Introduction
Acute myocardial infarction (AMI), a cardiovascular
ailment of severe its disabling and life-threatening implications,
is associated with high morbidity and sudden death rates (1,2).
Recent global prevalence statistics underscore its prevalence at
3.8% among individuals aged <60 years (sample size, 2,982,6717),
and escalating to 9.5% (sample size, 5,071,185) in those aged
>60 years (3). Amidst the
rapid aging of the Chinese population, Chinese healthcare
professionals will face new challenges in managing AMI. Early
therapeutic interventions such as pharmacological thrombolysis,
percutaneous coronary intervention, and surgical bypass grafting
effectively restore hemodynamics/reperfusion, ultimately mitigating
sudden death rates and improving patient prognosis. However,
reperfusion itself precipitates additional cardiomyocytes damage,
exacerbating cardiac dysfunction, which is termed
ischemia/reperfusion injury (I/RI) (4). Current understanding of the
pathophysiological mechanisms underlying I/RI encompasses
inflammatory responses, apoptosis, oxidative stress, autophagy,
ferroptosis (5-7), and others. Of note, multiple
moderated mortality patterns may occur independently, or there may
be simultaneous crosstalk or overlap between them (8). Meanwhile, autophagy may play the
role of a 'double-edged sword' in the different stages of ischemia
and reperfusion. During ischemic, ATP decreases, which activates
the mTOR/ULK1/PI3K pathway, thereby forming autophagic vesicles and
promoting ATP synthesis. However, upon the restoration of
reperfusion, increased ROS and Beclin1 overexpression can lead to
autophagy overactivation and cardiomyocyte death promotion
(9,10). ATP is the basis of several life
activities, including growth, proliferation, bio-metabolism, stress
and others. The activation of AMPK, which acts as the gatekeeper of
energy metabolism and mitochondrial homeostasis, restores energy
balance by promoting ATP-producing catabolic pathways and
inhibiting energy-consuming processes. However, mitochondrial
dysfunction can lead to reduced ATP production and cellular
dysfunction (11). Thus,
mitochondria play a critical role in cellular metabolism as the
powerhouses of mammalian energy. However, whether energy stress
regulates other non-apoptotic forms of regulatory cell death (RCD)
in myocardial I/RI is not known. Therefore, there is an urgent need
to evaluate the mechanisms of I/RI and find new therapeutic
approaches.
Ferroptosis, a novel iron-dependent phospholipid
peroxidation-driven mode of unique cell death, was first reported
by Dixon in 2012 (12). Over the
past decades, a preponderance of research has corroborated the view
that ferroptosis is the primary form of RCD in myocardial I/RI
(7,13,14). Recent investigation revealed that
pretreatment with tanshinone IIA effectively attenuated A/R injury
in H9c2 cardiomyocytes by modulating VDAC1-mediated ferroptosis and
apoptosis (15). Additionally,
puerarin has been shown to safeguard against I/RI in pressure
overload-induced heart failure by inhibiting ferroptosis (16). These studies indicated that
traditional Chinese medicine (TCM) may offer a new approach for
treating cardiovascular disease via ferroptosis modulation.
TCM, a time-honored treasure of China for 5,000
years, has significantly contributed to combating various ailments,
including the coronavirus disease 2019 pandemic, malaria and
cardiovascular diseases (17-19). Nevertheless, the precise role of
TCM in the prevention and treatment of certain diseases remains
unclear. Consequently, the search for safe and effective active
ingredients of TCM with clear molecular targets and low toxicity
and side effects is an important issue that needs to be addressed
urgently. Curcumin (Cur), a prominent TCM constituent, mainly
extracted from turmeric, has been implicated in modulating
apoptosis, oxidative stress, inflammatory responses and autophagy,
thereby safeguarding against I/RI across various organs (10). Studies have shown that Cur
opposed I/RI by activating the JAK2/STAT3 signaling (20), and attenuated I/R-induced lung
injury via the Notch2/Hairy and enhancer of split 1 (Hes1)
signaling (21). A prior study
revealed astragaloside IV modulation of HES1 and its ligand protein
VDAC1 to protect against myocardial I/RI, inhibiting apoptosis
through the Notch1/HES1 signaling pathway, thereby exerting
myocardial protection (22).
Notably, HES1 is an important target gene downstream of the Notch1
signaling pathway that acts as a transcriptional repressor encoding
the repressive basic helix-loop-helix (bHLH). A recent study has
shown that the Notch1/HES1 signaling pathway can activate the
PTEN/Akt signaling pathway through HES1 to reduce ROS generation,
stabilize ΔΨm and ultimately decrease apoptosis, thereby protecting
the myocardium (23).
Nonetheless, to the best of our knowledge, previous studies focused
only on inflammatory responses, oxidative stress level and
apoptosis, and it is unclear whether HES1 participates in autophagy
and ferroptosis after Cur treatment. The present study is the first
to suggest that Cur pretreatment attenuates myocardial I/RI by
inhibiting ferroptosis and autophagy via HES1. In addition, the
present study provides new insights into the crosstalk among
ferroptosis, autophagy and apoptosis in myocardial I/RI.
Therefore, in the present study, the H9c2
cardiomyocyte I/R model was established to i) confirm whether
myocardial I/R injury induces ferroptosis and excessive autophagy,
thereby damaging the myocardium; ii) investigate whether Cur
inhibits ferroptosis and excessive autophagy induced by myocardial
I/R injury; iii) evaluate whether HES1 mediates Cur inhibition of
myocardial ferroptosis and excessive autophagy; and iv) assess
whether the myocardial protective effect of Cur is related to the
inhibition of oxidative stress, maintenance of energy metabolism
homeostasis and maintenance of mitochondrial functional
homeostasis.
Materials and methods
Materials and chemicals
Cur, (purity ≥98%) was purchased from Chengdu Must
Bio-Technology Co., Ltd. Deferiprone (DFO, iron chelator),
ferrostatin-1 (Fer-1, ferroptosis suppressor), erastin,
3-methyladenine (3MA, autophagy inhibitor) and rapamycin (RA,
autophagy activator) were purchased from MedChemExpress. Compound C
(AMPK inhibitor) was purchased from MilliporeSigma. Adenoviral
pAD/HES1-short hairpin (sh) RNA and negative control (NC) were
acquired from Cyagen Biologicals Co., Ltd.
Culture of rat H9c2 cardiomyocytes and
development of an A/R injury model
A rat H9c2 cardiomyocyte cell line was obtained from
the Cell Bank/Stem Cell Bank of the Chinese Academy of Sciences.
Under standard conditions (95% humidity, 21% O2, and 5%
CO2), cells were cultured in a high-glucose Dulbecco's
modified Eagle's medium (H-DMEM; HyClone; Cytiva) enriched with 10%
fetal bovine serum (FBS) and 1% penicillin-streptomycin-glutamine
(PSG; 100X) (Gibco; Thermo Fisher Scientific, Inc.) at 37°C. A
previous research protocol was used to establish the in
vitro A/R model using the cellular A/R fluid method (15).
Preparation and transduction of
Adenovirus
The adenoviral vectors pAD/HES1, shRNA and NC were
prepared using a previously established method (15). The target sequences of
pAD/HES1-shRNA and NC are CAGACATTCTGGAAATGACAGTGAA and
TTCTCCGAACGTGTCACGT, separately. In brief, the pAD/HES1, shRNA, or
NC was introduced into rat H9c2 cells, which were cultured in fresh
H-DMEM enriched with 10% FBS ('multiplicative infection' of 80) and
incubated at 37°C, 95% O2, and 5% CO2 for 48
h for the subsequent experiments. Non-adenovirus transduced H9c2
cardiomyocytes were used as control. The successful transfection of
rat H9c2 cardiomyocytes with HES1 adenovirus was first validated at
the protein molecular level as illustrated in Fig. S1.
Treatment of rat H9c2 cardiomyocytes and
experimental design
Firstly, to assess weather prior treatment with Cur
can mitigate ferroptosis and autophagy, thereby safeguarding rat
H9c2 cardiomyocytes against injury caused by A/R, rat H9c2
cardiomyocytes were randomly divided into 9 groups: i) Control,
incubated in normal culture medium for 48 h; ii) erastin, rat H9c2
cardiomyocytes exposed to 10 μM erastin in 10% FBS for 24 h;
iii): Cur, H9c2 cardiomyocytes treated with 10 μM Cur in 10%
FBS for 48 h; iv) erastin + Cur, H9c2 cardiomyocytes pre-treated
with Cur in a 10% FBS for 24 h, followed by 24-h co-incubation with
10 μM erastin; v) A/R, H9c2 cardiomyocytes incubated for 48
h in normal culture medium and then exposure to A/R for 3 h/2 h;
vi): A/R + Cur, rat H9c2 cardiomyocytes pre-treated with 10
μM Cur under 10% FBS for 48 h; vii): A/R + Cur + erastin,
Cur pretreatment for 24 h and then co-incubation with 10 μM
erastin for 24 h; viii): A/R + DFO, DFO pretreatment for 24 h; and
ix): A/R + Fer-1, Fer-1 pretreatment for 2 h and then exposure to
A/R for 3 h/2 h.
Secondly, to investigate whether HES1 is involved in
ferroptosis and evaluate its mechanism of action in A/R, rat H9c2
cardiomyocytes were randomly divided into 8 groups: i) Control,
incubated for 48 h in normal culture medium; ii) erastin, rat H9c2
cardiomyocytes exposed to 10 μM erastin in 10% FBS for 24 h;
iii) erastin + pAD/HES1; iv) erastin + pAD/HES1-shRNA, rat H9c2
cardiomyocytes pretreated with pAD/HES1 or pAD/HES1-shRNA in 10%
FBS for 24 h and then co-treated with 10 μM erastin for 24
h; v) A/R, rat H9c2 cardiomyocytes cultured for 48 h and then
exposure to A/R for 3 h/2 h; vi) A/R + pAD/HES1; vii) A/R +
pAD/HES1-shRNA; and viii): A/R + NC; (rat H9c2 cardiomyocytes
pretreated with pAD/HES1, pAD/HES1-RNA, or NC in 10% FBS for 48 h
and exposed to A/R for 3 h/2 h).
Thirdly, to evaluate changes in autophagy after A/R
injury and the effect of Cur pretreatment on it, rat H9c2
cardiomyocytes were randomly divided into the groups below: I):
control; II): A/R; III): A/R + Cur. IV): A/R + Cur + RA; and V):
A/R + 3MA; (rat H9c2 cardiomyocytes pre-treated with 10 μM
Cur in 10% FBS for 48 h prior to A/R; 10 μM Cur +200 nM RA
co-incubated for 48 h; 5 mM 3MA pretreatment for 24 h and then
exposure to A/R for 3 h/2 h). The control, A/R, and A/R + Cur
groups were treated as per the aforementioned protocols.
Furthermore, to investigate the role of ferroptosis
and autophagy regulation during myocardial A/R injury as well as of
HES1 and Cur pretreatment, rat H9c2 cardiomyocytes were randomly
divided into the following groups: i) control; ii): A/R; iii): A/R
+ Cur; (the control, A/R and A/R + Cur groups were treated as per
aforementioned protocols); iv) A/R + Cur + pAD/HES1-shRNA; v) A/R +
pAD/HES1-shRNA; and vi) A/R + Cur + NC; (rat H9c2 cardiomyocytes
were pretreated with 10 μM Cur + pAD/HES1-shRNA,
pAD/HES1-shRNA, or NC in 10% FBS for 48 h prior to A/R and then
exposure to A/R for 3 h/2 h).
Ultimately, to explore how Cur pretreatment enhances
and maintains mitochondrial function and energy metabolism in rat
H9c2 cardiomyocytes induced by A/R injury, H9c2 cells were randomly
grouped as follows: i) Control; ii) A/R; iii) A/R + Cur; (the
control, A/R and A/R + Cur groups were treated as per
aforementioned methods; and iv): A/R + Cur + Compound C, H9c2
cardiomyocytes were co-incubated with 10 μM Cur + 5
μM Compound C in 10% FBS for 48 h prior to A/R and then
exposure to A/R for 3 h/2 h.
