Introduction
Myocardial ischemia refers to reduced cardiac
perfusion, which leads to abnormal oxygen reduction and energy
metabolism, thus resulting in cardiac pathology (1). Under normal conditions, the
myocardial oxygen uptake rate is as high as ~70%, in order to
guarantee normal myocardial function, whereas myocardial ischemia
occurs when the balance between myocardial blood supply and demand
is broken, which is caused by numerous factors, including coronary
heart disease (2). The prevalence
of myocardial ischemia has been reported to be increasing
worldwide, and it is a common and frequently encountered disease in
middle-aged and elderly individuals (2). Thrombolysis, interventional therapy
and coronary artery bypass grafting may be regularly applied for
the management of serious myocardial ischemia (3,4).
In addition, blood flow reperfusion leads to temporary survival of
cardiac cells in the ischemic region and recovery of damaged
tissue; however, the narrow therapeutic time window for these
therapies may result in the occurrence of ischemia/reperfusion
(I/R) injury. I/R injury may induce the disordered synthesis of
mitochondrial energy and Ca2+ homeostasis, and the
release of free radicals and inflammatory cytokines, eventually
leading to myocardial cell apoptosis and organ damage (5-7).
Due to its ability to induce irreversible damage to cardiac cells,
I/R injury is considered a critical issue in the treatment of
ischemic stroke.
In a previous study, anti-inflammatory agents were
reported to reduce tissue damage and protect cells from ischemia
(8). Triptolide (TP) is extracted
from the traditional Chinese medicinal plant Tripterygium
wilfordii Hook F, and is a bioactive ingredient with
anti-inflammatory activity, which may be used to treat numerous
disorders, including arthritis, pulmonary hypertension and
traumatic brain injury (9-12).
The inhibitory effects of TP on the production of inflammatory
cytokines in various cell lines has been determined in in
vitro studies, thus suggesting the potential use of TP in the
treatment of I/R injury (13-16). The protective effects of TP on
cardiac cells against I/R injury have rarely been reported.
Therefore, in the present study, a rat myocardial I/R model was
used to evaluate the protective effects of TP on I/R. Furthermore,
the H9C2 cardiac cell line was used to explore the potential
mechanism underlying the protective effects of TP on I/R
injury.
Materials and methods
Animal I/R model
Langendorff non-circulatory perfusion was employed
to evaluate the protective effects of TP against I/R in rat cardiac
tissues. Krebs-Henseleit (KH) buffer was equilibrated with 95%
O2 and 5% CO2 at pH 7.4, and was flushed
continually at 37°C. A total of 36 rats were randomly grouped into
six and were anesthetized via intraperitoneal injection of heparin
sodium (1,000 U/kg) and 10% chloral hydrate (350 mg/kg) for 20 min.
The hearts were rapidly excised and placed in ice-cold KH buffer.
The aorta of the control group was fixed with an infusion tube and
perfused at a constant perfusion pressure of 75 mmHg using a
Langendorff non-circulatory perfusion pump, for 170 min. The other
groups underwent ischemia for 30 min following perfusion for 80
min, and were then reperfused for 60 min. A fluid-filled balloon
was inserted into the left ventricle and attached to a pressure
transducer. A cardiac pacemaker was used to generate a heart rate
of 280 beats/min. The present study was approved by the
Institutional Animal Care and Use Committee
(IACUC-20130315-01).
Histology and terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) staining
Cardiac tissues were fixed in 10% formalin for 48 h.
Tissues were dehydrated using ethanol and were cleared with xylene,
after which they were embedded in paraffin and cut into 4-7
μm sections. Slides of the tissue sections were then
deparaffinized, dehydrated and stained with hematoxylin and eosin
(H&E). Infiltrating neutrophils were identified and visualized
under a microscope.
For TUNEL staining, the slides were deparaffinized
and dehydrated as aforementioned. The sections were digested for 40
min, and were then incubated with 50 μl TUNEL buffer at 37°C
for 1 h, and 50 μl POD at 37°C for 30 min, respectively.
Finally, the slides were stained with DAB for 3-10 min, sections
were visualized under a microscope and apoptotic rate was
calculated.
Cell culture
H9C2 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% fetal calf serum and 1%
×100 mycillin at 37°C in an atmosphere containing 5%
CO2. To study the effects of TP on I/R cells, H9C2 cells
were then digested, seeded into 96-well plates (3×103
cells/well) and divided into five groups in triplicate. The control
group was incubated in normal culture medium. The I/R group was
transferred into sugar- and serum-free DMEM, and was incubated at
37°C in an atmosphere containing 5% CO2 and 1%
O2 for 2 h, after which, cells were transferred into
normal DMEM and were cultured for 6 h. The TP groups were treated
with various concentrations of TP for 1 h, and underwent
hypoxia-reoxygenation as described in the I/R group. Furthermore,
cultured cells were treated with N-acetylcysteine (NAC) or
pyrrolidine dithiocarbamate (PDTC), in order to investigate the
mechanisms underlying the protective effects of TP against I/R
injury. To study the effects of TP on
H2O2-treated cells, cells were pretreated
with NAC, PDTC or TP for 1 h, and then treated with 100 μM
H2O2 for 24 h. Cultured and treated cells
were prepared for further experiments.
ELISA
The ELISA method was used to measure the expression
levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β and
IL-6 in cardiac tissues and H9C2 cells by determining the
absorbance at 450 nm. Briefly, ~10 mg cardiac tissues or 100
μl cultured cells were processed according to the
manufacturers' protocols. TNF-α, IL-1β and IL-6 concentrations were
calculated according to the standard curve, which was generated
from a series of known concentrations of standard bovine serum
albumin.
Biochemical analysis
Cardiac antioxidant status was determined by
measuring cardiac superoxide dismutase (SOD) content. Tissue
samples were homogenized in 100 mmol/l Tris-HCl buffer and were
centrifuged at 10,000 × g for 20 min. SOD, malondialdehyde (MDA)
and catalase (CAT) levels were assessed using commercial kits
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Total
protein concentration in heart homogenates was determined using the
Coomassie blue method.
Cell Counting kit (CCK)-8 assay
Cultured and treated H9C2 cells were treated with
100 μl serum-free DMEM containing 10% CCK-8 (Dojindo
Molecular Technologies, Inc., Kumamoto, Japan) at 37°C for 1 h in
an atmosphere containing 5% CO2. The optical density of
cell suspensions was measured at 450nm using a spectrophotometer,
in order to evaluate the viability of H9C2 cells.
Cell apoptosis assay
Cultured and treated H9C2 cells were treated with
0.25% trypsin, in order to form a single cell suspension, and were
washed with 10% PBS and centrifuged at 1,000 × g for 5 min. The
supernatant was discarded and cells were then incubated with the
Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit
(BD Biosciences, San Diego, CA, USA) for 10 min at room temperature
in the dark. Cell apoptotic rate was measured and data were
obtained using flow cytometry (FACSCalibur; BD Biosciences).
DNA fragmentation assay
Cultured cells were fixed with 3% paraformaldehyde
and incubated at room temperature for 5 min. Subsequently, the
cells were air-dried and stained with 10 ml Hoechst 33258 (Beyotime
Institute of Biotechnology, Jiangsu, China) for 10 min, followed by
the addition of 50% glycerol containing 20 mmol/l citric acid and
50 mmol/l orthophosphate. A fluorescence microscope was used to
evaluate nuclear morphology.
Reactive oxygen species (ROS) assay
Cultured cells were washed with PBS and digested
using trypsin following treatment with PA for 24 h; followed by
centrifugation at 1,500 rpm for 10 min. Cells were then collected
and resuspended in staining buffer containing 50 μM
dihydroethidium (Wegelasi Biotechnology Co., Ltd.). ROS was
measured by flow cytometry.
Western blot analysis
Briefly, 20 mg tissue samples were cut into pieces
and proteins were extracted using 250 μl
radioimmunoprecipitation assay buffer (Shanghai Solarbio Science
& Technology Co., Ltd., Shanghai, China) containing 0.01%
protease inhibitor cocktail (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). In addition, cultured cells were washed twice with 1X
PBS, and were lysed at 4°C. Furthermore, nuclear and plasma
proteins were extracted from tissue samples using the NE-PER kit
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). Subsequently,
samples were centrifuged at 12,000 × g for 15 min at 4°C and the
supernatants were collected. The bicinchoninic acid protein
quantification kit (Thermo Fisher Scientific, Inc.) was used to
quantify protein contents. Tissue and cell proteins were separated
by 15% (80 μg/well) or 10% (25 μg/well) SDS-PAGE,
respectively, and proteins were electrophoretically transferred to
a nitrocellulose filter membrane (EMD Millipore, Billerica, MA,
USA). Blots were blocked with 5% skim milk at room temperature for
1 h, and were then incubated with anti-B-cell lymphoma 2 (Bcl2) and
Bcl2-associated X protein (Bax) (Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA), caspase-3, lectin-like oxidized low-density
lipoprotein receptor-1 (LOX-1) and inducible nitric oxide synthase
(iNOS) (Abcam, Cambridge, UK), cyclooxygenase (COX)2, nuclear
factor (NF)-κB, phosphorylated (p)-NF-κB, NF-κB inhibitor α (IκBα),
p-IκBα, extracellular signal-regulated kinase (ERK)1/2, p-ERK1/2
and GAPDH antibodies (Cell Signaling Technology, Inc., Danvers, MA,
USA). and were incubated with goat anti-mouse or anti-rabbit
secondary antibodies (Beyotime Institute of Biotechnology).
