Introduction
The Aurora kinases, including Aurora A, B and C, are
serine/threonine kinases that play a central role in regulating
cell division and multiple signaling pathways. Aurora A functions
in the formation of a typical bipolar spindle (1), the maturation of centrosomes, which
is necessary for G2/M transition (2), and the formation and stimulation of
the cyclin B-CDK1 complex (3).
Moreover, Aurora A helps to increase both size and
microtubule-nucleating capacity just before mitotic entry (3). Aurora B plays a function in the
chromosome biorientation on the mitotic spindle. It mediates the
attachment of the microtubule to the kinetochores and regulates the
spindle assembly checkpoint (SAC) (4,5).
The improper attachment of kinetochores promotes Aurora B to
recruit and phosphorylate its substrates at the kinetochores to
depolymerize the uncorrected attachment, allowing other
microtubules to capture the unattached kinetochores. The inhibition
of Aurora B can impair the chromosome arrangement at the mitotic
spindle equator (6).
Furthermore, Aurora B phosphorylates histone H3 at
the serine 10 (H3S10ph) residue at the beginning of the prophase
and leads to a peak in H3S10ph at the prometaphase and metaphase.
This phosphorylation contributes to the active chromosome
conformation at the entry of mitosis (7). Other studies have reported that
H3S10ph may involve chromosome condensation and Aurora B
recruitment to the centromere (8,9).
Most notably, Aurora B is the only enzymatic member of the
chromosomal passenger protein complex (CPC). All members of CPC
share the co-localization during mitosis: They concentrate in the
kinetochore during the prophase, prometaphase and metaphase;
transfer to the midzone with anaphase onset; and remain in the
midbody in telophase and cytokinesis (10). The mislocalization of any CPC
members, including Aurora B, can lead to a defection in mitosis and
cytokinesis (10,11). Apart from the pivotal functions in
cell division, Aurora A and B kinases are also involved in tumor
angiogenesis. These enzymes phosphorylate MYCN, regulate vascular
endothelial growth factor (VEGF) production, and inhibit the
proliferation and tube formation of human endothelial cells
(12-14). Aurora C kinase has been found in
cells that undergo meiosis and has a unique physiological role in
spermatogenesis (15). The
limitation in understanding the role of Aurora C may stem from the
high sequence homology between this kinase and Aurora B, leading to
the overlapping in the function of these proteins (16). Aurora C can rescue the genetic
stability of the cells in case Aurora B is absent (17). Previously, it was demonstrated
that the overexpression of Aurora C induces abnormal cell division,
resulting in centrosome amplification and multinucleation in cells
(17).
The overexpression of Aurora kinases has been
observed in a broad range of human solid tumors, such as gliomas,
and colorectal, breast, ovarian and pancreatic cancer (18), as well as in liquid tumors such as
diffuse large B-cell lymphoma (19). Moreover, Aurora kinases have been
found to be associated with genetic instability and aneuploidy in
tumors (20). Hence, it is not
surprising that Aurora kinases have become attractive targets in
cancer treatment. The development of Aurora inhibitors has drawn
the attention of several scientists from academic institutes and
pharmaceutical companies. Over the first two decades of the 21st
century, a series of Aurora kinase inhibitors were produced, which
were Aurora A- or B-selective, or pan inhibitors. Although these
compounds exhibit preclinical and clinical efficacy, no Aurora
kinase inhibitor has yet been approved for clinical use due to
their poor outcomes (18). Thus,
there is an urgent need for the identification of novel small
molecule inhibitors.
Oxostephanine is a substance belonging to the group
of aporphine alkaloids isolated from several plants of the genus
Stephania. Previous studies have demonstrated that this
substance exerts a potent cytotoxic effect on several cancer cell
lines, such as KB (human epithelial carcinoma), HepG2 (human
hepatocellular carcinoma), GLC4/Adr (human small cell lung
adriamycin-resistant carcinoma), K562 (human chronic myelogenous
leukemia) and K562/Adr (human chronic myelogenous leukemia
resistant to adriamycin) (21),
whereas it has a minimal toxic effect on normal cells (MRC-5; human
fetal lung fibroblasts) (22). In
addition, oxostephanine has been shown to exhibit potent activity
against breast cancer cells and MOLT-3 acute lymphoblastic leukemia
cells (21). Moreover, Knockleby
et al (23) revealed that
oxostephanine inhibited the activity of Aurora kinase A and B by
the competition of ATP binding sites in an in vitro kinase
assay.
The aim of the present study was to examined the
effects of oxostephanine extracted from Vietnamese Stephania
dielsiana Y.C. Wu (S. dielsiana) as a novel Aurora
kinase inhibitor on an ovarian cancer cell line (OVCAR-8). As
demonstrated herein, S. dielsiana may prove to be a potent
Aurora kinase inhibitor, as well as an anti-angiogenic agent with
potential to be developed into an anticancer drug.
Materials and methods
Compound preparation
The stems and leaves of S. dielsiana were
collected in Ba Vi District, Hanoi, Vietnam in October, 2019 and
identified by the Department of Botany, Hanoi University of
Pharmacy, Hanoi, Vietnam. A voucher specimen (no. SD10/2019) has
been deposited at the Department of Botany and Pharmacognosy,
Vietnam University of Traditional Medicine, Hanoi, Vietnam. The
process used for the isolation and characterization of
oxostephanine from the leaves of S. dielsiana in Vietnam has
been previously published (22,23). In brief, the leaves of S.
dielsiana (7 kg) were extracted with 95% MeOH (3×15 liters, 3
days each) at room temperature. The extracts were concentrated
in vacuo to yield a MeOH extract (680 g), which was
suspended in H2O (2.5 liters) and adjusted to pH 3 with
10% HCl. The acidic aqueous phase was filtered off. The filtrate
was loaded on ion-exchange resin, eluted with 20% MeOH until the
eluate approached colorless to give the nonalkaloid parts, and then
eluted with 2% NaOH in 65% MeOH solution (five-fold of retention
volume) to yield the crude total alkaloids. The alkaloid-containing
solution was acidified to pH 5 with 10% HCl and partitioned with
EtOAc (3×2 liters) to yield the EtOAc extract (65 g).
The EtOAc-soluble portion was subjected to silica
gel column chromatography eluted with gradient systems of
CH2Cl2-MeOH (100:0, 100:10, 100:30 and
100:50, v/v). The eluted fractions were evaluated and pooled
according to thin layer chromatography (TLC) analysis, resulting in
six major fractions (SDE.1-SDE.6). The purification of SDE.6 over
Sephadex LH-20 (100% MeOH) was performed using the same
methodology, and subsequent preparative TLC, eluted with
CH2Cl2-MeOH (20:1) yielded oxostephanine (8.6
mg). The purification of oxostephanine by repeating
recrystallization in a mixture of methanol and ethanol yielded pure
oxostephanine compound as an amorphous yellow-orange powder (purity
99.0% as a percentage of the peak area using a HPLC-DA system
(Agilent 1260 Infinity II; Agilent Technologies, Inc.).
Cell lines and culture
OVCAR-8 (human ovarian carcinoma-8) and HeLa (Aurora
B-GFP) cells were grown in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc.). Human dermal
fibroblasts (hFBs) were cultured in DMEM/F12 medium (Gibco; Thermo
Fisher Scientific, Inc.). The media were supplemented with 10%
fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.),
100 units/ml penicillin and 100 µg/ml streptomycin (Gibco;
Thermo Fisher Scientific, Inc.). Human umbilical vein endothelial
cells (hUVECs) were cultured in EBM-2 medium (Lonza Group, Ltd.).
Umbilical cord-derived mesenchymal stem cells (UC-MSCs) were grown
on the surface of culture flasks coated by CELLstart™ CTS™
(CELLstart) in StemMACS™ MSC Expansion medium (StemMACS) (Miltenyi
Biotec). All the cells were cultured in an incubator at 37°C with
5% CO2. The hUVECs, hFBs and UC-MSCs were provided by
Vinmec Research Institute of Stem cell and Gene Technology, and
they were not immortalized cell lines. The protocols for cell
isolation were approved by the Ethics Committee of Vinmec
International Hospital (Document no. 40/2020/QD-Vinmec for hUVECs
and UC-MSCs, signed and dated on December 24, 2020; Document no.
311/2018/QD-Vinmec for hFBs, signed and dated on September 11,
2018). The HeLa (Aurora B-GFP) cells were kindly provided as a gift
from Professor Stefan Dimitrov at Institute Albert Bonniot (present
name is Institute for Advanced Biosciences) (11,24).
Cell viability assay
Cell viability was assessed using sulforhodamine B
(SRB) assay. The cells were seeded at a density of 3×103
cells/well in 96-well plates and incubated with oxostephanine for
24, 48 and 72 h at six concentrations differed by five from the
highest of 25 to 5, 1, 0.2 and 0.04 µM. Subsequently, the
medium was removed, and the cells were stained with 4% SRB
(Millipore, Sigma) for 10 min at room temperature after fixing with
10% TCA (MilliporeSigma) for 1 h at 4°C. The absorbance was
measured at 540 nm using a microplate reader (BioTech Power Wave
XS; BioTek Instruments, Inc.).
Real-time analysis of cell proliferation
using the xCELLigence system
The proliferation assay was performed using the
xCelligence system (ACEA Biosciences; Agilent Technologies, Inc.).
Media (100 µl/well) were added to each 96-well of an E-plate
(ACEA Biosciences; Agilent Technologies, Inc.) to take the
background reading for 15 min. In the meantime, the cells were
resuspended in medium, and 80 µl cell suspension were added
to yield a cell density of 3×103 cells/180
µl/well. Following incubation for 30 min at room
temperature, the E-plate was placed into the RTCA SP station in an
incubator. After 24 h, the cells were treated with oxostephanine
(125, 25, 5, 1 and 0.2 µM) and VX-680 (Vertex and Merck; 25,
5, 1, 0.2 and 0.04 µM). Dynamic cell proliferation was
monitored in 30-min intervals from the seeding point till the end
of the experiment with a total of >200 h. The electrical
impedance was measured using RTCA-integrated software of the
xCEL-Ligence system as a dimensionless parameter termed cell index
(CI). Normalized CI values were used to obtain the IC50
values, doubling times and other evaluations.