Assessment of cell viability and
cytotoxicity
Cell survival was measured using the Cell Counting
Kit-8 (CCK-8) colorimetric assay (cat. no. GK10001; GlpBio),
following the manufacturer's protocol. In brief, rat H9c2
cardiomyocytes were inoculated in the center of a 96-well plate at
a density of 1×104 cells/well along with per 100
μl culture medium/10 μl CCK-8 and incubated at 37°C
for 1.5 h. Then, absorbance levels were gauged at 450 nm utilizing
a microplate reader (Thermo Fisher Scientific, Inc.).
The supernatant of the treated cells from each group
was collected and the lactate dehydrogenase (LDH) level was
measured using an LDH assay kit (cat. no. C0017; Beyotime
Biotechnology) according to the manufacturer's protocol. Briefly,
60 μl LDH assay working solution was added to per 120
μl of supernatant, thoroughly mixed, and then incubated at
25°C for 30 min in the absence of light. Absorbance was measured at
490 nm using the aforementioned apparatus.
Quantification of malondialdehyde (MDA),
superoxide dismutase (SOD), total iron, glutathione peroxidase
(GSH-Px) and glutathione (GSH)/glutathione disulfide (GSSG)
After various treatments, cell lysate supernatants
were collected. An MDA assay kit (cat. no. S0131M), SOD assay kit
(cat no. S0101M), GSH and GSSG assay kit (cat no. S0053; all from
Beyotime Biotechnology), GSH-Px Assay kit (cat. no. A005-1-2;
Nanjing Jiancheng Bioengineering Institute) and total iron ion
colorimetric assay kit (cat. no. E1042-100; Applygen Technologies,
Inc.) were used for measuring MDA, SOD, GSH/GSSG, GPX and total
iron ion levels, respectively.
Determination of Caspase-3 activity
After various treatments, cell lysate supernatants
were collected. The Caspase-3 activity was quantified using a
caspase-3 assay kit (cat. no. C1115; Beyotime Biotechnology),
following the manufacturer's protocol.
Western blot analysis
After the treatment of rat cardiomyocytes, total
protein was extracted from the cells of each group using western
and IP cell lysates (cat. no. P0013; Beyotime Biotechnology)
following the manufacturer's protocol, followed by bicinchoninic
acid protein assay kit (cat. no. P0012; Beyotime Biotechnology) for
protein concentration quantification. Proteins were denatured by
adding an appropriate amount of sodium
dodecyl-sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
protein sampling buffer (6X; cat. no. P0015F; Beyotime
Biotechnology) and boiling at 100°C for 10 min in a metal heater.
Then, a 40-μg protein sample was added to each lane for
separation using 10 or 12% SDS-PAGE. The separated proteins were
transferred to polyvinylidene fluoride membranes, which were sealed
with 5% non-fat dry milk at room temperature for 2 h in a
three-buffer brine containing 0.1% Tween-20. The membranes were
then incubated overnight in a low-speed shaker maintained at 4°C
with primary antibodies against the following: HES1 (1:500; cat.
no. sc-5392; Santa Cruz Biotechnology, Inc.), PTGS2 (1:1,000; cat.
no. 12375-1-AP; Proteintech Group, Inc.), GPX4 (1:800; cat. no.
381958; ZEN-BIO), P62 (1:5,000; cat. no. 18420-1-AP; Proteintech
Group, Inc.), LC3B (1:800; cat. no. 381544; ZEN-BIO), NDUFB8
(1:800; cat. no. 383060; ZEN-BIO), UQCRC2 (1:800; cat. no. 382096;
ZEN-BIO), Bcl-2 (1:800; cat. no. 381702; ZEN-BIO), Bax (1:800; cat.
no. 380709; ZEN-BIO), phosphorylated (p-) AMPK (1:800; cat. no.
381164; ZEN-BIO), AMPKα (1:1,000; cat. no. AF6195; Beyotime
Biotechnology) and β-actin (1:2,000; cat. no. 20536-1-AP;
ProteinTech Group, Inc.). On the next day, the membranes underwent
five washes, each lasting 6 min, followed by incubation with a
secondary Goat Anti-Rabbit/Mouse IgG H&L-conjugated antibody
(1:5,000; cat. nos. 511203 and 511103; ZEN-BIO) for 1.5 h at room
temperature. Finally, the membranes were washed three times for 6
min/each before being visualized using the BeyoECL Plus kit (cat.
no. P0018S; Beyotime Biotechnology). The β-actin was used as a
loading control for normalization. The intensity of gray values in
the protein lanes was measured using the ImageJ software 1.8.0
(National Institutes of Health).
Quantification of intracellular ferrous
iron content
Intracellular Fe2+ levels were determined
using the FerroOrange kit (cat. no. F374; Dojindo Laboratories,
Inc.) according to the manufacturer's protocol. Briefly, following
the H9c2 cardiomyocyte treatment, the cells were incubated with 1
μM FerroOrange for 30 min at 37°C under light protection.
Excess FerroOrange was removed by rinsing once with HBSS (cat. no.
G4204; Servicebio), and ferrous iron content was assessed under an
inverted fluorescence microscope (Olympus Corporation;
magnification, ×200).
Detection of intracellular ROS
Intracellular ROS level was measured using a DCFH-DA
kit (cat. no. S0033S; Beyotime Biotechnology) as described in the
manufacturer's protocol.
Evaluation of lysosomes
The reagents were incubated with the LysoTracker Red
fluorescent dye (cat. no. C1046; Beyotime Biotechnology) for 45 min
at 37°C in the dark following the reagent manufacturer's protocol.
The relevant changes were observed under an inverted fluorescence
microscope (Olympus Corporation; magnification, ×200).
Assessment of apoptosis
Apoptosis was detected using the Annexin V-FITC/PI
apoptosis detection kit (cat. no. BB-4101; BestBio) as per the
manufacturer's protocol. Briefly, 1×106 cells/tube were
collected after treatment, and resuspended in 400 μl 1X
Annexin binding buffer, gently vortexed with 5 μl Annexin
V-FITC and 8 μl PI in succession and incubated for 15 and 5
min, respectively, at 6°C in the dark, followed by immediately flow
cytometric analysis using Agilent NovoCyte Advanteon flow cytometer
(NovoCyte; Agilent Technologies, Inc.) to detect apoptosis
(Agilent; excitation 488 nm; emission 578 nm). The total apoptotic
rate of positive cells was calculated as the rate of early
apoptotic cells Q2 + rate of late apoptotic cells Q3. Flow
cytometric data were analyzed using the FlowJo software (V.10; Tree
Star, Inc.).
Evaluation of mitochondrial permeability
transition pore (MPTP) opening and mitochondrial membrane potential
(MMP)
Cellular MPTP and MMP were assessed using the MPTP
assay kit (cat. no. BB-48122; BestBio) and MMP assay kit (cat no.
BB-4105; BestBio), respectively, adhering to the manufacturer's
protocol. In brief, 1×106 cells/tube were collected
after treatment and subjected to BBcellProbe M61 assay (which
involved the addition of BBcellProbe M61 working solution and
quencher, followed by incubation at 37°C for 15 min in the dark,
centrifugation at 600 × g to remove the supernatant, and cell
resuspension in 600 μl HBSS) and the JC-1 method (which
included the addition of JC-1 staining working solution, followed
by incubation at 37°C for 20 min protected from light, washing
twice with PBS, and resuspending the cells in 600 μl PBS),
respectively. These cells were then immediately analyzed using
Agilent NovoCyte Advanteon flow cytometer for in real time. The
FlowJo software was used to analyze the flow cytometric data.
Assessment of mitochondrial
ultrastructure using transmission electron microscopy (TEM)
Briefly, after completion of the experimental model,
H9c2 cardiomyocytes were collected from each group, fixed
(incubated with 2.5% glutaraldehyde at 25°C for 2 h), washed,
dehydrated with ascending ethanol, embedded with Epon 812 at 65°C
for 48 h, ultrathin sectioned (50 nm), stained with 2% uranyl
acetate and 2.6% lead citrate at 37°C for 10 min, and observed via
TEM (Hitachi, Ltd.). The degree of damage to mitochondrial
ultrastructure was assessed using the Flameng score (24).
Statistical analysis
The data were statistically analyzed using GraphPad
Prism 9.0 (Dotmatics). Each experiment was performed in triplicate,
and the results are presented as the mean ± standard deviation.
Group comparisons, involving groups of three or more, were
performed using one-way ANOVA, followed by Tukey's post-hoc test.
P≤0.05 was considered to indicate a statistically significant
difference.
Results
Cur pretreatment protects against
A/R-induced cardiomyocyte injury
To verify whether Cur pretreatment safeguards
cardiomyocytes from I/R damage, an A/R model was established using
rat H9c2 cardiomyocytes. Initially, the dose-response of Cur
protection was investigated. In the A/R model of rat H9c2
cardiomyocytes, CCK-8 and LDH assays were used to measure cell
viability and toxicity in cells pretreated with different Cur
concentrations (2.5, 5, 10, 20 and 40 μM), and the results
revealed that the effect of Cur on cells was
concentration-dependent (Fig. 1A and
B). In stark contrast to the control group, A/R injury
prominently decreased cell viability and elevated LDH activity;
whereas compared with A/R, pretreatment with 10 μM Cur
significantly bolstered A/R-induced H9c2 cell viability and
decreased LDH activity, without eliciting any notable cytotoxicity.
Therefore, 10 μM Cur was selected as the optimal
concentration for subsequent experiments.
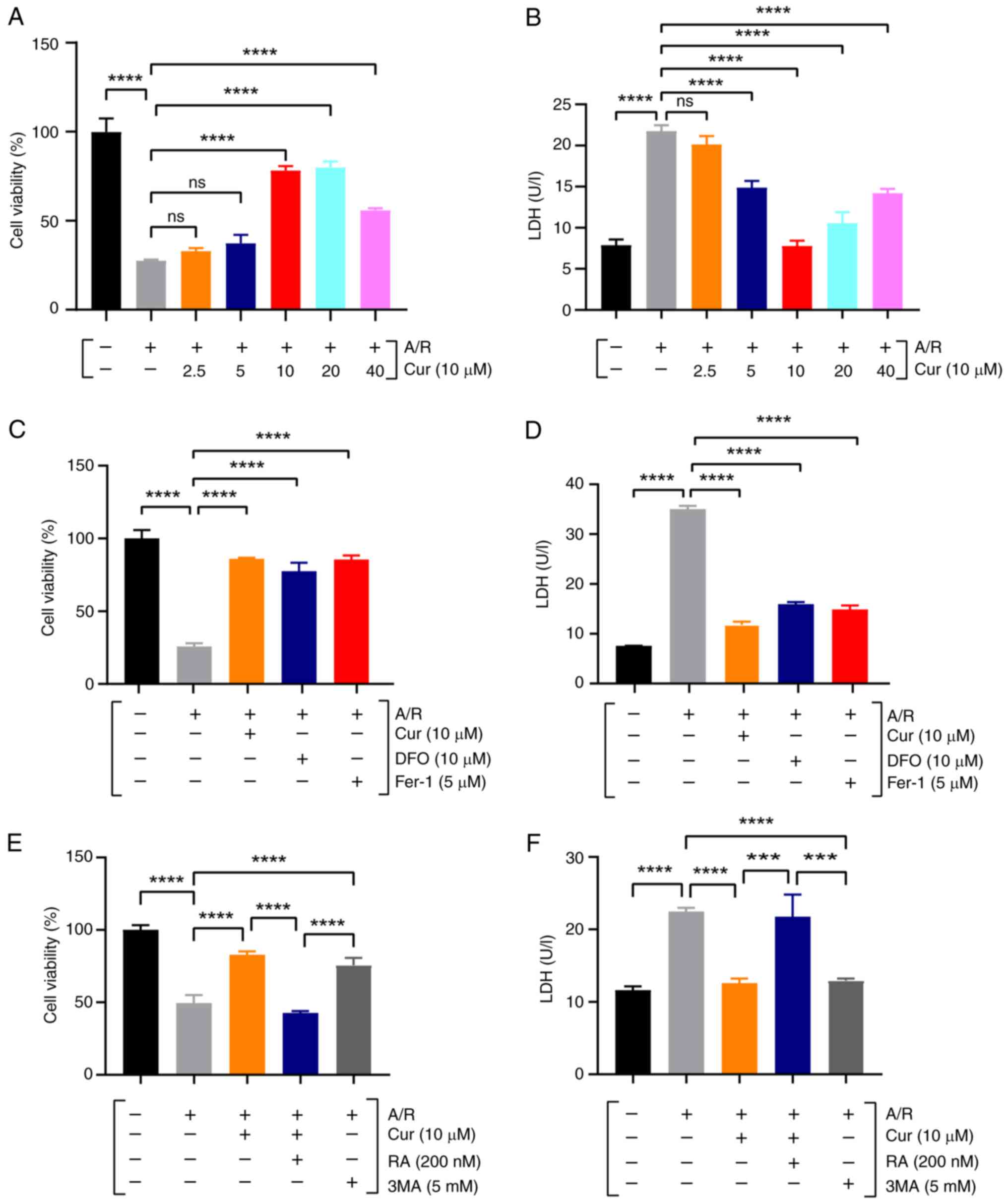 | Figure 1Cur protects rat H9c2 cardiomyocytes
from A/R injury. (A, C and E) Cell Counting Kit-8 assay of
A/R-triggered H9c2 cardiomyocyte viability after Cur, DFO, Fer-1,
RA and 3MA pretreatment. (B, D and F) LDH. Data are presented as
the mean ± SD (n=3). ***P<0.001 and
****P<0.0001. Cur, curcumin; A/R,
anoxia/reoxygenation; LDH, lactate dehydrogenase; DFO, deferiprone;
Fer-1, ferrostatin-1, RA, rapamycin; 3MA, 3-methyladenine, ns, not
significant. |
Moreover, the cardioprotective potency of Cur
pretreatment was found to be commensurate with that observed with
DFO, Fer-1, or 3MA. However, the beneficial effects of 10 μM
Cur were notably attenuated by RA (Fig. 1C-F). These findings indicated the
protective role of Cur against I/R injury in cardiomyocytes.