Enhanced chemiluminescence (Thermo Fisher Scientific, Inc.) was
used to visualize the blots.
Statistical analysis
Data are presented as the means ± standard deviation
of at least three independent replicates. The results were analyzed
using analysis of variance followed by the Tukey multiple
comparisons test. GraphPad Prism 5.0 software (GraphPad Software,
Inc., La Jolla, CA, USA) was used to perform data analysis.
P<0.05 was considered to indicate a statistically significant
difference.
Results
TP inhibits rat myocardial cell
apoptosis
H&E and TUNEL staining were performed to
evaluate the effects of I/R on control and TP-treated cardiac
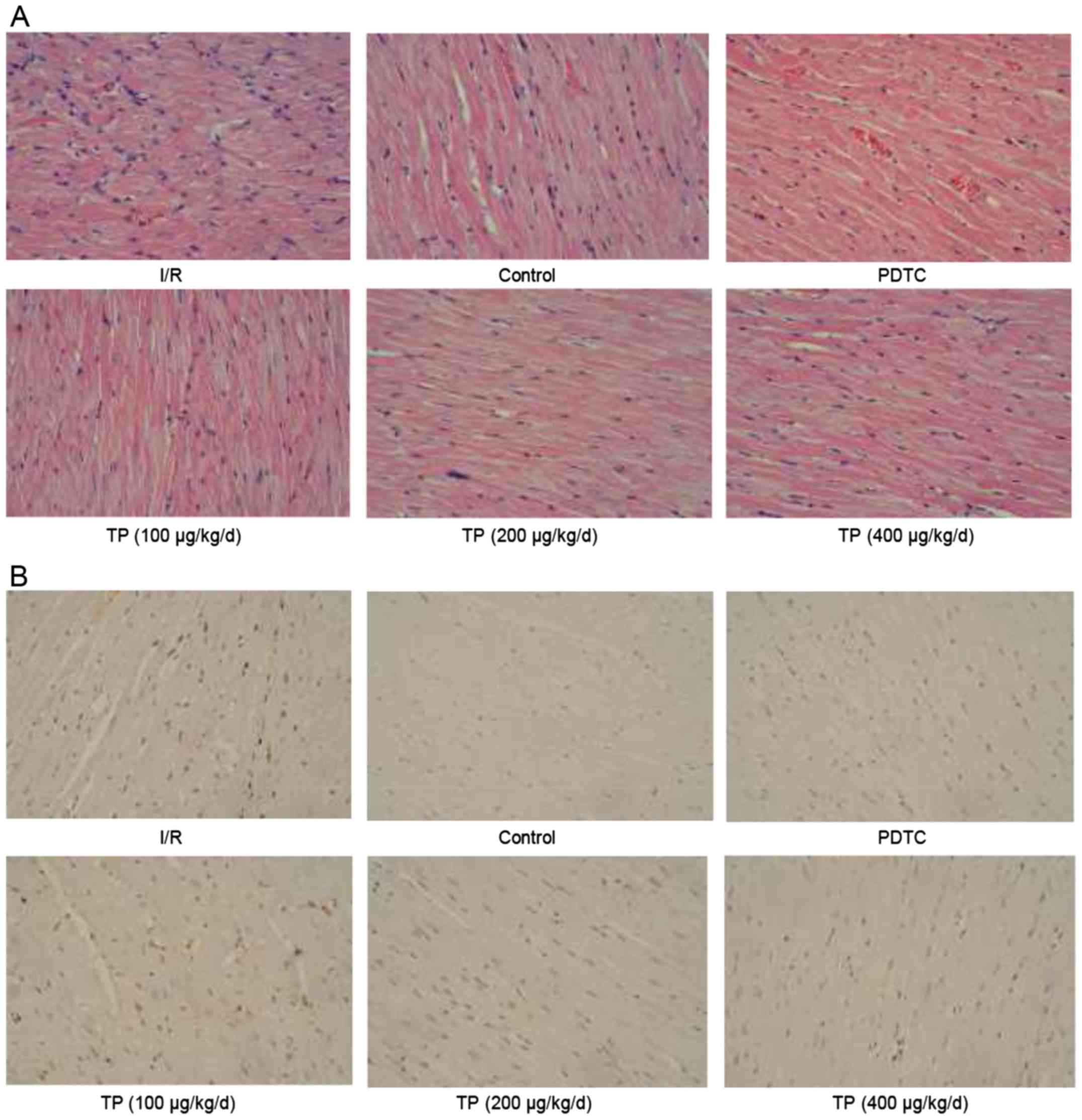
tissues. As shown in Fig. 1A,
cells in the I/R group exhibited swelling, necrosis and
degeneration, alongside marked neutrophil infiltration. Conversely,
less severe cell necrosis was determined, and stained cells
exhibited normal architecture in the PDTC- and TP-treated groups,
thus indicating that I/R induced cell damage, whereas TP exerted
protective effects against I/R. TUNEL staining demonstrated that
I/R induced cell apoptosis (Fig.
1B); apoptotic cells were stained brown in the I/R group. The
number of positively stained cells was reduced in the TP-treated
groups, thus indicating the inhibitory effects of TP against
I/R-induced cell apoptosis. In addition, Bcl2 was increased and Bax
was decreased, as determined by western blot analysis, indicating
that cell apoptosis was inhibited by TP compared with in the I/R
group (Fig. 2A and B).
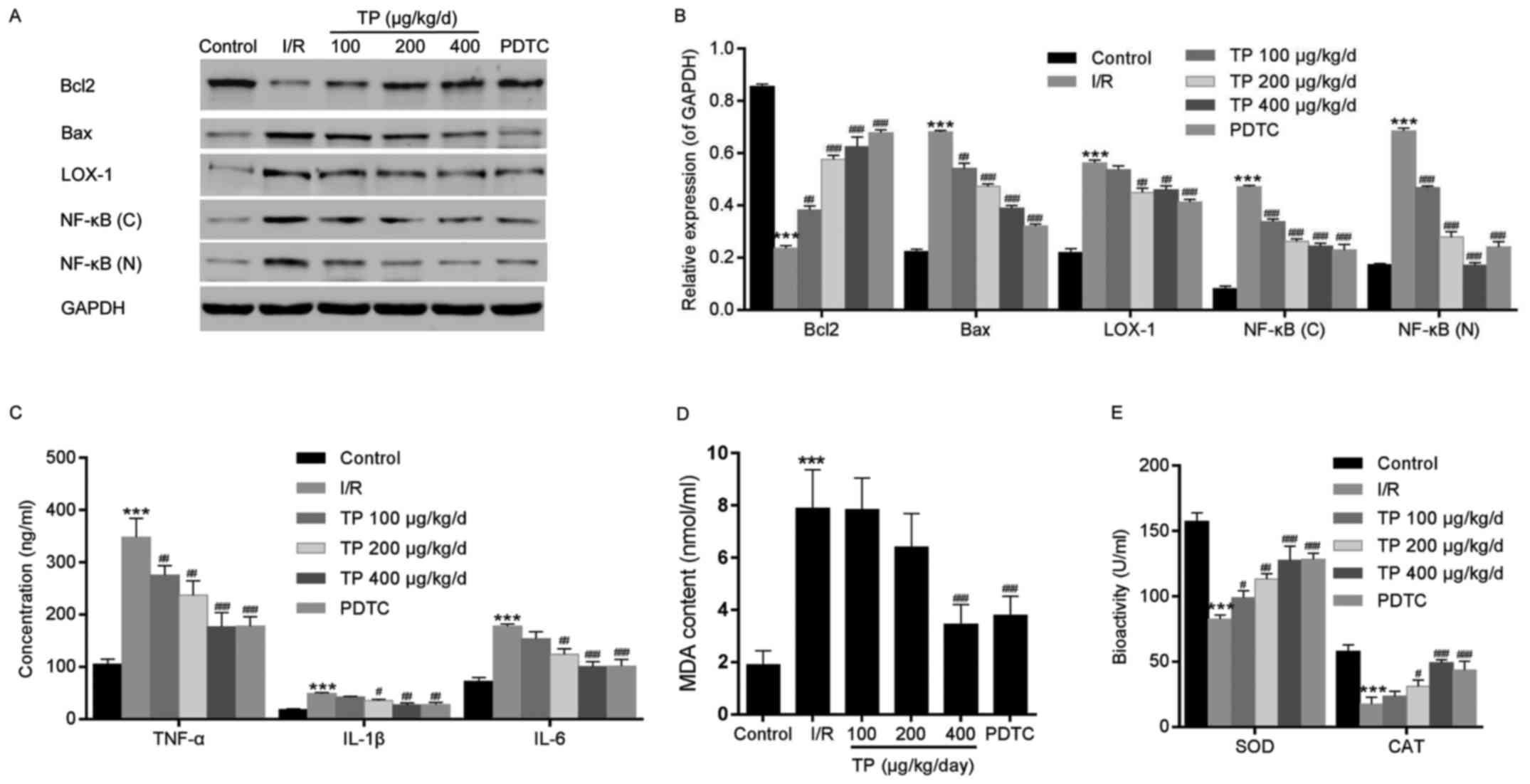 | Figure 2Western blot analysis and ELISA were
performed to investigate the mechanisms underlying the protective
effects of TP against I/R in cardiac tissues. (A and B) Western
blot analysis was used to measure the protein expression levels of
Bcl2, Bax, LOX-1 and NF-κB. (C) ELISA analysis was used to
determine the inhibitory effects of TP on the expression of
inflammation-associated proteins, TNF-α, IL-1β and IL-6. (D and E)
Biochemical analyses revealed the alterations in SOD, MDA and CAT.
