Introduction
Ubiquitylation, in which the highly conserved
76-residue polypeptide ubiquitin is covalently attached to a lysine
residue of substrate proteins, mediates the targeted destruction of
ubiquitylated proteins by the ubiquitin-proteasome system (1–4).
The ubiquitin-mediated protein degradation pathway serves a crucial
role in the efficient and specific removal of misfolded proteins
and certain key regulatory proteins (5,6).
Ubiquitin and other ubiquitin-like proteins, including
autophagy-related protein 8, Ubiquitin-like protein ISG15, NEDD8
conjugating enzyme UBE2F and SUMO-conjugating enzyme UBC9, may all
be targeted to the same lysine residue, and in this way they are
involved in regulating a number of cellular processes including
transcription, the cell cycle and signal transduction pathways
(1,3,7,8).
Modified lysine residues, covalently attached not only to ubiquitin
but also to other ubiquitin-like proteins, may be identified by
high-throughput techniques (9,10).
In the present study, the term ‘ubiquitylation’ is used to refer to
ubiquitin and other ubiquitin-like protein modifications.
The lysine residues that are targeted for
ubiquitylation may also be modified by acetylation, and so
cross-talk between ubiquitylation and acetylation may control the
stability of target proteins (11). It is also known that the
cross-talk between ubiquitylation and phosphorylation may modulate
various regulatory networks (12). For example, ubiquitylation may
positively or negatively regulate various protein kinases, either
with or without their subsequent degradation (12–14).
A large number of genetic changes have accumulated
during human evolution and are responsible for the acquisition of
different human phenotypes (15,16). These genetic modifications include
the de novo generation of genes (17), emergence of novel transcript
isoforms (18) and loss of gene
function (19–21). The gain of novel phosphorylation
sites in proteins may result in the reorganization of the
regulatory circuits (22,23). The gain of lysine residues that
are subject to ubiquitylation during human evolution may alter the
timing of protein degradation, subcellular localization and the
activity of the target proteins (24). Similarly, the gain or loss of
N-glycosylation modification sites in conserved proteins may have
an effect on protein folding and trafficking, and on adhesion and
cell signaling (25,26).
In the present study, we hypothesized that the loss
of conserved ubiquitylation sites in highly conserved proteins
during evolution may modify the ubiquitin-mediated regulatory
network, which potentially results in the acquisition of novel
phenotypes. Mouse ubiquitylation data compiled in the Mammalian
Ubiquitination Site Database (mUbiSiDa) (27) and multiple sequence alignments of
orthologous proteins from 62 mammalian species were analyzed to
identify the lost conserved ubiquitylation sites in the Euarchonta
lineage leading to humans. The timepoint during evolution when the
ancestral ubiquitylated lysine residues may have disappeared was
then determined.
Materials and methods
Mouse ubiquitylation site data
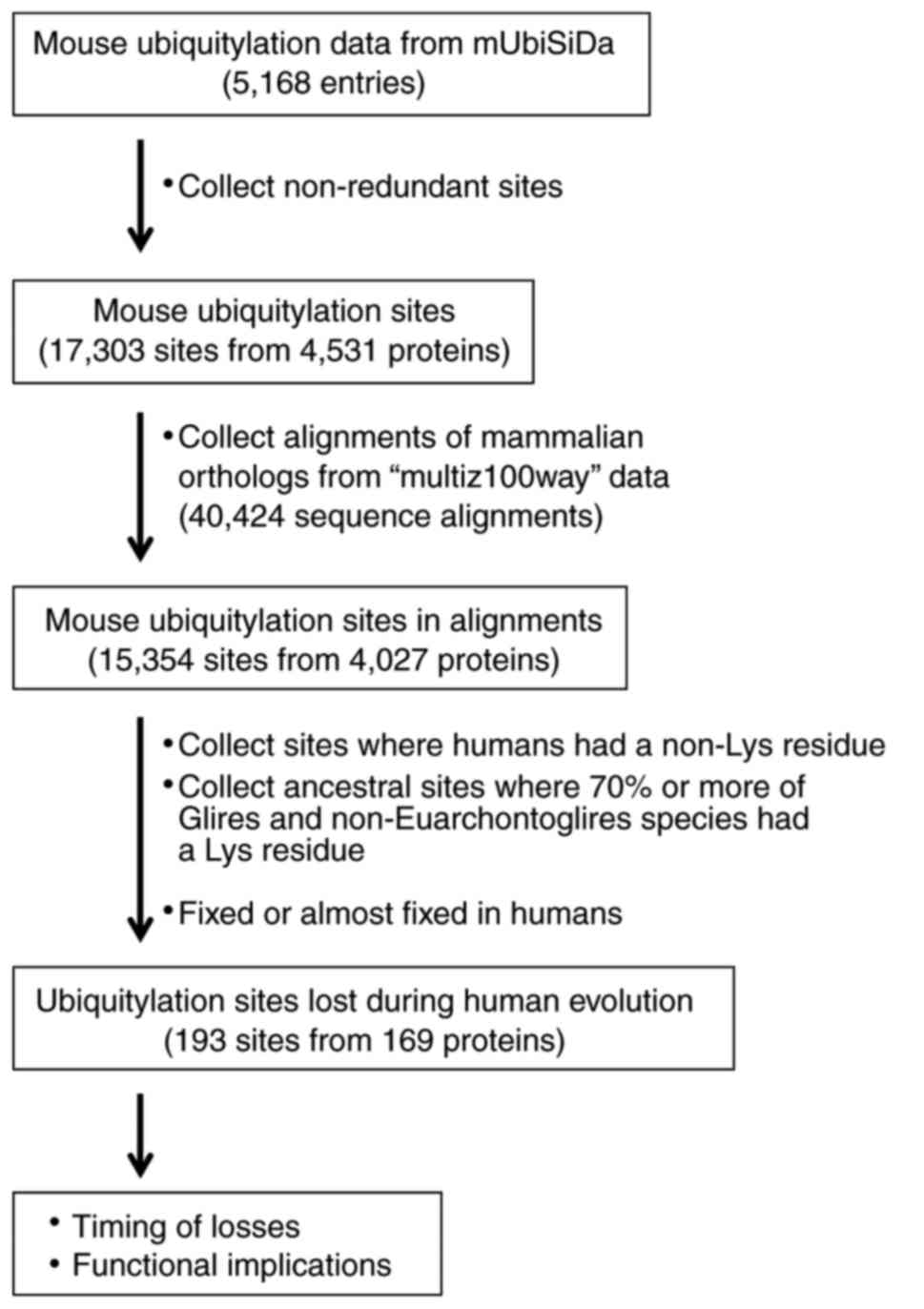
The mouse ubiquitylation site data were obtained
from the mUbiSiDa database (http://reprod.njmu.edu.cn/mUbiSiDa) to create a
non-human ubiquitylation site dataset (27). The database file
‘data_2013_10_22.txt’ was parsed to extract the mouse
ubiquitylation data entries; in total, there were 5,168 entries for
mouse proteins (Fig. 1). Each
entry contained one or more ubiquitylation sites from a protein
isoform sequence, which made the dataset highly redundant. Unique
ubiquitylation sites were obtained by comparing 13-amino acid (aa)
-long peptide sequences with the ubiquitylation site at the center.