Cur pretreatment ameliorates A/R or
erastin injury-induced ferroptosis in cardiomyocytes
Iron overload, ROS and lipid peroxidation constitute
the triad of factors pivotal to ferroptosis (13). Thus, iron content, ROS and lipid
oxidation-related parameters were measured in H9c2 cells. Lipid
oxidation metabolites such as ROS, MDA and total and labile iron
pools, were significantly increased in the A/R group relative to
the control group, whereas these levels were significantly
decreased following pretreatment with 10 μM Cur; similar
results were obtained with 10 μM DFO and 5 μM Fer-1
pretreatment (Fig. 2A-H).
GSH/GPX4 is a component of the classical regulatory mechanism of
ferroptosis (25). Following A/R
treatment, the activity of antioxidant enzymes such as SOD,
GSH/GSSG ratio and GSH-Px was significantly reduced, which were
reversed via pretreatment with Cur, DFO and Fer-1 (Fig. 2I-K). The molecular levels of
ferroptosis-related proteins were further examined by western
blotting, and it was found that pretreatment with Cur, DFO and
Fer-1 significantly inhibited PTGS2 protein expression and
upregulated GPX4 protein expression, respectively (Fig. 2L and M). These results suggested
that Cur may be involved in ferroptosis and oxidative stress after
A/R injury.
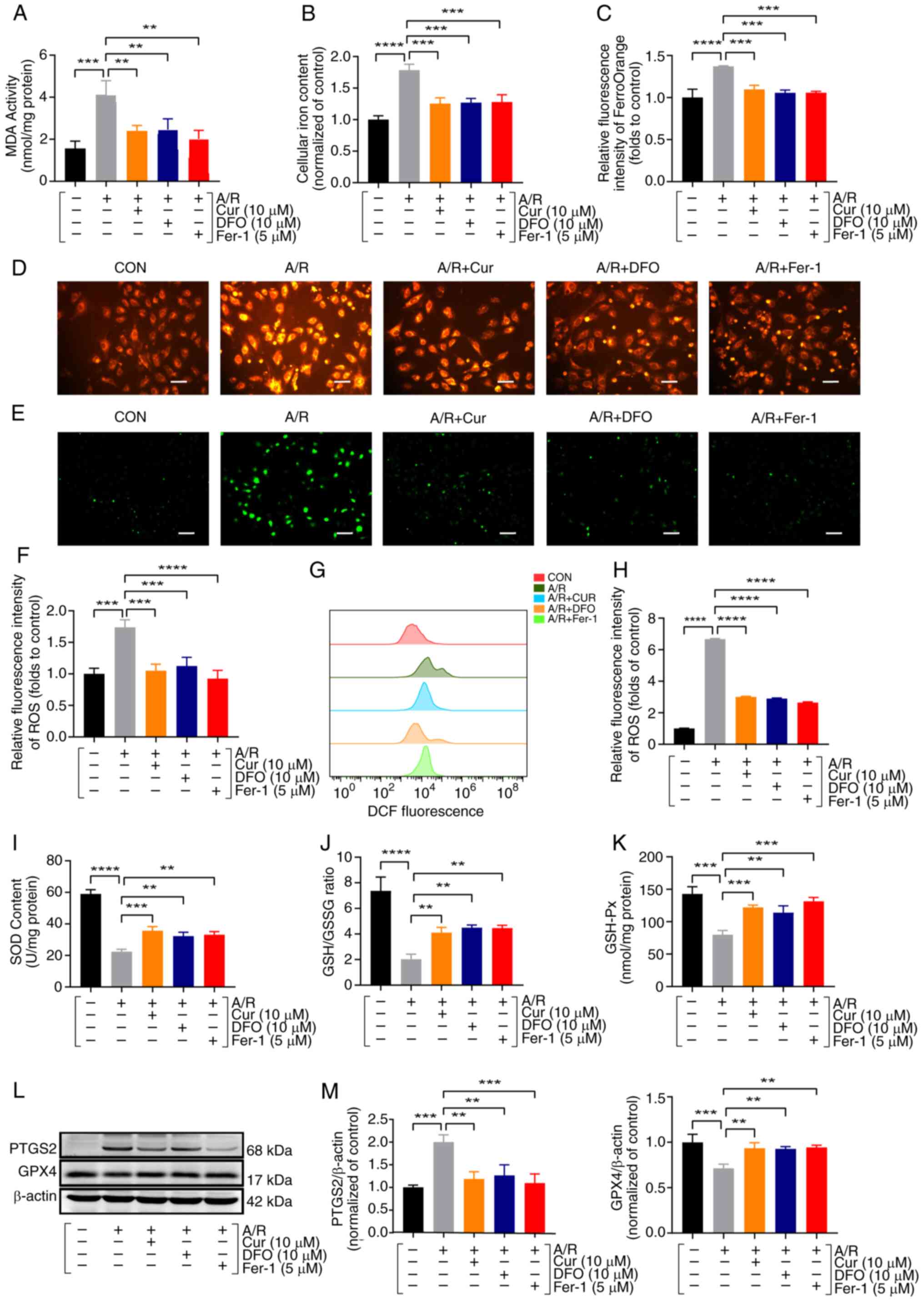 | Figure 2Cur pretreatment ameliorates A/R
injury-triggered ferroptosis in cardiomyocytes. (A) MDA assay of
A/R-triggered H9c2 cardiomyocyte after Cur, DFO and Fer-1
pretreatment. (B) Total iron content. (C and D) Ferrous ions
(magnification, ×200; scale bar, 100 μm). (E and F) ROS was
detected using a quantification kit in A/R-triggered cells after
Cur, DFO, or Fer-1 pretreatment (magnification, ×100; scale bar,
200 μm). (G and H) DCFH-DA assay for ROS with flow
cytometry. (I) SOD. (J) GSH/GSSG ratio. (K) GSH-Px. (L and M)
Protein expression of PTGS2 and GPX4 was determined using western
blot analysis in cell lysates after A/R-triggered following
pre-treatment with Cur, DFO, or Fer-1. Data are presented as the
mean ± SD (n=3). **P<0.01, ***P<0.001
and ****P<0.0001. Cur, curcumin; DFO, deferiprone;
Fer-1, ferrostatin-1; A/R, anoxia/reoxygenation; MDA,
malondialdehyde; GSH/GSSH, glutathione/glutathione disulfide;
GSH-Px, glutathione peroxidase; SOD, superoxide dismutase; ROS,
reactive oxygen species; PTGS2, prostaglandin-endoperoxide synthase
2; GPX4, glutathione peroxidase 4. |
To corroborate that Cur mitigates A/R-associated
injury by inhibiting ferroptosis and oxidative stress, the present
study further investigated the protective effect of Cur on
erastin-related injury. Upon erastin treatment, a significant
reduction in cell viability, SOD activity and the GSH/GSSG ratio
was observed, accompanied by an elevation in LDH, MDA levels and
MPTP opening. Intriguingly, pretreatment with 10 μM Cur
significantly attenuated these erastin-mediated injuries (Fig. S2A-E and L). However, in the A/R
model, when Cur and erastin co-treatment were performed in
cardiomyocytes, erastin counteracted the protective effect of Cur
(Fig. S2F-J and M). Western
blot analysis illuminated that Cur pretreatment robustly
upregulated HES1 and GPX4 protein expression levels after
erastin-induced injury (Fig.
S2K). These results suggested that Cur ameliorates A/R
injury-induced ferroptosis by inhibiting oxidative stress and that
HES1 may be involved in ferroptosis after A/R injury.
In addition, to investigate whether HES1 is involved
in erastin or A/R-induced ferroptosis, H9c2 cardiomyocytes were
transfected with high and low HES1-expressing adenoviruses before
A/R or erastin treatment. Unlike the control group, the erastin or
A/R group exhibited significantly reduced cell viability and
elevated LDH activity, whereas adenoviral transfection along with
pAD/HES1 overexpression attenuated cell viability reduction and LDH
elevation due to erastin or A/R injury. However, adenoviral
transfection along with pAD/HES1-shRNA further significantly
aggravated the erastin- or A/R injury-induced reduced cell
viability, elevated LDH and MDA level. Moreover, relative to the
control, SOD activity and GSH/GSSG ratio were significantly
decreased and MPTP was over-opened after erastin or A/R treatment.
Of note, although pAD/HES1-shRNA aggravated erastin or A/R injury,
pAD/HES1 pretreatment reversed the changes. Meanwhile, the
detection of relevant proteins in cell lysates showed that HES1
expression was reduced in the erastin or A/R group compared with
the control group and that it was further significantly reduced
after pAD/HES1-shRNA pretreatment. However, HES1 expression was
significantly increased after pAD/HES1 pretreatment. In addition,
PTGS2 expression was significantly increased in the erastin or A/R
group and further increased after pAD/HES1-shRNA pretreatment,
whereas GPX4 expression was reduced in the erastin or A/R group and
further significantly reduced after pAD/HES1-shRNA pretreatment.
However, pAD/HES1 pretreatment reversed these changes (Fig. S3A-N). These findings indicated
the potential role of HES1 in ferroptosis triggered by erastin or
A/R. However, additional research is required to clarify the exact
mechanism.
Cur pretreatment attenuates A/R-induced
excessive autophagy in cardiomyocytes
Autophagy, a lysosome-dependent process, is pivotal
in maintaining intracellular homeostasis and survival by degrading
abnormal or damaged macromolecules and organelles. When
cardiomyocytes are exposed to certain external stimuli, such as
ischemia and inflammation, ATP depletion inhibits the mTOR pathway
and induces ULK1 activation, which stimulates autophagic vesicle
formation and promotes ATP synthesis. Upon stimulus exacerbation
such as in reperfusion or sepsis, the increased overexpression of
Beclin1 and excessive accumulation of ROS can lead to the
overactivation of autophagy, which promotes cardiomyocyte death
(9,26,27). Fascinatingly, the current study
revealed that prior treatment with Cur influenced autophagy-related
marker expression, as evidenced by a significant increase in P62
expression and LC3II/LC3I expression ratios in Cur-treated H9c2
cells (Fig. 3A-C). This finding
indicated that the aforementioned safeguarding effect of Cur
pretreatment might be linked to the suppression of autophagy
overstimulation.