***P<0.001, compared with the control group.
#P<0.05, ##P<0.01,
###P<0.001, compared with the I/R group (n=6). Bax,
Bcl2-associated X protein; Bcl2, B-cell lymphoma 2; (C),
cytoplasmic; CAT, catalase; IL, interleukin; I/R,
ischemia/reperfusion; LOX-1, lectin-like oxidized low-density
lipoprotein receptor-1; MDA, malondialdehyde; (N), nuclear; NF-κB,
nuclear factor-κB; PDTC, pyrrolidine dithiocarbamate; SOD,
superoxide dismutase; TNF-α, tumor necrosis factor-α; TP,
triptolide. |
TP inhibits the inflammatory response in
rat myocardial cells
TNF-α, IL-1β and IL-6 were evaluated using ELISA, in
order to investigate the inflammatory response following I/R in rat
myocardial cells. Significant increases in the expression levels of
TNF-α, IL-1β and IL-6 were detected in the I/R group, whereas these
increased levels were attenuated in the TP-treated groups in a
dose-dependent manner (Fig. 2C).
Similarly, the relative nuclear and plasma protein expression
levels of NF-κB were decreased in the TP-treated groups compared
with in the I/R group (Fig. 2A and
B).
TP protects rat myocardial cells from
peroxidation
The levels of cardiac enzymes, SOD, MDA and CAT,
were measured to evaluate the oxidative damage caused by I/R
(Fig. 2D and E). Although higher
levels of SOD and MDA were detected in the I/R and TP-treated
groups compared with in the control group, the enzymatic activities
of SOD and MDA were reduced in the TP-treated groups compared with
in the I/R group in a dose-dependent manner. In addition, CAT
activity exhibited almost no alteration between the I/R and
TP-treated groups. Furthermore, LOX-1 was measured by western blot
analysis; the protein expression levels of LOX-1 were attenuated
compared with in the I/R group (Fig.
2A and B), thus suggesting that TP exerts protective effects
against I/R-induced peroxidation.
TP improves the viability of H9C2
cells
In order to investigate the possible mechanisms
underlying the protective effects of TP against I/R injury in
cardiac cells, an in vitro study was performed using H9C2
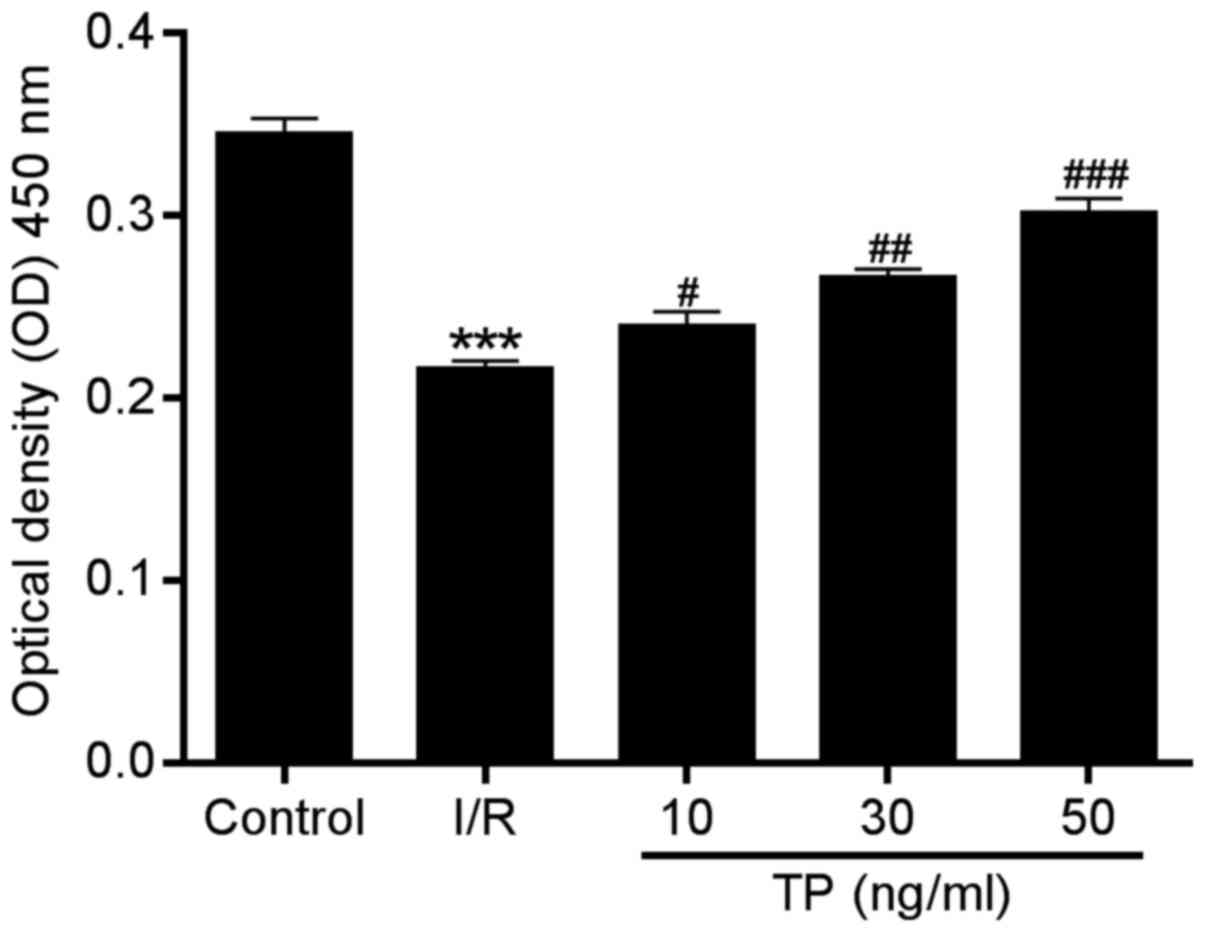
cells. The CCK-8 assay was performed to evaluate the viability of
H9C2 cells after 2 h of ischemia and 6 h of reperfusion. Compared
with in the I/R group, cell proliferation in the TP-treated groups
was increased in a dose-dependent manner (Fig. 3), thus indicating the improved
viability of H9C2 cells following TP treatment.
TP reduces inflammation in H9C2
cells
ELISA was used to measure the expression levels of
TNF-α, IL-1β and IL-6 in H9C2 cells after I/R. Similar to the
expression levels of TNF-α, IL-1β and IL-6 detected in cardiac
tissues, the expression levels of these proteins were significantly
increased in the I/R group compared with in the control group, and
were decreased in the TP-treated groups compared with in the I/R
group in a dose-dependent manner (Fig. 4).
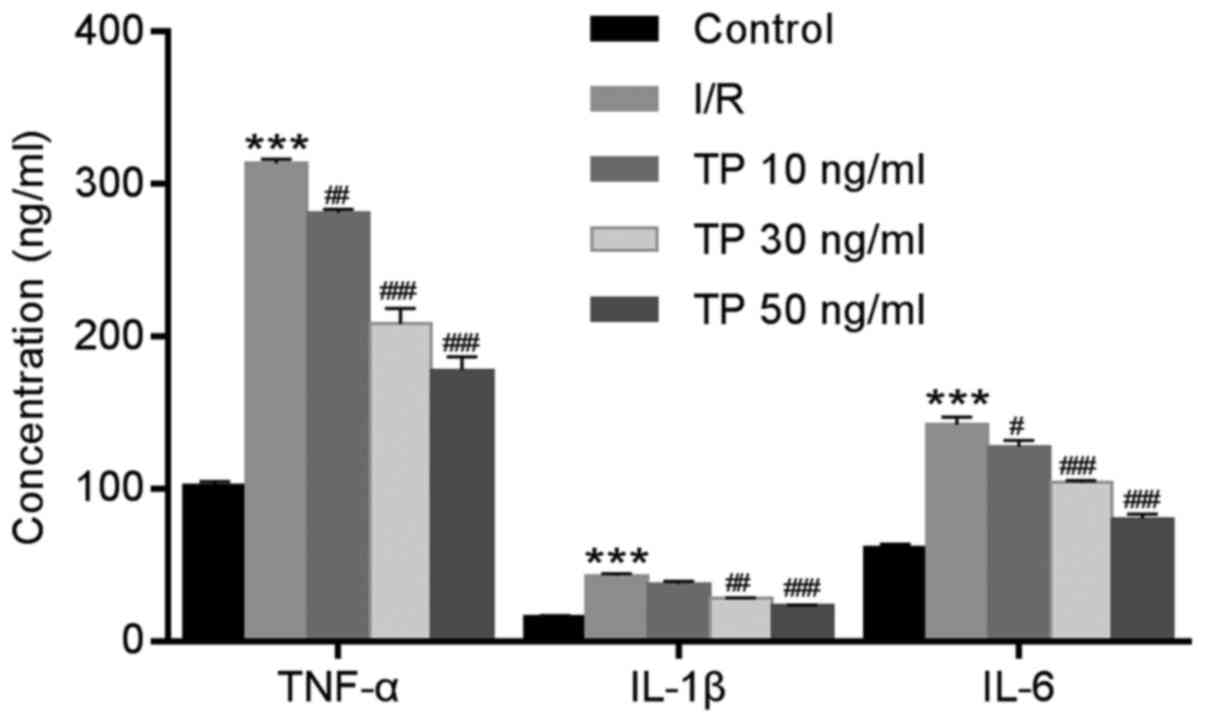 | Figure 4Expression of inflammatory factors in
H9C2 cells was determined using ELISA after 2 h of ischemia and 6 h
of reperfusion. TP inhibited the expression of inflammatory
factors, TNF-α, IL-1β and IL-6, in a dose-dependent manner.