Consequently, 17,303 non-redundant ubiquitylation sites were
identified from 4,531 mouse proteins.
Mammalian orthologous proteins
Multiple sequence alignments of mammalian proteins,
including from humans, were obtained from the University of
California Santa Cruz Genome Browser Database (http://genome.ucsc.edu) (28). The ‘CDS FASTA alignment from
multiple alignment’ data of the human hg38 genome track were
downloaded using the Table Browser tool. These alignment datasets
were derived from the ‘multiz100way’ alignments (http://hgdownload.cse.ucsc.edu/goldenPath/hg38/multiz100way)
(29), and included 62 mammalian
species: Humans (Homo sapiens) and 12 other Euarchonta
[chimpanzees (Pan troglodytes), gorillas (Gorilla gorilla
gorilla), orangutans (Pongo pygmaeus abelii), gibbons
(Nomascus leucogenys), rhesus macaques (Macaca
mulatta), crab-eating macaques (Macaca fascicularis),
baboons (Papio hamadryas), green monkeys (Chlorocebus
sabaeus), marmosets (Callithrix jacchus), squirrel
monkeys (Saimiri boliviensis), bush babies (Otolemur
garnettii) and Chinese treeshrews (Tupaia chinensis)],
13 Glires [squirrels (Spermophilus tridecemlineatus), lesser
Egyptian jerboas (Jaculus jaculus), prairie voles
(Microtus ochrogaster), Chinese hamsters (Cricetulus
griseus), golden hamsters (Mesocricetus auratus), mice
(Mus musculus), rats (Rattus norvegicus), naked
mole-rats (Heterocephalus glaber), guinea pigs (Cavia
porcellus), chinchillas (Chinchilla lanigera),
brush-tailed rats (Octodon degus), rabbits (Oryctolagus
cuniculus) and pikas (Ochotona princeps)], 25
Laurasiatheria [pigs (Sus scrofa), alpacas (Vicugna
pacos), Bactrian camels (Camelus ferus), dolphins
(Tursiops truncatus), killer whales (Orcinus orca),
Tibetan antelopes (Pantholops hodgsonii), cows (Bos
taurus), sheep (Ovis aries), domestic goats (Capra
hircus), horses (Equus caballus), white rhinoceroses
(Ceratotherium simum), cats (Felis catus), dogs
(Canis lupus familiaris), ferrets (Mustela putorius
furo), pandas (Ailuropoda melanoleuca), Pacific walruses
(Odobenus rosmarus divergens), Weddell seals
(Leptonychotes weddellii), black flying-foxes (Pteropus
alecto), megabats (Pteropus vampyrus), David's myotis
bats (Myotis davidii), microbats (Myotis lucifugus),
big brown bats (Eptesicus fuscus), hedgehogs (Erinaceus
europaeus), shrews (Sorex araneus) and star-nosed moles
(Condylura cristata)], 6 Afrotheria [elephants (Loxodonta
africana), cape elephant shrews (Elephantulus edwardii),
manatees (Trichechus manatus latirostris), cape golden moles
(Chrysochloris asiatica), tenrecs (Echinops telfairi)
and aardvarks (Orycteropus afer afer)], 1 Xenarthra
[armadillos (Dasypus novemcinctus)], 3 Marsupialia [opossums
(Monodelphis domestica), Tasmanian devils (Sarcophilus
harrisii) and wallabies (Macropus eugenii)], and 1
Monotremata [platypuses (Ornithorhynchus anatinus)].
Consequently, a total of 40,424 sequence alignments were
downloaded.
Identification of lost ubiquitylation
sites and timing of loss
The 13-aa sequences of the 17,303 unique mouse
ubiquitylation sites obtained from the mUbiSiDa database were
mapped to the mouse sequences within the 40,424 mammalian protein
sequence alignments; 15,354 sites were identified in 4,027
proteins. Each position that aligned with a mouse ubiquitylation
site was examined using a series of custom scripts in Perl
programming language (version 5.22.1) (https://www.perl.org/). Firstly, sites where the human
protein contained a non-lysine residue were collected, as it
indicated that the modifiable lysine residue was lost in the
lineage leading to humans. Following this, putative ancestral
ubiquitylation sites where ≥70% of Glires and non-Euarchontoglires
species contained a lysine residue were obtained; this condition
allowed independent losses in up to 30% of Glires or
non-Euarchontoglires species. As a result, a total of 193 sites
from 169 proteins were identified to be lost in the Euarchonta
lineage leading to humans following its divergence from the Glires
lineage. The timing of the loss of the ubiquitylated lysine residue
was determined by examining the multiple sequence alignment and the
phylogenetic tree of the mammals included in the present study.
Examination of human single nucleotide
polymorphisms
The dbSNP data (build 150) track available in the
UCSC Genome Browser was examined to identify potential
polymorphisms at the lost ubiquitylation sites of human proteins.
Allele frequency information was obtained from dbSNP (https://www.ncbi.nlm.nih.gov/snp) and Exome
Aggregation Consortium (ExAC; http://exac.broadinstitute.org) databases.