Immunofluorescence
The cells were grown on glass coverslips for 24 h
before being treated with either oxostephanine (5 µM) or
VX-680 (0.2 µM) with or without paclitaxel (0.035 µM;
Millipore, Sigma) and incubated for 15 h in an incubator at 37°C
with 5% CO2. Paclitaxel was used to synchronize the
cells to the M phase in the cell cycle, in order to obtain dividing
cells. The cells were then fixed with 4% paraformaldehyde and 2%
sucrose for 15 min at 37°C, permeabilized with 0.2% Triton X-100
for 10 min, blocked with 5 mg/ml BSA, and incubated with primary
antibodies for 2 h at room temperature. Phosphorylated histone H3
was detected using a polyclonal rabbit antibody (ab183626, Abcam),
at a dilution of 1:500. Aurora B was detected using mouse
monoclonal antibodies (36-5200, Invitrogen; Thermo Fisher
Scientific, Inc.), at a dilution of 1:250. DNA was visualized with
5 µg/ml Hoechst 33342 (Invitrogen; Thermo Fisher Scientific,
Inc.) or 2 µg/ml propidium iodide (PI; Thermo Fisher
Scientific, Inc.). Images were collected using a ZEISS 510 Laser
Scanning Confocal (LSM) microscope with 40X or 63X objectives (Carl
Zeiss AG). For the HeLa (Aurora B-GFP), the cells were grown on a
Lab-Tek chamber coverglass (Nalge Nunc International). Following 24
h of treatment with the compounds at concentrations of
oxostephanine (5 µM) or VX-680 (0.2 µM), cells were
observed without fixing.
As regards the cell nuclear morphological
examination, the cells were incubated with either oxostephanine (5
µM) or VX-680 (0.2 µM) for 48 h. The cells were then
fixed with 4% paraformaldehyde and 2% sucrose for 15 min at 37°C,
permeabilized with 0.2% Triton X-100 for 10 min and stained with 5
µg/ml Hoechst 33342. Following incubation for 15 min, the
cells were collected, washed with phosphate-buffered saline (PBS;
Millipore, Sigma), and observed using a LSM microscope. Images were
analyzed using LSM Image Browser (Carl Zeiss AG).
Apoptosis assay
Apoptosis assay was performed using the Alexa Fluor
488 Annexin V/dead cell apoptosis kit (Invitrogen; Thermo Fisher
Scientific, Inc.). As mentioned in the kit, Annexin V is a
phospholipid binding protein, and it specifically binds to
negatively charged phosphatidylserine molecules exposure on the
surface of apoptotic cells. Following treatment of the cells with
either 0.5 µM oxostephanine or 0.2 µM VX-680 for 48
h, the cells were harvested and prepared for apoptosis analysis.
Briefly, the cells were washed with PBS, then suspended in
Annexin-binding buffer to obtain a density of 106
cells/ml. The cell solution was then incubated with 5 µl
Alexa Fluor® 488-Annexin V and 100 µl PI working
solution for 15 min at room temperature. Subsequently, 400
µl Annexin-binding buffer were gently mixed into the
solution with and the cell solution was analyzed on a FACS Canto II
System (BD Biosciences). For the visualization of apoptotic marker
expression, following 24 h of treatment with the compounds, the
cells were incubated with Alexa Fluor® 488-Annexin V for
30 min and observed under a LSM microscope.
Multicellular tumor spheroid assay
OVCAR-8 spheroids were created using the hanging
drop method as previously described (25). A total of 15 µl of the
medium that contained 5×103 cells were added to each
circle on the inverted cover of a 96-well plate to create one
spheroid. The cover was then placed upside down on the plate coated
with sterile agarose 1.5% (w/v) containing 200 µl complete
medium. Following 48 h of incubation in a humidified chamber with
5% CO2 at 37°C, spheroids were transferred from the
cover into each well of the agarose-coated plate and further
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.).
Spheroids were treated with oxostephanine under two conditions: i)
The compound was added to the cell preparation before making the
hanging drop; and ii) the compound was added after transferring the
formed spheroids into the culture wells. Two concentrations at 5
and 1 µM of Oxostephanine were used in both conditions.
Images were obtained using an Axiovert 40CFL microscope (Carl Zeiss
AG) with Powershot G9 camera. These images were analyzed using Axio
version 4.5 software (Carl Zeiss AG) to determine the spheroid
diameter. The approximated volume (V) of each spheroid was
calculated as follows: V= (4/3) x π x (D1/2) x (D2/2)2,
where D1 and D2 were the longest and shortest diameters,
respectively (26).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from the five cell lines
using the RNeasy Mini kit (Qiagen GmbH) according to the
manufacturer's instructions. A total of 1 µg total RNA from
each sample was converted into cDNA using the M-MLV cDNA Synthesis
kit (Enzynomics, Inc.). The reaction was performed at 25°C for 10
min, 42°C for 60 min, 95°C for 5 min, and held at 4°C on a
SimpliAmp™ Thermal Cycler (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The cDNA products from each sample were used to
perform qPCR. A total of 1 µl five-time diluted cDNA was
used for qPCR, and reagents were mixed followed by PCR using the
SensiFAST SYBR® Lo-ROX kit (Bioline Pty Ltd, Meridian
Bioscience, Inc.). The primers used are listed in Table I. β-actin mRNA was used as an
internal control gene to normalize the data. RT-qPCR was performed
for the initial activation at 95°C for 20 sec, followed by 40
cycles at 95°C for 10 sec, 63°C for 30 sec, and 70°C for 1 sec. The
melting curve was analyzed using the instrument default setting.
The assays were performed in triplicate on a Light
Cycle® 96 system (Roche Diagnostics). The DDCq method
(31) was used for the
quantification of mRNA expression.
 | Table ISequences of specific primers used
for RT-qPCR. |
Table I
Sequences of specific primers used
for RT-qPCR.
| Gene | Accession no. | Primer
sequence | Amplicon size
(bp) | (Refs.) |
|---|
| Aurora A | NM_198433.3 | Fw
5′-TTCCAGGAGGACCACTCTCTGT-3′
Rv 5′-TGCATCCGACCTTCAA TCATT-3′ | 69 | (27) |
| Aurora B | NM_001313950.2 | Fw
5′-CGCAGAGAGATCGAAATCCAG-3′
Rv 5′-AGATCCTCCTCCGGTCATAAAA-3′ | 85 | (28) |
| VEGF | NM_001025366.3 | Fw
5′-AGGAGGAGGGCAGAATCATCAC-3′;
Rv 5′-ATGTCCACCAGGGTCTCGATTG-3′ | 90 | (29) |
| β-actin | NM_001101.5 | Fw
5′-ACAGAGCCTCGCCTTTG-3′
Rv 5′-CCTTGCACATGCCGGAG-3′ | 110 | (30) |
Wound healing assay
The hUVECs and hFBs were cultured in EGM-2
endothelial cell growth medium-2 Bulletkit (Lonza Bioscience) and
DMEM/F12 (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% FBS, respectively, to reach the completed confluence in 24-well
plates. The cells were then supplemented with mitomycin C (5
µg/ml) to inhibit cell proliferation. Thereafter, the cells
were cultured in serum-free medium for 24 h (hUVECs) and 48 h
(hFBs). Scratches were created using cell scrapers SPLScar (SPL
Life Sciences Co., Ltd.), and floating cells were removed by
washing the wells twice with PBS. Oxostephanine was incubated with
the cells at three concentrations of 25, 5 and 1 µM for 24 h
(hUVECs) and 48 h (hFBs). Images were captured every 6 h (Olympus
IX73 Inverted Microscope, Olympus Corporation) from the scars
created. The cell migration ability was analyzed using ImageJ
software (version 1.53e, National Institutes of Health).
Colony formation assay
The hUVECs and hFBs were seeded in a six-well plate
at a density of 1×103 cells/well and treated with
oxostephanine at four different concentrations (25, 5, 1 and 0.2
µM) for 24 h. The medium was refreshed, and the cells were
then incubated in a humidified incubator with 5% CO2 at
37°C for a further 10 days. The cells were then stained with Giemsa
(Millipore, Sigma) for 5 min at room temperature after fixing with
70% methanol for 10 min at room temperature. The formation of
colony units of endothelial cells (CFU-ECs) and fibroblasts
(CFU-Fs) was observed, photographed and counted using an Axiovert
40 Inverted Microscope (Carl Zeiss AG) (magnification, ×4). The
number of colonies was determined per 1,000 cells at seeding.
Growth factor analysis using luminex
assay
Growth factors, including VEGF-A, fibroblast growth
factor-2 (FGF-2) and hepatocyte growth factor (HGF), were analyzed
using Luminex assay with ProcartaPlexTM Multiplex Immunoassays
(Human Custom ProcartaPlex 4-Plex kit; Thermo Fisher Scientific,
Inc.). The conditioned media was prepared by culturing cells to 90%
confluency in an appropriate medium without supplement or FBS for
48 h. The conditioned medium was then collected and kept on ice
prior to use. Reagents and procedures were processed following the
manufacturer's instructions. The luminescent signals of the growth
factors were detected using a LuminexTM 100/200TM system equipped
with the xPONENT 3.1 software (Luminex Co., Ltd).
Tube formation assay
The tube formation assay was performed using
Angiogenesis Assay kit (ab204726, Abcam). Briefly, extracellular
matrix solution (Matrigel, supplied with the kit, Abcam) was added
to a 96-well plate and incubated for 1 h at 37°C to allow the
solution to form a gel. hVUECs were seeded at 1.5×104
cells/well (three replicates per group) on the gel and incubated
with oxostephanine at two concentrations of 5 and 1 µM. For
the background control wells, no Matrigel was added. Suramin
(supplied with the kit, Abcam) was used as an angiogenesis
suppressor control. Following 8 h of incubation at 37°C in the
incubator, the tube formation was examined using an inverted
microscope. The total tube length, total branching points and mean
tube length were analyzed using Wimasis software (Web-based
version, wimasis.com).
Statistical analysis
All statistical analyses were performed using R
software version 3.4.4. The differences between groups were
assessed using an unpaired t-test, two-way ANOVA and Tukey's HSD
tests. A P-value <0.05 was considered to indicate a
statistically significant difference. All data are presented as the
mean ± SD.
