Introduction
Ischemic stroke, accounting for ~85% of all cases of
strokes (1), is a leading cause
of mortality and disability (2,3).
Diabetes mellitus (DM), characterized by hyperglycemia, is an
important risk factor for ischemic stroke, and ~30% of patients
with stroke have diabetes (4).
Hyperglycemia can cause microvascular lesions, and the blood-brain
barrier (BBB), which acts as a semipermeable barrier between
peripheral blood and the central nervous system (CNS), is damaged
during the acute phase of diabetic stroke (5-7).
In addition, increased vascular permeability and the spillover of
macromolecules and pro-inflammatory factors can exacerbate
post-stroke injury and lead to hemorrhagic transformation, posing
additional therapeutic challenges (8,9).
At present, tissue plasminogen activator is the only drug used for
thrombolytic therapy after a stroke, but its therapeutic time
window is extremely narrow, and the risk of bleeding is significant
(10). Even with thrombolytic
therapy, the mortality and disability rates in patients who have
suffered a stroke remain high (11). Therefore, it is necessary to
develop new treatments for ischemic stroke in hyperglycemic
patients.
Autophagy is a physiological process characterized
by the degradation and recycling of cellular components. Autophagy
is activated as a survival response in hypoxic conditions, low
nutrient availability and other forms of stress to remove damaged
proteins and dysfunctional organelles (12). Below a certain threshold,
autophagy is beneficial; however, when this threshold is exceeded,
it can lead to cell death (13-15). BBB damage is a key event in the
secondary CNS damage following stroke, which is often associated
with the aberrant autophagy of brain microvascular endothelial
cells (16). Studies have
revealed that excessive autophagy can lead to cell dysfunction and
death, as shown by the activation of the autophagy pathway
following oxygen-glucose deprivation (OGD), the loss of tight
junction (TJ) protein expression and the increase in endothelial
cell permeability (17,18). Furthermore, it has been revealed
that the inhibition of autophagy significantly reduces endothelial
cell permeability. Similarly, a recent study revealed that,
following autophagy inhibitor intervention in a rat model of middle
cerebral artery occlusion (MCAO), Evans blue (EB) leakage and TJ
protein expression were increased, which protected BBB integrity
(16). In a recent study,
autophagy was induced in a pMCAO mouse model of ischemic stroke and
oxygen-glucose deprivation intervention was performed in
vitro, following which a significant amount of pericytes
(important constituent cells of the BBB) underwent cell death and
BBB integrity was destroyed. The inhibition of autophagy could also
significantly promote the survival of pericytes and effectively
reduce BBB damage (19). Studies
have revealed that autophagic activity is significantly increased
and the BBB is damaged in diabetic mice following cerebral
ischemia/reperfusion (I/R) injury, and that the inhibition of
autophagy can attenuate brain injury (20,21). For example, a recent study
revealed that apigenin attenuates brain injury by inhibiting
autophagy in cerebral vascular endothelial cells, exerting a
protective effect against I/R injury (22). In addition, icariside II can
inhibit autophagy, reduce MMP9 and MMP2 levels, significantly
reduce EB leakage following cerebral I/R in rats and effectively
alleviate BBB injury (1). The
PI3K/AKT/mTOR signaling pathway has been revealed to regulate
several biological processes, such as proliferation,
differentiation and apoptosis (23). The PI3K/AKT/mTOR pathway has also
been revealed to be closely associated with autophagy (24,25). In addition, Liu et al
(26) demonstrated that the
PI3K/AKT signaling pathway was inhibited in a hyperglycemic
environment, Autophagy-related circular RNA can attenuate autophagy
and ROS production in Schwann cells subjected to hyperglycemia by
promoting the activation of the PI3K/AKT/mTOR pathway (27).
Selenium (symbol Se) is an essential trace element
that is necessary for animal and plant health (28,29). Selenium plays an important role
in antioxidation by affecting the activity of glutathione
peroxidase (30,31). In addition to its antioxidative
activity, selenium has become the focus of intensive research for
its potential role in the regulation of autophagy. Selenium can
reverse the inhibitory effect of citreoviridin on mTOR2 and inhibit
autophagy in cardiomyocytes (32). Selenium can also inhibit lactate
dehydrogenase (LDH) release and cadmium-induced autophagy by
regulating calcium homeostasis in leghorn male hepatoma cells
(33). Our previous study showed
that sodium selenite exerts a neuroprotective effect by modulating
autophagy-related proteins (34); however, whether it exerts a
protective effect on the BBB following cerebral I/R injury in
hyperglycemic conditions remains unknown.
In the present study, an in vivo MCAO model
of diabetic rats was established by co-culturing bEnd.3 mouse brain
microvascular endothelial and MA-h mouse astrocyte-hippocampal
cells to simulate the BBB in hyperglycemic conditions by subjecting
them to OGD/reoxygenation (OGD/R) in vitro. The purpose of
the present study was to explore the molecular mechanisms
underlying the protective effects of selenium on the BBB following
I/R injury in hyperglycemic rats.
Materials and methods
In vivo experiments
Animals
All animal experiments and procedures were conducted
in accordance with the Chinese Laboratory Animal Use Guidelines
(35) and followed the
Laboratory Animal Care and Use guidelines of Ningxia Medical
University (Yinchuan, China). All animal procedures were approved
by the Institutional Animal Care and Use Committee (IACUC) of the
Ningxia Medical University (Yinchuan, China). A total of 100 adult
male Sprague-Dawley rats (weight, 220-240 g) at 8 weeks of age were
provided by Ningxia Medical University (approval no.
IACUC-NYLAC-2019-064). Animals were housed under controlled
temperature (22-24°C) and humidity (60±5%) conditions in a 12-h
light/dark cycle and they were allowed free access to food and
water. A total of 4 weeks after the induction of diabetes with
streptozotocin (STZ), the rats were randomly divided into five
groups: The hyperglycemia Sham (Sham), hyperglycemia I/R (HIR),
sodium selenite-treated hyperglycemia I/R (Se), 3-methyladenine
(3-MA)-treated hyperglycemia I/R (3-MA) and sodium selenite plus
3-MA-treated hyperglycemia I/R (Se + 3-MA) groups (20 rats per
group). For the inhibition of autophagy, 3-MA (30 µg; Merck
KGaA) was dissolved in 10 µl saline (36) and injected into the ipsilateral
ventricle immediately after MCAO.
Induction of diabetes
Diabetes was induced in the rats by 12-h fasting
followed by intraperitoneal (i.p.) administration of STZ (60 mg/kg;
Merck KGaA) (37). Blood glucose
levels were assessed after 72 h, with levels of >16.8 mmol/l
considered hyperglycemic. Diabetic rats in the Se group received
i.p. injections of sodium selenite solution (0.4 mg/kg/day) and
cerebral ischemia was induced 4 weeks later (38).
Cerebral I/R injury
Cerebral I/R injury was induced by unilateral MCAO
(39). This was carried out
following the induction of diabetes and sodium selenite treatment
for 4 weeks. The animals were anesthetized by inhalation of 3%
isoflurane (induction) and maintained with 1.5-2.0% isoflurane
during surgical procedures, and the right internal carotid artery
was exposed and the thread bolt was inserted. Following MCAO (30
min), the thread bolt was removed to restore normal blood flow. The
sham group underwent the same procedure, but the middle cerebral
artery was only exposed instead of occluding it. Immediately after
the surgery, the 3-MA treatment group was intracerebroventricularly
(i.c.v.) injected with 3-MA (30 µg dissolved in 10 µl
saline) (36). Normal saline (10
µl) was injected in the Sham group. Successful induction was
defined as the appearance of hemiplegia 24 h after MCAO. Rats were
euthanized by an injection of excessive sodium pentobarbital (800
mg/kg) (40); successful
euthanasia was defined as the disappearance of reflex and
respiration, and cardiac arrest. Following euthanasia, the brain
tissues of 6 rats from each group were randomly selected for
triphenyltetrazolium chloride (TTC) staining, and those from the
other 10 rats were collected for follow-up biochemical tests.
Neurological deficit scoring
Two investigators who were blinded to the groups
conducted a neurological deficit evaluation 24 h after MCAO,
according to the Z-longa score grading criteria: 0, Asymptomatic;
1, left forelimb internal rotation; 2, left rotation; 3, left
rotation dumping; 4, no spontaneous activity and/or exhibiting
depressed levels of consciousness (41). Rats with scores of ≥2 were
selected for the subsequent experiment.
Brain water content determination
The rat brains were quickly collected following
anesthesia and weighed (wet weight). Next, the brains were dried at
100°C for 24 h and weighed (dry weight) (42). The formula for calculating the
brain water content was: (wet weight-dry weight)/wet weight
×100%.
Infarct volume assessment
A total of 24 h after MCAO, the brains were
harvested and cut into 2-mm thick coronal sections. The slices were
stained with 3-5-TTC (1.5%; Merck KGaA) for 15 min at 37°C and then
fixed with 4% paraformaldehyde for 12 h at room temperature. Normal
tissue appeared red, while the infarcted area was stained white.