Examination of multiple modification
sites
Lysine residues may be mutually exclusively modified
by multiple post-translational modifications (PTMs), including
ubiquitylation, ubiquitin-like protein modification, acetylation,
and succinylation. To examine whether the mouse ubiquitylation
sites may be modified by other PTMs, UniProt database (https://www.uniprot.org) records of mouse proteins
were analyzed. When a ubiquitylation site is associated with the
term ‘MOD_RES’ in the feature table, it was considered that the
site may be also modified by other PTMs.
Results and Discussion
Identification of ancestral
ubiquitylation sites that are lost in the Euarchonta lineage
leading to humans
It was identified that 193 ancestral ubiquitylation
sites were lost in 169 human proteins since the Euarchonta lineage
diverged from the Glires lineage. A total of 19 proteins lost >1
putative ancestral ubiquitylation sites: The protein Titin, which
is the largest known protein, lost 5 sites; 2 proteins
[achaete-scute homolog 1 and ribosome-binding protein 1 (RRBP1)]
lost 3 sites each; 16 proteins [acetyl-CoA acetyltransferase,
mitochondrial, cytoplasmic aconitate hydratase,
long-chain-fatty-acid-CoA ligase 5, cytosolic
10-formyltetrahydrofolate dehydrogenase, bifunctional purine
biosynthesis protein PURH, complement component 3, carbonic
anhydrase 3, clustered mitochondria protein homolog,
dimethylaniline monooxygenase (N-oxide-forming) 1, nebulin, PDZ
domain containing 1 (PDZK1), short-chain dehydrogenase/reductase
family 42E member 1 (SDR42E1), protein SEC13 homolog, solute
carrier family 12 member 1, multidrug and toxin extrusion protein
1, and urocanate hydratase] lost 2 sites each; and the remaining
151 proteins lost 1 site each.
The number of conserved ubiquitylation sites that
were lost in each branch is demonstrated in Fig. 2, and were as follows: Humans, 8;
Hominini (humans and chimpanzees), 3; African great apes, 16; great
apes, 6; apes, 15; catarrhines, 33; simians, 75; primates, 26; and
Euarchonta, 11. Of the 193 sites that were lost in the lineage
leading to humans, 9 events occurred in human proteins following
the human-chimpanzee divergence.
A total of 2 human-specific lost
ubiquitylation sites are polymorphic in humans
Each of the 8 proteins [betaine homocysteine
S-methyltransferase (BHMT), cyclin and CBS domain divalent metal
cation transport mediator 3 (CNNM3), epoxide hydrolase 2 (EPHX2),
hydroxy-δ-5-steroid dehydrogenase, 3 β- and steroid δ-isomerase 7
(HSD3B7), PDZK1, RRBP1, SDR42E1, and solute carrier family 37
member 4 (SLC37A4)] that lost 1 conserved ubiquitylated lysine
residue following the human-chimpanzee divergence are summarized in
Table I. Examination of the human
variant data in dbSNP and ExAC databases indicated that the loss of
the ubiquitylated lysine was fixed in 6 of these 8 proteins; the 6
proteins were BHMT, CNNM3, EPHX2, HSD3B7, SDR42E1, and SLC37A4.
 | Table IHuman-specific loss of ancestral
ubiquitylation sites. |
Table I
Human-specific loss of ancestral
ubiquitylation sites.
| Case no. | Human
| Mouse
| Gene | Definition |
|---|
| Accessiona | Positionb | Sequencec | Accessiond | Positione | Sequencef |
|---|
| 34 | NP_001704.2 | 98 |
EKISGQEVNEAAC | O35490 | 98 |
EKISGQKVNEAAC | BHMT |
Betaine-homocysteine
S-methyltransferase |
| 49 | NP_060093.3 | 491 |
DDEYKVTISPQLL | Q32NY4 | 497 |
DDEYKVKISPQLL | CNNM3 | Cyclin and CBS
domain divalent metal cation transport mediator 3 (transcript
variant 1) |
| 68 | NP_001970.2 | 505 |
LVPQMSQHMEDWI | P34914 | 504 |
LRPEMSKNMEKWI | EPHX2 | Epoxide hydrolase 2
(transcript variant 1) |
| 92 | NP_079469.2 | 266 |
YDGSPYRSYEDFN | Q9EQC1 | 266 |
YDKSPYKSYEDFN | HSD3B7 |
Hydroxy-delta-5-steroid dehydrogenase, 3
beta- and steroid delta-isomerase 7 (transcript variant 1) |
| 128 | NP_001188254.1 | 171 |
PQGVAMRAGVLAD | Q9JIL4 | 171 |
PQGVAMKAGVLAD | PDZK1 | PDZ domain
containing 1 (transcript variant 2) |
| 143 | NP_001036041.1 | 936 |
LGRAATRLQELLK | Q99PL5 | 1,564 |
LGRAAIKLQELLK | RRBP1 | Ribosome binding
protein 1 (transcript variant 1) |
| 148 | NP_660151.2 | 31 |
GCALNQNGVHVIL | Q9D665 | 31 |
GCALNQKGARVIL | SDR42E1 | Short chain
dehydrogenase/reductase family 42E, member 1 |
| 159 | NP_001157751.1 | 132 |
LDPMPSEGKKGSL | Q6WG34 | 205 |
LDPAPSKGKKGSS | SLC37A4 | Solute carrier
family 37 member 4 (transcript variant 3) |
For 2 of these proteins, PDZK1 and RRBP1, the amino
acid position where the ubiquitylated lysine was lost indicated a
polymorphism. In the case of PDZK1, the lost ubiquitylation site
(amino acid position 171) was polymorphic (dbSNP accession number
rs138296787): The ancestral allele (AAA codon for lysine) and the
derived allele (AGA codon for arginine) were identified. The
frequencies of the 2 alleles demonstrated that the human-specific
derived allele forms the majority (96.70%) and this allele is,
therefore, almost fixed in modern humans. Notably, the derived G
allele demonstrated slight differences in frequency distribution
across the human populations, with the lowest frequency in Africans
(92.63%) and the highest in South Asians (99.04%). It is not known
whether the difference in frequency of the derived allele is
associated with the adaptation of human populations to local
environments.