***P<0.001, compared with the control group.
#P<0.05, ##P<0.01 and
###P<0.001, compared with the I/R group (n=3). IL,
interleukin; I/R, ischemia/reperfusion; TNF-α, tumor necrosis
factor-α; TP, triptolide. |
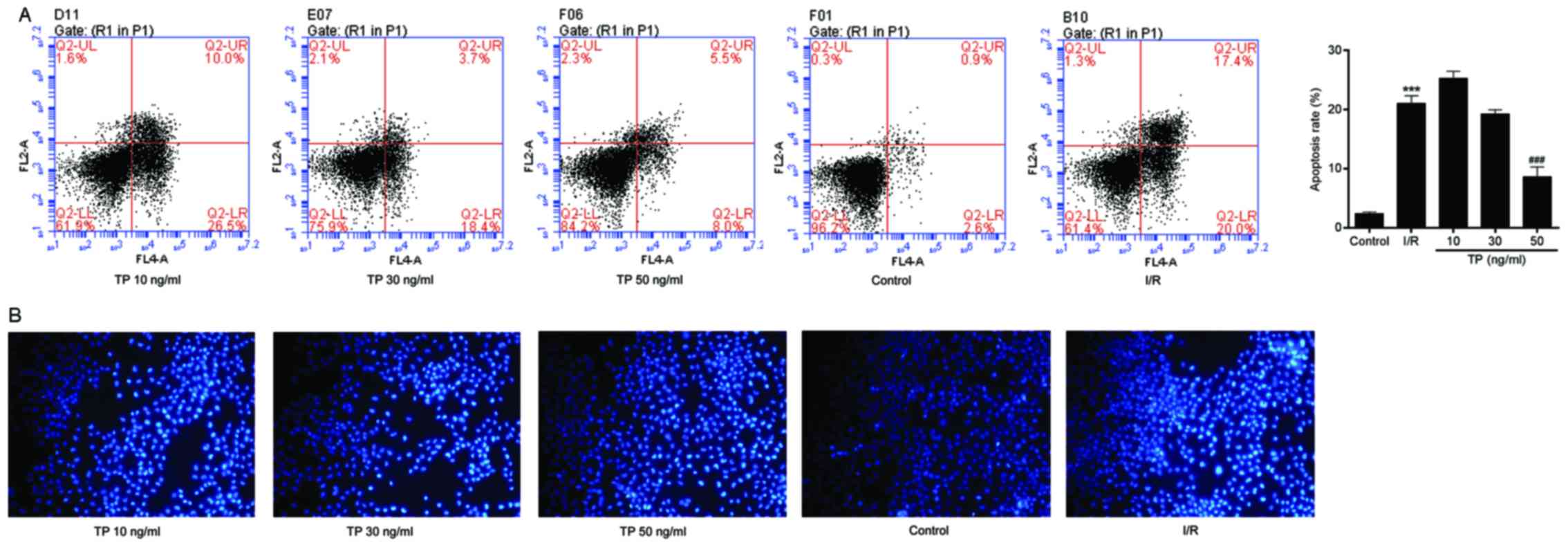
TP inhibits apoptosis of H9C2 cells
Flow cytometry was used to evaluate apoptosis in the
control and TP-treated H9C2 cells. I/R-induced cell apoptosis was
detected using Annexin V-FITC/propidium iodide (PI) double staining
(Fig. 5A). The apoptotic rate was
calculated from the percentage of early apoptotic cells presented
in the lower right quadrant of the histograms. The apoptotic rate
was dose-dependently reduced in the TP-treated H9C2 cells compared
with in the I/R group, thus revealing the inhibitory effect of TP
against I/R. Hoechst 33258 staining was also used to
morphologically detect apoptosis of H9C2 cells through fluorescence
staining (Fig. 5B). Nuclear
fragmentation and chromosomal condensation were improved in the
cells treated with various concentrations of TP compared with in
the I/R group. Furthermore, apoptosis of H9C2 cells was measured,
in order to evaluate the protective effects of TP against
H2O2-induced peroxidation. As shown in
Fig. 6A and B, the apoptotic rate
was increased in the H2O2 and I/R groups,
whereas the apoptotic rate was significantly reduced in the NAC-,
PDTC- and TP-treated groups.
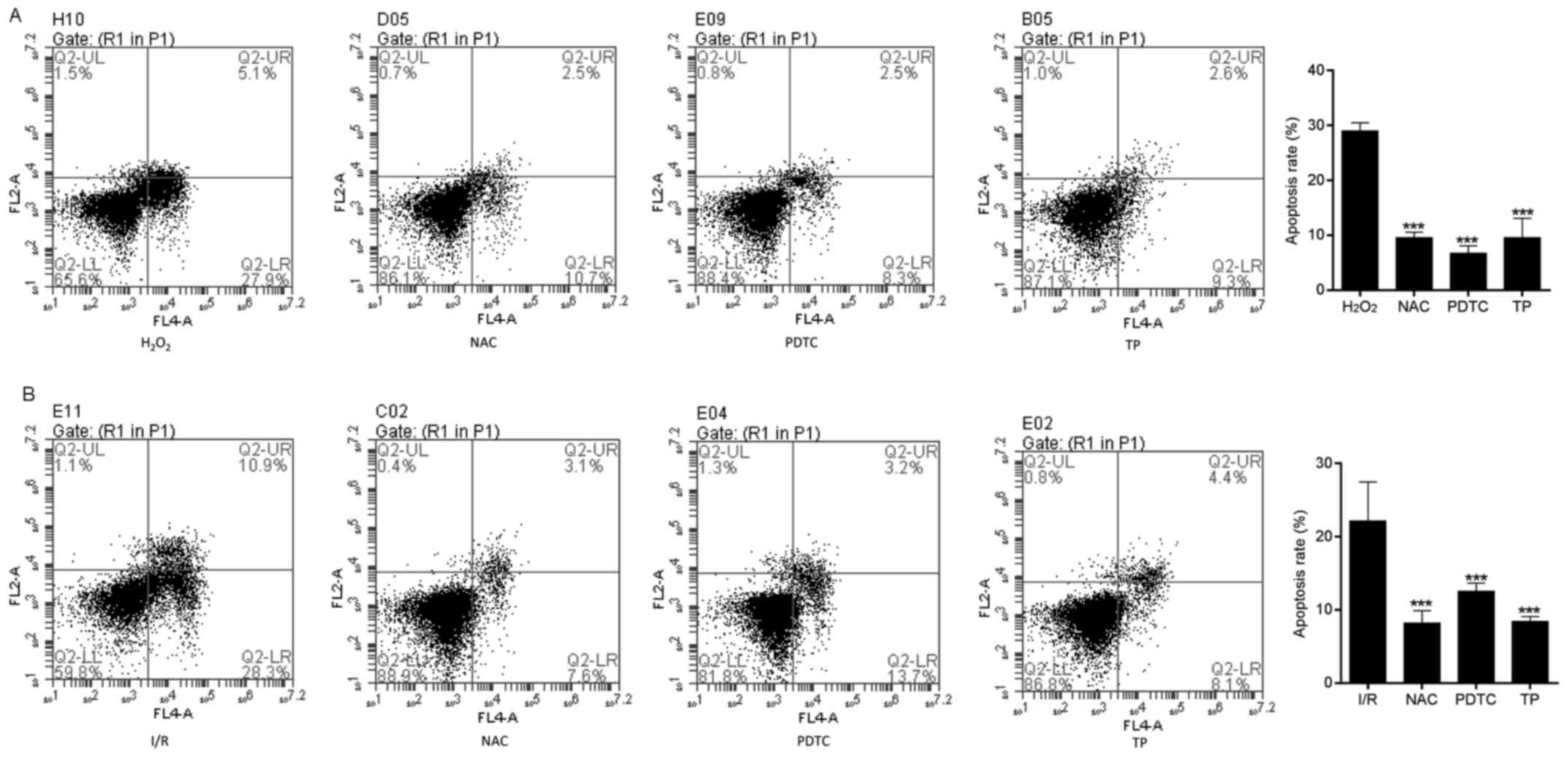 | Figure 6Cell apoptosis was measured by flow
cytometry, in order to evaluate the protective effects of TP
against H2O2. (A) Early apoptotic cells were
determined following treatment with H2O2 for
24 h using Annexin V/PI double staining, and are presented in the
lower right quadrant. ***P<0.001, compared with the
H2O2 group (n=3). (B) Early apoptotic cells
were determined using Annexin V/PI double staining after 2 h of
ischemia and 6 h of reperfusion. ***P<0.001, compared
with the I/R group. H2O2, hydrogen peroxide;
I/R, ischemia/reperfusion; NAC, N-acetylcysteine; PDTC,
pyrrolidine dithiocarbamate; PI, propidium iodide; TP,
triptolide. |
TP reduces ROS generation
ROS are produced by the mitochondrial electron
transport chain in cells, and are involved in cell apoptosis via
the regulation of caspases. ROS generation may be detected using
dihydroethidium, which has free access into live cells through the
cell membrane. As shown in Fig.