Brain sections were visualized by a digital camera and ImageJ 1.46
(National Institutes of Health) was used to assess the infarct area
and correct for edema (43). The
formula used for calculating the cerebral infarct volume is as
follows: Cerebral infarct volume=contralateral hemispheric
volume-(ipsilateral hemispheric volume-infarcted area volume).
BBB permeability evaluation
The animals were anesthetized 24 h after MCAO, and
EB (2%; Merck KGaA) was injected into the tail vein at a dose of 5
ml/kg. Each hemisphere was homogenized in 50% trichloroacetic acid,
followed by centrifugation at 12,000 × g for 15 min at 4°C. Next, 1
ml supernatant was collected and the optical density value was
measured at 632 nm using a fluorescence spectrophotometer (44).
Transmission electron microscopy
(TEM)
The penumbral cortex was collected following
cerebral I/R and fixed overnight in 2% glutaraldehyde at 4°C,
followed by three washes with 0.1 M sodium dimethyl arsenate
buffer. The cortex was then soaked for 2 h at 4°C in 1% osmic acid
and rinsed three times with dimethyl arsenic sodium buffer. Next,
the cortex was dehydrated by immersing in a gradient series of
alcohol (30, 50, 70, 80, 90 and 100%) for 10 min at each
concentration at room temperature. The sample was then embedded in
epoxy resin, polymerized, sliced into ultra-thin sections and
placed on the copper net. Following 1% uranium acetate and 0.2%
lead citrate double staining for 10 min respectively at room
temperature, the sample was observed using a transmission electron
microscope (magnification, ×8,000; model no. H7800; Hitachi,
Ltd.).
Immunofluorescence staining
Autophagy-associated proteins Beclin-1, LC3B and p62
were detected by immunofluorescence. Briefly, paraffin sections (4
µm thick) of brain tissue were incubated in 0.3% Triton
X-100 for 30 min at room temperature, and then incubated overnight
at 4°C with rabbit anti-Beclin-1 (1:50; product code ab62557;
Abcam), rabbit anti-LC3B (1:100; product code ab192890; Abcam) and
rabbit anti-p62 (1:50; product code ab109012; Abcam) primary
antibodies, followed by incubation at room temperature for 1 h with
tetramethylrhodamine/FITC-conjugated anti-rabbit secondary
antibodies (1:100; product code bsm-33179; BIOSS). Finally, samples
were fixed with DAPI sealant (10 µg/ml; Shanghai Yeasen
Biotechnology Co., Ltd.) at room temperature in the dark for 5 min
and analyzed under a fluorescence microscope (magnification, ×400;
Olympus Corporation).
In vitro experiments
Cell identification, co-culture and
treatments
The bEnd.3 mouse brain microvascular endothelial and
MA-h mouse astrocyte-hippocampal cell lines were purchased from The
Cell Bank of Type Culture Collection of The Chinese Academy of
Sciences. Mouse anti-CD34 (1:100; product code ab54208; Abcam) and
mouse anti-glial fibrillary acid protein (GFAP; 1:100; product code
ab4648; Abcam) were used for bEnd.3 and MA-h cell identification,
respectively; the method is similar to tissue immunofluorescence.
Cells were co-cultured in vitro to simulate the damage to
the BBB caused by hyperglycemic conditions and I/R injury (45,46). Briefly, bEnd.3 and MA-h cells
were first cultured separately in DMEM/F12 (Cytiva) in an incubator
(94% relative humidity, 5% CO2 and 37°C). Next,
poly-lysine-coated Transwell (0.4 µm; Corning, Inc.) inserts
were removed, and the MA-h cell line (5×105 cells/ml)
was seeded into the outer chamber, cultured for 6 h, and then the
chamber was turned over to be cultured for another 6 h. Next, the
bEnd.3 cell line (density, 5×105 cells/ml) was seeded on
the insert and incubation was continued for an additional 12 h.
D-glucose (50 mM; control) and hyperglycemia + OGD/R
(H + O) were used to simulate the BBB injury induced by
hyperglycemic conditions and I/R injury. Prior to OGD/R, the
high-sugar medium was replaced with sugar-free medium and cells
were cultured in a hypoxic chamber (oxygen concentration, 1%) for 1
h (38). After 24 h of
reoxygenation, cells were collected for analysis. Sodium selenite
(100 nM) was added 24 h before OGD/R induction. To determine
whether selenium inhibited autophagy and to determine the
underlying mechanisms, 3-MA (10 nmol; Merck KGaA), wortmannin
(Wort; 10 nmol; Merck KGaA; a specific PI3K inhibitor) and
insulin-like growth factor 1 (IGF-1; 100 nmol ng/ml; Merck KGaA; a
PI3K activator) were added to the culture medium 2 h before sodium
selenite treatment. All reagents were dissolved in PBS (47) (vehicle). First, the cells were
divided into the following groups: Control (hyperglycemia), H + O
(hyperglycemia + OGD/R), Se (hyperglycemia + OGD/R + sodium
selenite), 3-MA (hyperglycemia + OGD/R + 3-MA) and Se + 3-MA
(hyperglycemia + OGD/R + sodium selenite + 3-MA) groups. Then the
cells were divided into the following groups: Vehicle
(hyperglycemia + OGD/R + vehicle), Se (hyperglycemia + OGD/R +
sodium selenite), IGF-1 (hyperglycemia + OGD/R + IGF-1), Wort
(hyperglycemia + OGD/R + wortmannin) and Se + Wort (hyperglycemia +
OGD/R + sodium selenite + wortmannin) groups. All tests were
repeated three times.
Cell viability assessment
Cell viability was determined using a Cell Counting
Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc.),
according to the manufacturer's instructions. Briefly, bEnd.3 and
MA-h cells were plated into a 96-well plate (5,000 cells/well) and
treated with different reagents (25, 50, 100, 200 and 400 nmol
sodium selenite, and 17.5, 30, 40, 50, 60 and 70 mM D-glucose). In
the control group, 17.5-mM glucose diluted in DMEM/F12 medium was
used. Following the addition of CCK-8 (10 µl/well, incubated
at 37°C for 2 h), the absorbance was measured at 450 nm using a
microplate reader.
In vitro BBB permeability
The in vitro permeability of the BBB was
evaluated using fluorescein sodium dye (cat. no. F6377; Merck KGaA)
(48). After the co-cultured
cells had fused, they were subjected to hyperglycemia (D-glucose,
50 mM) and hyperglycemia + OGD/R (H + O), and treated with sodium
selenite (100 nM) and fluorescein sodium dye for 24 h. Next, 5
µg/ml fluorescein sodium was added to the upper Transwell
chamber, followed by incubation at 37°C for 30 min. Finally, 100
µl medium was collected from the basal chamber and the
absorbance was measured at 485/535 nm using a
spectrophotometer.
Western blot analysis
Co-cultured cells and rat ischemic penumbral tissue
were collected and lysed with RIPA lysis buffer (Beyotime Institute
of Biotechnology). Protein concentration was determined using the
BCA protein analysis kit (Beyotime Institute of Biotechnology).
Equivalent amounts of protein samples (30 µg/well) were
separated by electrophoresis via 6-15% SDS-PAGE, transferred to
PVDF membranes (EMD Millipore), blocked with 5% skimmed milk for 70
min at 4°C and incubated overnight with primary antibodies at 4°C.
The antibodies used were as follows: Anti-zonula occludens-1 (ZO-1;
1:1,000; cat. no. bs-1329R; BIOSS), anti-claudin-5 (1:1,000;
product code ab131259; Abcam), anti-occludin (1:1,000; product code
ab216327; Abcam), anti-Beclin-1 (1:2,000), anti-LC3B (1:1,000),
anti-p62 (1:1,000; product code ab109012; Abcam),
anti-phosphorylated (p)-PI3K (1:1,000; cat. no. bs-6417R; BIOSS),
anti-PI3K (1:1,000; product code ab191606; Abcam), anti-p-AKT
(1:2,000; cat. no. 4060S; Cell Signaling Technology, Inc.),
anti-AKT (1:2,000; cat. no. 4685S; Cell Signaling Technology,
Inc.), anti-p-mTOR (1:1,000; cat. no. 5536S; Cell Signaling
Technology, Inc.) and anti-mTOR (1:1,000; cat. no. 2983S; Cell
Signaling Technology, Inc.). The membranes were washed with
TBS-Tween-20 (1:1,000) and incubated with secondary antibodies
(1:5,000; cat. no. 33101ES60; Shanghai Yeasen Biotechnology Co.,
Ltd.) for 1 h at room temperature. The bands were visualized by ECL
(EMD Millipore). The ImageJ software (version 1.47; National
Institutes of Health) was used for densitometric analysis.