Similarly, for the RRBP1 protein, position 936 also
indicated a polymorphism at the ubiquitylation site (rs2229887):
The derived allele (AGA codon for arginine) is the major allele
with a frequency of 99.70% compared to the ancestral allele (AAA
codon for lysine) and is, therefore, almost fixed in modern humans.
The frequency of the derived G allele was lowest in Africans
(97.01%), while it was 99.81–100.00% in other populations.
Therefore, human-specific losses of ubiquitylation
sites are fixed or almost fixed in modern humans. Presently, to the
best of our knowledge, there are no studies describing the
phenotypes associated with these polymorphisms in PDZK1 or
RRBP1.
Representative cases of human-specific
loss of a ubiquitylation site
Among the 8 cases in which a conserved ubiquitylated
lysine was lost in humans following the human-chimpanzee
divergence, 4 representative cases are demonstrated in Fig. 3. These are BHMT, CNNM3, RRBP1 and
SLC37A4.
The human BHMT protein (NCBI accession number
NP_001704.2) lost a conserved lysine at aa position 98 (case no.
34) subsequent to the human-chimpanzee divergence (Fig. 3A). Of the 62 mammalian species
analyzed in the present study, the lysine residue that was
demonstrated to be ubiquitylated in mice was identified to be
conserved in 60 species. A high level of sequence conservation of
the lysine residue and surrounding region suggests that the
ubiquitylation modification may be shared by other mammals. Humans
exhibit a glutamic acid residue at this position. Wallabies (M.
eugenii), members of the Marsupial family, have a glycine
residue, implying that they lost the lysine independently. Notably,
the same lysine residue was identified to be succinylated in mice
(30). Therefore, if the
modification at this lysine is conserved among mammals, the human
BHMT protein would have lost 2 types of modifications
concomitantly. The BHMT protein is a betainehomocysteine
S-methyltransferase that converts homocysteine to methionine using
betaine (trimethylglycine) and S-methylmethionine (31,32). The enzyme has been demonstrated to
be crucial for a wide range of biological processes, including
inner cell mass development in embryo-genesis (33) and normal brain development and
function (34). At present, to
the best of our knowledge, there have been no studies examining the
molecular functions directly associated with this modifiable lysine
in the mouse BHMT protein. However, due to the high level of
sequence conservation of the lysine sequence in mammals and the 2
known alternative modifications in the mouse protein, it is highly
likely that the loss of lysine in the human BHMT protein may be
implicated in the acquisition of a novel phenotype in humans.
The human CNNM3 (NP_060093.3) protein specifically
lost the lysine residue at aa position 491 (case no. 49), compared
with its mouse counterpart, which possesses a lysine known to be a
target for ubiquitylation (Fig.
3B). Instead, humans exhibit a threonine residue at this
position, whereas the other 60 mammalian species have a conserved
lysine residue. Pikas (O. princeps) are the only other
species without this lysine as shown in Fig. 3B. CNNM3 is a member of a family of
membrane-bound metal transporters that serve a critical role in
magnesium homeostasis (35,36). CNNM3 interacts with members of the
protein tyrosine phosphatases 4A family of proteins, also known as
the phosphatase of regenerating liver family, namely PTP4A2 (PRL2)
and PTP4A3 (PRL3), via its CBS domain (36,37). This interaction has been
implicated in the promotion of tumor formation and invasiveness in
animals (38,39). It would be valuable to investigate
whether the loss of the conserved ubiquitylation site at aa
position 491, which, according to the Pfam domain database, follows
the CBS domain at 382-446, affects the interaction between the
CNNM3 and PTP4A proteins.
During evolution, the human RRBP1 (NP_001036041.1)
lost 3 lysine residues that are ubiquitylated in mice: The loss at
aa position 936 (case no. 143) is human-specific, whereas those at
the other 2 positions are shared with other primates (case nos. 141
and 142). With respect to aa position 936, humans have an arginine
residue instead of a lysine (Fig.
3C). The lysine residue is conserved in all 56 mammalian
species in which the RRBP1 protein sequence is present; the
remaining 5 species did not contain an RRBP1 sequence, potentially
due to the incompleteness of their genomic sequences. RRBP1 is a
membrane-bound ribosome-binding protein identified on the rough
endoplasmic reticulum (ER) (40)
and is implicated in the regulation of the unfolded protein
response (UPR) (41,42). Overexpression of the RRBP1 protein
has been demonstrated in numerous types of cancer and is correlated
with the progression and survival of these types of cancer
(41,43–45). A molecular functional study
focusing on the loss of this ubiquitylation site in human RRBP1 may
provide a means of identifying potential human-specific traits
associated with the UPR and tumorigenesis.
During evolution, following the human-chimpanzee
divergence, the human protein SLC37A4 (NP_001157751.1) lost a
ubiquitylation site at aa position 132 (case no. 159; (Fig. 3D). At this position, humans have a
glutamic acid, whereas 58 other mammals have a conserved lysine.
Black flying foxes (P. alecto) and megabats (P.
vampyrus) have also independently lost this lysine and have an
arginine residue instead. SLC37A4 is the glucose 6-phosphate
transporter localized in the ER membrane, and is responsible for
transporting glucose-6-phosphate from the cytoplasm into the ER
lumen (46). SLC37A4 serves a key
role in the maintenance of blood glucose homeostasis, and its
inactivation causes glycogen storage disease types Ib and Ic
(47,48). It has also been identified as
being a key regulator in the initiation step for autophagy
(49). It would be valuable to
explore if the loss of this conserved ubiquitylation site in the
human SLC37A4 protein is involved with the emergence of a novel
human-specific trait associated with glucose homeostasis.