Results
Real-time analysis of the effects of
oxostephanine on OVCAR-8 cancer cells
The present study performed a cytotoxicity analysis
of oxostephanine using the OVCAR-8 cell line with the xCELLigence
RTCA system. During >200 h of incubation, the viability, number,
morphology and adherent ability of the cells were recorded and
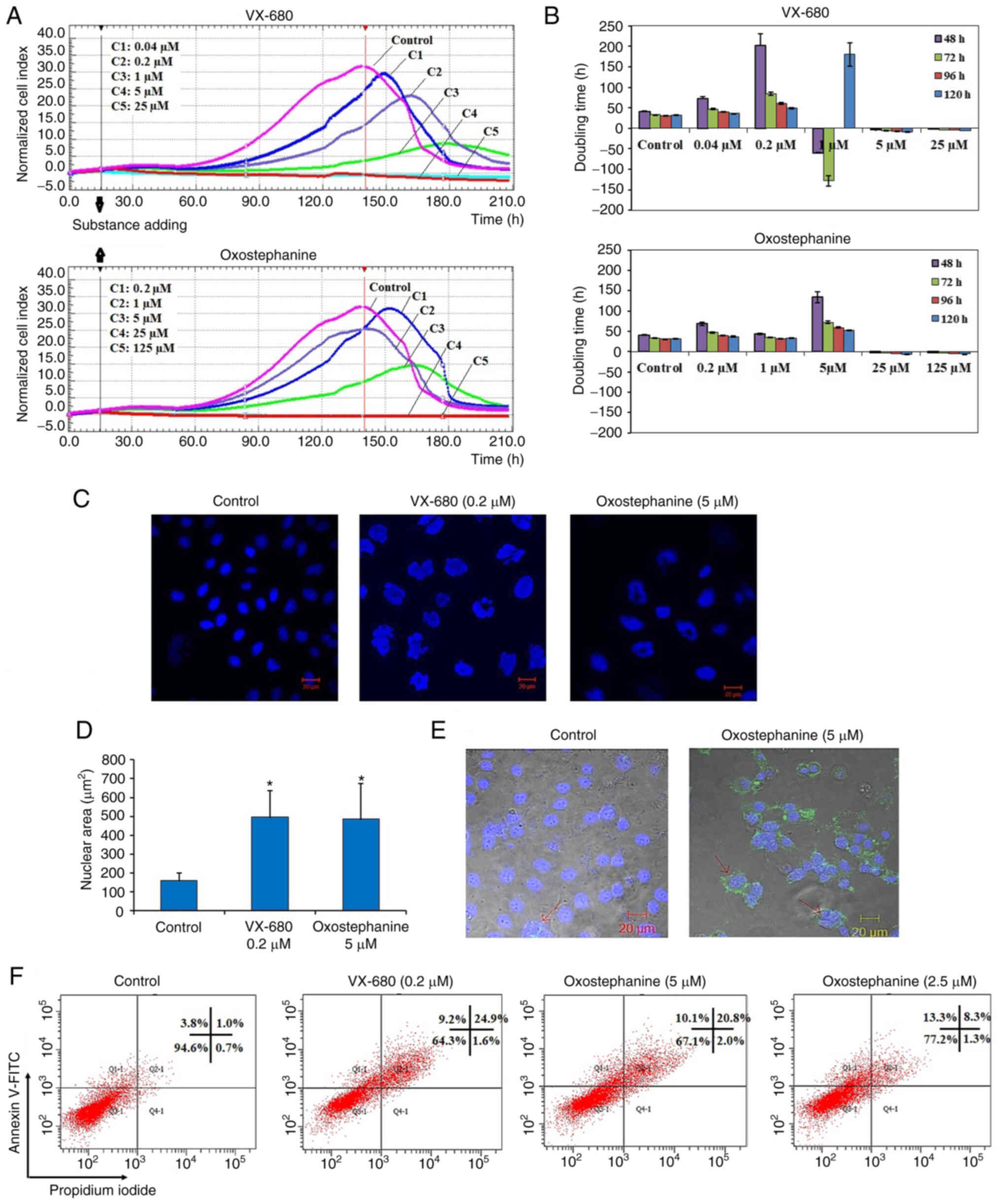
visualized as a graph (Fig. 1A).
The utilities in the RTCA Control Unit software allowed for the
creation of a dose-response curve and the calculation of the
IC50 value of the drug at different time points. The
results revealed that oxostephanine and VX-680 exerted a similar
effect on cell proliferation; the higher concentrations of the
compounds the greater the inhibitory effects on cell growth. In the
control wells, the cell index values gradually increased and peaked
at the time point of 140 h, with a CI of 32 (Fig. 1A). In the wells treated with the
two highest concentrations of 125 and 25 µM oxostephanine,
cell proliferation was entirely inhibited compared to the control
with the CI values decreasing after 3 h of incubation, indicating
that the cells could not grow, but were killed. At the
oxostephanine concentration of 5 µM, the cell proliferation
rate was approximately half that of the control, with the time to
get the peak of CI values was 165 h. At the oxostephanine
concentration of 1 µM, the peak was reached at the same time
but with a smaller value equivalent to 78% of the control. At the
smallest concentration of 0.2 µM oxostephanine, the cell
proliferation was lower than that of the control. For the wells
treated with VX-680, while all cells were killed at the two highest
concentrations, the CI values at the peaks associated with the
other concentrations were smaller and were observed at later time
points than those of the control (Fig. 1A). Using RTCA software, the
IC50 values at different time points of incubation from
24 to 120 h were calculated. The IC50 values were from
3.8-7.3 µM for oxostephanine and 0.2-0.6 µM for
VX-680 (Table II).
| Table IIIC50 values of
oxostephanine and VX-680 in OVCAR-8 cancer cells with different
incubation times. |
Table II
IC50 values of
oxostephanine and VX-680 in OVCAR-8 cancer cells with different
incubation times.
| Compound | Incubation time (h)
|
|---|
| 24 | 48 | 72 | 96 | 120 |
|---|
| VX-680
(µM) | 0.3±0.02 | 0.6±0.06 | 0.2±0.04 | 0.2±0.07 | 0.2±0.05 |
| Oxostephanine
(µM) | 7.3±1.5 | 6.6±0.9 | 5.6±0.7 | 4.6±0.8 | 3.8±0.5 |
The doubling time of the OVCAR-8 cells was also
affected by these two compounds. Following treatment with VX-680,
the cells did not grow and died rapidly following the addition of
the substance expressed by the minus values of the doubling time at
the three highest concentrations. In terms of oxostephanine, the
majority of the doubling time was higher compared to the controls,
indicating that the proliferation of cells was inhibited (Fig. 1B). Of note, a change in the size
of the cells treated with oxostephanine and VX-680 at low
concentrations was observed. The cells increased their size
following the incubation time. Not only the cell size, but the
immunostaining of these cells also indicated that there was a
significant increase in the nuclei area (Fig. 1C). Additionally, the morphology of
the cell nuclei was changed, with the nuclei becoming
heterogeneous, multi-lobed and enlarged, that were not homogeneous
or oval-shaped as in the controls (Fig. 1C). Using the LSM image browser
software, the nuclear area was measured. The data indicated that
the nucleic size of the cells treated with oxostephanine or VX-680
was three-fold larger than that in the control group (Fig. 1D). Taken together, these results
demonstrated that oxostephanine inhibited the proliferation of
OVCAR-8 cells in the micromolar range. The real-time effects of
oxostephanine were comparable to those of VX-680, an Aurora kinase
inhibitor.
Apoptosis induction is a characteristic of Aurora
kinase inhibitors (18,23). Hence, the present study examined
whether oxostephanine could induce the apoptosis of OVCAR-8 cancer
cells. At the oxostephanine concentration of 5 µM, we
observed the expression of phosphatidylserine molecule, an
apoptosis marker, that binding to Annexin-V on the cell surface
after 24 h of incubation (Fig.
1E). The rate of cells positive with Annexin-V was calculated
from the sum of Q1-1 (early apoptosis) and Q2-1 (late apoptosis)
quadrants in the flow cytometry plots (Fig. 1F). Accordingly, the percentage of
oxostephanine (5 µM)-treated cells positive for Annexin-V
was 30.4±6.8%, which was 7.4-fold higher than that of the control
(4.1±0.8%). Moreover, a 33.7±5.1% cell population was positive for
Annexin-V when treated with 0.2 µM VX-680 (Fig. 1F).