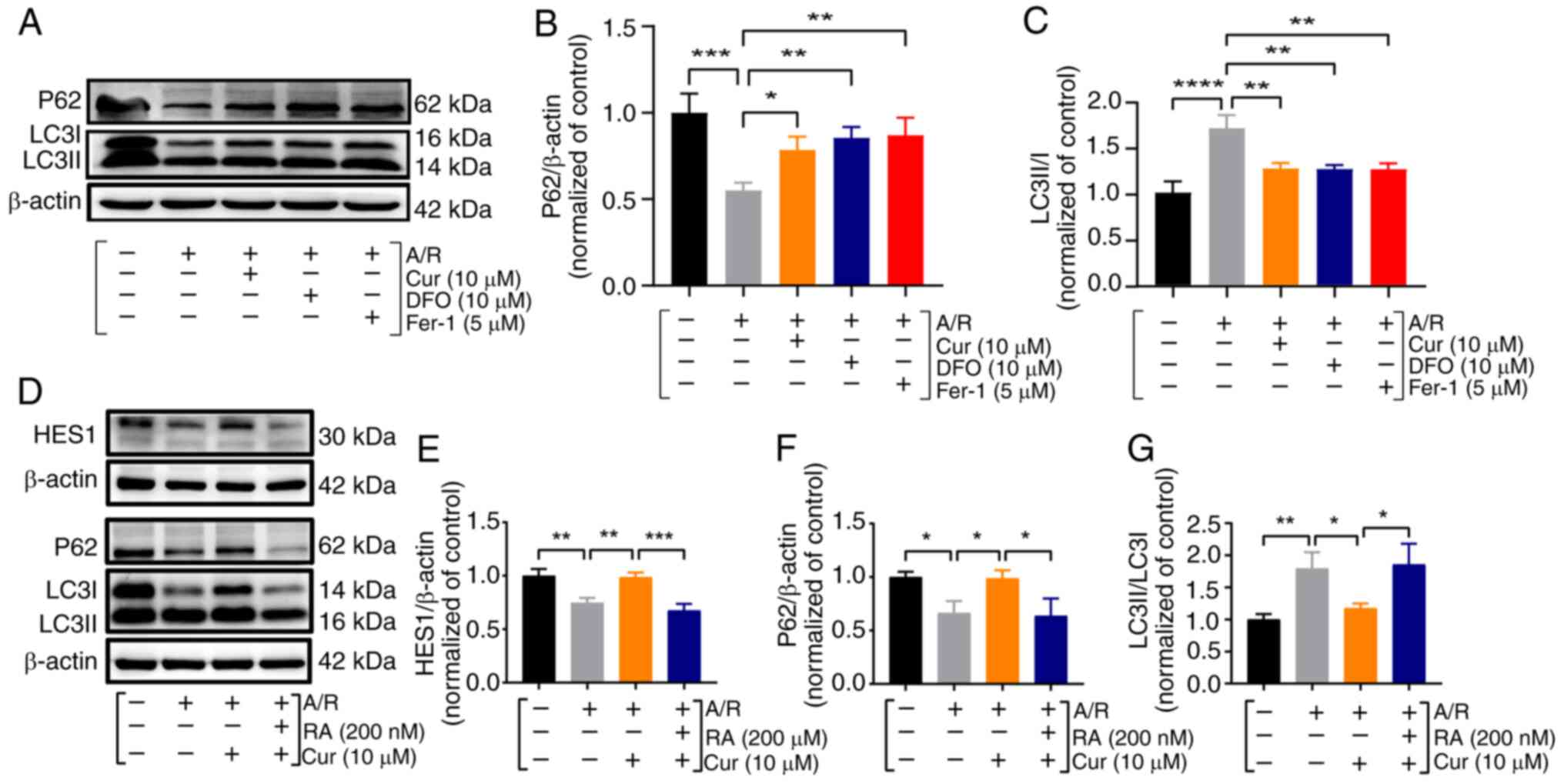 | Figure 3Cur pretreatment inhibits
A/R-triggered excessive autophagy in cardiomyocytes. (A-G)
Expression and quantification of HES1, P62 and LC3II/I proteins in
A/R-triggered cells were determined using western blot analysis
after pretreatment with Cur, DFO, Fer-1 and RA. Data are presented
as the mean ± SD (n=3). *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. Cur, curcumin; DFO, deferiprone; Fer-1,
ferrostatin-1; RA, rapamycin; A/R, anoxia/reoxygenation. HES1,
hairy and enhancer of split 1; P62, Sequestosome 1; LC3II/I,
microtubule-associated protein 1 light chain 3. |
To confirm that Cur preconditioning plays a
protective role against myocardial A/R injury by affecting
autophagy, RA (autophagy activator) and 3MA (autophagy inhibitor)
were used to observe their effects on autophagy. Analysis of cell
lysates revealed pertinent protein alterations. Compared with the
A/R group, there was a significant increase in the HES1 level and
P62 level, substantial decrease in LC3II/LC3I ratio, significant
reduction in the ferroptosis indicator PTGS2, and significant
increase in GPX4 level following pretreatment with 10 μM Cur
and 5 mM 3MA; whereas in the A/R + Cur + RA group, RA reversed
favorable changes when the myocardium was co-incubated with Cur and
RA (Figs. 3D-G and S4A-F). These results indicated that
autophagy and ferroptosis probably overlap or crosstalk and that
HES1 may be involved in the Cur-based amelioration of A/R injury in
cardiomyocytes by inhibiting excessive autophagy activation and
ferroptosis.
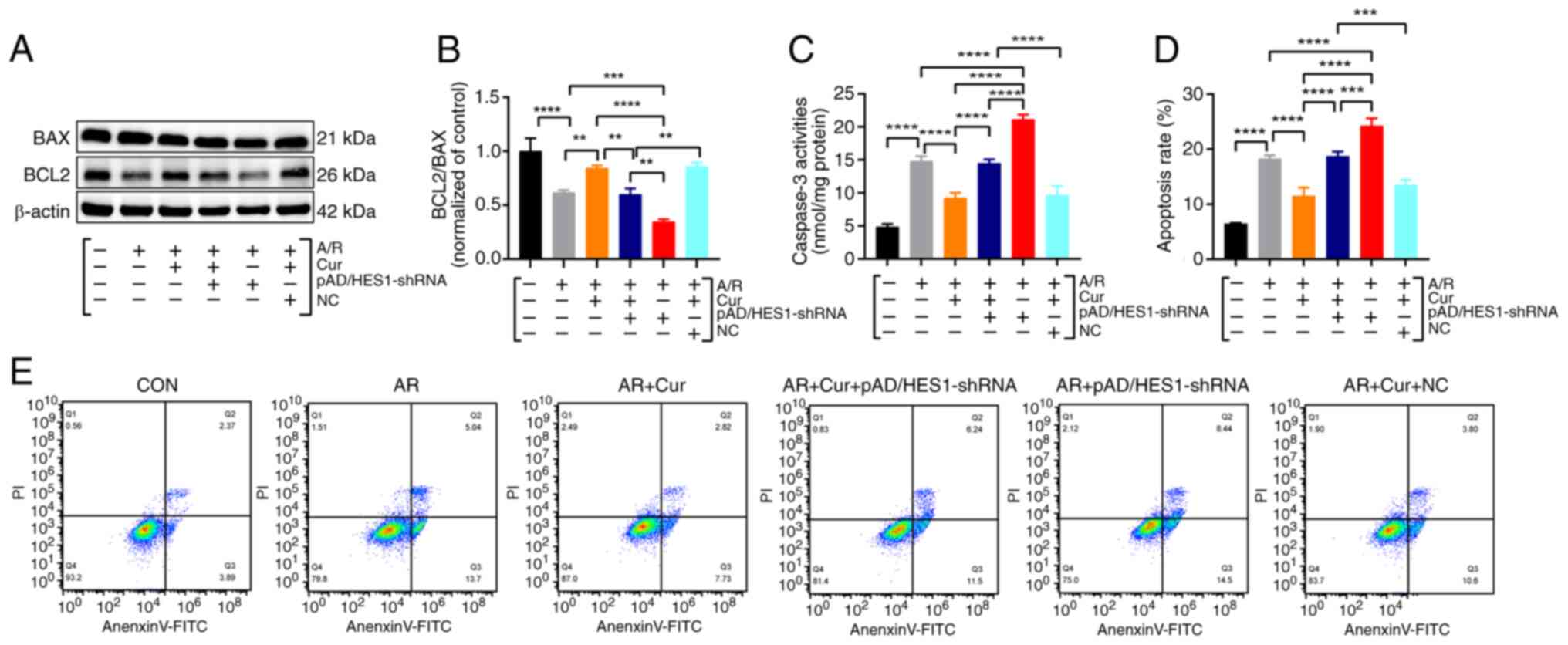
Cur pretreatment ameliorates A/R-induced
apoptosis in cardiomyocytes
In the present investigation of the intricate
interplay between A/R-induced ferroptosis, autophagy, and the
prophylactic role of Cur pretreatment, a novel aspect was
fortuitously uncovered: Cur pretreatment modulated the expression
of proteins implicated in apoptosis. Specifically, the BCL2/BAX
ratio was significantly reduced in the A/R group compared with the
control, whereas Cur pretreatment exerted a restorative influence,
reversing this trend (Fig. 4A and
B). Additionally, Cur pretreatment significantly attenuated
A/R-stimulated caspase 3 levels, further substantiating its
anti-apoptotic effect (Fig. 4C).
Utilizing flow cytometry, apoptosis, MPTP and MMP were analyzed.
The present findings revealed that A/R injury led to a significant
increase in apoptotic rates, accompanied by exaggerated MPTP
opening and MMP disruption. Notably, pretreatment with Cur, DFO and
Fer-1 effectively alleviated A/R-mediated damage, as evidenced by
reduced apoptotic indices and mitigated mitochondrial dysfunction
(Fig. 4D-I). These observations
underscore the potential of Cur pretreatment to mitigate A/R injury
by inhibiting apoptosis, ferroptosis and autophagy. Furthermore,
protein analyses demonstrated that Cur pretreatment significantly
enhanced the expression of the HES1 protein (Fig. 4J and K). These findings indicated
that Cur pretreatment can inhibit ferroptosis, autophagy and
apoptosis by upregulating HES1.