Certain lost ubiquitylated lysines are
target sites for other PTMs
Lysine residues are target sites for other PTMs,
including acetylation, methylation, succinylation and malonylation
(50). Therefore, the loss of a
lysine residue may affect regulation not only by ubiquitylation,
but also by other modifications. To determine whether the
ubiquitylation sites lost in human proteins were potential targets
for other modifications, the mouse sequences in the UniProt
database were additionally examined. This analysis revealed that 17
ubiquitylated lysines were identified to be also modified by
acetylation or succinylation in mice (Table II): 4 sites were modified by
either acetylation or succinylation; 5 sites by acetylation alone;
and 8 sites by succinylation alone.
| Table IIOther modifications at the mouse
ubiquitylated lysine residues that were lost during human
evolution. |
Table II
Other modifications at the mouse
ubiquitylated lysine residues that were lost during human
evolution.
| Case no. | Gene | Human RefSeq | Human position | Clade | Mouse UniProt | Mouse position | Modification |
|---|
| 7 | ACADM | NP_001120800.1 | 83 | Simians | P45952 | 79 | Acetylation |
| 8 | ACAT1 | NP_000010.1 | 245 | Catarrhines | Q8QZT1 | 242 | Acetylation,
Succinylation |
| 12 | ACO2 | NP_001089.1 | 517 | Catarrhines | Q99KI0 | 517 | Acetylation,
Succinylation |
| 23 | ALDH2 | NP_000681.2 | 441 | Simians | P47738 | 443 | Acetylation |
| 31 | ATP5J | NP_001003696.1 | 84 | Great apes | P97450 | 84 | Acetylation,
Succinylation |
| 33 | BAAT | NP_001692.1 | 40 | Catarrhines | Q91X34 | 40 | Succinylation |
| 34 | BHMT | NP_001704.2 | 98 | Humans | O35490 | 98 | Succinylation |
| 40 | CAT | NP_001743.1 | 13 | African great
apes | P24270 | 13 | Succinylation |
| 51 | CPS1 | NP_001866.2 | 603 | Primates | Q8C196 | 603 | Acetylation,
Succinylation |
| 53 | CYB5R3 | NP_001123291.1 | 27 | Simians | Q9DCN2 | 50 | Acetylation |
| 63 | EHHADH | NP_001957.2 | 331 | Euarchonta | Q9DBM2 | 329 | Succinylation |
| 65 | ENO3 | NP_443739.3 | 71 | African great
apes | P21550 | 71 | Acetylation |
| 68 | EPHX2 | NP_001970.2 | 505 | Humans | P34914 | 504 | Succinylation |
| 77 | GLYCTK | NP_660305.2 | 200 | Catarrhines | Q8QZY2 | 200 | Acetylation |
| 82 | GOT1 | NP_002070.1 | 318 | Catarrhines | P05201 | 318 | Succinylation |
| 86 | HADHA | NP_000173.2 | 620 | Simians | Q8BMS1 | 620 | Succinylation |
| 147 | SCP2 | NP_002970.2 | 40 | Simians | P32020 | 40 | Succinylation |
Although it must be confirmed experimentally, it is
highly likely that the same PTMs may occur in other species, as the
lysine residues are highly conserved. Therefore, these lysines may
be target sites for multiple alternative PTMs, which may be
involved in refining the molecular functions of the target proteins
so that they modulate a wide range of biological processes. Loss of
these multiple modification sites may result in rewiring of the
associated protein regulatory networks.
Of the 17 sites identified as being targets for
multiple PTMs, 2 sites were lost in the human protein [BHMT (case
no. 34) and EPHX2 (case no. 68)] following the divergence of humans
and chimpanzees. These are the human BMHT protein aa position 98
(NP_001704.2, case no. 34) and the EPHX2 protein aa position 505
(NP_001970.2, case no. 68). The corresponding positions in the
mouse proteins were demonstrated to be subject to succinylation
(30). Molecular functional
studies of these 2 cases may reveal instances of rearrangement of
the protein regulatory networks during human evolution.
Certain lost ubiquitylation sites may be
compensated for by novel sites
A previous study indicated that ubiquitylation sites
may move from their original position to a nearby lysine that
appears during the evolutionary process (51). This means that the existence of a
ubiquitylation site within a segment, rather than the exact
position, is important for the regulation of protein function.
Therefore, a modification site may move around over time, within a
small window of the peptide sequence. This phenomenon has been
commonly identified to occur for sites modified by phosphorylation
(52). A previous study on the
gain of novel ubiquitylation sites during human evolution have
indicated that certain conserved lysines disappeared and novel
ubiquitylated sites appeared along the human lineage, suggesting
that the ubiquitylation site may have shifted during human
evolution (24).
To assess if a potential turnover event occurred
along with the loss of a ubiquitylation site, a 13-aa window with
the lost site at the center was used in the present study. When a
novel lysine residue appeared at a nearby flanking site, it was
considered a turnover event. There were 8 such cases, in which a
novel lysine was identified in the human protein at a site up to 6
residues away from the lost site: These were case nos. 83, 85, 98,
100, 119, 138, 147 and 162.
In 3 cases (nos. 83, 100 and 119), the gain of the
novel lysine coincided with the loss of the ancestral lysine,
markedly implying a turnover of the ubiquitylation site. The
G-protein coupled receptor 39 protein lost an ancestral lysine at
the position corresponding to the human protein aa position 253
(NP_001499.1) in catarrhines, but a novel lysine appeared at the
next position (+1 position) in the same clade (case no. 83). At aa
position 146 (NP_005359.1) in the human MB protein, a lysine
disappeared in apes, but a novel lysine appeared at the -5 position
concomitantly (case no. 100). The human nitric oxide synthase,
brain protein lacked a lysine at aa position 221 (NP_001191147.1)
that was lost in simians, but a novel lysine was present at the +3
position in all simians (case no. 119). The coincidence of the loss
and gain of lysines in these cases suggests that the novel lysine
may compensate for the lost one.
In two cases (nos. 138 and 162), the gain of a novel
lysine residue preceded the loss of the ancestral lysine. The ape
regulator of microtubule dynamics protein 2 proteins lacked an
ancestral lysine at the position corresponding to aa position 41 in
the human protein (NP_001164263.1), but a novel lysine at the -3
position appeared in the ancestral catarrhine, which preceded this
loss (case no. 138). At aa position 495 in the human sodium/glucose
cotransporter 1 protein (NP_001243243.1), the ancestral lysine was
lost in catarrhines, but a new lysine at +4 position was present in
all simians (case no. 162). Therefore, it is possible that these
proteins gained a novel ubiquitylation site that replaced the
function of the ancestral site and was subsequently lost.