RNA isolation and reverse
transcription-quantitative (RT-q) PCR
Total RNA was extracted from brain ischemic penumbra
tissues and co-culture cells with TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and first strand cDNA
was synthesized using a reverse transcription kit (PrimeScript™ RT
Reagent kit-Perfect Real-Time; Takara Bio, Inc.). The kit was used
according to the manufacturer's protocol. GAPDH was used as the
control. The SYBR Green PCR Master Mix kit (Takara Biotechnology
co., Ltd.) was used for RT-qPCR analysis. The thermocycling
conditions were as follows: Initial denaturation at 95°C for 8 min,
40 cycles of denaturation at 95°C for 15 sec, annealing at 55°C for
30 sec and extension at 70°C for 25 sec. The relative expression of
mRNA was analyzed using the 2−ΔΔCq method (49). The primers used were as follows:
Beclin-1 forward, 5′-CTG CAT TGG CTA CCA GCC CAG-3′ and reverse,
5′-TCC TCA CCA CTC GGG CTC A-3′; LC3B forward, 5′-AGT CTC GAT AAA
GTG CCA CG-3′ and reverse, 5′-GGG CAC CAT ATC GGC CAT CA-3′; p62
forward, 5′-TGC AGG GCT ACC TCA CAC TC-3′ and reverse, 5′-AAC CTC
ACT CAG CGT GTC CA-3′; and GAPDH forward, 5′-AGT GCC TCG GTC GTC
TCA TA-3′ and reverse, 5′-ATG AAG GGG TGC AGG ATG GC-3′.
Statistical analysis
The data are expressed as the mean ± SD. Statistical
analysis was performed using SPSS 20.0 (IBM Corp.). To analyze the
difference between means, one-way ANOVA and Kruskal Wallis test
with a least significant difference post hoc test was performed,
such as Dunnett's and Tukey's post hoc tests. P<0.05 was
considered to indicate a statistically significant difference. Each
experiment was repeated at least three times.
Results
Selenium attenuates cerebral I/R injury
in diabetic rats
The flow diagram of the in vivo experiment is
illustrated in Fig. 1A. To
investigate whether selenium exerted a protective effect on rats
during I/R injury in hyperglycemic conditions, diabetic rats were
subjected to MCAO, and the neurological deficit score, infarct
volume, brain water content and pathological changes were analyzed
24 h later. The neurological deficit score of the HIR group was
significantly higher than that of the Sham group after 24 h of I/R.
Conversely, neurological deficit scores in the Se, 3-MA and Se +
3-MA groups were significantly lower than those of the HIR group
(Fig. 1B). Furthermore, selenium
significantly reduced the brain water content (Fig. 1C) and infarct volume (Fig. 1D and E) compared with the HIR
group, indicating that selenium therapy can reduce cerebral I/R
injury in hyperglycemic conditions.
 | Figure 1Selenium attenuates cerebral I/R
injury in diabetic rats. (A) Flow diagram of the in vivo
experiment. (B) Neurological deficit scores and (C) brain water
content after 24 h of MCAO. (D) Representative triphenyltetrazolium
chloride-stained slices. Pale areas correspond to infarcted
tissues. (E) Bar-graph revealing infarct volumes (n=4 per group).
Experiments were repeated 3 times for each condition. Scale bar,
1.0 cm. Results are expressed as the mean ± SD.
*P<0.05 and **P<0.01 vs. the sham
group; #P<0.05 vs. the HIR group. I/R,
ischemia/reperfusion; MCAO, middle cerebral artery occlusion; HIR,
hyperglycemia ischemia/reperfusion; TTC, triphenyltetrazolium
chloride; STZ, streptozotocin; WB, western blotting; IF,
immunofluorescence; TEM, transmission electron microscopy; 3-MA,
3-methyladenine; Se, sodium selenite. |
Selenium protects BBB integrity
To further investigate the protective effects of
selenium, BBB permeability was assessed by measuring EB leakage. As
anticipated, hyperglycemic animals subjected to MCAO revealed
increased EB leakage after 24 h of reperfusion. Conversely, the Se
and 3-MA groups revealed reduced EB leakage when compared with the
HIR group (Fig. 2A).
The TJ proteins ZO-1, claudin-5 and occludin are
important determinants of the integrity and paracellular
permeability of the BBB (50).
Their expression was assessed by western blot analysis, and it was
revealed that TJ protein expression in the HIR group was
significantly reduced when compared with the Sham group.
Conversely, a significant increase in TJ protein expression was
observed in the Se, 3-MA and Se + 3-MA groups when compared with
the HIR group (Fig. 2B-E).
I/R injury in hyperglycemic conditions
increases autophagy but selenium reverses this effect
To investigate the role of autophagy in I/R injury
in hyperglycemic conditions, the expression of the
autophagy-associated proteins Beclin-1, LC3B and p62 was assessed
by western blot analysis and immunofluorescence (n=4 per group).
Immunofluorescence staining revealed an increased expression of
Beclin-1 and LC3B, but a decreased expression of p62 (Fig. 3A-D) in the HIR group compared
with the Sham group. Of note, selenium administration significantly
decreased the expression of Beclin-1 and LC3B, but increased that
of p62 (Fig. 3A-D). As revealed
in Fig. 3E-I, the western blot
analysis results confirmed the immunofluorescence data, since
selenium significantly reduced the expression of Beclin-1 and LC3B,
but increased that of p62.
BBB ultrastructural changes and autophagy were also
evaluated by TEM. It was revealed that, in the Sham group, the BBB
was intact, the mitochondrial morphology and plasma membrane were
normal, and mitochondrial crests were arranged regularly. In the
other four groups, the integrity of the BBB was compromised: The
mitochondria exhibited different degrees of crest rupture,
dissolution and vacuolation, and varying numbers of autophagosomes
could be observed. The integrity of the BBB was damaged the most,
and mitochondrial autophagosomes were most abundant in the HIR
group. Conversely, the integrity of the BBB was only slightly
compromised and the number of autophagosomes was decreased in the
selenium-treated and 3-MA groups (Fig. 4A).
The expression of proteins associated with the PI3K
pathway was also evaluated. A lower expression of p-PI3K/PI3K,
p-AKT/AKT and p-mTOR/mTOR was observed in the HIR group compared
with the Sham group. Of note, selenium reversed these changes
(Fig. 4B-E). These results
indicated that selenium has the same effects as 3-MA, suggesting
that selenium reduces BBB damage following cerebral I/R injury in
hyperglycemic conditions by inhibiting excessive autophagy.
In vitro, selenium increases cell
viability and protects the BBB, suppressing autophagy following
OGD/R in hyperglycemic conditions
CD34 and GFAP are specific markers of brain
microvascular endothelial cells and astrocytes, respectively
(51,52). Immunofluorescence staining was
used for bEnd.3 and MA-h cell identification (Fig. 5A and B). To determine the optimal
concentrations of glucose and sodium selenite for the in
vitro assays, cell viability was assessed using a CCK-8 assay.
Co-cultured cells were treated with different concentrations of
D-glucose for 24 h. The results revealed that the co-cultured cells
were significantly damaged when the concentration of D-glucose
reached 50 mM (Fig. 6A cell
viability: 56.4±2.6%; P<0.05 vs. the control). Therefore, 50 mM
D-glucose was used for the subsequent experiments. As revealed in
Fig. 6B, selenium exerted a
protective effect under hyperglycemic conditions and after OGD/R
injury (cell viability in 100 nmol selenium: 89.2±2.7%, P<0.05
vs. the H + O group). There was no significant difference in cell
viability between the 100 nmol selenium and control groups,
indicating that selenium itself had no toxic effects on the
co-cultured cells. The protective effects of selenium peaked at 100
nmol, and thus, this concentration was used for the subsequent
experiments.