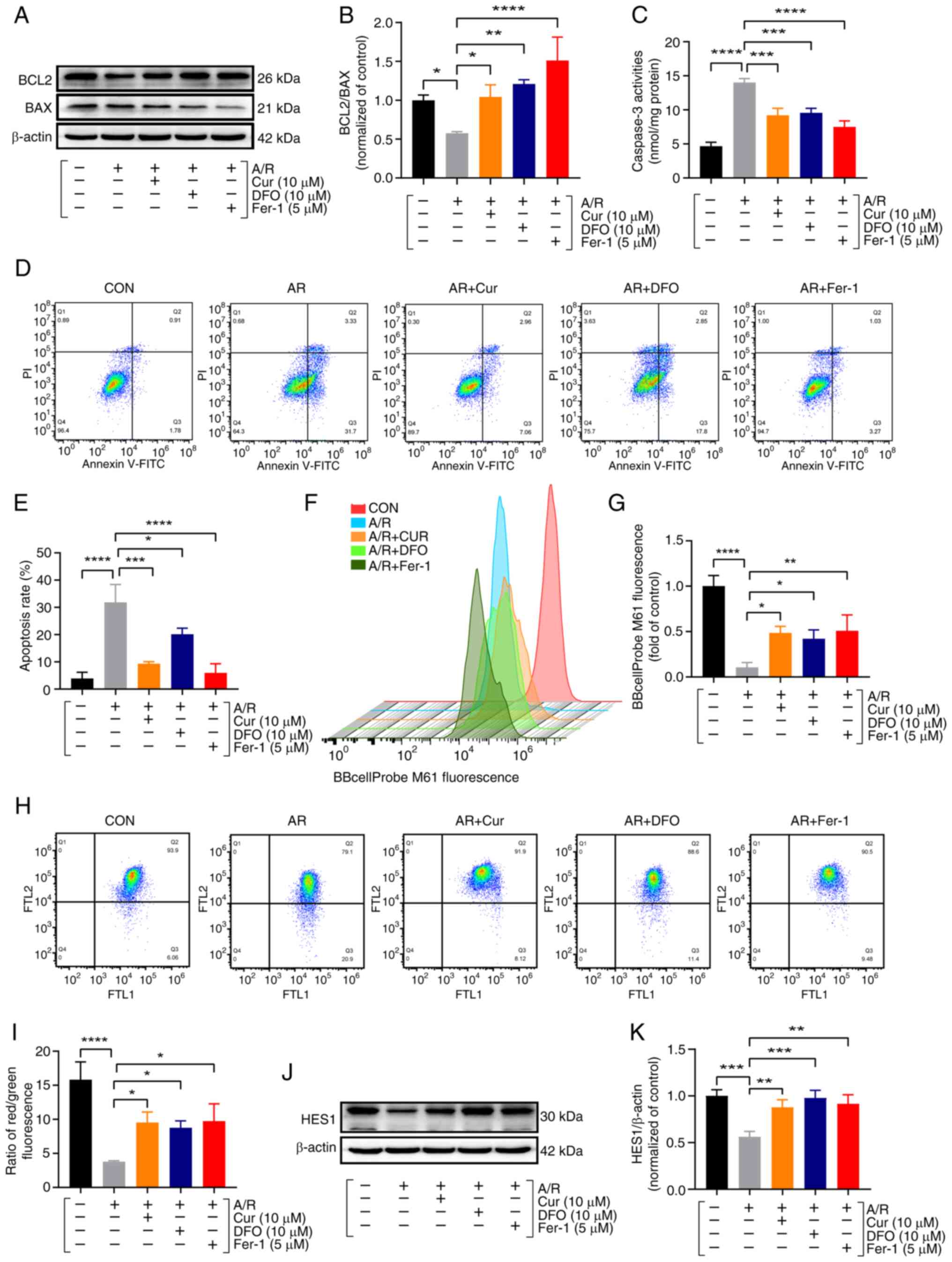 | Figure 4Cur pretreatment ameliorates
A/R-triggered apoptosis in cardiomyocytes. (A and B) Protein
expression of BCL2 and BAX was determined using western blot
analysis in cell lysates after A/R-triggered following
pre-treatment with Cur, DFO, or Fer-1. (C) Caspase-3 activity. (D
and E) Annexin V-FITC/PI assay for apoptosis with flow cytometry.
(F and G) BBcellProbe M61 assay for cellular MPTP. (H and I) JC-1
assay for cellular MMP. (J and K) Protein expression of HES1. Data
are presented as the mean ± SD (n=3). *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. Cur, curcumin; DFO, deferiprone; Fer-1,
ferrostatin-1; A/R, anoxia/reoxygenation; BCL2, B-cell lymphoma2;
BAX, BCL2-associated X protein; MPTP, mitochondrial permeability
transition pore; MMP, mitochondrial membrane potential. |
Cur pretreatment inhibits A/R-induced
ferroptosis in H9c2 cardiomyocytes via mediating HES1
To robustly substantiate the HES1-dependency of Cur
pretreatment in safeguarding against myocardial A/R damage through
ferroptosis suppression, experiments were conducted using H9c2
cardiomyocytes transfected with an adenovirus (pAD/HES1-shRNA or
NC). After A/R treatment, MDA, SOD, GSH/GSSG ratio, GSH-Px and
total intracellular iron content were detected in cell lysates. The
results revealed that H9c2 cardiomyocytes subjected to A/R injury
exhibited heightened MDA and total intracellular iron levels,
alongside diminished SOD activity, GSH/GSSG ratio and GSH-Px
activity, compared with control. Notably, 10 μM Cur
pretreatment significantly ameliorated these detrimental effects.
By contrast, pAD/HES1-shRNA treatment abolished the protective
effect of Cur pretreatment and increased the susceptibility of
cardiomyocytes to A/R injury (Fig.
5A-D). ROS and ferrous iron deposition acted as primary drivers
of ferroptosis (14). Thus,
these factors were quantified in H9c2 cells using
immunofluorescence and flow cytometry, and the obtained results
were consistent with the aforementioned findings (Fig. 5E-K). Therefore, it was
hypothesized that Cur preconditioning alleviates I/RI-associated
ferroptosis by regulating HES1. Additionally, the relevant protein
molecules of cell lysates were also detected. Compared with the A/R
group, a significant reduction in the expression of the ferroptosis
marker PTGS2 and a significant increase in GPX4 expression after 10
μM Cur pretreatment were revealed. By contrast, Cur +
pAD/HES1-shRNA co-incubation significantly downregulated GPX4
expression and upregulated PTGS2 compared with the Cur pretreatment
group alone, and pAD/HES1-shRNA exacerbated A/R-induced damage
(Fig. 5L and M), highlighting
the counteractive effect of HES1 knockdown.
 | Figure 5Cur pretreatment inhibits
A/R-triggered ferroptosis in H9c2 cardiomyocytes via mediating
HES1. (A) MDA. (B) SOD. (C) GSH/GSSG ratio. (D) GSH-Px. (E and F)
Immunofluorescence for the detection and quantitative analysis of
ROS (magnification, ×100; scale bar, 200 μm), (G and H)
DCFH-DA assay for ROS with flow cytometry. (I) Total iron content.
(J and K) Immunofluorescence for the detection of ferrous ions
(magnification, ×200; scale bar, 100 μm). (L and M) Western
blot analysis for the detection of protein expression and
quantitative analysis of PTGS2 and GPX4 in cell lysates after
treatment. Data are presented as the mean ± SD (n=3).
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. Cur,
curcumin; A/R, anoxia/reoxygenation; ROS, reactive oxygen species;
MDA, malondialdehyde; SOD, superoxide dismutase; GSH/GSSH,
glutathione/glutathione disulfide; GSH-Px, glutathione peroxidase;
GPX4, glutathione peroxidase 4; NC, negative control; shRNA, short
hairpin RNA. |
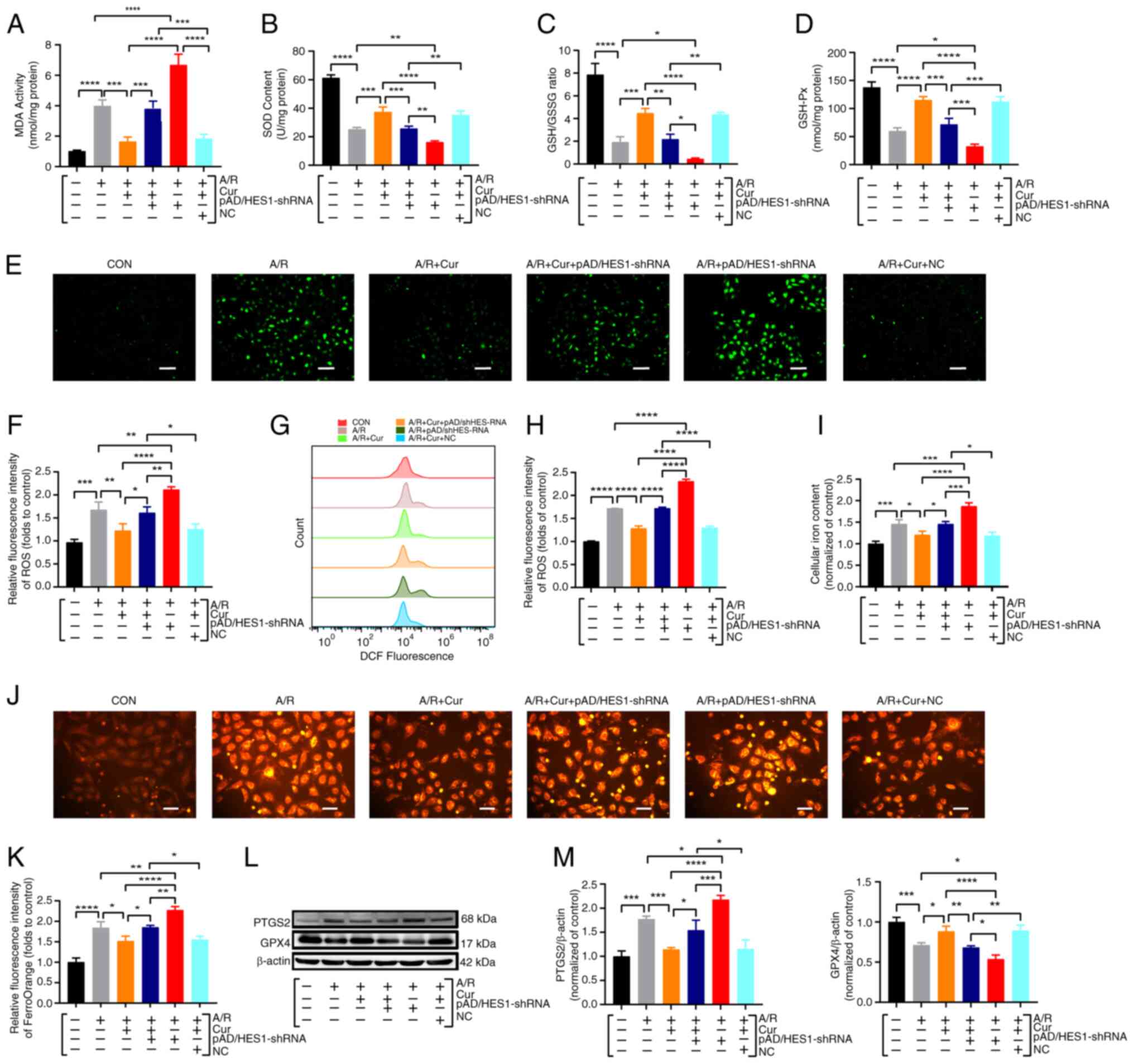
Collectively, Cur pretreatment effectively
ameliorated HES1-mediated I/RI-associated ferroptosis, while
pAD/HES1-shRNA blocked the protective effect and exacerbated
A/R-induced damage. These results indicated that HES1 may be
involved in the Cur-based amelioration of cardiomyocyte A/R via
ferroptosis inhibition.
Cur preconditioning inhibits A/R-induced
excessive autophagy in H9c2 cardiomyocytes via mediating HES1
The aforementioned experimental observations
illuminated that Cur upregulated HES1 expression after A/R injury,
thereby mitigating excessive autophagy activation. To confirm that
Cur preconditioning depends on active HES1 expression by inhibiting
autophagic hyperactivation, based on the upregulation of HES1
expression, HES1 expression was silenced the present study by
pAD/HES1-shRNA. The results demonstrated that compared with the A/R
group, HES1 expression was significantly increased, the LC3 II/LC3
I expression ratio was significantly decreased, and P62 expression
was significantly upregulated in the 10 μM Cur pretreatment
group. Conversely, co-incubation with Cur + pAD/HES1-shRNA
significantly decreased HES1 and P62 expression while upregulating
the LC3 II/LC3 I ratio. Notably, pAD/HES1-shRNA exacerbated A/R
injury (Fig. 6A-D). The
LysoTracker Red dye is used to observe autophagy lysosomes. In
A/R-injured cardiomyocytes, the red fluorescent spot was more
intense, which was significantly reduced by Cur pretreatment.
However, co-incubation with Cur + pAD/HES1-shRNA significantly
increased the red fluorescence intensity (Fig. 6E-H). Although Cur pretreatment
effectively attenuated HES1-mediated autophagy
hyperactivation-associated cell death, pAD/HES1-shRNA counteracted
this protective effect. These results suggested that HES1 is
involved in the Cur-based amelioration of cardiomyocyte I/RI by
inhibiting autophagic hyperactivation.