In the remaining 3 cases (nos. 85, 98, and 147), the
emergence of the novel lysine appeared to occur much later,
following the loss of the ancestral lysine. For example, the
primate 3-hydroxyanthranilate 3,4-dioxygenase protein lacked an
ancestral lysine at the position corresponding to aa position 8 in
the human protein (NP_036337.2), but humans exhibited a novel
lysine residue at the +4 position (case no. 85). Concurrently, in
the catarrhine ancestor, the plastin-2 protein lost a lysine at the
position corresponding to aa position 530 in the human protein
(NP_002289.2), but humans gained a novel lysine at the +3 position
(case no. 98). As these 2 sites in these human proteins were not
shared with other primates, these events cannot be considered as a
timely turnover event. However, it is possible that humans restored
a ubiquitylation site in the corresponding segments of these
proteins. In the case of the non-specific lipid-transfer protein,
the ancestral lysine at the position corresponding to aa position
40 in the human protein (NP_002970.2) was absent in simians, but a
novel lysine was present in apes at the +4 position (case no.
147).
Loss of ubiquitylation sites may lead to
the development of novel phenotypes
The gain or loss of PTM sites in proteins may lead
to changes in the activity of a protein and may contribute to the
development to a novel phenotype (53,54). In this regard, certain novel
ubiquitylation sites that have evolved during human evolution have
been suggested to be associated with the development of novel
beneficial traits (24). For
example, a novel ubiquitylation site in the human general
transcription and DNA repair factor IIH helicase subunit XPD
protein is associated with enhanced DNA repair activity in humans,
which may have developed to provide extra protection for skin cells
from ultraviolet light-induced DNA damages when ancestral humans
lost their body hair and started to live in open plains (55).
Ubiquitylation serves crucial roles in numerous
biological processes, including innate and adaptive immune
responses, by regulating pattern-recognition receptor signaling and
activating dendritic cells (56–58). Mutations of ubiquitin ligases and
disruption of ubiquitylation modifications of proteins have
frequently been observed in immune disorders, cancer, and
neurodegenerative diseases including Parkinson’s disease and
Huntington’s disease (7,59–;62). There are numerous previous studies
indicating that natural or experimental mutations of lysines within
PTMs may disrupt normal protein function, which results in disease
phenotypes (63–67). However, the present study
demonstrated that the 8 ubiquitylation sites that were lost during
human evolution were completely or almost fixed in modern humans,
and the remaining 185 cases were shared in humans and other
primates, implying that these losses do not confer a disease
phenotype in extant primates. The loss of a ubiquitylation site may
be compensated for by other genetic changes or be selected for when
it increases the fitness of the species.
Therefore, the loss of a ubiquitylation site during
organismal evolution may lead to the emergence of an advantageous
phenotype and may be fixed by selection. Molecular functional
studies of the conserved mammalian ubiquitylation sites that are
lost in human proteins may reveal potential beneficial phenotypes
associated with the loss. The ancestral ubiquitylation sites
identified in the present study may serve as a good resource for
investigating the association between the loss of ubiquitylation
sites and the emergence of novel phenotypes during the evolution
towards modern humans.
Acknowledgments
Not applicable.
Abbreviations:
|
aa
|
amino acid
|
|
ER
|
endoplasmic reticulum
|
|
mUbiSiDa
|
mammalian ubiquitination site
database
|
|
PTM
|
post-translational modification
|
|
SNP
|
single nucleotide polymorphism
|
|
UPR
|
unfolded protein response
|
Funding
The present study was supported by the National
Research Foundation of Korea funded by the Government of Korea
(grant no., 2017R1A1B4005866).
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request. The complete list and detailed sequence alignments of the
193 lost ubiquitylation sites identified in the present study are
available through the Figshare repository (https://figshare.com/s/2f6d03011988cf071a1c and
https://figshare.com/s/af4ab-d53ed0a9bad36f2,
respectively).
Authors’ contributions
YH conceived and designed the experiments; DP, CJG,
HK, JSL and YH analyzed data. YH wrote the paper.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kerscher O, Felberbaum R and Hochstrasser
M: Modification of proteins by ubiquitin and ubiquitin-like
proteins. Annu Rev Cell Dev Biol. 22:159–180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Komander D and Rape M: The ubiquitin code.
Annu Rev Biochem. 81:203–229. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Swatek KN and Komander D: Ubiquitin
modifications. Cell Res. 26:399–422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kwon YT and Ciechanover A: The ubiquitin
code in the ubiquitin-proteasome system and autophagy. Trends
Biochem Sci. 42:873–886. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Konstantinova IM, Tsimokha AS and
Mittenberg AG: Role of proteasomes in cellular regulation. Int Rev
Cell Mol Biol. 267:59–124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Preston GM and Brodsky JL: The evolving
role of ubiquitin modification in endoplasmic reticulum-associated
degradation. Biochem J. 474:445–469. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Popovic D, Vucic D and Dikic I:
Ubiquitination in disease pathogenesis and treatment. Nat Med.
20:1242–1253. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stojkovic K, Wing SS and Cermakian N: A
central role for ubiquitination within a circadian clock protein
modification code. Front Mol Neurosci. 7:692014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim W, Bennett EJ, Huttlin EL, Guo A, Li
J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al: Systematic
and quantitative assessment of the ubiquitin-modified proteome. Mol
Cell. 44:325–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wagner SA, Beli P, Weinert BT, Nielsen ML,
Cox J, Mann M and Choudhary C: A proteome-wide, quantitative survey
of in vivo ubiquitylation sites reveals widespread regulatory
roles. Mol Cell Proteomics. 10:M1110132842011. View Article : Google Scholar
|
|
11
|
Caron C, Boyault C and Khochbin S:
Regulatory cross-talk between lysine acetylation and
ubiquitination: Role in the control of protein stability.