7A, compared with in the control group, the fluorescence
intensity of ROS was increased in H9C2 cells in the I/R group.
However, there was a significant dose-dependent decrease in ROS
generation in the TP-treated groups compared with in the I/R group.
In addition, reductions in SOD, CAT and MDA expression in the
TP-treated groups indicated the antioxidative effects of TP
(Fig. 7B and C). As presented in
Fig. 8, TP exerted protective
effects against H2O2-induced peroxidation,
according to the decreased fluorescence intensity of ROS in NAC-,
PDTA- and TP-treated groups.
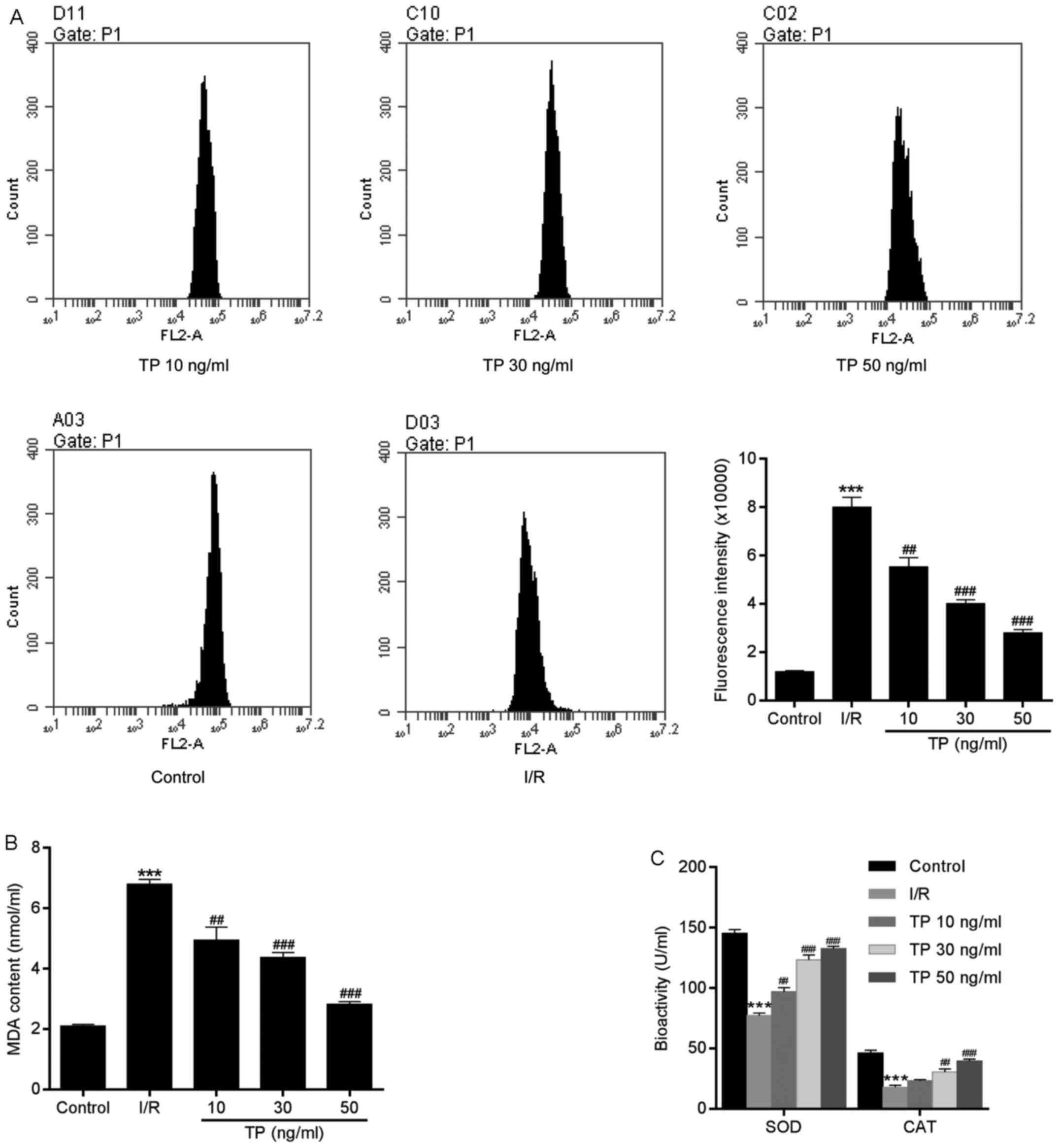 | Figure 7Generation of ROS in H9C2 cells
during the process of cell apoptosis. (A) Compared with in the
control group, ROS, as measured using dihydroethidium, was
significantly decreased in TP-treated cells in a dose-dependent
manner. (B and C) TP exerted protective effects against I/R via
SOD, MDA and CAT. ***P<0.001, compared with the
control group. ##P<0.01, ###P<0.001,
compared with the I/R group (n=3). CAT, catalase; I/R,
ischemia/reperfusion; MDA, malondialdehyde; ROS, reactive oxygen
species; SOD, superoxide dismutase; TP, triptolide. |
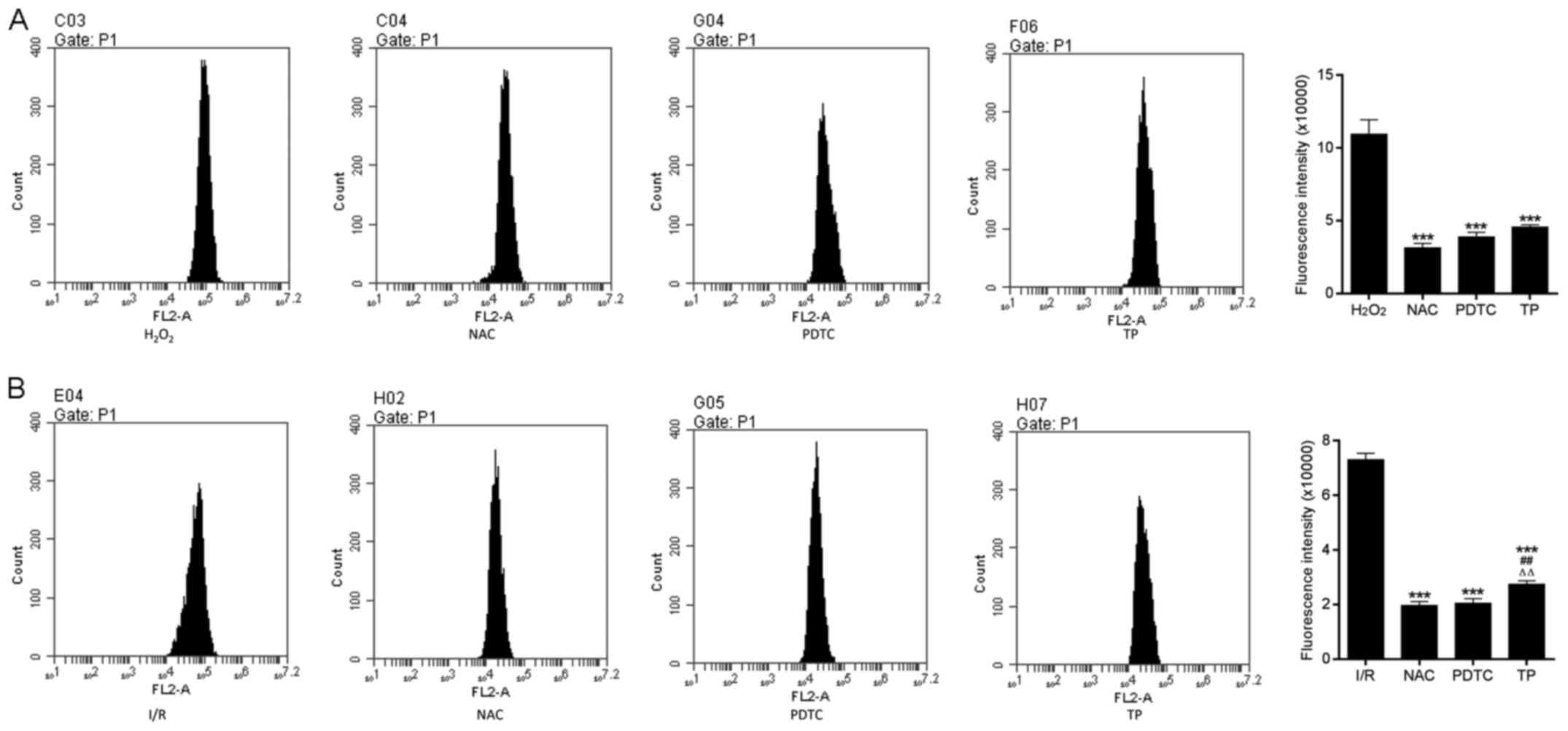 | Figure 8ROS generation in H9C2 cells during
the process of cell apoptosis. (A) ROS generation was measured in
H9C2 cells treated with H2O2 for 24 h.