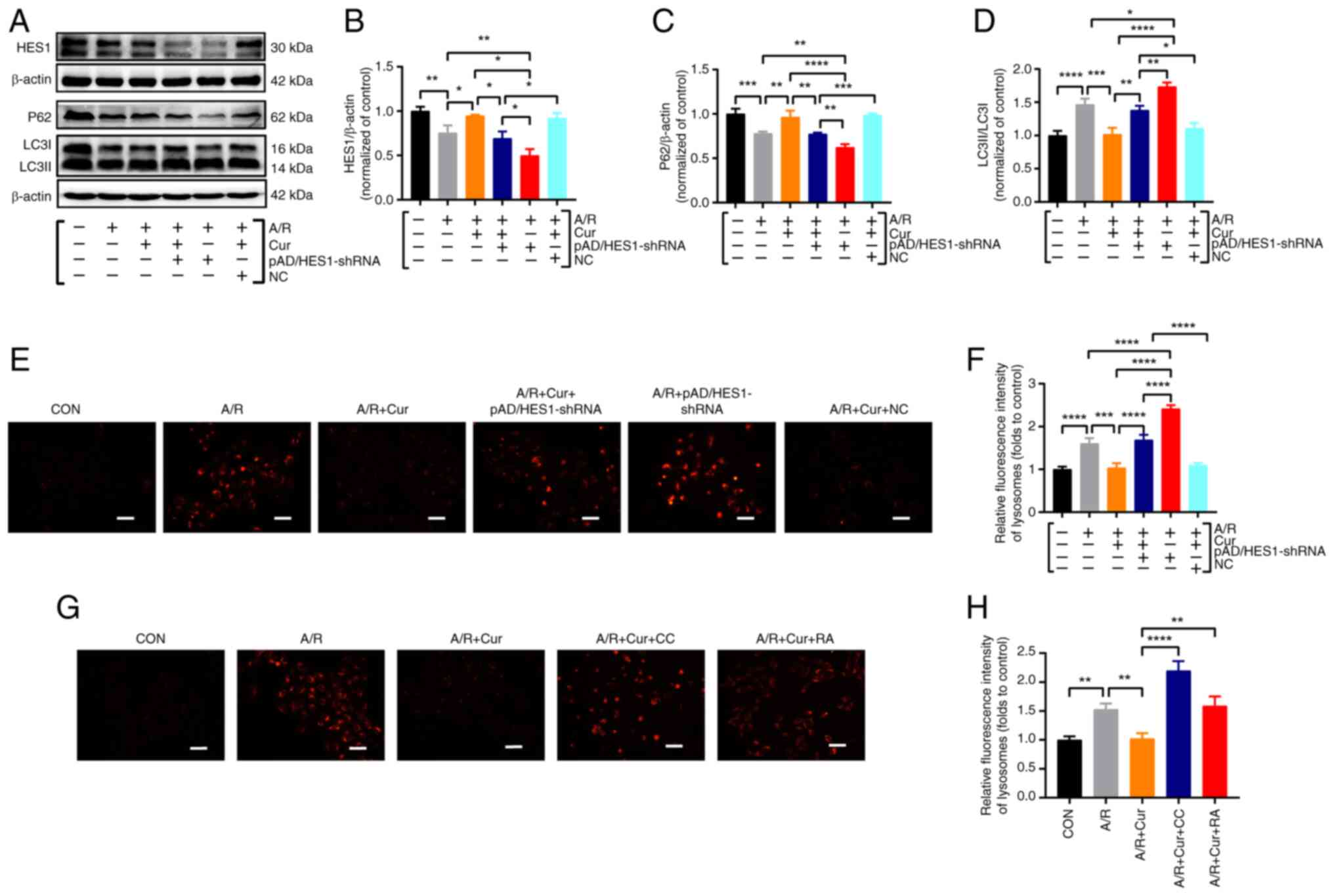 | Figure 6Cur pretreatment inhibits excessive
autophagy of A/R-triggered cardiomyocytes via adjusting HES1. (A-D)
Western blot analysis for the detection of protein expression and
quantitative analysis of P62 and LC3II/I in cell lysates after
treatment. (E-H) Fluorescent probe LysoTracker Red for detecting
lysosomes and quantitative analysis following treatment
(magnification, ×200; scale bar, 100 μm). Data are presented
as the mean ± SD (n=3). *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. Cur, curcumin; A/R,
anoxia/reoxygenation; P62, Sequestosome 1; LC3II/I,
microtubule-associated protein 1 light chain 3; CC, compound C; RA,
rapamycin; NC, negative control; shRNA, short hairpin RNA. |
Cur preconditioning inhibits A/R-induced
apoptosis in H9c2 cardiomyocytes via mediating HES1
In addition, interestingly, alterations in
apoptosis-related indices were also observed. Specifically, A/R
injury significantly downregulated BCL2/BAX ratio, augmented
Caspase 3 levels, and increased the total apoptotic rate (early +
late apoptosis). Cur pretreatment reversed A/R injury-induced
apoptosis, yet pAD/HES1-shRNA blocked this protective effect
(Fig. 7A-E). These results
indicated that in I/RI, there may be a mutual overlap and crosstalk
among ferroptosis, apoptosis and autophagy, thereby together
affecting the homeostasis of the internal environment and final
state of cardiomyocytes. However, this requires an in-depth
investigation of the pathways and mechanisms among them.
Cur pretreatment ameliorates
mitochondrial dysfunction via adjusting HES1 and upregulates AMPK
maintained homeostasis of energy metabolism in A/R injured
cardiomyocytes
MPTP and MMP, one of the essential components of
mitochondria, may be involved in the exchange of mitochondrial
components during cell death. They play a significant part in cell
survival, apoptosis and ferroptosis, which are associated with
several functions, such as tumor progression and
ischemia/reperfusion (15,28). Based on the upregulation of HES1
expression, the present study aimed to investigate the effect of
Cur pretreatment on mitochondrial function and morphological
changes in H9c2 cardiomyocytes with A/R injury. For a visual
assessment of mitochondrial morphological changes, TEM was used to
observe the changes among the different groups. Mitochondria from
A/R-treated H9c2 cells were largely wrinkled, with an obvious
distortion of the internal structure, reduced or broken cristae,
and a significantly higher Flameng score. By contrast, 10 μM
Cur pretreatment attenuated the A/R-induced effects, whereas
pAD/HES1-shRNA reversed the effects, blocked the protective effect
of Cur, and increased the sensitivity of H9c2 to A/R injury
(Fig. 8A and B). Furthermore,
the results revealed that Cur or Cur + NC pretreatment
significantly inhibited MPTP opening as well as elevated MMP in
H9c2 cardiomyocytes after A/R injury, thereby maintaining
mitochondrial function and homeostasis. However, pAD/HES1-shRNA
pretreatment blocked the protective effect of Cur (Fig. 8C-F).
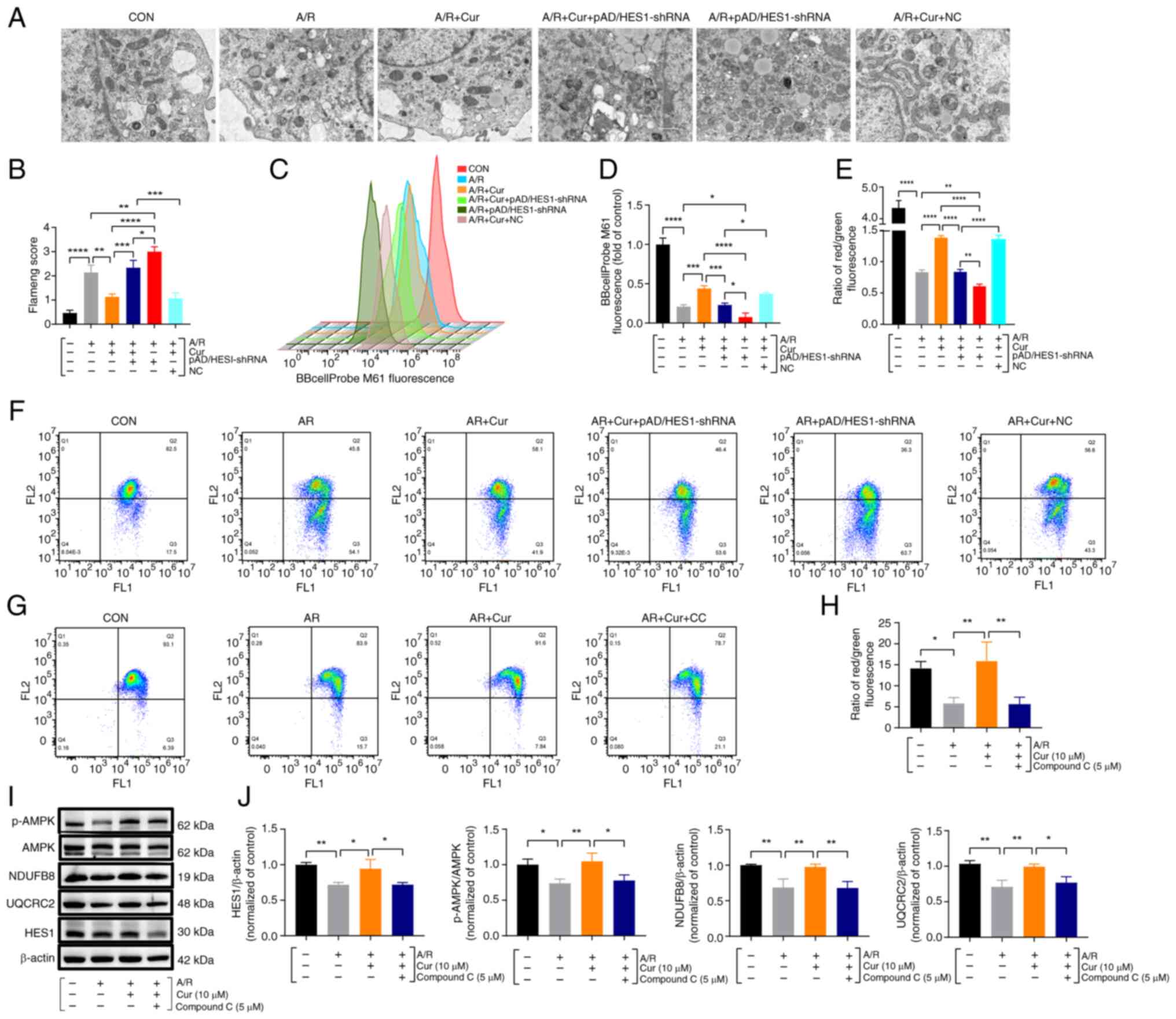 | Figure 8Cur preconditioning ameliorates
mitochondrial dysfunction in A/R injured cardiomyocytes via
adjusting HES1 and upregulates AMPK, maintaining energy metabolism
homeostasis. (A and B) Transmission electron microscopy for
assessing mitochondrial ultrastructure and Flameng scoring
(magnification, ×8000; scale bar, 1 μm). (C and D)
BBcellProbe M61 assay for cellular MPTP by Flow cytometry. (E-H)
JC-1 assay for cellular MMP. (I and J) Protein expression and
quantification of HES1, p-AMPK, AMPK, NDUFB8 and UQCRC2 in the
lysates of exposed A/R cells after Cur or Compound C pretreatment
using western blotting. Data are presented as the mean ± SD (n=3).
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. Cur,
curcumin; A/R, anoxia/reoxygenation; MPTP, mitochondrial
permeability transition pore; MMP, mitochondrial membrane
potential; NDUFB8, NADH dehydrogenase [ubiquinone] 1 beta
subcomplex subunit 8; UQCRRC2, cytochrome b-c1 complex subunit 2;
NC, negative control; shRNA, short hairpin RNA; p-,
phosphorylated. |
Energy is the basis of several life activities,
including growth, proliferation, bio-metabolism and stress, among
others. The activation of AMPK, the gatekeeper of energy metabolism
and mitochondrial homeostasis, restores energy balance by
stimulating ATP-generating catabolic routes and suppressing
energy-consuming processes (29). Compared with the control, A/R
injury significantly decreased the red/green fluorescence ratio and
decreased MMP, Cur pretreatment significantly increased MMP and
maintained MMP homeostasis, and Compound C (AMPK inhibitor)
reversed these changes (Fig. 8G and
H). Protein detection in cell lysates revealed that Cur
pretreatment significantly upregulated the p-AMPK/AMPK ratio and
NDUFB8, UQCRC2 and HES1 expression compared with the A/R group and
that Compound C blocked the protective effect of Cur (Fig. 8I and J). Thus, it was
hypothesized that HES1 is involved in the Cur-based regulation of
the maintenance of cellular energy metabolism homeostasis via AMPK
to ameliorate I/RI-induced mitochondrial dysfunction.
