depend
on tissues and glycotypes. J Cell Biochem. 114:2690–2698. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ning L, Guo J, Jin N, Liu H and Yao X: The
role of Cys179-Cys214 disulfide bond in the stability and folding
of prion protein: Insights from molecular dynamics simulations. J
Mol Model. 20:21062014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maiti NR and Surewicz WK: The role of
disulfide bridge in the folding and stability of the recombinant
human prion protein. J Biol Chem. 276:2427–2431. 2001. View Article : Google Scholar
|
|
11
|
Singh N, Singh A, Das D and Mohan ML:
Redox control of prion and disease pathogenesis. Antioxid Redox
Signal. 12:1271–1294. 2010. View Article : Google Scholar :
|
|
12
|
An R, Dong C, Lei Y, Han L, Li P, Chen J,
Wang G, Shi Q, Gao C, Jiang H, et al: PrP mutants with different
numbers of octarepeat sequences are more susceptible to the
oxidative stress. Sci China C Life Sci. 51:630–639. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang FP, Zhang J, Zhou W, Zhang BY, Hung
T and Dong XP: Expression of PrP(C) as HIS-fusion form in a
baculovirus system and conversion of expressed PrP-sen to PrP-res
in a cell-free system. Virus Res. 87:145–153. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee S and Eisenberg D: Seeded conversion
of recombinant prion protein to a disulfide-bonded oligomer by a
reduction-oxidation process. Nat Struct Biol. 10:725–730. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dietz KJ and Hell R: Thiol switches in
redox regulation of chloroplasts: Balancing redox state, metabolism
and oxidative stress. Biol Chem. 396:483–494. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ckless K: Redox proteomics: From bench to
bedside. Adv Exp Med Biol. 806:301–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rana A, Gnaneswari D, Bansal S and Kundu
B: Prion metal interaction: Is prion pathogenesis a cause or a
consequence of metal imbalance? Chem Biol Interact. 181:282–291.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu F, Davies P, Thompsett AR, Kelly SM,
Tranter GE, Hecht L, Isaacs NW, Brown DR and Barron LD: Raman
optical activity and circular dichroism reveal dramatic differences
in the influence of divalent copper and manganese ions on prion
protein folding. Biochemistry. 47:2510–2517. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim NH, Choi JK, Jeong BH, Kim JI, Kwon
MS, Carp RI and Kim YS: Effect of transition metals (Mn, Cu, Fe)
and deoxycholic acid (DA) on the conversion of PrPC to
PrPres. FASEB J. 19:783–785. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cingaram PK, Nyeste A, Dondapati DT, Fodor
E and Welker E: Prion protein does not confer resistance to
hippocampus-derived zpl cells against the toxic effects of
Cu2+, Mn2+, Zn2+ and
Co2+ not supporting a general protective role for PrP in
transition metal induced toxicity. PLoS One. 10:e01392192015.
View Article : Google Scholar
|
|
21
|
Riesner D, Kellings K, Post K, Wille H,
Serban H, Groth D, Baldwin MA and Prusiner SB: Disruption of prion
rods generates 10-nm spherical particles having high alpha-helical
content and lacking scrapie infectivity. J Virol. 70:1714–1722.
1996.PubMed/NCBI
|
|
22
|
Lu BY and Chang JY: Rapid and irreversible
reduction of protein disulfide bonds. Anal Biochem. 405:67–72.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yuan Z, Yang L, Chen B, Zhu T, Hassan MF,
Yin X, Zhou X and Zhao D: Protein misfolding cyclic amplification
induces the conversion of recombinant prion protein to PrP
oligomers causing neuronal apoptosis. J Neurochem. 133:722–729.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shin JY, Shin JI, Kim JS, Yang YS, Shin
YK, Kim KK, Lee S and Kweon DH: Disulfide bond as a structural
determinant of prion protein membrane insertion. Mol Cells.
27:673–680. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou X, Bi H, Wong J, Shimoji M, Wang Y,
Yuan J, Xiao X, Wang GX and Zou WQ: Alkylating antitumor drug
mechlorethamine conceals a structured PrP domain and inhibits in
vitro prion amplification. J Toxicol Environ Health A.
74:1493–1503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dong CF, Wang XF, An R, Chen JM, Shan B,
Han L, Lei YJ, Han J and Dong XP: Interaction analysis between
various PrP fusion proteins and the tubulin in vitro. Bing Du Xue
Bao. 23:28–32. 2007.(In Chinese).PubMed/NCBI
|
|
27
|
Dong CF, Shi S, Wang XF, An R, Li P, Chen
JM, Wang X, Wang GR, Shan B, Zhang BY, et al: The N-terminus of PrP
is responsible for interacting with tubulin and fCJD related PrP
mutants possess stronger inhibitive effect on microtubule assembly
in vitro. Arch Biochem Biophys. 470:83–92. 2008. View Article : Google Scholar
|
|
28
|
Mitteregger G, Vosko M, Krebs B, Xiang W,
Kohlmannsperger V, Nölting S, Hamann GF and Kretzschmar HA: The
role of the octarepeat region in neuroprotective function of the
cellular prion protein. Brain Pathol. 17:174–183. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li XL, Dong CF, Wang GR, Zhou RM, Shi Q,
Tian C, Gao C, Mei GY, Chen C, Xu K, et al: Manganese-induced
changes of the biochemical characteristics of the recombinant
wild-type and mutant PrPs. Med Microbiol Immunol (Berl).
198:239–245. 2009. View Article : Google Scholar
|