Oxostephanine inhibits the growth of
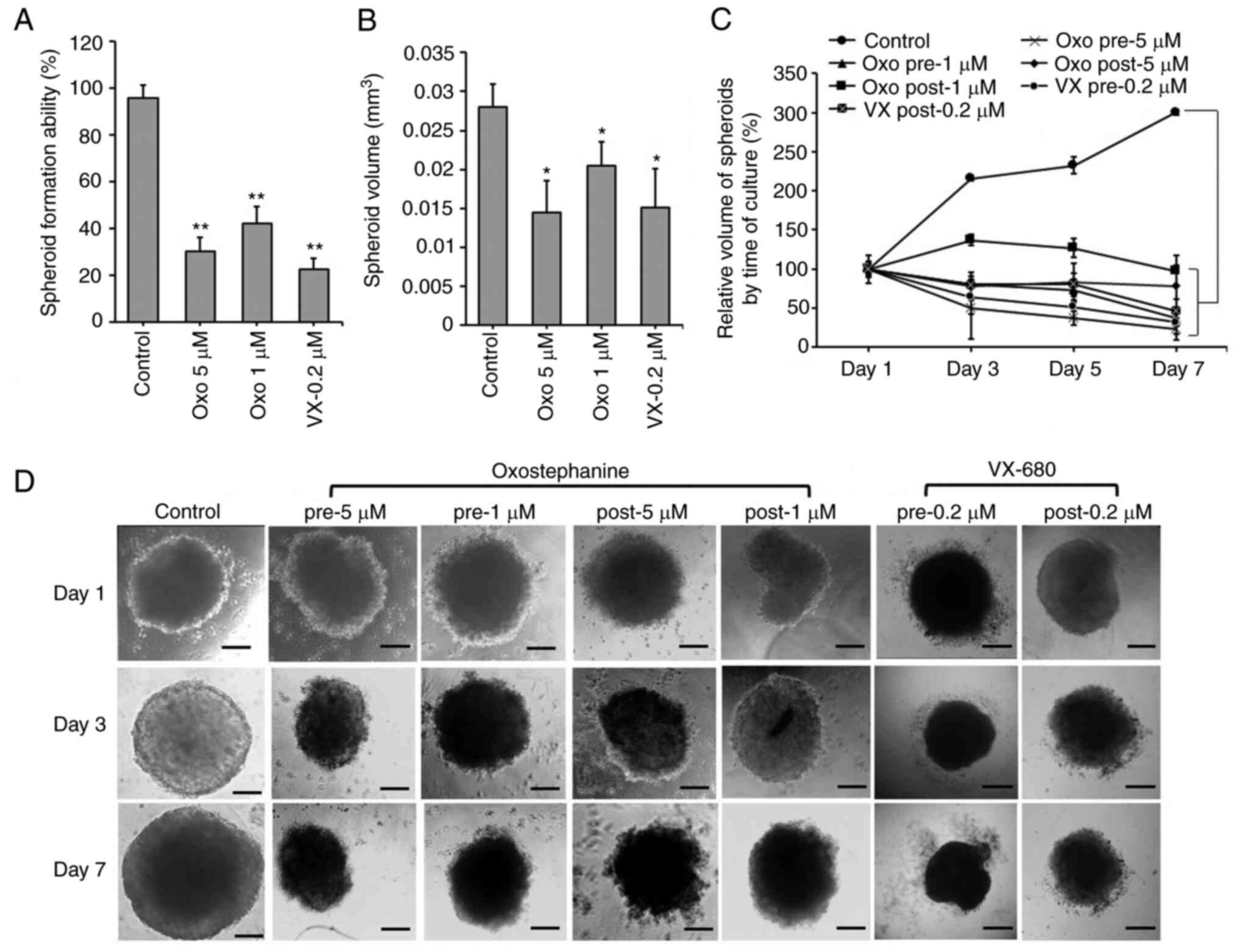
OVCAR-8 spheroids
The effects of oxostephanine on the growth of
OVCAR-8 cells in 3D culture were investigated. When adding the
substance at the time of spheroid preparation, this compound
prevented 70% spheroid formation at 5 µM and 58% spheroid
formation at 1 µM. A similar result was obtained with
VX-680; only 22.5% of spheroids could be formed at the
concentration of 0.2 µM (Fig.
2A). Moreover, the volume of the formed spheroids was smaller
than that of the control (Fig.
2B). After transferring the spheroids into agar plates, the
growth was unaltered at the concentration of 1 µM, whereas
this decreased at the concentration of 5 µM following the
time of culture even with the absence of the compound (Fig. 2C). For the other treatments,
oxostephanine was added and maintained in the medium after the
spheroids were transferred into the agar plate. Under this
condition, after 7 days, the substance inhibited the growth of
spheroids, with the size decreasing 4.3-fold at 5 µM and
2.7-fold at 1 µM. The effect of oxostephanine on spheroid
growth was even more prominent than that of VX-680 at 0.2
µM, with a decrease of 2.1-fold in the volume on day 7 of
treatment. Moreover, the control increased the spheroid volume
3-fold on day 7 of culture on agar (Fig. 2C). Furthermore, the morphology of
the treated spheroids was also changed into loose cell clusters
with numerous cells separately surrounded, in contrast to the tight
and impact control spheroids (Fig.
2D).
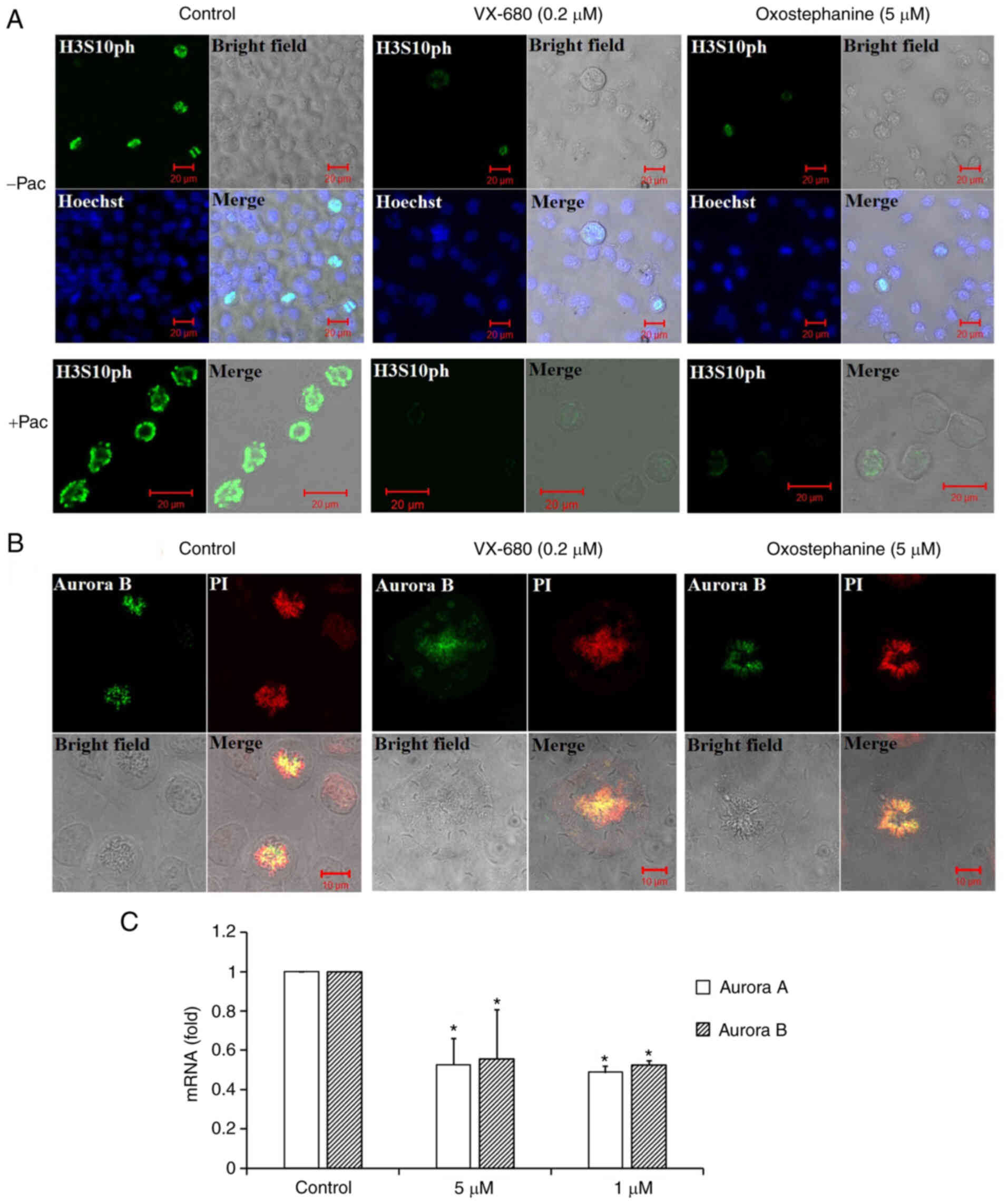
Oxostephanine inhibits Aurora kinase
expression and activity
To characterize oxostephanine as a novel Aurora
kinase inhibitor, the effect of this compound on the
phosphorylation of H3S10ph was evaluated in OVCAR-8 cancer cells.
To collect cells at the mitotic phase, the cell population was
synchronized by the addition of paclitaxel followed by incubation
with oxostephanine and VX-680 at concentrations of 5 and 0.2
µM, respectively. The images revealed that the fluorescence
signal of H3S10ph was markedly decreased in mitotic cells incubated
with oxostephanine and VX-680, even with or without paclitaxel
(Fig. 3A).
In addition, the distribution of Aurora B was
affected by these compounds. In mitotic OVCAR-8 cells, this protein
was not expressed at the centromere, but was diffused on the whole
chromosomes at the metaphase. Moreover, Aurora B presented as
bright dots in the centromere in the control cell group (Fig. 3B). Additionally, RT-qPCR revealed
that the mRNA expression of Aurora B was decreased following
incubation with oxostephanine in OVCAR-8 cells (Fig. 3C).