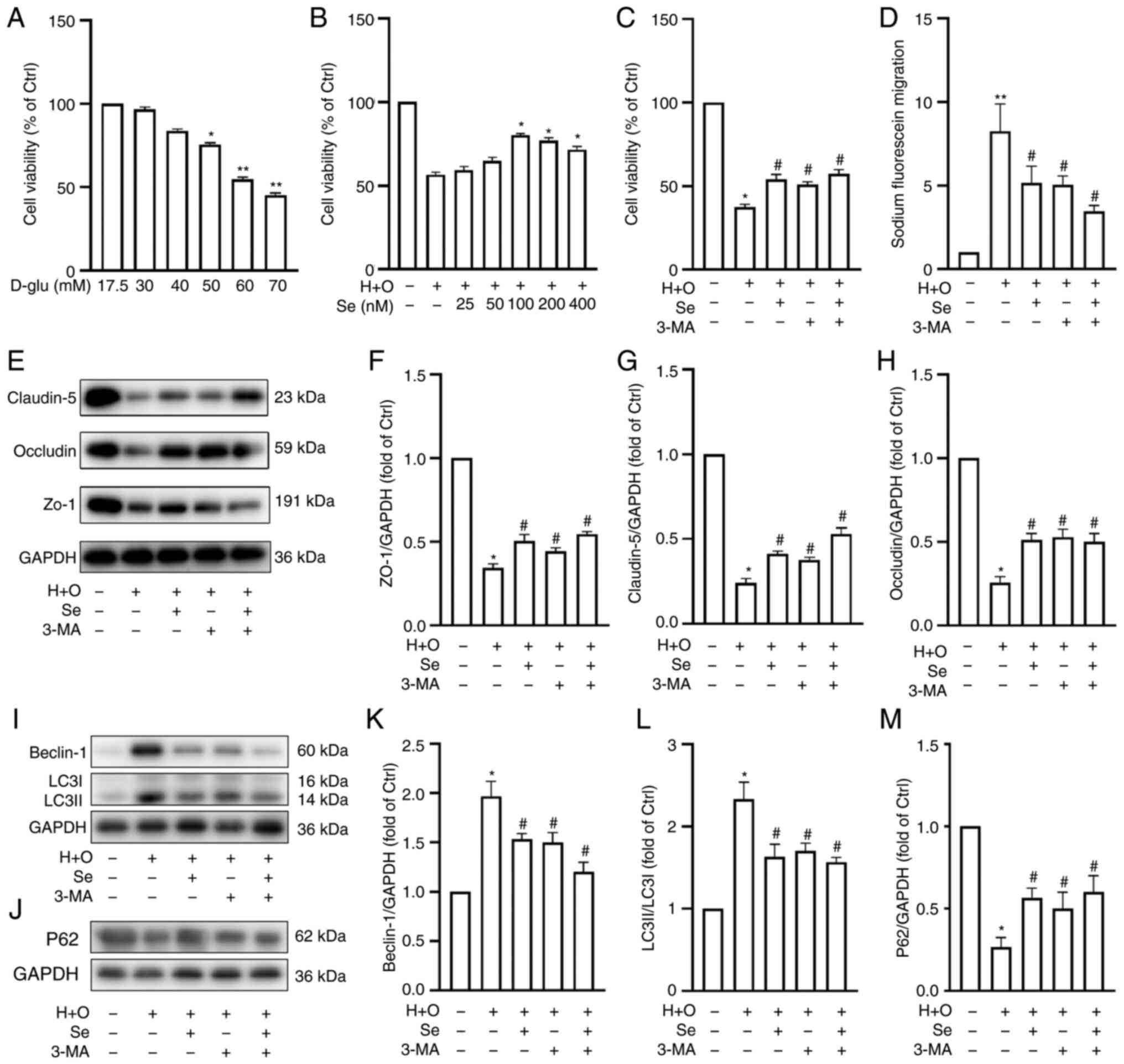 | Figure 6Effects of selenium on cell
viability, fluorescein leakage and expression of TJ and
autophagy-related proteins in vitro. Cell viability under
different (A) D-glucose and (B) selenium concentrations, and (C)
after 24 h of reoxygenation following 1 h of OGD plus 50 mM glucose
(H + O). Sodium selenite, 100 nM; and 3-methyladenine, 50 nM. (D)
Fluorescein leakage after OGD/R in hyperglycemic conditions. (E)
Representative western blots revealing TJ protein expression. (F-H)
Western blot analysis semi-quantification of TJ proteins. (I and J)
Western blots revealing Beclin-1, LC3B and p62 expression. (K-M)
Western blot analysis semi-quantification of Beclin-1, LC3B and
p62. Experiments were repeated 3 times for each condition and each
time-point. Results are expressed as the mean ± SD.
*P<0.05 and **P<0.01 vs. the control
group; #P<0.05 vs. the H + O group. TJ, tight
junction; OGD/R, oxygen-glucose deprivation/reoxygenation; 3-MA,
3-methyladenine; Se, sodium selenite. |
To further investigate the protective effects of
selenium against BBB damage, cell viability and BBB permeability
were assessed in the control, H + O, Se, 3-MA and Se + 3-MA groups.
Hyperglycemia and OGD/R decreased cell viability (Fig. 6C) and increased permeability to
sodium fluoride (Fig. 6D) when
compared with the control group. Conversely, selenium increased
cell viability (Fig. 6C) and
decreased permeability to sodium fluoride following OGD/R under
hyperglycemic conditions (Fig.
6D).
Furthermore, ZO-1, occludin and claudin-5 expression
was significantly decreased following OGD/R under hyperglycemic
conditions, as determined by western blot analysis. Conversely,
selenium increased the levels of expression of these TJ proteins
compared with the H + O group (Fig.
6E-H). These results indicated that selenium protects the BBB
following OGD/R under hyperglycemic conditions in vitro.
Conversely, selenium reduced Beclin-1 and LC3B
expression but increased that of p62 compared with the H + O group
(Fig. 6I-M). These results were
consistent with the in vivo results and indicated that
selenium may protect the integrity of the BBB by inhibiting
autophagy.
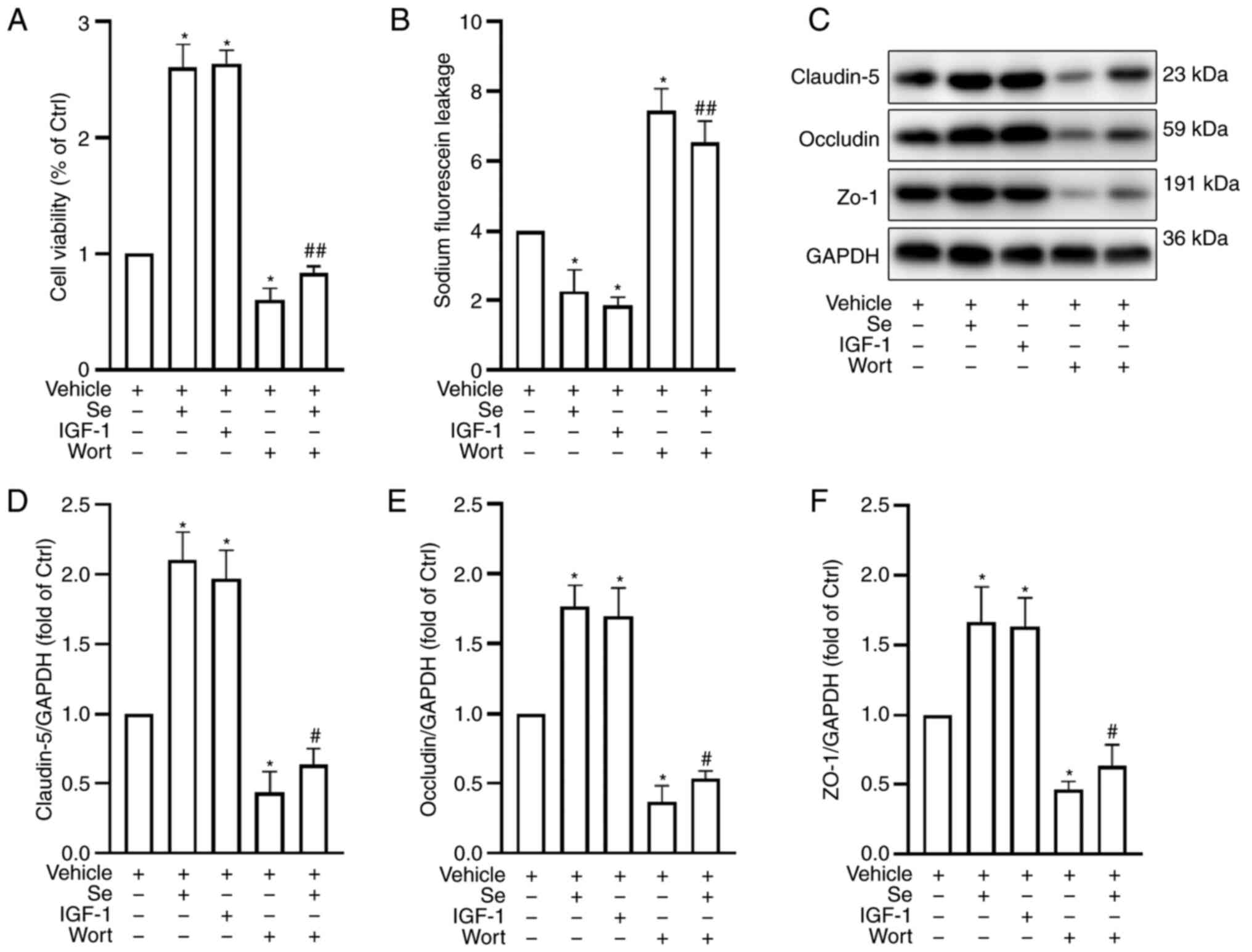
PI3K pathway inhibition reduces the
protective effects of selenium
To explore the mechanisms underlying the protective
effects of selenium on the BBB, cell viability, fluorescein leakage
and TJ protein expression were assessed following the addition of
Wort (a specific PI3K inhibitor) and insulin-like growth factor 1
(IGF-1; a PI3K activator). The cells were divided into the
following groups: Vehicle (H + O + vehicle), Se, IGF-1, Wort and Se
+ Wort groups. Following treatment with selenium, cell viability
increased (Fig. 7A) and
permeability to sodium fluoride decreased (Fig. 7B) compared with the vehicle
group, which was similar to the result following treatment with
IGF-1. Conversely, Wort counteracted the beneficial effects of
selenium.
In addition, TJ protein expression in the Wort group
decreased significantly compared with the Se and IGF-1 groups
(Fig. 7C-F). These results
indicated that selenium improved BBB function through the PI3K
signaling pathway.