BioEssays. 27:408–415. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hunter T: The age of crosstalk:
Phosphorylation, ubiquitination, and beyond. Mol Cell. 28:730–738.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen ZJ: Ubiquitin signalling in the
NF-kappaB pathway. Nat Cell Biol. 7:758–765. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Al-Hakim AK, Zagorska A, Chapman L, Deak
M, Peggie M and Alessi DR: Control of AMPK-related kinases by USP9X
and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem
J. 411:249–260. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li WH and Saunders MA: News and views: The
chimpanzee and us. Nature. 437:50–51. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Varki A and Altheide TK: Comparing the
human and chimpanzee genomes: Searching for needles in a haystack.
Genome Res. 15:1746–1758. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ruiz-Orera J, Hernandez-Rodriguez J, Chiva
C, Sabidó E, Kondova I, Bontrop R, Marqués-Bonet T and Albà MM:
Origins of de novo genes in human and chimpanzee. PLoS Genet.
11:e10057212015. View Article : Google Scholar
|
|
18
|
Kim DS and Hahn Y: Identification of
human-specific transcript variants induced by DNA insertions in the
human genome. Bioinformatics. 27:14–21. 2011. View Article : Google Scholar
|
|
19
|
Hahn Y, Jeong S and Lee B: Inactivation of
MOXD2 and S100A15A by exon deletion during human evolution. Mol
Biol Evol. 24:2203–2212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu J, Sanborn JZ, Diekhans M, Lowe CB,
Pringle TH and Haussler D: Comparative genomics search for losses
of long-established genes on the human lineage. PLoS Comput Biol.
3:e2472007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oh HJ, Choi D, Goh CJ and Hahn Y: Loss of
gene function and evolution of human phenotypes. BMB Rep.
48:373–379. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lynch VJ, May G and Wagner GP: Regulatory
evolution through divergence of a phosphoswitch in the
transcription factor CEBPB. Nature. 480:383–386. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim DS and Hahn Y: Identification of novel
phosphorylation modification sites in human proteins that
originated after the human-chimpanzee divergence. Bioinformatics.
27:2494–2501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim DS and Hahn Y: Gains of ubiquitylation
sites in highly conserved proteins in the human lineage. BMC
Bioinformatics. 13:3062012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim DS and Hahn Y: The acquisition of
novel N-glycosylation sites in conserved proteins during human
evolution. BMC Bioinformatics. 16:292015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim DS, Choi D and Hahn Y: Loss of
ancestral N-glycosylation sites in conserved proteins during human
evolution. Int J Mol Med. 36:1685–1692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen T, Zhou T, He B, Yu H, Guo X, Song X
and Sha J: mUbi-SiDa: A comprehensive database for protein
ubiquitination sites in mammals. PLoS One. 9:e857442014. View Article : Google Scholar
|
|
28
|
Rosenbloom KR, Armstrong J, Barber GP,
Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo
L, Haeussler M, et al: The UCSC genome browser database: 2015
update. Nucleic Acids Res. 43:D670–D681. 2015. View Article : Google Scholar :
|
|
29
|
Blanchette M, Kent WJ, Riemer C, Elnitski
L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green
ED, et al: Aligning multiple genomic sequences with the threaded
blockset aligner. Genome Res. 14:708–715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park J, Chen Y, Tishkoff DX, Peng C, Tan
M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, et al:
SIRT5-mediated lysine desuccinylation impacts diverse metabolic
pathways. Mol Cell. 50:919–930. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Finkelstein JD, Harris BJ and Kyle WE:
Methionine metabolism in mammals: Kinetic study of
betaine-homocysteine methyl-transferase. Arch Biochem Biophys.
153:320–324. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garrow TA: Purification, kinetic
properties, and cDNA cloning of mammalian betaine-homocysteine
methyltransferase. J Biol Chem. 271:22831–22838. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee MB, Kooistra M, Zhang B, Slow S,
Fortier AL, Garrow TA, Lever M, Trasler JM and Baltz JM: Betaine
homocysteine meth-yltransferase is active in the mouse blastocyst
and promotes inner cell mass development. J Biol Chem.
287:33094–33103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Prieur EAK, Pjetri E, Zeisel SH and
Jadavji NM: Reduced brain volume and impaired memory in betaine
homocysteine S-methyltransferase knockout mice. Appl Physiol Nutr
Metab. 42:1228–1231. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Islam Z, Hayashi N, Inoue H, Umezawa T,
Kimura Y, Doi H, Romero MF, Hirose S and Kato A: Identification and
lateral membrane localization of cyclin M3, likely to be involved
in renal Mg2+ handling in seawater fish. Am J Physiol
Regul Integr Comp Physiol. 307:R525–R537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gulerez I, Funato Y, Wu H, Yang M, Kozlov
G, Miki H and Gehring K: Phosphocysteine in the PRL-CNNM pathway
mediates magnesium homeostasis. EMBO Rep. 17:1890–1900. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang H, Kozlov G, Li X, Wu H, Gulerez I
and Gehring K: PRL3 phosphatase active site is required for binding
the putative magnesium transporter CNNM3. Sci Rep. 7:482017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hardy S, Uetani N, Wong N, Kostantin E,
Labbé DP, Bégin LR, Mes-Masson A, Miranda-Saavedra D and Tremblay
ML: The protein tyrosine phosphatase PRL-2 interacts with the
magnesium transporter CNNM3 to promote oncogenesis. Oncogene.
34:986–995. 2015. View Article : Google Scholar
|
|
39
|
Kostantin E, Hardy S, Valinsky WC,
Kompatscher A, de Baaij JH, Zolotarov Y, Landry M, Uetani N,
Martínez-Cruz LA, Hoenderop JG, et al: Inhibition of PRL-2-CNNM3
protein complex formation decreases breast cancer proliferation and
tumor growth. J Biol Chem. 291:10716–10725. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Savitz AJ and Meyer DI: Identification of
a ribosome receptor in the rough endoplasmic reticulum. Nature.
346:540–544. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tsai HY, Yang YF, Wu AT, Yang CJ, Liu YP,
Jan YH, Lee CH, Hsiao YW, Yeh CT, Shen CN, et al: Endoplasmic
reticulum ribo-some-binding protein 1 (RRBP1) overexpression is
frequently found in lung cancer patients and alleviates
intracellular stress-induced apoptosis through the enhancement of
GRP78. Oncogene. 32:4921–4931. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hyde M, Block-Alper L, Felix J, Webster P
and Meyer DI: Induction of secretory pathway components in yeast is
associated with increased stability of their mRNA. J Cell Biol.