***P<0.001, compared with the
H2O2 group. (B) ROS generation in H9C2 cells
after 2 h of ischemia and 6 h of reperfusion.
***P<0.001, compared with the I/R group.
##P<0.01, compared with the NAC group.
△△P<0.01, compared with the PDTC group (n=3).
H2O2, hydrogen peroxide; I/R,
ischemia/reperfusion; NAC, N-acetylcysteine; PDTC,
pyrrolidine dithiocarbamate ROS, reactive oxygen species; TP,
triptolide. |
Effects of TP on protein expression in
H9C2 cells
The expression levels of inflammatory-associated
proteins, NF-κB and IκBα, were measured by western blot analysis
(Fig. 9A and B), as was the
expression of iNOS (Fig. 9A and
C). IκBα was downregulated in the I/R group, whereas NF-κB was
upregulated. Conversely, the expression levels of NF-κB were
decreased and IκBα was increased in the TP-treated groups in a
dose-dependent manner; similar findings were detected in the NAC
and PDTC groups. The phosphorylation of IκBα and NF-κB was
increased in the I/R group, but decreased in the TP-treated groups
(Fig. 10). In addition,
upregulation of iNOS was detected following I/R, whereas in the
TP-treated groups iNOS was decreased in a dose-dependent manner,
thus indicating the inhibitory effects of TP during I/R.
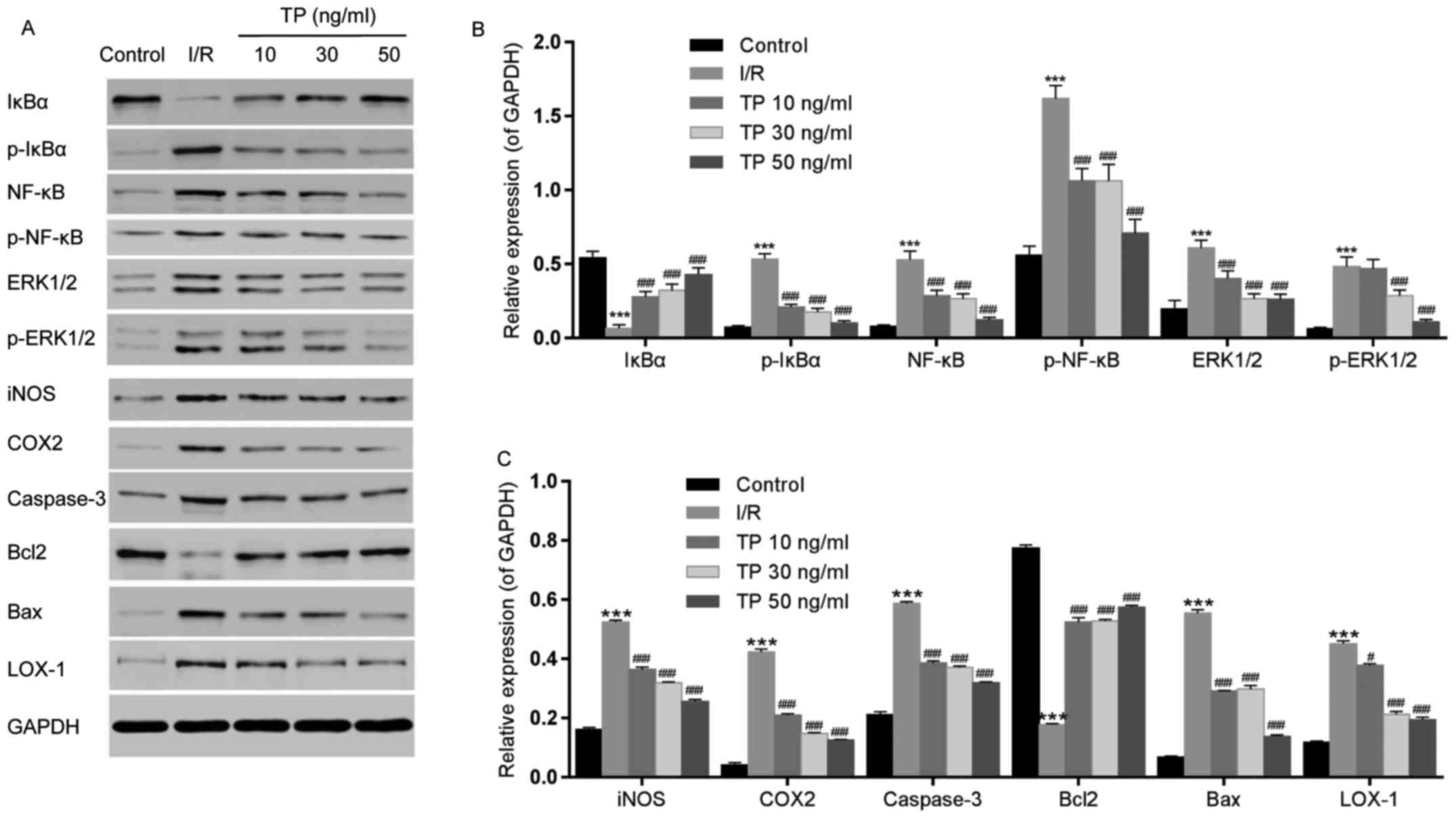 | Figure 9Effects of TP on the phosphorylation
of IκBα, NF-κB and ERK1/2, and the expression of iNOS, COX2,
caspase-3, Bcl2, Bax and LOX-1, as determined by western blot
analysis. (A) Relative expression levels of IκBα, NF-κB, ERK1/2,
iNOS, COX2, caspase-3, Bcl2, Bax and LOX-1 were determined. (B)
Semi-quantification of western blot analysis was used to measure
the phosphorylation of IκBα, NF-κB and ERK1/2. (C) Expression
levels of iNOS, COX2, caspase-3, Bcl2, Bax and LOX-1 were analyzed
by semi-quantification of western blot analysis.
***P<0.001, compared with the control group.
#P<0.05, ###P<0.001, compared with the
I/R group (n=3). Bax, Bcl2-associated X protein; Bcl2, B-cell
lymphoma 2; COX2, cyclooxygenase 2; ERK1/2, extracellular
signal-regulated kinase 1/2; IκBα, NF-κB inhibitor α; iNOS,
inducible nitric oxide synthase; I/R, ischemia/reperfusion; LOX-1,
lectin-like oxidized low-density lipoprotein receptor-1; NF-κB;
nuclear factor-κB; p-, phosphorylated; TP, triptolide. |
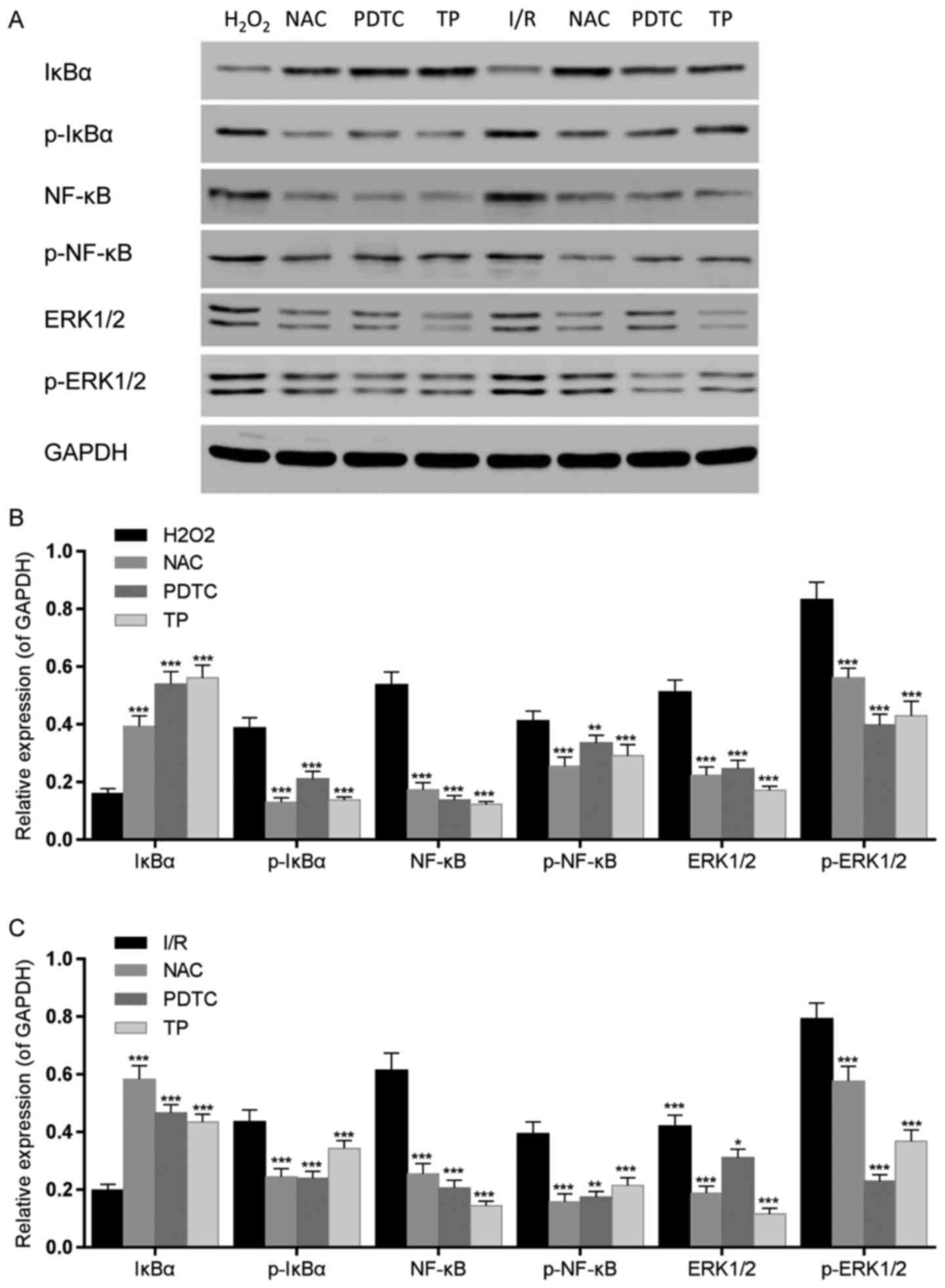 | Figure 10Effects of TP on IκBα, NF-κB and
ERK1/2 phosphorylation in H9C2 cells, as evaluated by western blot
analysis. (A) IκBα, NF-κB and ERK1/2 phosphorylation was detected
by western blotting. (B) IκBα, NF-κB and ERK1/2 phosphorylation was
semi-quantified after western blot analysis following treatment
with H2O2 for 24 h. ** ***
P<0.01, P<0.001, compared with the H2O2
group. (C) IκBα, NF-κB and ERK1/2 phosphorylation was
semi-quantified after western blot analysis following 2 h of
ischemia and 6 h of reperfusion. *P<0.05,
**P<0.01, ***P<0.001, compared with the
I/R group (n=3). ERK1/2, extracellular signal-regulated kinase 1/2;
H2O2, hydrogen peroxide; IκBα, NF-κB
inhibitor α; I/R, ischemia/reperfusion; NAC,
N-acetylcysteine; NF-κB; nuclear factor-κB; p-,
phosphorylated; PDTC, pyrrolidine dithiocarbamate; TP,
triptolide. |
COX2 and LOX-1 were measured using western blot
analysis, in order to evaluate the peroxidative damage caused by
I/R (Fig. 9A and C). COX2 and
LOX-1 expression levels were increased following I/R, whereas in
the TP-treated groups COX2 and LOX-1 were decreased in a
dose-dependent manner.