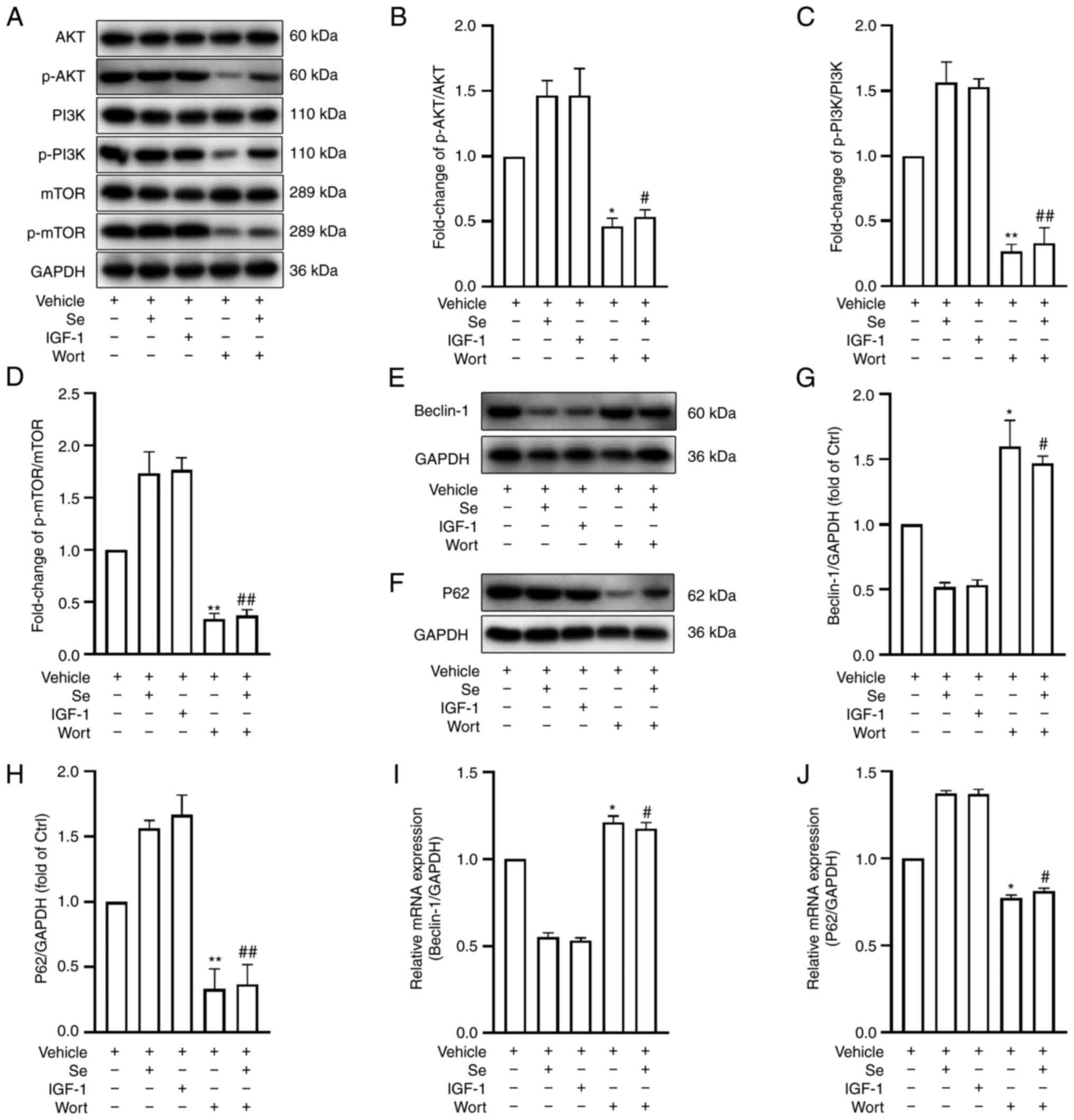
Pharmacologic modulation of the PI3K
signaling pathway enhances or inhibits autophagy following OGD/R
under hyperglycemic conditions
To gain further insights into the mechanisms
underlying the protective effects of selenium on the BBB, the PI3K
signaling pathway was pharmacologically modulated and the
expression of autophagy-related proteins was evaluated by western
blot analysis. It was revealed that IGF-1 markedly increased the
p-PI3K/PI3K, p-AKT/AKT and p-mTOR/mTOR ratios compared with the
vehicle group. (Fig. 8A-D).
Conversely, PI3K pathway protein expression was decreased following
the addition of Wort (Fig.
8A-D). Furthermore, the expression levels of the autophagy
proteins Beclin-1 and p62 were assessed by western blot analysis
and RT-qPCR. The results revealed that IGF-1 decreased the
expression of Beclin-1, but increased that of p62 (Fig. 8E-H), while Wort had the opposite
effects. Similar results were observed in terms of Beclin-1 and p62
mRNA expression (Fig. 8I and J).
In conclusion, the activation or inhibition of the PI3K pathway was
revealed to downregulate or upregulate autophagy, respectively.
Selenium inhibits autophagy by activating
the PI3K/AKT/mTOR signaling pathway
To verify whether selenium inhibits autophagy
through the activation of the PI3K/AKT/mTOR signaling pathway, PI3K
signaling pathway protein expression was assessed by western blot
analysis following the addition of Wort. A clear reduction was
identified in the Se + Wort group when compared with the Se group
(Fig. 8A-D). Furthermore, the
addition of Wort significantly increased the expression of Beclin-1
but decreased that of p62 (Fig.
8E-H). Consistent with the western blot analysis results,
Beclin-1 mRNA levels were significantly upregulated and p62 levels
were downregulated (Fig. 8I and
J). In conclusion, the pharmacological inhibition of the PI3K
pathway neutralized the selenium-induced effects on autophagy,
suggesting that selenium acts through the PI3K signaling
pathway.
Discussion
To the best of our knowledge, this is the first
study demonstrating that selenium significantly reduces cerebral
I/R injury-induced BBB damage in diabetic rats by inhibiting
autophagy through the PI3K/AKT/mTOR signaling pathway.
Ischemic stroke is one of the main causes of
disability worldwide. High glucose is an important risk factor for
ischemic stroke. Acute hyperglycemia and chronic diabetes can
aggravate this condition (53).
High glucose levels can lead to microangiopathy, including
thickening of the capillary basement membrane, changes in the
intercellular charges and an increase in vascular permeability
(54). Collectively, these
factors can lead to a high incidence of ischemic stroke.
Experimental studies have revealed that, compared with non-diabetic
ischemic stroke, diabetic stroke can lead to more serious vascular
dysfunction and inflammatory response and result in damage to the
brain parenchyma (55,56). Currently, the treatment
modalities for stroke in diabetic patients are relatively limited,
whereas the disability and fatality rates in these patients are
significantly increased (11).
As a physical diffusion barrier, the BBB prevents
harmful substances from entering the brain tissue through the
blood. The destruction of the BBB is a common clinical
manifestation of CNS diseases, including stroke, and one of the
causes of secondary damage to the brain parenchyma (57). During a stroke, the hypoxic
microenvironment is formed by the whole penumbra after cerebral
ischemia injury, resulting in BBB damage and increased permeability
due to the interruption of the blood supply; BBB injury occurs in
the acute phase after a stroke and is one of the first steps in the
aggravation of stroke damage (4). However, there are few studies on
the role of BBB in the pathology of stroke. Therefore, regulating
the permeability of the BBB to protect the CNS from secondary
injury may be a potential strategy for the treatment of CNS
diseases, including stroke. Several experimental studies have
revealed that damage to the BBB leads to increased permeability and
hemorrhagic transformation in diabetic animals (55,58,59). The BBB is composed of endothelial
cells, astrocytic terminal processes, pericytes and a basement
membrane, which prevent microscopic and neurotoxic substances from
entering the CNS (60). TJs
between the endothelial cells form a diffusion barrier. These TJs
are formed by proteins such as ZO-1, occludin and claudin-5
(61). TJs in mice with type 2
diabetes were revealed to be damaged following a stroke, increasing
the infarcted area and aggravating the neurological function
defects (62). The results of
the present study revealed that the integrity of the BBB was
compromised following I/R injury in hyperglycemic conditions. Both
in vivo and in vitro experiments revealed that the
expression of TJ proteins decreased, while the permeability of the
BBB increased. The expression of TJs was decreased in diabetic rats
after cerebral I/R (P<0.01), whereas the expression of TJs was
increased after selenium intervention (P<0.05) in vitro.
Selenium treatment could increase the expression of TJ proteins,
reduce EB leakage and improve the cell viability of brain
microvascular endothelial cells, thereby reducing damage to the
BBB. It is possible that in vitro, the co-cultured cells
could not completely simulate BBB injury after HIR in vivo.
The construction of BBB in vitro will be further improved in
our future studies to render it more consistent with the ischemic
microenvironment in vivo.