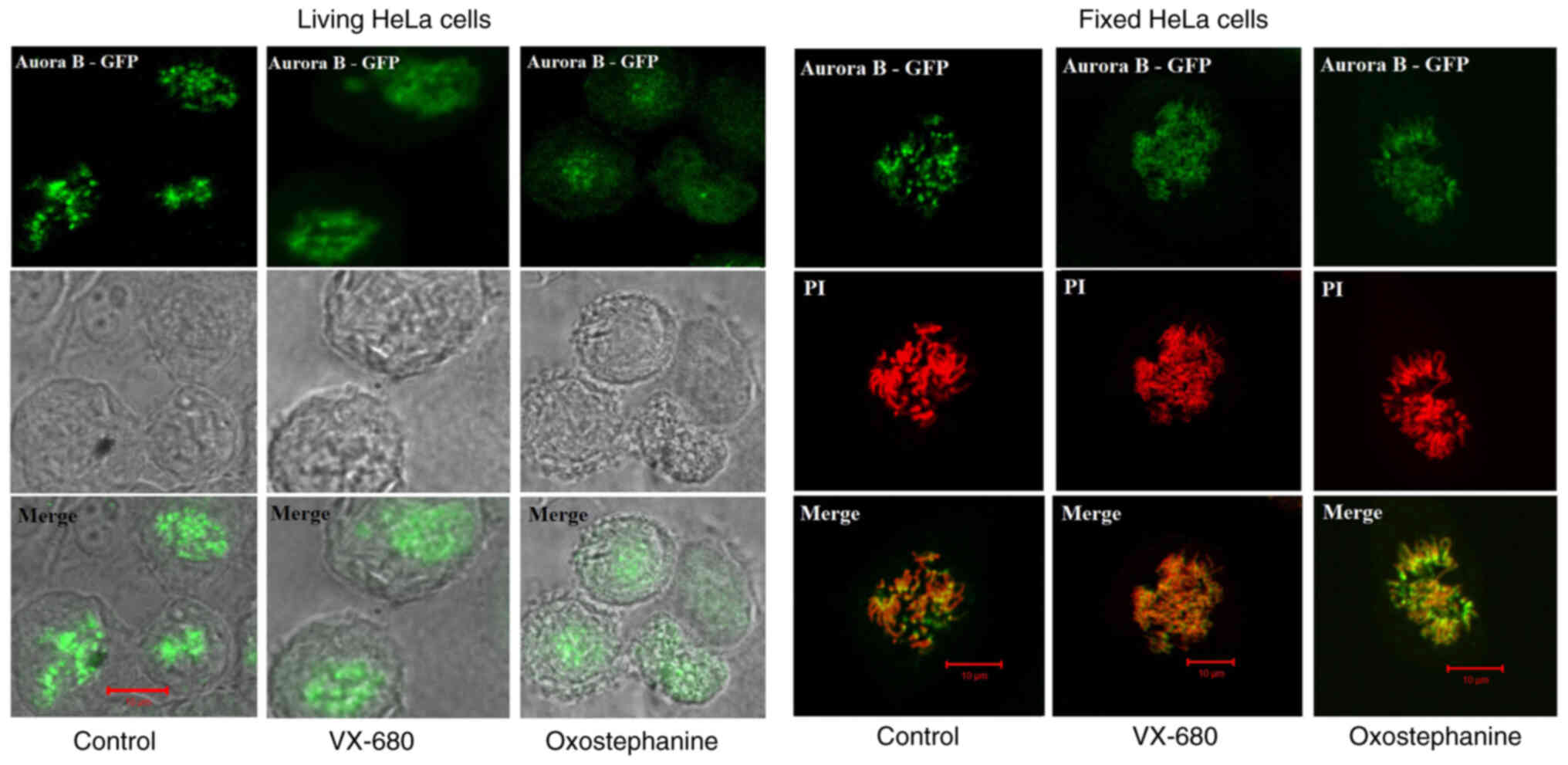
To determine the effects of oxostephanine on the
localization of Aurora B kinase, HeLa cells stably expressing
Aurora kinase B-GFP were used. Notably, the diffusion of Aurora B
was observed in both living and fixed HeLa cells (Fig. 4). Furthermore, in mitotic cells
treated with oxostephanine and VX-680, Aurora B-GFP was observed on
the entire chromosomes when the cells were at metaphase.
In summary, these data illustrated that the
treatment of the cells with oxostephanine affected the behavior of
Aurora B during the cell cycle in a similar manner to VX-680, but
with a lower efficiency.
Oxostephanine is selectively cytotoxic on
different cell types
The present study also selected three cell lines,
including human UC-MSCs, hUVECs and hFBs for the examination of
oxostephanine cytotoxicity. Firstly, the expression of Aurora A and
Aurora B kinase genes relative to the actin gene control was
examined in normal and cancer cells. The results revealed that
these genes were highly expressed at the mRNA level, with the
highest levels observed in the hUVECs and OVCAR-8 cells, and the
lowest in hFBs (Fig. 5A).
Secondly, the cells were incubated with
oxostephanine for the analysis of cell death. Following 24 h of
incubation with oxostephanine, the death of hUVECs was observed at
the two highest concentrations. After 48 and 72 h, the cell death
number increased continuously in these wells containing hUVECs.
Similar results were detected in UC-MSCs. On the other hand, in the
wells of hFBs, no cell death was observed (Fig. 5B). Additionally, the
IC50 values were consistent with these observations. The
IC50 values from the hUVECs were 7.9±0.6, 3.1±0.5 and
1.9±0.5 µM after 24, 48 and 72 h of incubation,
respectively. However, the IC50 values from the hFBs
could not be determined after 24 and 48 h, but were 17.1±0.8
µM after 72 h of incubation. Notably, the cytotoxicity
effect of oxostephanine on UC-MSCs was lower than that on hUVECs,
but higher than that on hFBs, with IC50 values at 48 and
72 h were 4.7±0.8 and 5.1±0.7 µM, respectively (Fig. 5C). These data, as well as the
results of the mRNA levels indicated that the oxostephanine may be
more toxic to OVCAR-8 cancer cells and hUVECs, but less on hFBs and
UC-MSCs.
Oxostephanine reduces colony formation
and growth factor secretion by hUVECs and hFBs
The effects of Oxostephanine on the capacity of
endothelial progenitor cells and fibroblast precursor cells to form
colonies were then examined. As shown in Fig. 6A, both the number of colonies and
the density of cells/colonies were reduced in the treated wells
compared to the controls. The numbers of CFU-ECs and CFU-Fs were
significantly decreased with the two highest concentrations (25 and
5 µM) (P<0.01). Colony formation was also disrupted with
the lower concentrations of oxostephanine, with a smaller number of
colonies compared to the control in hUVECs (P<0.05) (Fig. 6B, left panel. In addition, the
inhibitory effects of oxostephanine on colony formation were more
prominent in hUVECs than in hFBs, with a smaller number of CFUs
relative to the control (%) in the endothelial cells compared to
that in the fibroblasts (P<0.05) (Fig. 6B, right panel).
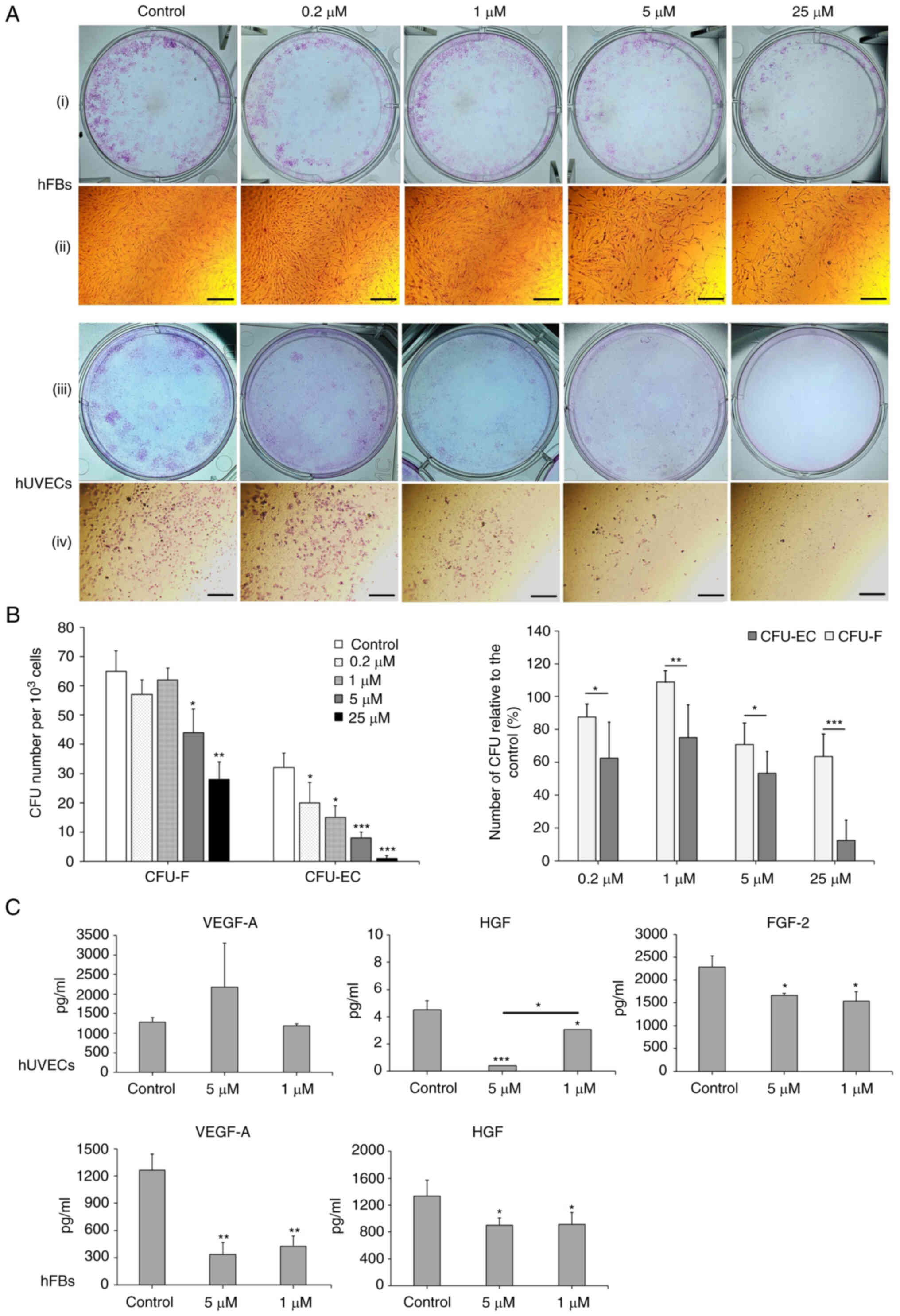 | Figure 6Oxostephanine reduces the colony
formation and growth factor secretion by hUVECs and hFBs. (A)
Images of CFU-Fs (hFBs) and CFU-ECs (hUVECs) and cell morphology in
each type of CFU. CFUs were reduced in both the number of CFU and
the number of cells per CFU. (i and iii) Macroscopic images of hFBs
and hUVECs culture plates, respectively, following Giemsa staining;
(ii and iv) microscopic of a single stained colony in hFBs and
hUVECs, respectively. Scale bars, 200 µM. (B) The colony
formation ability of hUVECs and hFBs treated with the indicated
concentrations of oxostephanine. (C) The secretion of three types
of growth factors (VEGF-A, HGF and FGF-2) in the presence of
oxostephanine at various concentrations. *P<0.05,
**P<0.01 and ***P<0.001, vs. control.
hUVECs, human umbilical vein endothelial cells; hFB, human dermal
fibroblasts; CFU, colony-forming units. |
Three types of growth factors, including VEGF-A,
FGF-2 and HGF, were measured in the cell culture medium after
treating the cells with oxostephanine at 1 and 5 µM. The
data indicated that the secretion of these proteins was differed
between the cell types. In the controls, both the hUVECs and hFBs
secreted HGF with values of 4.5, and 1,333±243.2 µg/ml,
respectively (Fig. 6C).