ERK1/2 phosphorylation was also detected by western
blot analysis (Fig. 9A and B).
The phosphorylation of ERK1/2 was increased in the I/R group, but
decreased in the TP-treated groups. Furthermore, the expression
levels of caspase-3 and Bax were increased in the I/R group
compared with in the control group, whereas Bcl2 was decreased
(Fig. 9A and C). Conversely,
caspase-3 and Bax expression were decreased by TP in a
dose-dependent manner, whereas Bcl2 was increased in the TP-treated
groups, thus indicating the inhibitory effects of TP against cell
apoptosis.
Discussion
In the present study, the protective effects of TP
were determined against a rat model of I/R and H9C2 cardiac cells
undergoing I/R. The results of the present study revealed the
mechanisms underlying myocardial I/R injury. Conversely, TP-treated
cardiac cells exhibited reduced cell swelling, necrosis and
degeneration following I/R, and the production of proinflammatory
cytokines, including TNF-α, IL-1β and IL-6, was reduced, as was
NF-κB expression. In addition, attenuation of ROS generation
induced by TP reduced the lipid peroxidation damage caused by I/R,
thus suggesting the anti-oxidative effects of TP. Furthermore,
ERK1/2 and caspase-3 were inhibited by TP, thus suggesting that TP
suppressed the ERK1/2 pathway and inhibited cell apoptosis.
NF-κB, which is regulated by IκBα, is a
transcription factor that controls the production and expression of
numerous cytokines and chemokines (17). In addition, NF-κB mediates cell
apoptosis and inflammation in response to immunological stimuli.
NF-κB was previously reported to be significantly activated in
cerebral I/R (18,19). Activated NF-κB may upregulate the
expression of proinflammatory cytokines, including TNF-α, IL-1β and
IL-6, thus resulting in inflammation. In the present study,
phosphorylation of NF-κB and IκBα, and the expression of TNF-α,
IL-1β and IL-6, was measured in response to cardiac I/R. p-IκBα
accompanied by activated NF-κB induced inflammation in H9C2 cells,
which was relieved following TP treatment. NF-κB deactivation was
detected in the TP-treated groups, thus suggesting that TP
inhibited activation of NF-κB, which was followed by suppression of
proinflammatory cytokines, TNF-α, IL-1β and IL-6. Furthermore,
similar results were obtained following administration of PDTC,
which was confirmed to act as an NF-κB inhibitor. These data
suggested that TP exerted similar effects as NF-κB inhibitor. iNOS
expression is involved in early inflammation, and is considered a
response to tissue damage (20,21). In the present study, the
expression levels of iNOS were increased during I/R and were
attenuated in TP-pretreated H9C2 cells, thus suggesting the
anti-inflammatory effects of TP. Furthermore, the percentage of
TUNEL-positive cells was significantly reduced following treatment
with TP. Therefore, these findings demonstrated the detrimental
role of NF-κB in myocardial ischemia, and the inhibitory effects of
TP on NF-κB.
MDA is a sensitive indicator of ROS-mediated lipid
peroxidation, which contributes to I/R injury, whereas SOD and CAT
are endogenous antioxidant enzymes that are developed to inhibit
the production of ROS (22,23). It has previously been suggested
that the development of ROS serves an important role in tissue
damage caused by I/R. ROS in tissues that experience I/R are
metabolized to prostaglandins and leukotrienes, which result in the
chemotaxis of leukocytes, and control of vascular endothelial cell
function, thus indicating the possible role of ROS-mediated lipid
peroxidation in I/R injury. LOX-1 is an indicator of oxidative
stress, which has been reported to present increased expression
during tissue damage, whereas COX2 is the essential enzyme in
prostaglandin synthesis (24-27). In the present study, elevated ROS
levels were detected in tissue samples and H9C2 cells. The results
obtained from biochemical analyses in rat tissues and H9C2 cells
demonstrated that MDA levels were significantly increased, whereas
SOD and CAT activity was decreased in response to I/R. These
effects were markedly attenuated following treatment with TP in a
dose-dependent manner, thus indicating that I/R induces injury via
ROS-mediated lipid peroxidation. Furthermore, the expression levels
of LOX-1 and COX2 were assessed by western blot analysis; LOX-1 and
COX2 were increased during I/R, whereas their expression was
attenuated by TP, thus indicating that I/R induced damage via lipid
peroxidation and TP exerted protective effects against I/R. In
addition, in order to explore the mechanism underlying ROS-induced
damage and the mechanism underlying the protective effects of TP
against I/R injury, H9C2 cells were treated with NAC, and ROS
generation, MDA levels, and the activity levels of SOD and CAT,
were assessed in response to H2O2-induced
peroxidation damage. NAC, as a ROS inhibitor, exerted inhibitory
effects on ROS-mediated lipid peroxidation caused by
H2O2. ROS generation was increased in H9C2
cells following treatment with H2O2; however,
ROS levels were attenuated in NAC- and TP-treated groups. These
findings support the hypothesis that ROS-mediated lipid
peroxidation causes cardiac cell injury during I/R. The present
study indicated that tissues may be damaged by I/R through
ROS-mediated lipid peroxidation, whereas TP pretreatment could
alleviate I/R injury by enhancing the activities of endogenous
antioxidant enzymes, including CAT and SOD, in order to inhibit
lipid peroxidation.
TUNEL and Annexin V/PI staining revealed that cell
apoptosis was increased in response to I/R injury. Conversely,
apoptotic rate was significantly decreased following TP
pretreatment. In order to explore the mechanisms underlying
I/R-induced cell apoptosis, numerous apoptosis-associated proteins,
including Bcl2, Bax and caspase-3, were detected by western blot
analysis. Caspase-3 is activated by apoptotic signals acts on
peptide chain and split substrate, resulting in cell apoptosis.
Caspase-3 serves a direct role in cell apoptosis as an effector
caspase. It has been reported that caspase-3 induces activation of
caspase-activated deoxyribonuclease (CAD), which is associated with
DNA degradation (28). When
apoptosis occurs, CAD is released by the inhibitor of CAD, which is
activated by caspase-3. In addition, nuclear lamina, which is
responsible for the stability of chromatin, may be cut by caspase-3
at a single site, thus resulting in chromatin degradation (29,30). Bax and Bcl2 belong to the Bcl2
family, which possess numerous highly conserved fragments that are
mainly distributed in the nuclear membrane, endoplasmic reticulum
and mitochondrial membrane (31).
Bcl2 exerts an anti-apoptotic function by inhibiting the release of
apoptosis-promoting substances from the mitochondria and
suppressing activation of proapoptotic proteins, caspase-3 and Bax
(32,33). Therefore, TP-induced Bcl2
upregulation, and caspase-3 and Bax downregulation, suggested that
TP may suppress cell apoptosis. In addition, p-ERK1/2 was also
assessed in the present study. It has been reported that reduced
activation of the phosphoinositide 3-kinase-protein kinase B
pathway in myocardial I/R may be accompanied by decreased ERK1/2
expression (34). The results of
the present study corresponded with those in previous studies.