Autophagy is a physiological process that maintains
the stability of the intracellular environment that degrades and
recovers damaged proteins and organelles through the lysosomal
pathway. Autophagy plays a central role in several physiological
and pathological processes (63,64). The autophagic process can be
activated by nutritional deprivation or other stressful conditions.
Previous studies have revealed that autophagy is upregulated
following cerebral I/R in diabetes (2,65,66). Autophagy is characterized by the
formation of autophagosomes that can engulf cytosolic organelles.
Beclin-1, LC3B and p62 are the main autophagy-related proteins
(67,68). Beclin-1 triggers autophagy and
LC3B forms autophagosomes, whereas p62 is negatively correlated
with autophagic activity (69).
Growing evidence has indicated that overactivated autophagy under
pathological conditions can trigger cell death and BBB damage; for
example, following a stroke in diabetic mice, the expression of
autophagy-related proteins was increased, while that of the TJ
protein occludin was decreased, and 3-MA reversed these effects
(70,71). Wu et al reported that
Dl-3n-butylphthalide can exert neuroprotective effects by
inhibiting autophagy and blocking TJ protein loss (72). Our previous study also revealed
that hyperglycemia increased LC3 II/I expression following cerebral
I/R injury (34). In the present
study, it was revealed that the expression levels of Beclin-1 and
LC3 were increased in diabetic rats after an ischemic stroke,
whereas that of p62 was decreased, which was consistent with our
previous results. However, selenium significantly attenuated these
changes. In the in vivo experiment, the expression of the
autophagy-related proteins Beclin-1 and LC3 II/I was significantly
decreased, whereas that of p62 was significantly increased
following selenium treatment. Similarly, following the use of the
autophagy inhibitor 3-MA, selenium reduced autophagy and diabetic
cerebral I/R injury. Selenium inhibited the expression of
autophagy-related proteins in vitro. Furthermore, the
expression of the PI3K/AKT/mTOR autophagy-related pathway proteins
was also detected in vivo, and it was revealed that the
expression of PI3K/AKT/mTOR-related pathway proteins was increased
following treatment with sodium selenite. These findings indicated
that selenium may inhibit autophagy through the PI3K signaling
pathway.
The PI3K/AKT/mTOR signaling pathway regulates
several biological processes, such as cell proliferation,
differentiation and apoptosis, and is closely associated with
autophagy (73). Nicotine can
inhibit autophagy in brain microvascular endothelial cells by
activating the PI3K/AKT/mTOR pathway (24). Homocysteine plays a
neuroprotective role by inhibiting the excessive autophagy of
neural stem cells following OGD/R through the PI3K/AKT/mTOR
signaling pathway (74). In the
in vivo results of the present study, it was revealed that
the phosphorylation levels of the PI3K, AKT and mTOR proteins were
significantly decreased following cerebral I/R injury under high
glucose conditions, but selenium significantly inhibited these
effects. However, it was unclear whether selenium affected
autophagy through the PI3K/Akt/mTOR signaling pathway. Therefore,
an in vitro study was designed and conducted, using the
PI3K/Akt/mTOR signaling pathway specific activator, IGF-1, and the
specific inhibitor Wort, to further determine whether there was a
causal relationship between selenium and the PI3K/Akt/mTOR
signaling pathway. The selenium-mediated effects were accompanied
by corresponding changes in the expression of autophagy-related
proteins. Of note, Wort, a PI3K inhibitor, counteracted the
beneficial effects of selenium following BBB damage. These results
indicated that selenium attenuated cerebral I/R injury-induced BBB
damage under hyperglycemic conditions by inhibiting autophagy
through the PI3K/AKT/mTOR signaling pathway.
The present study had certain limitations. Although
it was demonstrated herein that selenium can reduce damage to the
BBB during the acute phase of cerebral I/R injury under
hyperglycemic conditions, whether selenium can also protect the BBB
in the subacute and chronic phases after stroke needs to be
evaluated in future studies.
In conclusion, the results of the present study
revealed that selenium significantly ameliorated I/R injury-induced
BBB damage under hyperglycemic conditions, both in vivo and
in vitro, by activating the PI3K/AKT/mTOR pathway to inhibit
overactive autophagy.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BY, YL and YM contributed to the conception and
design of this study. BY, XZ and LY performed the experiments. XS
and LJ analysed the data and wrote the manuscript. JZ reviewed the
manuscript for important intellectual content. BY and LJ confirmed
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All animal procedures were approved by the
Institutional Animal Care and Use Committee (IACUC) of the Ningxia
Medical University (Yinchuan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
References
|
1
|
Liu MB, Wang W, Gao JM, Li F, Shi JS and
Gong QH: Icariside II attenuates cerebral
ischemia/reperfusion-induced blood-brain barrier dysfunction in
rats via regulating the balance of MMP9/TIMP1. Acta Pharmacol Sin.
41:1547–1556. 2020. View Article : Google Scholar
|
|
2
|
Gao J, Long L, Xu F, Feng L, Liu Y, Shi J
and Gong Q: Icariside II, a phosphodiesterase 5 inhibitor,
attenuates cerebral ischaemia/reperfusion injury by inhibiting
glycogen synthase kinase-3β-mediated activation of autophagy. Br J
Pharmacol. 177:1434–1452. 2020. View Article : Google Scholar
|
|
3
|
Wu C, Chen J, Yang R, Duan F, Li S and
Chen X: Mitochondrial protective effect of neferine through the
modulation of nuclear factor erythroid 2-related factor 2
signalling in ischaemic stroke. Br J Pharmacol. 176:400–415. 2019.
View Article : Google Scholar
|
|
4
|
Venkat P, Chopp M and Chen J: Blood-brain
barrier disruption, vascular impairment, and ischemia/reperfusion
damage in diabetic stroke. J Am Heart Assoc. 6:e0058192017.
View Article : Google Scholar
|
|
5
|
Shou J, Zhou L, Zhu S and Zhang X:
Diabetes is an independent risk factor for stroke recurrence in
stroke patients: A meta-analysis. J Stroke Cerebrovasc Dis.
24:1961–1968. 2015. View Article : Google Scholar
|
|
6
|
Chen J, Cui X, Zacharek A, Cui Y, Roberts
C and Chopp M: White matter damage and the effect of matrix
metalloproteinases in type 2 diabetic mice after stroke. Stroke.
42:445–452. 2011. View Article : Google Scholar
|
|
7
|
Zhang X, Wei M, Fan J, Yan W, Zha X, Song
H, Wan R, Yin Y and Wang W: Ischemia-induced upregulation of
autophagy preludes dysfunctional lysosomal storage and associated
synaptic impairments in neurons. Autophagy. 17:1519–1542. 2021.
View Article : Google Scholar
|
|
8
|
Chen J, Ye X, Yan T, Zhang C, Yang XP, Cui
X, Cui Y, Zacharek A, Roberts C, Liu X, et al: Adverse effects of
bone marrow stromal cell treatment of stroke in diabetic rats.
Stroke. 42:3551–3558. 2011. View Article : Google Scholar
|
|
9
|
Wang W, Li M, Chen Q and Wang J:
Hemorrhagic transformation after tissue plasminogen activator
reperfusion therapy for ischemic stroke: Mechanisms, models, and
biomarkers. Mol Neurobiol. 52:1572–1579. 2015. View Article : Google Scholar
|
|
10
|
Karatas H, Eun Jung J, Lo EH and van Leyen
K: Inhibiting 12/15-lipoxygenase to treat acute stroke in permanent
and tPA induced thrombolysis models. Brain Res. 1678:123–128. 2018.
View Article : Google Scholar
|
|
11
|
Muir KW: Stroke in 2015: The year of
endovascular treatment. Lancet Neurol. 15:2–3. 2016. View Article : Google Scholar
|
|
12
|
Zhang Y, Cao Y and Liu C: Autophagy and
ischemic stroke. Adv Exp Med Biol. 1207:111–134. 2020. View Article : Google Scholar
|
|
13
|
Puyal J, Vaslin A, Mottier V and Clarke
PG: Postischemic treatment of neonatal cerebral ischemia should
target autophagy. Ann Neurol. 66:378–389. 2009. View Article : Google Scholar
|
|
14
|
Wei K, Wang P and Miao CY: A double-edged
sword with therapeutic potential: An updated role of autophagy in
ischemic cerebral injury. CNS Neurosci Ther. 18:879–886. 2012.
View Article : Google Scholar
|
|
15
|
Feng D, Wang B, Wang L, Abraham N, Tao K,
Huang L, Shi W, Dong Y and Qu Y: Pre-ischemia melatonin treatment
alleviated acute neuronal injury after ischemic stroke by
inhibiting endoplasmic reticulum stress-dependent autophagy via
PERK and IRE1 signalings. J Pineal Res. 62:2017. View Article : Google Scholar
|
|
16
|
Yang Z, Lin P, Chen B, Zhang X, Xiao W, Wu
S, Huang C, Feng D, Zhang W and Zhang J: Autophagy alleviates
hypoxia-induced blood-brain barrier injury via regulation of CLDN5
(claudin 5). Autophagy. 1–20. 2020.Online ahead of print.
|
|
17
|
Li H, He Y, Zhang C, Ba T, Guo Z, Zhuo Y,
He L and Dai H: NOX1 down-regulation attenuated the autophagy and
oxidative damage in pig intestinal epithelial cell following
transcriptome analysis of transport stress. Gene. 763:1450712020.