Additionally, both the hFbs and hUVECs produced VEGF-A into the
medium at a concentration of around ~1,270 pg/ml. The hUVECs
secreted a high amount of FGF-2 (2,285.8±240.1 pg/m). Following
incubation with oxostephanine, the capacity of growth factor
secretion by the cells was consistent with the control regarding
the factor component that only hUVECs could secrete all three
factors (VEGF-A, HGF and FGF-2) and hFBs secreted only VEGF-A and
HGF. However, the amount of all tested growth factors decreased
(P<0.05), apart from VEGF-A secreted by hUVECs treated with 5
µM oxostephanine (Fig.
6C). These results demonstrated that oxostephanine affected the
secretion of growth factors by cells.
Oxostephanine inhibits the migration of
hUVECs and hFBs
Fibroblast and endothelial cell migration is a
critical step in the wound healing and angiogenesis processes
(32). Thus, in the present
study, a wound healing assay was performed to examine the capacity
of oxostephanine to regulate the migration of endothelial cells and
fibroblasts. In the control group, both hUVECs and hFBs expressed
their ability to migrate to close the gap at a more rapid rate; the
hUVECs exhibited a greater migratory ability (covering 100% of the
wound after 24 h) compared to the hFBs (covering 46.1% of the wound
after 24 h) (Fig. 7). When the
cells were treated with oxostephanine, a significant decrease in
the migration of hUVECs and hFBs were observed (P<0.05; Fig. 7). As regards the hUVECs, the
percentage of the wound covered by cells treated with oxostephanine
at the concentrations of 25 and 5 µM was ~11% compared to
100% of that in the control group after 24 h, which indicated that
the compound inhibited the migration of hUVECs >10-fold
(Fig. 7). This inhibitory effect
was less prominent in hFBs at the two highest concentrations
(5.7-fold decrease at 25 µM and 3.2-fold decrease at 5
µM at 48 h). However, at the concentration of 1 µM,
the compound exerted more prominent inhibitory effects on the
migration of hFBs than that of hUVECs. These results demonstrated
that oxostephanine significantly inhibited the migration of hUVECs
and hFBs.
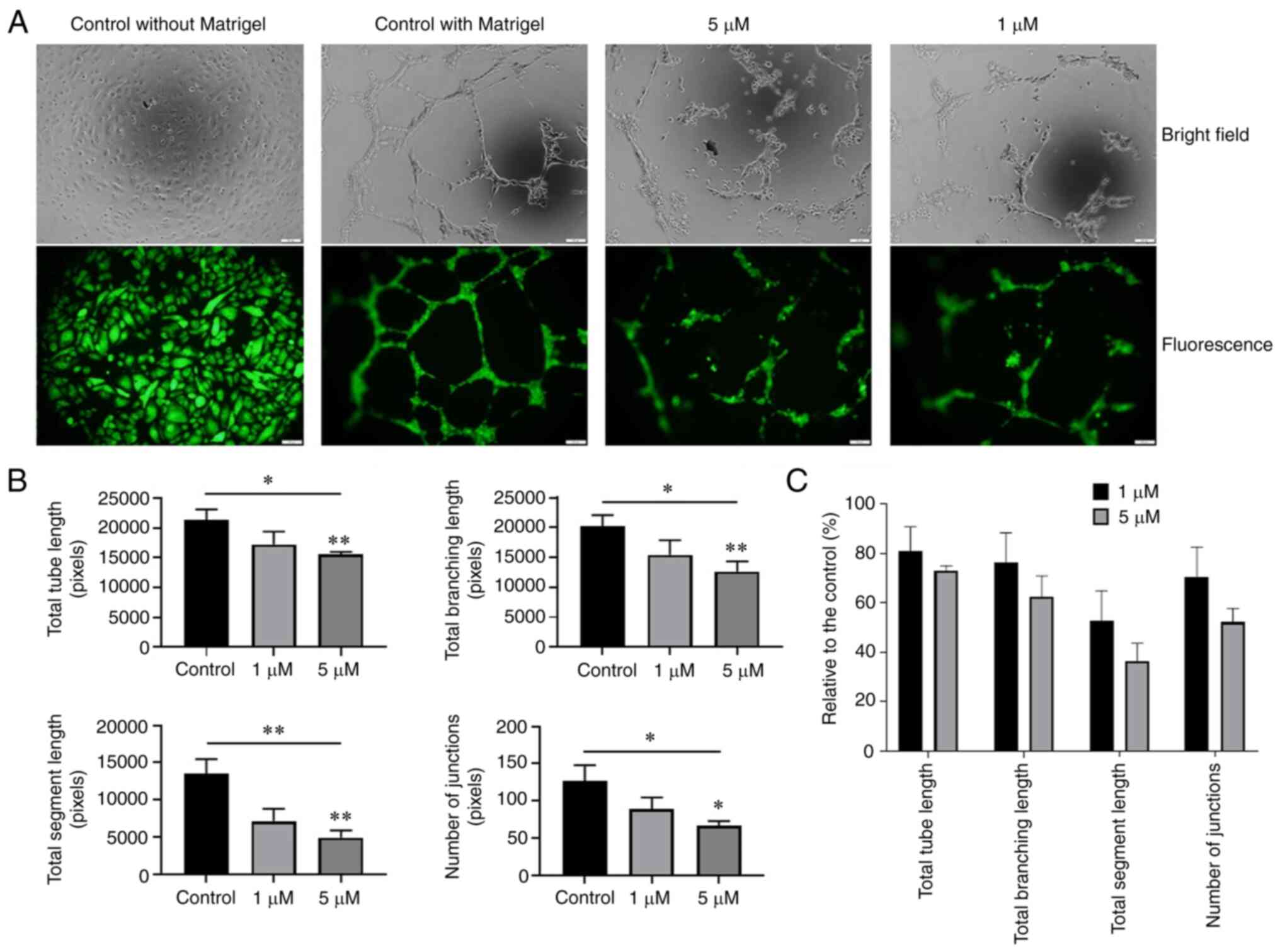
Oxostephanine suppresses angiogenesis in
vitro
The effect of oxostephanine on the angiogenesis of
hUVECs was examined using tube formation assay. As shown in
Fig. 8A, the hUVECs formed a
capillary-like network on the Matrigel, with the highest number of
total tube lengths and tube branching points after 10 h. By
contrast, the tube-formation capacity significantly decreased when
the cells were treated with 5 µM oxostephanine (P<0.05)
(Fig. 8B). The total tube length,
tube branching, tube segments and the number of junctions were
72.9±2.1, 62.5±8.4, 36.4±7.2, and 52.1±5.6%, respectively, compared
to the control group. The majority of hUVECs clustered, and very
few tube-like structures were observed. When the cells were treated
with 1 µM oxostephanine, the percentage of total tube
length, branching, segments, and number of junctions reached
80.8±10, 76.2±12, 52.7±12.2, and 70.3±12.3%, respectively, compared
to the control (Fig. 8C). These
findings suggested that oxostephanine suppressed angiogenesis in
vitro.
Discussion
The crucial role of Aurora kinases, particularly
Aurora A and B, in cell division, as well as the overexpression of
these kinases in a wide range of cancers, renders them a potential
target in cancer treatment (18).
Oxostephanine extracted from the Stephania plant was first
reported by Makarasen et al (21) for its activity in inhibiting the
growth of a variety of cancer cell lines. The present study first
aimed to characterize oxostephanine, extracted from S.
dielsiana leaves in Vietnam, as a novel Aurora inhibitor by
comparing the real-time effects of this substance on cancer cells
to those of VX-680, a well-known Aurora kinase inhibitor (33). An ovarian cancer cell line
(OVCAR-8), was used to examine the effects of oxostephanine, since
Aurora kinase has been reported to be overexpressed in epithelial
ovarian cancer, in addition to two recent clinical trials that have
used Aurora kinase inhibitors to treat ovarian cancer (34-36). In the present study, the analysis
using the xCelligence system revealed similar responses of the
OVCAR-8 cells to both compounds (oxostephanine and VX-680) in
real-time growth dose-response curves, cell population doubling
time and cellular size change.
Of note, at low tested concentrations of
oxostephanine (<5 µM) and VX-680 (1 µM), the cells
became aneuploidy with an increase in their size, but not in their
number. Previous research has indicated that when Aurora kinase
activity is inhibited, the mitotic SAC is activated, which leads to
mitotic arrest. However, this SAC could be overridden, which causes
the mitotic slippage of cells in the presence of Aurora kinase
inhibitors. This phenomenon eventually led to cells becoming
aneuploidy or apoptotic (37). In
the present study, OVCAR-8 cells treated with oxostephanine and
VX-680 at low concentrations expressed enlarged and lobed nuclei.
Moreover, as shown by immunofluorescence assay, both compounds
downregulated the phosphorylation of protein histone H3 at serine
10 in cancer cells. These data are consistent with those of the
study by Knockleby et al (23), demonstrating that oxostephanine
inhibited H3S10ph in HeLa cells (23). As the phosphorylation of histone
H3 at serine 10 is considered a marker of activated Aurora B kinase
(7,8), hence oxostephanine could prevent the
function of this kinase.
Previous studies have indicated that the activity of
Aurora B is associated with auto-phosphorylation and centromere
distribution (5,23). Under normal conditions, Aurora B
must concentrate at the kinetochore to phosphorylate some proteins
in the conserved outer kinetochore KNL1/Mis12 complex/Ndc80 complex
(KMN) network, which plays a role in the kinetochore-microtubule
attachment (4-6). The present study demonstrated that
oxostephanine affected the normal localization of Aurora B kinase;
thus, it may inhibit the auto-phosphorylation activity of this
enzyme. In the presence of oxostephanine and VX-680, Aurora B
diffused to all chromosome arms and in the cytoplasm. This
phenomenon ocurred in all fixed and living OVCAR-8 and HeLa cells.
By observing living HeLa cells that express Aurora B-GFP, it was
noted that the cells that have chromosomes with diffused Aurora B
remained longer in metaphase and eventually became aneuploidy. This
phenomenon of Aurora B has been mentioned with the other inhibitors
(24,25). This could be explained by the fact
that Aurora B did not concentrate at the kinetochore, leading to an
effect on the correct attachment of the chromosome to the
microtubule and subsequently activating the SAC, consequently
leading to mitotic slippage, as discussed above. Moreover,
oxostephanine decreased the expression of both Aurora A and Aurora
B at the mRNA level as did VX-680. The reduction in the levels of
these proteins contributed to defects in cell division functions.