Discussion
Owing to the rapidly aging society of China, the
count of patients suffering from cardiovascular conditions,
especially AMI, has been escalating annually (30). Consequently, this poses
formidable challenges for medical personnel while presenting a
unique, once-in-a-century opportunity. Preventing and treating
patients with AMI in an improved and faster manner and reducing
morbidity, death and disability have become important issues to be
solved. While early hematopoiesis/reperfusion therapy represents an
effective therapeutic strategy to reduce the rate of sudden death
and improve the prognosis of patients, reperfusion itself
aggravates cardiomyocyte damage and cardiac tissue dysfunction in
patients with AMI, that is, I/RI (4). The pathophysiological mechanisms of
I/RI are complex and involve multiple forms of RCD, encompassing
inflammatory responses, oxidative stress, apoptosis, pyroptosis,
ferroptosis and autophagy, among others. These may occur
independently or in cross-talk with each other, occasionally
overlapping (5-8). Therefore, the exploration and
elucidation of the underlying pathophysiological mechanisms is
critical for the discovery and development of more effective
molecular drug targets against I/RI. The present study results
revealed that after A/R injury, LDH levels were significantly
elevated and cell viability was suppressed; MDA, total iron and
free iron levels were enhanced; and SOD activity and GSH/GSSG ratio
were inhibited compared with the control group, indicating that
H9c2 cardiomyocytes were significantly injured in the I/RI model
in vitro. Furthermore, the protein expression of BCL2, BAX,
PTGS2, GPX4, LC3II/I and P62 was altered, suggesting that
apoptosis-, ferroptosis- and autophagy-based regulatory mechanisms
are involved in I/RI in vitro (Fig. 9).
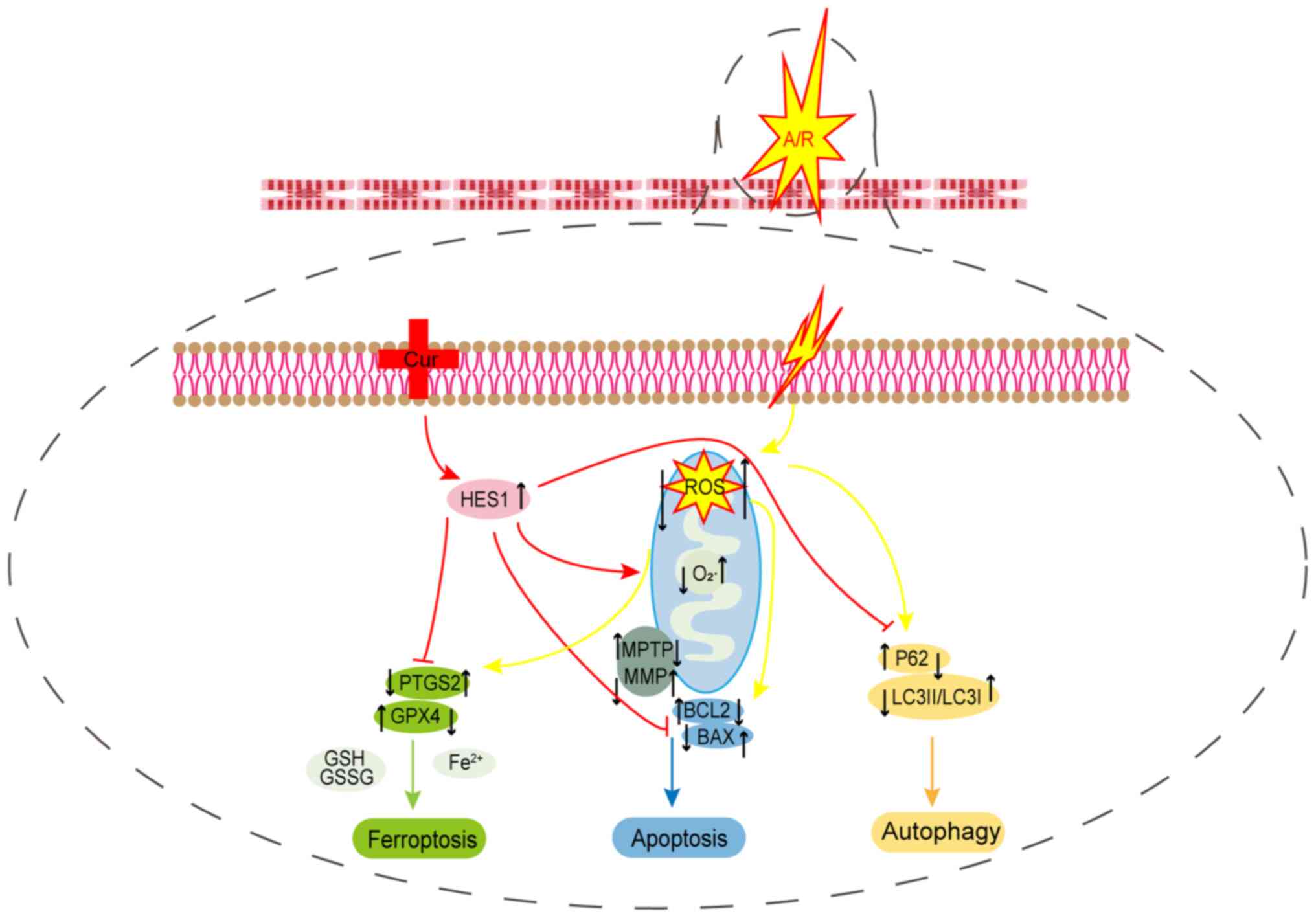 | Figure 9Potential mechanism of Cur in
A/R-triggered injury. Pre-treatment with Cur could inhibit
oxidative stress, ferroptosis, apoptosis and autophagy while
ameliorating mitochondrial function to protect cardiomyocytes from
A/R injury by adjusting HES1. Cur, curcumin; A/R,
anoxia/reoxygenation; HES1, hairy and enhancer of split 1; PTGS2,
prostaglandin-endoperoxide synthase 2; GPX4, glutathione peroxidase
4; P62, Sequestosome 1; LC3II/I, microtubule-associated protein 1
light chain 3; ROS, reactive oxygen species; GSH/GSSH,
glutathione/glutathione disulfide; BCL2, B-cell lymphoma2; BAX,
BCL2-associated X protein; MPTP, mitochondrial permeability
transition pore; MMP, mitochondrial membrane potential. |
Hypoxic/ischemic preconditioning, as its
nomenclature suggests, entails the alleviation of I/RI through the
employment of diverse salutary preconditioning protocols before
hypoxia/ischemia. In recent years, there has been a surge of
research interest in various forms of preconditioning, notably
cardiac ischemic preconditioning, remote ischemic preconditioning
and pharmacological preconditioning (31). In particular, pharmacological
preconditioning to improve I/RI is of great clinical utility owing
to the simplicity of protocols and their ease of implementation.
Illustrative instances include dihydrotanshinone I preconditioning
protects the myocardium from ischemic injury via PKM2 glutathione
sialylation; naringenin improves mitochondrial dysfunction in I/RI
via the AMPK-SIRT3 pathway; and HHQ16, a Flavin IV derivative,
ameliorates myocardial infarction by degrading lncRNA4012/9456
(32-34). Thus, it was hypothesized that Cur
is a candidate phytochemical for treating I/RI.
Cur, a class of naturally occurring polyphenolic
compounds primarily extracted from turmeric rhizomes within TCM,
has emerged as a pivotal agent exhibiting protective function
against sepsis, Alzheimer's disease and I/RI, as evidenced by
studies (21,35,36). Hu et al showed that Cur
could prevent and treat myocardial infarction via the Epac1-Akt
pathway to alleviate A/R injury (37). Additionally, Kim et al
(38) revealed that Cur could
prevent and treat myocardial infarction through TLR2 inhibition.
Furthermore, Cur has been shown to regulate the Notch2/HES1
pathway, leading to reduced I/R-induced lung injury (39). HES1, a vital gene downstream of
the Notch signaling pathway, encodes a repressive bHLH that acts as
a transcriptional repressor. HES1 proteins regulate numerous
biological processes in the organism, including cell proliferation,
apoptosis and stress (40).
Previous studies revealed that the upregulation of HES1 protein
contributed to the alleviation of I/RI and improvement of AMI
(41,42). In our recent study, it has been
revealed that ischemic preconditioning/post-ischemic treatment
attenuates A/R damage via the Notch1/HES1/VDAC1 axis (43). However, whether Cur can improve
I/RI by regulating HES1 expression remains unknown. Therefore,
further in-depth studies are required to investigate the regulatory
mechanism of Cur. The present study delves into this knowledge gap
and demonstrated that Cur pretreatment significantly enhanced cell
survival rate, decreased LDH activity, inhibited oxidative stress,
reduced iron level, and ameliorated the in vitro I/RI injury
mimicry by upregulating HES1 expression. Of note, Cur may have
effects similar to those of the small molecule drugs DFO, Fer-1 and
3MA, highlighting its therapeutic potential.
Ferroptosis is an iron-dependent form of
non-apoptotic cell death that involves lipid hydroperoxide
accumulation, which distinguishes it from other RCDs (12,13). Over the past decade, ferroptosis
has emerged as a pivotal mechanism underlying the initiation and
progression of various cardiovascular disease subtypes,
encompassing actinomycin- or sepsis-induced cardiomyopathy,
atherosclerosis, myocardial I/RI, arrhythmias and diabetic
cardiomyopathy (7,44,45). Iron, an essential trace element,
plays pivotal roles in numerous biological processes, spanning
growth, development and energy metabolism across life forms.
Notably, iron serves as a central player in the occurrence of
ferroptosis, emphasizing the significance of maintaining iron
homeostasis for preserving cardiac physiological function (46). Deviations from iron homeostasis,
such as deficiency, can precipitate heart failure in humans
(47). Iron overload, either
secondary or primary, particularly unstable ferrous ions, can lead
to cardiac damage through the action of oxidative stress (48,49). However, the mechanisms regulating
these phenomena are currently unknown. In addition to altering iron
homeostasis, excessive ROS accumulation directly damages
cardiomyocytes via oxidative lipid metabolism (50). Apart from iron metabolism, ROS
and lipid metabolism, the glutathione-dependent antioxidant system,
which is the most classical anti-ferroptosis pathway, has been
shown to prevent and treat cardiovascular disease. Meanwhile,
cysteine deficiency, glutathione depletion and inactivation of the
phospholipid hydroperoxide GPX4 have been identified to promote
ferroptosis (51). In the
present study, Cur pretreatment significantly decreased the overall
iron level inside cells and ferrous ion level in the unstable iron
pool, decreased lipid metabolism indices (such as MDA), increased
SOD, and GSH/GSSG ratios, and inhibited ROS overproduction. In
addition, Cur pretreatment significantly reduced PTGS2 expression
and increased GPX4 and HES1 expression. Of note, pAD/HES1-shRNA
counteracted the effect of Cur pretreatment on A/R injury.
Therefore, it was hypothesized that Cur inhibits ferroptosis
against A/R injury by upregulating HES1.
Autophagy, a pivotal mechanism, enables organisms to
respond to various external stimuli, maintain homeostasis of the
internal environment, and adapt for survival through the
phagocytosis of abnormal molecules or organelles (52). However, research focusing on the
impact of autophagy on I/RI has yielded contrasting findings.