View Article : Google Scholar
|
|
18
|
Kim KA, Kim D, Kim JH, Shin YJ, Kim ES,
Akram M, Kim EH, Majid A, Baek SH and Bae ON: Autophagy-mediated
occludin degradation contributes to blood-brain barrier disruption
during ischemia in bEnd.3 brain endothelial cells and rat ischemic
stroke models. Fluids Barriers CNS. 17:212020. View Article : Google Scholar
|
|
19
|
Zhang Y, Zhang X, Wei Q, Leng S, Li C, Han
B, Bai Y, Zhang H and Yao H: Activation of sigma-1 receptor
enhanced pericyte survival via the interplay between apoptosis and
autophagy: Implications for blood-brain barrier integrity in
stroke. Transl Stroke Res. 11:267–287. 2020. View Article : Google Scholar
|
|
20
|
Wei N, Yu SP, Gu XH, Chen DD, Whalin MK,
Xu GL, Liu XF and Wei L: The involvement of autophagy pathway in
exaggerated ischemic brain damage in diabetic mice. CNS Neurosci
Ther. 19:753–763. 2013.
|
|
21
|
Che H, Li H, Li Y, Wang YQ, Yang ZY, Wang
RL and Wang LH: Melatonin exerts neuroprotective effects by
inhibiting neuronal pyroptosis and autophagy in STZ-induced
diabetic mice. FASEB J. 34:14042–14054. 2020. View Article : Google Scholar
|
|
22
|
Fu B, Zeng Q, Zhang Z, Qian M, Chen J,
Dong W and Li M: Epicatechin gallate protects HBMVECs from
ischemia/reperfusion injury through ameliorating apoptosis and
autophagy and promoting neovascularization. Oxid Med Cell Longev.
2019:78246842019. View Article : Google Scholar
|
|
23
|
Wei R, Liu H, Chen R, Sheng Y and Liu T:
Astragaloside IV combating liver cirrhosis through the
PI3K/Akt/mTOR signaling pathway. Exp Ther Med. 17:393–397.
2019.
|
|
24
|
Wu C, Yang M, Liu R, Hu H, Ji L, Zhang X,
Huang S and Wang L: Nicotine reduces human brain microvascular
endothelial cell response to Escherichia coli K1 infection by
inhibiting autophagy. Front Cell Infect Microbiol. 10:4842020.
View Article : Google Scholar
|
|
25
|
Wang M, Li YJ, Ding Y, Zhang HN, Sun T,
Zhang K, Yang L, Guo YY, Liu SB, Zhao MG and Wu YM: Silibinin
prevents autophagic cell death upon oxidative stress in cortical
neurons and cerebral ischemia-reperfusion injury. Mol Neurobiol.
53:932–943. 2016. View Article : Google Scholar
|
|
26
|
Liu YP, Shao SJ and Guo HD: Schwann cells
apoptosis is induced by high glucose in diabetic peripheral
neuropathy. Life Sci. 248:1174592020. View Article : Google Scholar
|
|
27
|
Liu Y, Chen X, Yao J and Kang J: Circular
RNA ACR relieves high glucose-aroused RSC96 cell apoptosis and
autophagy via declining microRNA-145-3p. J Cell Biochem. Dec
30–2019.Online ahead of print. View Article : Google Scholar
|
|
28
|
Arias-Borrego A, Callejón-Leblic B,
Calatayud M, Gómez-Ariza JL, Collado MC and Garcia-Barrera T:
Insights into cancer and neurodegenerative diseases through
selenoproteins and the connection with gut microbiota-current
analytical methodologies. Expert Rev Proteomics. 16:805–814. 2019.
View Article : Google Scholar
|
|
29
|
Yang J, Hamid S, Cai J, Liu Q, Xu S and
Zhang Z: Selenium deficiency-induced thioredoxin suppression and
thioredoxin knock down disbalanced insulin responsiveness in
chicken cardiomyocytes through PI3K/Akt pathway inhibition. Cell
Signal. 38:192–200. 2017. View Article : Google Scholar
|
|
30
|
Zhang Z, Liu M, Guan Z, Yang J, Liu Z and
Xu S: Disbalance of calcium regulation-related genes in broiler
hearts induced by selenium deficiency. Avian Pathol. 46:265–271.
2017. View Article : Google Scholar
|
|
31
|
Zhang Q, Zhang C, Ge J, Lv MW, Talukder M,
Guo K, Li YH and Li JL: Ameliorative effects of resveratrol against
cadmium-induced nephrotoxicity via modulating nuclear xenobiotic
receptor response and PINK1/Parkin-mediated mitophagy. Food Funct.
11:1856–1868. 2020. View Article : Google Scholar
|
|
32
|
Feng C, Li D, Chen M, Jiang L, Liu X, Li
Q, Geng C, Sun X, Yang G, Zhang L and Yao X: Citreoviridin induces
myocardial apoptosis through PPAR-γ-mTORC2-mediated autophagic
pathway and the protective effect of thiamine and selenium. Chem
Biol Interact. 311:1087952019. View Article : Google Scholar
|
|
33
|
Zhang C, Wang LL, Cao CY, Li N, Talukder M
and Li JL: Selenium mitigates cadmium-induced crosstalk between
autophagy and endoplasmic reticulum stress via regulating calcium
homeostasis in avian leghorn male hepatoma (LMH) cells. Environ
Pollut. 265:1146132020. View Article : Google Scholar
|
|
34
|
Lu C, Guo Y, Zhang Y, Yang L, Chang Y,
Zhang JW, Jing L and Zhang JZ: Coenzyme Q10 ameliorates cerebral
ischemia reperfusion injury in hyperglycemic rats. Pathol Res
Pract. 213:1191–1199. 2017. View Article : Google Scholar
|
|
35
|
MacArthur Clark JA and Sun D: Guidelines
for the ethical review of laboratory animal welfare People's
Republic of China national standard GB/T 35892-2018 (issued 6
February 2018 effective from 1 September 2018). Animal Model Exp
Med. 3:103–113. 2020. View Article : Google Scholar
|
|
36
|
Sun X, Wang D, Zhang T, Lu X, Duan F, Ju
L, Zhuang X and Jiang X: Eugenol Attenuates cerebral
ischemia-reperfusion injury by enhancing autophagy via
AMPK-mTOR-P70S6K pathway. Front Pharmacol. 11:842020. View Article : Google Scholar
|
|
37
|
Fan X, Qiu J, Yu Z, Dai H, Singhal AB, Lo
EH and Wang X: A rat model of studying tissue-type plasminogen
activator thrombolysis in ischemic stroke with diabetes. Stroke.
43:567–570. 2012. View Article : Google Scholar
|
|
38
|
Yang L, Ma YM, Shen XL, Fan YC, Zhang JZ,
Li PA and Jing L: The involvement of mitochondrial biogenesis in
selenium reduced hyperglycemia-aggravated cerebral ischemia injury.
Neurochem Res. 45:1888–1901. 2020. View Article : Google Scholar
|
|
39
|
Liu L, Cen J, Man Y, Li J, Zhang D, Wang
F, Li J, Ma J, Wang X and Ji B: Transplantation of human umbilical
cord blood mononuclear cells attenuated ischemic injury in MCAO
Rats via inhibition of NF-κB and NLRP3 inflammasome. Neuroscience.
369:314–324. 2018. View Article : Google Scholar
|
|
40
|
Yu Q, Li Q, Yang X, Liu Q, Deng J, Zhao Y,
Hu R and Dai M: Dexmedetomidine suppresses the development of
abdominal aortic aneurysm by downregulating the mircoRNA-21/PDCD 4
axis. Int J Mol Med. 47:902021. View Article : Google Scholar
|
|
41
|
Xiong D, Deng Y, Huang B, Yin C, Liu B,
Shi J and Gong Q: Icariin attenuates cerebral ischemia-reperfusion
injury through inhibition of inflammatory response mediated by
NF-κB, PPARα and PPARγ in rats. Int Immunopharmacol. 30:157–162.
2016. View Article : Google Scholar
|
|
42
|
Tureyen K, Bowen K, Liang J, Dempsey RJ
and Vemuganti R: Exacerbated brain damage, edema and inflammation
in type-2 diabetic mice subjected to focal ischemia. J Neurochem.
116:499–507. 2011. View Article : Google Scholar
|
|
43
|
Zhang G, Zhang T, Li N, Wu L, Gu J, Li C,
Zhao C, Liu W, Shan L, Yu P, et al: Tetramethylpyrazine nitrone
activates the BDNF/Akt/CREB pathway to promote post-ischaemic
neuroregeneration and recovery of neurological functions in rats.
Br J Pharmacol. 175:517–531. 2018. View Article : Google Scholar
|
|
44
|
Fang L, Li X, Zhong Y, Yu J, Yu L, Dai H
and Yan M: Autophagy protects human brain microvascular endothelial
cells against methylglyoxal-induced injuries, reproducible in a
cerebral ischemic model in diabetic rats. J Neurochem. 135:431–440.