156:993–1001. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Telikicherla D, Marimuthu A, Kashyap MK,
Ramachandra YL, Mohan S, Roa JC, Maharudraiah J and Pandey A:
Overexpression of ribosome binding protein 1 (RRBP1) in breast
cancer. Clin Proteomics. 9:72012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liang X, Sun S, Zhang X, Wu H, Tao W, Liu
T, Wei W, Geng J and Pang D: Expression of ribosome-binding protein
1 correlates with shorter survival in Her-2 positive breast cancer.
Cancer Sci. 106:740–746. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pan Y, Cao F, Guo A, Chang W, Chen X, Ma
W, Gao X, Guo S, Fu C and Zhu J: Endoplasmic reticulum
ribosome-binding protein 1, RRBP1, promotes progression of
colorectal cancer and predicts an unfavourable prognosis. Br J
Cancer. 113:763–772. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gerin I, Veiga-da-Cunha M, Achouri Y,
Collet JF and Van Schaftingen E: Sequence of a putative glucose
6-phosphate translocase, mutated in glycogen storage disease type
Ib. FEBS Lett. 419:235–238. 1997. View Article : Google Scholar
|
|
47
|
Veiga-da-Cunha M, Gerin I, Chen YT, de
Barsy T, de Lonlay P, Dionisi-Vici C, Fenske CD, Lee PJ, Leonard
JV, Maire I, et al: A gene on chromosome 11q23 coding for a
putative glucose- 6-phosphate translocase is mutated in
glycogen-storage disease types Ib and Ic. Am J Hum Genet.
63:976–983. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chou JY and Mansfield BC: The SLC37 family
of sugar-phosphate/phosphate exchangers. Curr Top Membr.
73:357–382. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ahn HH, Oh Y, Lee H, Lee W, Chang JW, Pyo
HK, Nah do H and Jung YK: Identification of glucose-6-phosphate
transporter as a key regulator functioning at the autophagy
initiation step. FEBS Lett. 589:2100–2109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Azevedo C and Saiardi A: Why always
lysine? The ongoing tale of one of the most modified amino acids.
Adv Biol Regul. 60:144–150. 2016. View Article : Google Scholar
|
|
51
|
Hagai T, Toth-Petroczy A, Azia A and Levy
Y: The origins and evolution of ubiquitination sites. Mol Biosyst.
8:1865–1877. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Moses AM, Liku ME, Li JJ and Durbin R:
Regulatory evolution in proteins by turnover and lineage-specific
changes of cyclin-dependent kinase consensus sites. Proc Natl Acad
Sci USA. 104:17713–17718. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gulzar N, Dingerdissen H, Yan C and
Mazumder R: Impact of nonsynonymous single-nucleotide variations on
post-translational modification sites in human proteins. Methods
Mol Biol. 1558:159–190. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lu L, Li Y, Liu Z, Liang F, Guo F, Yang S,
Wang D, He Y, Xiong J, Li D and He F: Functional constraints on
adaptive evolution of protein ubiquitination sites. Sci Rep.
7:399492017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lehmann AR: DNA repair-deficient diseases,
xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy.
Biochimie. 85:1101–1111. 2003. View Article : Google Scholar
|
|
56
|
Hu H and Sun SC: Ubiquitin signaling in
immune responses. Cell Res. 26:457–483. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zohaib A, Duan X, Zhu B, Ye J, Wan S, Chen
H, Liu X and Cao S: The role of ubiquitination in regulation of
innate immune signaling. Curr Issues Mol Biol. 18:1–10. 2016.
|
|
58
|
Liu X, Wang Q, Chen W and Wang C: Dynamic
regulation of innate immunity by ubiquitin and ubiquitin-like
proteins. Cytokine Growth Factor Rev. 24:559–570. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chakraborty J, Basso V and Ziviani E: Post
translational modification of Parkin. Biol Direct. 12:62017.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fulda S, Rajalingam K and Dikic I:
Ubiquitylation in immune disorders and cancer: From molecular
mechanisms to therapeutic implications. EMBO Mol Med. 4:545–556.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Narayan S, Bader GD and Reimand J:
Frequent mutations in acetylation and ubiquitination sites suggest
novel driver mechanisms of cancer. Genome Med. 8:552016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ortega Z and Lucas JJ:
Ubiquitin-proteasome system involvement in Huntington’s disease.
Front Mol Neurosci. 7:772014. View Article : Google Scholar
|
|
63
|
McClurg UL, Cork DMW, Darby S, Ryan-Munden
CA, Nakjang S, Mendes Côrtes L, Treumann A, Gaughan L and Robson
CN: Identification of a novel K311 ubiquitination site critical for
androgen receptor transcriptional activity. Nucleic Acids Res.
45:1793–1804. 2017. View Article : Google Scholar :
|
|
64
|
Bonacci T, Audebert S, Camoin L, Baudelet
E, Iovanna JL and Soubeyran P: Regulation of NUB1 activity through
non-proteolytic Mdm2-mediated ubiquitination. PLoS One.
12:e01699882017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Glorian V, Allègre J, Berthelet J,
Dumetier B, Boutanquoi PM, Droin N, Kayaci C, Cartier J, Gemble S,
Marcion G, et al: DNA damage and S phase-dependent E2F1
stabilization requires the cIAP1 E3-ubiquitin ligase and is
associated with K63-poly-ubiquitination on lysine 161/164 residues.
Cell Death Dis. 8:e28162017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tessadori F, Giltay JC, Hurst JA, Massink
MP, Duran K, Vos HR, van Es RM; Deciphering Developmental Disorders
Study; Scott RH, van Gassen KLI, et al: Germline mutations
affecting the histone H4 core cause a developmental syndrome by
altering DNA damage response and cell cycle control. Nat Genet.
49:1642–1646. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kim OJ: A single mutation at lysine 241
alters expression and trafficking of the D2 dopamine receptor. J
Recept Signal Transduct Res. 28:453–464. 2008. View Article : Google Scholar : PubMed/NCBI
|