ERK1/2 was downregulated in the I/R group, thus suggesting that the
ERK1/2 pathway is inhibited during I/R; however, ERK1/2 expression
was improved in TP-pretreated H9C2 cells, thus suggesting that
ERK1/2 may be activated by TP so as to inhibit cell apoptosis
during I/R injury.
Three different dosages of TP were used in the
present study to investigate the association between the protective
effects and dosage of TP, in order to provide a theoretical basis
for clinical study. Data obtained from high-dose TP presented a
significant difference compared with in the control group. The
difference between the TP-treated groups and the control group may
be due to the rapid absorption time and short half-life of TP.
Reduced bioactivity of TP may affect the protective effects against
I/R injury, which should be taken into account in future studies.
Regardless, TP exerted efficient protective effects against I/R
damage in cardiac cells.
In conclusion, the present study demonstrated that
TP exerted protective effects on cardiac cells during I/R in
vivo and in vitro. In addition, the mechanisms
underlying the protective effects of TP, including
anti-inflammatory action, antioxidation and apoptotic resistance,
were investigated.
Acknowledgments
Not applicable.
Notes
[1]
Funding
The present study was funded by a grant from the
National Natural Science Foundation of China (grant no. 81500285),
Natural Science Foundation of Shanxi Province (grant no.
2011011041-1) and the Doctor Start-up Fund of Shanxi Medical
University (grant no. 03201409).
[2] Availability
of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
BY and BL conceived and designed the study. BY, PY,
GZY, HLC and FW performed the experiments. BY and BL wrote the
manuscript. All authors read and approved the manuscript.
[4] Ethics
approval and consent to participate
The present study was approved by the Institutional
Animal Care and Use Committee (IACUC-20130315-01).
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Durukan A and Tatlisumak T: Acute ischemic
stroke: Overview of major experimental rodent models,
pathophysiology, and therapy of focal cerebral ischemia. Pharmacol
Biochem Behav. 87:179–197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Donnan GA, Fisher M, Macleod M and Davis
SM: Stroke. Lancet. 71:1612–1623. 2008. View Article : Google Scholar
|
|
3
|
Molina CA and Alvarez-Sabín J:
Recanalization and reperfusion therapies for acute ischemic stroke.
Cerebrovasc Dis. 27(Suppl 1): 162–167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Peralta C, Bulbena O, Xaus C, Prats N,
Cutrin JC, Poli G, Gelpi E and Roselló-Catafau J: Ischemic
preconditioning: A defense mechanism against the reactive oxygen
species generated after hepatic ischemia reperfusion.
Transplantation. 73:1203–1211. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang J, Upadhyay UM and Tamargo RJ:
Inflammation in stroke and focal cerebral ischemia. Surg Neurol.
66:232–245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Esterbauer H and Cheeseman KH:
Determination of aldehydic lipid peroxidation products:
Malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 186:407–421.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tuttolomondo A, Di Sciacca R, Di Raimondo
D, Renda C, Pinto A and Licata G: Inflammation as a therapeutic
target in acute ischemic stroke treatment. Curr Top Med Chem.
9:1240–1260. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Faul JL, Nishimura T, Berry GJ, Benson GV,
Pearl RG and Kao PN: Triptolide attenuates pulmonary arterial
hypertension and neointimal formation in rats. Am J Respir Crit
Care Med. 162:2252–2258. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hao M, Li X, Feng J and Pan N: Triptolide
protects against ischemic stroke in rats. Inflammation.
38:1617–1623. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hoyle GW, Hoyle CI, Chen J, Chang W,
Williams RW and Rando RJ: Identification of triptolide, a natural
diterpenoid compound, as an inhibitor of lung inflammation. Am J
Physiol Lung Cell Mol Physiol. 298:L830–L836. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu C, Xia Y, Wang P, Lu L and Zhang F:
Triptolide protects mice from ischemia/reperfusion injury by
inhibition of IL-17 production. Int Immunopharmacol. 11:1564–1572.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He JK, Yu SD, Zhu HJ, Wu JC and Qin ZH:
Triptolide inhibits NF-kappaB activation and reduces injury of
donor lung induced by ischemia/reperfusion. Acta Pharmacol Sin.
28:1919–1923. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin N, Liu C, Xiao C, Jia H, Imada K, Wu H
and Ito A: Triptolide, a diterpenoid triepoxide, suppresses
inflammation and cartilage destruction in collagen-induced
arthritis mice. Biochem Pharmacol. 73:136–146. 2007. View Article : Google Scholar
|
|
15
|
Lu Y, Liu Y, Fukuda K, Nakamura Y, Kumagai
N and Nishida T: Inhibition by triptolide of chemokine,
proinflammatory cytokine, and adhesion molecule expression induced
by lipopolysaccharide in corneal fibroblasts. Invest Ophthalmol Vis
Sci. 47:3796–3800. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matta R, Wang X, Ge H, Ray W, Nelin LD and
Liu Y: Triptolide induces anti-inflammatory cellular responses. Am
J Transl Res. 1:267–282. 2009.PubMed/NCBI
|
|
17
|
Chandel NS, Trzyna WC, McClintock DS and
Schumacker PT: Role of oxidants in NF-kappa B activation and
TNF-alpha gene transcription induced by hypoxia and endotoxin. J
Immunol. 165:1013–1021. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lakhan SE, Kirchgessner A and Hofer M:
Inflammatory mechanisms in ischemic stroke: Therapeutic approaches.
J Transl Med. 7:972009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin XQ, Ye F, Zhang JJ, Zhao Y and Zhou
XL: Triptolide attenuates cerebral ischemia and reperfusion injury
in rats through the inhibition the nuclear factor kappa B signaling
pathway. Neuropsychiatr Dis Treat. 11:1395–1403. 2015.PubMed/NCBI
|
|
20
|
Hur GM, Ryu YS, Yun HY, Jeon BH, Kim YM,
Seok JH and Lee JH: Hepatic ischemia/reperfusion in rats induces
iNOS gene transcription by activation of NF-kappaB. Biochem Biophys
Res Commun. 261:917–922. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim ID, Sawicki E, Lee HK, Lee EH, Park
HJ, Han PL, Kim KK, Choi H and Lee JK: Robust neuroprotective
effects of intranasally delivered iNOS siRNA encapsulated in
gelatin nanoparticles in the postischemic brain. Nanomedicine.
12:1219–1229. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Murakami K, Kondo T, Kawase M, Li Y, Sato
S, Chen SF and Chan PH: Mitochondrial susceptibility to oxidative
stress exacerbates cerebral infarction that follows permanent focal
cerebral ischemia in mutant mice with manganese superoxide
dismutase deficiency. J Neurosci. 18:205–213. 1998.PubMed/NCBI
|
|
23
|
Wu C, Wang P, Rao J, Wang Z, Zhang C, Lu L
and Zhang F: Triptolide alleviates hepatic ischemia/reperfusion
injury by attenuating oxidative stress and inhibiting NF-κB
activity in mice. J Surg Res. 166:e205–e213. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shiraki T, Aoyama T, Yokoyama C, Hayakawa
Y, Tanaka T, Nishigaki K, Sawamura T and Minatoguchi S: LOX-1 plays
an important role in ischemia-induced angiogenesis of limbs. PLoS
One. 9:e1145422014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mao X, Xie L and Greenberg DA: Effects of
flow on LOX-1 and oxidized low-density lipoprotein interactions in
brain endothelial cell cultures. Free Radic Biol Med. 89:638–641.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ozturk H, Gezici A and Ozturk H: The
effect of celecoxib, a selective COX-2 inhibitor, on liver
ischemia/reperfusion-induced oxidative stress in rats. Hepatol Res.
34:76–83. 2006. View Article : Google Scholar
|
|
27
|
Du Y, Zhu Y, Teng X, Zhang K, Teng X and
Li S: Toxicological effect of manganese on NF-κB/iNOS-COX-2
signaling pathway in chicken testes. Biol Trace Elem Res.
168:227–234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weng C, Li Y, Xu D, Shi Y and Tang H:
Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis
in Jurkat leukemia T cells. J Biol Chem. 280:10491–10500. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lavrik IN, Golks A and Krammer PH:
Caspases: Pharmacological manipulation of cell death. J Clin
Invest. 115:2665–2672. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stennicke HR and Salvesen GS: Biochemical
characteristics of caspases-3, -6, -7, and -8. J Biol Chem.
272:25719–25723. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hsu YT, Wolter KG and Youle RJ:
Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during
apoptosis. Proc Natl Acad Sci USA. 94:3668–3672. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nechushtan A, Smith CL, Hsu YT and Youle
RJ: Conformation of the Bax C-terminus regulates subcellular
location and cell death. EMBO J. 18:2330–2341. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shi Y, Chen J, Weng C, Chen R, Zheng Y,
Chen Q and Tang H: Identification of the protein-protein contact
site and interaction mode of human VDAC1 with Bcl-2 family
proteins. Biochem Biophys Res Commun. 305:989–996. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Duan M, Wang ZC, Wang XY, Shi JY, Yang LX,
Ding ZB, Gao Q, Zhou J and Fan J: TREM-1, an inflammatory
modulator, is expressed in hepatocellular carcinoma cells and
significantly promotes tumor progression. Ann Surg Oncol.
22:3121–3129. 2015. View Article : Google Scholar
|