2015. View Article : Google Scholar
|
|
45
|
Tian X, Peng J, Zhong J, Yang M, Pang J,
Lou J, Li M, An R, Zhang Q, Xu L and Dong Z: β-Caryophyllene
protects in vitro neurovascular unit against oxygen-glucose
deprivation and re-oxygenation-induced injury. J Neurochem.
139:757–768. 2016. View Article : Google Scholar
|
|
46
|
Mysiorek C, Culot M, Dehouck L, Derudas B,
Staels B, Bordet R, Cecchelli R, Fenart L and Berezowski V:
Peroxisome-proliferator-activated receptor-alpha activation
protects brain capillary endothelial cells from oxygen-glucose
deprivation-induced hyperpermeability in the blood-brain barrier.
Curr Neurovasc Res. 6:181–193. 2009. View Article : Google Scholar
|
|
47
|
Liu M, Pi H, Xi Y, Wang L, Tian L, Chen M,
Xie J, Deng P, Zhang T, Zhou C, et al: KIF5A-dependent axonal
transport deficiency disrupts autophagic flux in trimethyltin
chloride-induced neurotoxicity. Autophagy. 17:903–924. 2021.
View Article : Google Scholar
|
|
48
|
Rastogi M and Singh SK: Zika virus NS1
affects the junctional integrity of human brain microvascular
endothelial cells. Biochimie. 176:52–61. 2020. View Article : Google Scholar
|
|
49
|
Livak K and Schmittgen T: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
50
|
Manthari RK, Tikka C, Ommati MM, Niu R,
Sun Z and Wang J, Zhang J and Wang J: Arsenic-induced autophagy in
the developing mouse cerebellum: Involvement of the blood-brain
Barrier's tight-junction proteins and the PI3K-Akt-mTOR signaling
pathway. J Agric Food Chem. 66:8602–8614. 2018. View Article : Google Scholar
|
|
51
|
Franke H, Galla H and Beuckmann C: Primary
cultures of brain microvessel endothelial cells: A valid and
flexible model to study drug transport through the blood-brain
barrier in vitro. Brain Res Brain Res Protoc. 5:248–256. 2000.
View Article : Google Scholar
|
|
52
|
Hamprecht B and Löffler F: Primary glial
cultures as a model for studying hormone action. Comparative Study.
109:341–345. 1985.
|
|
53
|
Lee KJ, Lee JS and Jung KH: Interactive
effect of acute and chronic glycemic indexes for severity in acute
ischemic stroke patients. BMC Neurol. 18:1052018. View Article : Google Scholar
|
|
54
|
Ji Q, Han J, Wang L, Liu J, Dong Y, Zhu K
and Shi L: MicroRNA-34a promotes apoptosis of retinal vascular
endothelial cells by targeting SIRT1 in rats with diabetic
retinopathy. Cell Cycle. 19:2886–2896. 2020. View Article : Google Scholar
|
|
55
|
Tu Y, Guo C, Song F, Huo Y, Geng Y, Guo M,
Bao H, Wu X and Fan W: Mild hypothermia alleviates diabetes
aggravated cerebral ischemic injury via activating autophagy and
inhibiting pyroptosis. Brain Res Bull. 150:1–12. 2019. View Article : Google Scholar
|
|
56
|
Kumari R, Bettermann K, Willing L, Sinha K
and Simpson IA: The role of neutrophils in mediating stroke injury
in the diabetic db/db mouse brain following hypoxia-ischemia.
Neurochem Int. 139:1047902020. View Article : Google Scholar
|
|
57
|
Okada T, Suzuki H, Travis ZD and Zhang JH:
The stroke-induced blood-brain barrier disruption: Current progress
of inspection technique, mechanism, and therapeutic target. Curr
Neuropharmacol. 18:1187–1212. 2020. View Article : Google Scholar
|
|
58
|
Li W, Qu Z, Prakash R, Chung C, Ma H, Hoda
MN, Fagan SC and Ergul A: Comparative analysis of the neurovascular
injury and functional outcomes in experimental stroke models in
diabetic Goto-Kakizaki rats. Brain Res. 1541:106–114. 2013.
View Article : Google Scholar
|
|
59
|
Venkat P, Zacharek A, Landschoot-Ward J,
Wang F, Culmone L, Chen Z, Chopp M and Chen J: Exosomes derived
from bone marrow mesenchymal stem cells harvested from type two
diabetes rats promotes neurorestorative effects after stroke in
type two diabetes rats. Exp Neurol. 334:1134562020. View Article : Google Scholar
|
|
60
|
Nian K, Harding IC, Herman IM and Ebong
EE: Blood-brain barrier damage in ischemic stroke and its
regulation by endothelial mechanotransduction. Front Physiol.
11:6053982020. View Article : Google Scholar
|
|
61
|
Tietz S and Engelhardt B: Brain barriers:
Crosstalk between complex tight junctions and adherens junctions. J
Cell Biol. 209:493–506. 2015. View Article : Google Scholar
|
|
62
|
Cui X, Chopp M, Zacharek A, Ye X, Roberts
C and Chen J: Angiopoietin/Tie2 pathway mediates type 2 diabetes
induced vascular damage after cerebral stroke. Neurobiol Dis.
43:285–292. 2011. View Article : Google Scholar
|
|
63
|
Shao Z, Dou S, Zhu J, Wang H, Xu D, Wang
C, Cheng B and Bai B: The role of mitophagy in ischemic stroke.
Front Neurol. 11:6086102020. View Article : Google Scholar
|
|
64
|
Xu W, Ocak U, Gao L, Tu S, Lenahan CJ,
Zhang J and Shao A: Selective autophagy as a therapeutic target for
neurological diseases. Cell Mol Life Sci. 78:1369–1392. 2021.
View Article : Google Scholar
|
|
65
|
Chen L, Zhang B and Toborek M: Autophagy
is involved in nanoalumina-induced cerebrovascular toxicity.
Nanomedicine. 9:212–221. 2013. View Article : Google Scholar
|
|
66
|
Xiao Y, Fan M, Jin W, Li WA, Jia Y, Dong
Y, Jiang X, Xu J, Meng N and Lv P: Lithium chloride ameliorated
spatial cognitive impairment through activating mTOR
phosphorylation and inhibiting excessive autophagy in the repeated
cerebral ischemia-reperfusion mouse model. Exp Ther Med.
20:1092020. View Article : Google Scholar
|
|
67
|
Menzie-Suderam JM, Modi J, Xu H, Bent A,
Trujillo P, Medley K, Jimenez E, Shen J, Marshall M, Tao R, et al:
Granulocyte-colony stimulating factor gene therapy as a novel
therapeutics for stroke in a mouse model. J Biomed Sci. 27:992020.
View Article : Google Scholar
|
|
68
|
Fu C, Zhang X, Lu Y, Wang F, Xu Z, Liu S,
Zheng H and Liu X: Geniposide inhibits NLRP3 inflammasome
activation via autophagy in BV-2 microglial cells exposed to
oxygen-glucose deprivation/reoxygenation. Int Immunopharmacol.
84:1065472020. View Article : Google Scholar
|
|
69
|
Zhao B, Yuan Q, Hou JB, Xia ZY, Zhan LY,
Li M, Jiang M, Gao WW and Liu L: Inhibition of HDAC3 ameliorates
cerebral ischemia reperfusion injury in diabetic mice in vivo and
in vitro. J Diabetes Res. 2019:85208562019. View Article : Google Scholar
|
|
70
|
Pan Q, Liu Y, Wang G, Wen Z and Wang Y:
MTMR14 protects against cerebral stroke through suppressing
PTEN-regulated autophagy. Biochem Biophys Res Commun.
529:1045–1052. 2020. View Article : Google Scholar
|
|
71
|
Nazarinia D, Aboutaleb N, Gholamzadeh R,
Nasseri Maleki S, Mokhtari B and Nikougoftar M: Conditioned medium
obtained from human amniotic mesenchymal stem cells attenuates
focal cerebral ischemia/reperfusion injury in rats by targeting
mTOR pathway. J Chem Neuroanat. 102:1017072019. View Article : Google Scholar
|
|
72
|
Wu F, Xu K, Xu K, Teng C, Zhang M, Xia L,
Zhang K, Liu L, Chen Z, Xiao J, et al: Dl-3n-butylphthalide
improves traumatic brain injury recovery via inhibiting
autophagy-induced blood-brain barrier disruption and cell
apoptosis. J Cell Mol Med. 24:1220–1232. 2020. View Article : Google Scholar
|
|
73
|
Heras-Sandoval D, Pérez-Rojas JM,
Hernández-Damián J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014. View Article : Google Scholar
|
|
74
|
Wang M, Liang X, Cheng M, Yang L, Liu H,
Wang X, Sai N and Zhang X: Homocysteine enhances neural stem cell
autophagy in in vivo and in vitro model of ischemic stroke. Cell
Death Dis. 10:5612019. View Article : Google Scholar
|