Taken together, these data demonstrated that oxostephanine was an
Aurora kinase inhibitor, and this compound was cytotoxic to OVCAR-8
cells in both monolayer culture and tumor spheroids. It is worth
noting that Knockleby et al (23) indicated the effect of
Oxostephanine on both Aurora A and Aurora B in the kinase assay.
The present study first focused on Aurora B in OVCAR-8 cells. In
future studies, the authors aim to continue to test the effects of
oxostephanine on Aurora A kinase in cell culture.
Cancer-associated mesenchymal stem cells and
fibroblasts have been proven to facilitate tumor progression.
Recent research has revealed the function of mesenchymal stem cells
in glioblastoma resistant to Aurora kinase inhibitor, leading to
the recurrence of tumors (38).
In acute myeloid leukemia (AML), one mechanism of mesenchymal stem
cells used to protect leukemic cells from chemotherapeutic agents
is activating Aurora A by increasing IL-6 secretion (39). In a co-culture system, fibroblasts
have been shown to induce the upregulation of Aurora A in non-small
cell lung carcinoma to protect the cancer cells from gefitinib
treatment (40,41). Fibroblasts can be activated by
Aurora B through Wilms tumor 1 signaling, leading to an induction
of fibrogenesis (42). Moreover,
the downregulation of Aurora B stimulates cellular senescence in
hFBs (43). Aurora kinase and
stromal cells exert synergistic effects on the development of
cancer cells. Moreover, angiogenesis is necessary for the
progression of tumors (44).
Hence, in the present study examined the effects of oxostephanine
on four cell types (UC-MSCs, hFBs, hUVECs and OVCAR-8). Firstly, it
was found that all tested cells highly expressed Aurora A and B,
with the highest expression level observed in OVCAR-8 cells and
hUVECs, followed by UC-MSCs, and finally hFBs. Accordingly, the
IC50 values of oxostephanine in these cell lines were
the lowest in the OVCAR-8 cells and hUVECs, higher in the MSCs, and
highest in the hFBs. Moreover, the reduction in the colony-forming
units indicated that oxostephanine could inhibit the proliferation
of endothelial progenitor cells and fibroblasts. One limitation of
the present study was that the presentation of colonies needed
improvement as the location of the closely clustered colonies could
not be seen. However, the number of colonies could still be
counted. At the concentration of 5 µM, oxostephanine
significantly inhibited the colony formation of hUVECs; however,
the colony-forming inhibitory effect was less prominent in hFBs
(~30% CFUs). Additionally, in the wound healing assay,
oxostephanine also exerted a more prominent inhibitory effect on
the migration of hUVECs than hFBs. These results demonstrated the
selective activity of oxostephanine toward hUVECs. The targeting of
the compound to different cell types may result from the expression
of Aurora kinase in these cells. Higher levels of Aurora kinase are
associated with a more prominent effect of oxostephanine on the
cells. Apart from cell growth, the function of hUVECs in
angiogenesis was also disrupted by oxostephanine. These cells could
not successfully form tubes in Matrigel in the presence of 5
µM oxostephanine. The anti-angiogenic effect of Aurora
kinase inhibitors has been previously reported (13) through their involvement in a
signaling pathway that enhances angiogenesis (45) and stabilizes N-Myc, which is a
well-known oncogene (46,47). These results indicate that
oxostephanine functions as a suppressor of angiogenesis.
Furthermore, the data indicated that oxostephanine
decreased the production of VEGF-A, HGF and FGF-2, which functions
in the proliferation, migration and tube formation processes
(48-51), by both hUVECs and hFBs. Notably,
in the present study, in hUVECs, the mRNA expression of VEGF-A in
cells treated with oxostephanine was not considerably altered;
however, the expression of FGF-2 was significantly decreased
compared to the control. This activity of oxostephanine differed
from VX-680, which has been shown to inhibit VEGF-A expression
(13). Nonetheless, the decrease
in the levels of FGF-2 and HFG was sufficient to inhibit the growth
and function of hUVECs.
Of note, the effects of oxostephanine one growth
factor secretion by cells have not yet been clarified. In addition,
the involvement of Aurora kinases in angiogenesis have not yet been
elucidated. However, it can be hypothesized that Aurora kinase
inhibitors, such as oxostephanine, are cytotoxic toward ovarian
cancer cells and endothelial cells, which leads to the inhibition
of tumor angiogenesis. Furthermore, even though this compound was
less cytotoxic to the other stromal cells such as hFBs and UC-MSCs,
it prevented the cell functions that can result in stromal cells
being inefficient in supporting tumor growth. This hypothesis was
encouraged by a published study on the antitumor activity of the
methanol fractional extraction from S. dielsiana roots on
Swiss mice bearing Sarcoma-180 tumors, which reported a 4-fold
decrease in tumor volume in the treated mine (52). It is necessary to examine the
effects of oxostephanine in vivo using animal models
transplanted with human tumor cells. The authors aim to perform
such experiments in the future.
In conclusion, the findings of the present study
indicate that oxostephanine is a potential Aurora kinase inhibitor.
It inhibited the proliferation of ovarian cancer OVCAR-8 cells and
multicellular tumor spheroids. Moreover, oxostephanine exhibited
selective cytotoxicity to normal cells by inducing a high
expression of Aurora kinase A and B. Furthermore, this compound
downregulated the expression of growth factors, prevented the
migration of hUVECs and hFBs, and reduced tube formation. However,
further studies are required for oxostephanine to be developed as
an anticancer drug. This compound needs to be tested on other
ovarian cancer cell lines, particularly primary cell lines, to
confirm its effects on ovarian cancer. In addition, the expression
of Aurora A and B in different cell types needs to be quantified
using effective methods, such as western blot analysis, in order to
determine to the association of Aurora kinase expression and the
effects of oxostephanine. More importantly, in the long term,
experiments using in vivo tumor models need be performed to
confirm the efficiency of oxostephanine.
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
THTT and LDBV were involved in the study
experimental design and performance, data analysis and in the
writing of the manuscript. XPTD, LDD and HBP were involved in
performing the experiments and in data analysis. UTTT and THTP were
involved in the guidance of the experimental design and in
manuscript revision. KVTL and TPN were involved in the guidance of
the experimental operations. MNTH and HQN were involved in the
conceptualization of the study, in the provision of resources, in
the experimental design, data analysis and in the writing and
revision of the manuscript.
Ethics approval and consent to
participate
The hUVECs, hFBs and UC-MSCs were provided by the
Vinmec Research Institute of Stem cell and Gene Technology, and
they were not immortalized cell lines. The protocols for cell
isolation were approved by the Ethics Committee of Vinmec
International Hospital (Document no. 40/2020/QD-Vinmec for hUVECs
and UCMSCs, signed and dated on December 24, 2020; Document no.
311/2018/QD-Vinmec for hFBs, signed and dated on September 11,
2018).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was funded by the Administration of Science
Technology and Training-Ministry of Health-Vietnam (according to
Decision no. 2721/QD-BYT, dated June 28, 2019, and Contract no.
09/HD-K2DT, dated September 18, 2019).
References
|
1
|
Cowley DO, Rivera-Pérez JA, Schliekelman
M, He YJ, Oliver TG, Lu L, O'Quinn R, Salmon ED, Magnuson T and Van
Dyke T: Aurora-A kinase is essential for bipolar spindle formation
and early development. Mol Cell Biol. 29:1059–1071. 2009.
View Article : Google Scholar :
|
|
2
|
Barretta ML, Spano D, D'Ambrosio C,
Cervigni RI, Scaloni A, Corda D and Colanzi A: Aurora-A recruitment
and centrosomal maturation are regulated by a Golgi-activated pool
of Src during G2. Nat Commun. 7:117272016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carmena M, Ruchaud S and Earnshaw WC:
Making the Auroras glow: Regulation of Aurora A and B kinase
function by inter-acting proteins. Curr Opin Cell Biol. 21:796–805.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gurden MD, Anderhub SJ, Faisal A and
Linardopoulos S: Aurora B prevents premature removal of spindle
assembly checkpoint proteins from the kinetochore: A key role for
Aurora B in mitosis. Oncotarget. 9:19525–19542. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shimada M, Goshima T, Matsuo H, Johmura Y,
Haruta M, Murata K, Tanaka H, Ikawa M, Nakanishi K and Nakanishi M:
Essential role of autoactivation circuitry on Aurora B-mediated
H2AX-pS121 in mitosis. Nat Commun. 7:120592016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lan W, Zhang X, Kline-Smith SL, Rosasco
SE, Barrett-Wilt GA, Shabanowitz J, Hunt DF, Walczak CE and
Stukenberg PT: Aurora B phosphorylates centromeric MCAK and
regulates its localization and microtubule depolymerization
activity. Curr Biol. 14:273–286. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mallm JP and Rippe K: Aurora kinase B
regulates telomerase activity via a centromeric RNA in stem cells.
Cell Rep. 11:1667–1678. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rosasco-Nitcher SE, Lan W, Khorasanizadeh
S and Stukenberg PT: Centromeric Aurora-B activation requires
TD-60, microtubules, and substrate priming phosphorylation.
Science. 319:469–472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang F, Dai J, Daum JR, Niedzialkowska E,
Banerjee B, Stukenberg PT, Gorbsky GJ and Higgins JMG: Histone H3
Thr-3 phosphorylation by Haspin positions Aurora B at centromeres
in mitosis. Science. 330:231–235. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vader G, Medema RH and Lens SMA: The
chromosomal passenger complex: Guiding Aurora-B through mitosis. J
Cell Biol. 173:833–837. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delacour-Larose M, Thi MNH, Dimitrov S and
Molla A: Role of survivin phosphorylation by aurora B in mitosis.
Cell cycle. 6:1878–1885. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Otto T, Horn S, Brockmann M, Eilers U,
Schüttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R,
Christiansen H, et al: Stabilization of N-myc is a critical
function of aurora A in human neuroblastoma. Cancer Cell. 15:67–78.
2009. View Article : Google Scholar
|
|
13
|
Sun X, Niu S, Zhang Z, Wang A, Yang C, Guo
Z, Hao Y, Li X and Wang X: Aurora kinase inhibitor VX-680
suppresses the proliferation and migration of HUVECs and
angiogenesis. Mol Med Rep. 19:3841–3847. 2019.PubMed/NCBI
|
|
14
|
Romain CV, Paul P, Lee S, Qiao J and Chung
DH: Targeting Aurora kinase A inhibits hypoxia-mediated
neuroblastoma cell tumorigenesis. Anticancer Res. 34:2269–2274.
2014.PubMed/NCBI
|
|
15
|
Tang CJC, Lin CY and Tang TK: Dynamic
localization and functional implications of Aurora-C kinase during
male mouse meiosis. Dev Biol. 290:398–410. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Balboula AZ and Schindler K: Selective
disruption of aurora C kinase reveals distinct functions from
aurora B kinase during meiosis in mouse oocytes. PLoS Genet.
10:e10041942014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Quartuccio SM and Schindler K: Functions
of Aurora kinase C in meiosis and cancer. Front Cell Dev Biol.
3:502015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bavetsias V and Linardopoulos S: Aurora
kinase inhibitors: Current status and outlook. Front Oncol.
5:2782015. View Article : Google Scholar
|
|
19
|
Inamdar KV, O'Brien S, Sen S, Keating M,
Nguyen MH, Wang X, Fernandez M, Thomazy V, Medeiros LJ and
Bueso-Ramos CE: Aurora-A kinase nuclear expression in chronic
lymphocytic leukemia. Mod Pathol. 21:1428–1435. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giet R, Petretti C and Prigent C: Aurora
kinases, aneuploidy and cancer, a coincidence or a real link?
Trends Cell Biol. 15:241–250. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Makarasen A, Sirithana W, Mogkhuntod S,
Khunnawutmanotham N, Chimnoi N and Techasakul S: Cytotoxic and
antimicrobial activities of aporphine alkaloids isolated from
stephania venosa (Blume) spreng. Planta Med. 77:1519–1524. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thien DD, Thuy TT, Huy NQ, Van TH, Duong
LTT and Tam NT: Cytotoxic alkaloids from stephania dielsiana. Chem
Nat Compd. 54:613–616. 2018. View Article : Google Scholar
|
|
23
|
Knockleby J, Pradines B, Gendrot M,
Mosnier J, Nguyen TT, Trinh TT, Lee H and Le PM: Cytotoxic and
anti-plasmodial activities of stephania dielsiana Y.C. Wu extracts
and the isolated compounds. Molecules. 25:37552020. View Article : Google Scholar :
|
|
24
|
Hoang TMN, Favier B, Valette A, Barette C,
Nguyen CH, Lafanechère L, Grierson DS, Dimitrov S and Molla A:
Benzo[e] pyridoindoles, novel inhibitors of the aurora kinases.
Cell Cycle. 8:765–772. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hoang NTM, Phuong TT, Nguyen TTN, Tran
YTH, Nguyen ATN, Nguyen TL and Bui KTV: In vitro characterization
of derrone as an aurora kinase inhibitor. Biol Pharm Bull.
39:935–945. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McMillan KS, McCluskey AG, Sorensen A,
Boyd M and Zagnoni M: Emulsion technologies for multicellular
tumour spheroid radiation assays. Analyst. 141:100–110. 2016.
View Article : Google Scholar
|
|
27
|
Lin YS, Su LJ, Yu CT, Wong FH, Yeh HH,
Chen SL, Wu JC, Lin WJ, Shiue YL, Liu HS, et al: Gene expression
profiles of the aurora family kinases. Gene Expr. 13:15–26. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He J, Qi Z, Zhang X, Yang Y, Liu F, Zhao G
and Wang Z: Aurora kinase B inhibitor barasertib (AZD1152) inhibits
glucose metabolism in gastric cancer cells. Anticancer Drugs.
30:19–26. 2019. View Article : Google Scholar
|
|
29
|
Romain C, Paul R, Kim KW, Lee S, Qiao J
and Chung DH: Targeting Aurora kinase-A downregulates cell
proliferation and angiogenesis in neuroblastoma. J Pediatr Surg.
49:159–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Roy JG, McElhaney JE and Verschoor CP:
Reliable reference genes for the quantification of mRNA in human
T-cells and PBMCs stimulated with live influenza virus. BMC
Immunol. 21:42020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Bi H, Li H, Zhang C, Mao Y, Nie F, Xing Y,
Sha W, Wang X, Irwin DM and Tan H: Stromal vascular fraction
promotes migration of fibroblasts and angiogenesis through
regulation of extracellular matrix in the skin wound healing
process. Stem Cell Res Ther. 10:3022019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, Zhang ZF, Chen J, Huang D, Ding Y,
Tan MH, Qian CN, Resau JH, Kim H and The BT: VX680/MK-0457, a
potent and selective Aurora kinase inhibitor, targets both tumor
and endothelial cells in clear cell renal cell carcinoma. Am J
Transl Res. 2:296–308. 2010.PubMed/NCBI
|
|
34
|
Pérez-Fidalgo JA, Gambardella V, Pineda B,
Burgues O, Piñero O and Cervantes A: Aurora kinases in ovarian
cancer. ESMO Open. 5:e0007182010. View Article : Google Scholar
|
|
35
|
Cervantes A, Elez E, Roda D, Ecsedy J,
Macarulla T, Venkatakrishnan K, Roselló S, Andreu J, Jung J,
Sanchis-Garcia JM, et al: Phase I pharmacokinetic/pharmacodynamic
study of MLN8237, an investigational, oral, selective Aurora a
kinase inhibitor, in patients with advanced solid tumors. Clin
Cancer Res. 18:4764–4774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Falchook G, Coleman RL, Roszak A, Behbakht
K, Matulonis U, Ray-Coquard I, Sawrycki P, Duska LR, Tew W,
Ghamande S, et al: Alisertib in combination with weekly paclitaxel
in patients with advanced breast cancer or recurrent ovarian
cancer: A randomized clinical trial. JAMA Oncol. 5:e1837732019.
View Article : Google Scholar :
|
|
37
|
Brito DA, Yang Z and Rieder CL:
Microtubules do not promote mitotic slippage when the spindle
assembly checkpoint cannot be satisfied. J Cell Biol. 182:623–629.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Willems E, Lombard A, Dedobbeleer M,
Goffart N and Rogister B: The unexpected roles of aurora A kinase
in gliobastoma recurrences. Target Oncol. 12:11–18. 2017.
View Article : Google Scholar
|
|
39
|
Wang JD, Zhang W, Zhang JW, Zhang L, Wang
LX, Zhou HS, Long L, Lu G, Liu Q and Long ZJ: A novel aurora kinase
inhibitor attenuates leukemic cell proliferation induced by
mesenchymal stem cells. Mol Ther Oncolytics. 18:491–503. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu CC, Yu CTR, Chang GC, Lai JM and Hsu
SL: Aurora-A promotes gefitinib resistance via a NF-κB signaling
pathway in p53 knockdown lung cancer cells. Biochem Bioph Res
Commun. 405:168–172. 2011. View Article : Google Scholar
|
|
41
|
Chen J, Lu H, Zhou W, Yin H, Zhu L, Liu C,
Zhang P, Hu H, Yang Y and Han H: AURKA upregulation plays a role in
fibroblast-reduced gefitinib sensitivity in the NSCLC cell line
HCC827. Oncol Rep. 33:1860–1866. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kasam RK, Ghandikota S, Soundararajan D,
Reddy GB, Huang SK, Jegga AG and Madala SK: Inhibition of Aurora
kinase B attenuates fibroblast activation and pulmonary fibrosis.
EMBO Mol Med. 12:e121312020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim HJ, Cho JH, Quan H and Kim JR:
Down-regulation of Aurora B kinase induces cellular senescence in
human fibro-blasts and endothelial cells through a p53-dependent
pathway. FEBS Lett. 585:3569–3576. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lugano R, Ramachandran M and Dimberg A:
Tumor angiogenesis: Causes, consequences, challenges and
opportunities. Cell Mol Life Sci. 77:1745–1770. 2020. View Article : Google Scholar :
|
|
45
|
Wang Z, Zhao Y, An Z and Li W: Molecular
links between angiogenesis and neuroendocrine phenotypes in
prostate cancer progression. Front Oncol. 9:14912020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Villaume K, Blanc M, Gouysse G, Walter T,
Couderc C, Nejjari M, Vercherat C, Cordier-Bussat M, Roche C and
Scoazec JY: VEGF secretion by neuroendocrine yumor cells is
inhibited by octreotide and by inhibitors of the PI3K/AKT/mTOR
pathway. Neuroendocrinology. 91:268–278. 2010. View Article : Google Scholar
|
|
47
|
Ton AT, Singh K, Morin H, Ban F, Leblanc
E, Lee J, Lallous N and Cherkasov A: Dual-inhibitors of N-Myc and
AURKA as potential therapy for neuroendocrine prostate cancer. Int
J Mol Sci. 21:82772020. View Article : Google Scholar :
|
|
48
|
Sedlář A, Trávníčková M, Matějka R, Pražák
Š, Mészáros Z, Bojarová P, Bačáková L, Křen V and Slámová K: Growth
factors VEGF-A165 and FGF-2 as multifunctional biomolecules
governing cell adhesion and proliferation. Int J Mol Sci.
22:18432021. View Article : Google Scholar
|
|
49
|
Cao R, Eriksson A, Kubo H, Alitalo K, Cao
Y and Thyberg J: Comparative evaluation of FGF-2-, VEGF-A-, and
VEGF-C-induced angiogenesis, lymphangiogenesis, vascular
fenestrations, and permeability. Circ Res. 94:664–670. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Grugan KD, Miller CG, Yao Y, Michaylira
CZ, Ohashi S, Klein-Szanto AJ, Diehl JA, Herlyn M, Han M, Nakagawa
H and Rustgi AK: Fibroblast-secreted hepatocyte growth factor plays
a functional role in esophageal squamous cell carcinoma invasion.
Proc Natl Acad Sci USA. 107:11026–11031. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sahni A and Francis CW: Stimulation of
endothelial cell proliferation by FGF-2 in the presence of
fibrinogen requires alphavbeta3. Blood. 104:3635–3641. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Huy NQ and Trang NTM: Evaluation the
anti-tumor activity of SM2 fraction extracted from Stephania
dielsiana Y.C.Wu on Swiss mice bearing S180 sarcoma tumor. Vietnam
Pharm J. 55:42–45. 2015.In Vietnamese.
|