Certain studies posit that augmenting autophagy mitigates I/RI,
whereas others contend that inhibiting excessive autophagy shields
the myocardium from I/RI (53-56). Notably, in acute myocardial I/RI,
autophagy is a 'double-edged sword'. If autophagic homeostasis is
disrupted, autophagy overactivation will degrade normal
intracellular proteins, subcellular organelles, membranes and other
substances, ultimately resulting in cell death. In the current
study, P62 expression and LC3II/LC3I ratio were significantly
downregulated in simulated I/RI in vitro, suggesting
excessive autophagy activation after reoxygenation. Notably, Cur
pretreatment reversed these alterations, indicating that Cur's
protective effect may stem from impeding excessive autophagy
activation. To confirm that Cur preconditioning may play a
protective role against myocardial A/R injury via autophagy
regulation. RA (autophagy activator) and 3MA (autophagy inhibitor)
were used to observe their effects on autophagy. The results showed
that 3-MA inhibited autophagy to attenuate I/RI, whereas RA
activated excessive autophagy to exacerbate I/RI, suggesting that
Cur has a similar effect to 3MA. Of note, with the autophagy tool
drug, P62 expression and LC3II/LC3I ratio were affected similarly
to PTGS2 and GPX4 expression, suggesting a possible overlap or
crosstalk between ferroptosis and autophagy. Nevertheless,
pAD/HES1-shRNA blocked the myocardial protective effect of Cur
preconditioning against A/R injury. The findings indicated that Cur
may play a role in mitigating A/R injury in cardiomyocytes by
inhibiting excessive autophagy activation and ferroptosis via
mediating HES1.
The excessive autophagy triggered by I/R linked to
myocardial mitochondrial homeostasis and energy metabolism. It has
been revealed that during cardiomyocyte ischemia, ATP depletion
activates the mTOR/ULK1/PI3K pathway, fostering autophagic vesicle
formation and ATP synthesis. Conversely, upon reperfusion, ROS
surplus elicits autophagy overactivation, exacerbating
cardiomyocyte demise (57).
Notably, mitochondrion is the main site of ATP production in
mammals, and ATP serves as the energy source for most life
activities, including growth and development, proliferation,
metabolism and stress, among others. In addition, the activation of
AMPK, which serves as a gatekeeper of energy metabolism and
mitochondrial homeostasis, helps restore energy balance by
facilitating catabolic routes for ATP production while inhibiting
energy overconsumption. However, mitochondrial dysfunction can lead
to reduced ATP production, ROS overproduction and cellular
dysfunction (11,29). Moreover, MMP and MPTP, one of the
key components of the mitochondrial membrane, may be engaged in the
exchange of mitochondrial and cytoplasmic intercellular components
during cell proliferation, apoptosis, ferroptosis and autophagy in
several diseases, such as septic cardiomyopathy, tumors, and
myocardial ischemia/reperfusion (28,58,59). In the present study, Cur
preconditioning increased NDUFB8 and UQCRC2 protein expression and
p-AMPK/AMPK protein ratio, whereas Compound C (an AMPK inhibitor)
inhibited the defensive impact of Cur preconditioning against A/R
damage. In addition, Cur pretreatment prevented A/R-induced MPTP
over-opening and MMP reduction, whereas pAD/HES1-shRNA blocked this
effect. Thus, it was hypothesized that Cur preconditioning
attenuates A/R injury by participating in the maintenance of
mitochondrial functional homeostasis and energy biogenesis.
The findings of present study indicated that I/R
causes significant harm to the myocardium by triggering
ferroptosis, apoptosis and excessive autophagy. Notably, Cur can
regulate ferroptosis and apoptosis-related protein expression by
upregulating HES1 expression. Furthermore, the current results
demonstrated that Cur pretreatment increased the expression of P62
and the LC3II/I ratio, reduced ROS generation, stopped excessive
MPTP opening, stabilized MMP levels, upregulated pAMPK/AMPK ratio,
and maintained mitochondrial function. Collectively, HES1-mediated
Cur preconditioning protected the myocardium from I/RI by
inhibiting ferroptosis, apoptosis, excessive autophagy and
oxidative stress; ameliorating mitochondrial dysfunction; and
maintaining energy homeostasis.
In the present study, the molecular protective
mechanism of Cur was explored using only ferroptosis
inhibitor/activator and autophagy inhibitor/activator in an in
vitro A/R model of H9c2 cells. To understand the potential
mechanism of action, transgenic or knockdown-based
HES1-overexpressing I/RI animal models need to be established. In
addition, although these results suggested that ferroptosis,
apoptosis, autophagy crosstalk, or overlap may exist in A/R, clear
hub molecular targets need to be identified using more in-depth
experiments.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SQL and JCL conceived and designed the study. YY and
HH performed cell experiments, data analysis and visualization. TH
and CCZ performed cell experiments and data curation. YMQ and MF
developed methodology. All authors wrote the original draft. All
authors read and approved the final version of the manuscript. SQL
and JCL revised the manuscript. SQL and HH supervised the study and
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of Jiangxi (grant nos. 82070303 and 82360057) and
Jiangxi Provincial Natural Science Foundation (grant nos.
20224ACB206002, 20232BAB206009 and 20232BAB206010).
References
|
1
|
Anderson JL and Morrow DA: Acute
myocardial infarction. N Engl J Med. 376:2053–2064. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Algoet M, Janssens S, Himmelreich U, Gsell
W, Pusovnik M, Van den Eynde J and Oosterlinck W: Myocardial
ischemia-reperfusion injury and the influence of inflammation.
Trends Cardiovasc Med. 33:357–366. 2023. View Article : Google Scholar
|
|
3
|
Salari N, Morddarvanjoghi F, Abdolmaleki
A, Rasoulpoor S, Khaleghi AA, Hezarkhani LA, Shohaimi S and
Mohammadi M: The global prevalence of myocardial infarction: A
systematic review and meta-analysis. BMC Cardiovasc Disord.
23:2062023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martí-Pàmies Í, Thoonen R, Morley M,
Graves L, Tamez J, Caplan A, McDaid K, Yao V, Hindle A, Gerszten
RE, et al: Brown adipose tissue and BMP3b decrease injury in
cardiac ischemia-reperfusion. Circ Res. 133:353–365. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xue Y, Fu W, Yu P, Li Y, Yu X, Xu H and
Sui D: Ginsenoside Rc alleviates myocardial ischemia-reperfusion
injury by reducing mitochondrial oxidative stress and apoptosis:
Role of SIRT1 activation. J Agric Food Chem. 71:1547–1561. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gu S, Tan J, Li Q, Liu S, Ma J, Zheng Y,
Liu J, Bi W, Sha P, Li X, et al: Downregulation of LAPTM4B
contributes to the impairment of the autophagic flux via unopposed
activation of mTORC1 signaling during myocardial
ischemia/reperfusion injury. Circ Res. 127:e148–e165. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cai W, Liu L, Shi X, Liu Y, Wang J, Fang
X, Chen Z, Ai D, Zhu Y and Zhang X: Alox15/15-HpETE aggravates
myocardial ischemia-reperfusion injury by promoting cardiomyocyte
ferroptosis. Circulation. 147:1444–1460. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental mechanisms of regulated cell death and
implications for heart disease. Physiol Rev. 99:1765–1817. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sciarretta S, Maejima Y, Zablocki D and
Sadoshima J: The role of autophagy in the heart. Annu Rev Physiol.
80:1–26. 2018. View Article : Google Scholar
|
|
10
|
Chen X, Xie Q, Zhu Y, Xu J, Lin G, Liu S,
Su Z, Lai X, Li Q, Xie J, et al: Cardio-protective effect of
tetrahydrocurcumin, the primary hydrogenated metabolite of curcumin
in vivo and in vitro: Induction of apoptosis and autophagy via
PI3K/AKT/mTOR pathways. Eur J Pharmacol. 911:1744952021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Steinberg GR and Hardie DG: New insights
into activation and function of the AMPK. Nat Rev Mol Cell Biol.
24:255–272. 2023. View Article : Google Scholar
|
|
12
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fang X, Ardehali H, Min J and Wang F: The
molecular and metabolic landscape of iron and ferroptosis in
cardiovascular disease. Nat Rev Cardiol. 20:7–23. 2023. View Article : Google Scholar
|
|
14
|
Xing G, Meng L, Cao S, Liu S, Wu J, Li Q,
Huang W and Zhang L: PPARα alleviates iron overload-induced
ferroptosis in mouse liver. EMBO Rep. 23:e522802022. View Article : Google Scholar
|
|
15
|
Hu T, Zou HX, Le SY, Wang YR, Qiao YM,
Yuan Y, Liu JC, Lai SQ and Huang H: Tanshinone IIA confers
protection against myocardial ischemia/reperfusion injury by
inhibiting ferroptosis and apoptosis via VDAC1. Int J Mol Med.
52:109 [pii]2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu B, Zhao C, Li H, Chen X, Ding Y and Xu
S: Puerarin protects against heart failure induced by pressure
overload through mitigation of ferroptosis. Biochem Biophys Res
Commun. 497:233–240. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cao JF, Gong Y, Wu M, Xiong L, Chen S,
Huang H, Zhou X, Peng YC, Shen XF, Qu J, et al: Molecular docking
and molecular dynamics study Lianhua Qingwen granules (LHQW) treats
COVID-19 by inhibiting inflammatory response and regulating cell
survival. Front Cell Infect Microbiol. 12:10447702022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Poespoprodjo JR, Douglas NM, Ansong D, Kho
S and Anstey NM: Malaria. Lancet. 402:2328–2345. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang ZK, Chen RR, Li JH, Chen JY, Li W,
Niu XL, Wang FF, Wang J and Yang JX: Puerarin protects against
myocardial ischemia/reperfusion injury by inhibiting inflammation
and the NLRP3 inflammasome: The role of the SIRT1/NF-κB pathway.
Int Immunopharmacol. 89:1070862020. View Article : Google Scholar
|
|
20
|
Duan W, Yang Y, Yan J, Yu S, Liu J, Zhou
J, Zhang J, Jin Z and Yi D: The effects of curcumin post-treatment
against myocardial ischemia and reperfusion by activation of the
JAK2/STAT3 signaling pathway. Basic Res Cardiol. 107:2632012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu P, Yang M, He H, Kuang Z, Liang M, Lin
A, Liang S, Wen Q, Cheng Z and Sun C: Curcumin attenuates
hypoxia/reoxygenation-induced cardiomyocyte injury by
downregulating Notch signaling. Mol Med Rep. 20:1541–1550.
2019.PubMed/NCBI
|
|
22
|
Huang H, Lai S, Wan Q, Qi W and Liu J:
Astragaloside IV protects cardiomyocytes from anoxia/reoxygenation
injury by upregulating the expression of Hes1 protein. Can J
Physiol Pharmacol. 94:542–553. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu L, Li F, Zhao G, Yang Y, Jin Z, Zhai M,
Yu W, Zhao L, Chen W, Duan W, et al: Protective effect of berberine
against myocardial ischemia reperfusion injury: role of
Notch1/Hes1-PTEN/Akt signaling. Apoptosis. 20:796–810. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Flameng W, Borgers M, Daenen W and
Stalpaert G: Ultrastructural and cytochemical correlates of
myocardial protection by cardiac hypothermia in man. J Thorac
Cardiovasc Surg. 79:413–424. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li FJ, Long HZ, Zhou ZW, Luo HY, Xu SG and
Gao LC: System X(c) (-)/GSH/GPX4 axis: An important antioxidant
system for the ferroptosis in drug-resistant solid tumor therapy.
Front Pharmacol. 13:9102922022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cai C, Guo Z, Chang X, Li Z, Wu F, He J,
Cao T, Wang K, Shi N, Zhou H, et al: Empagliflozin attenuates
cardiac microvascular ischemia/reperfusion through activating the
AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 52:1022882022.
View Article : Google Scholar
|
|
27
|
Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci
B, Xie Y, Carlson D, Rothermel BA, Sun Y, et al: Beclin-1-dependent
autophagy protects the heart during sepsis. Circulation.
138:2247–2262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ye T, Yang W, Gao T, Yu X, Chen T, Yang Y,
Guo J, Li Q, Li H and Yang L: Trastuzumab-induced cardiomyopathy
via ferroptosis-mediated mitochondrial dysfunction. Free Radic Biol
Med. 206:143–161. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Herzig S and Shaw RJ: AMPK: Guardian of
metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol.
19:121–135. 2018. View Article : Google Scholar :
|
|
30
|
Zhao D, Liu J, Wang M, Zhang X and Zhou M:
Epidemiology of cardiovascular disease in China: Current features
and implications. Nat Rev Cardiol. 16:203–212. 2019. View Article : Google Scholar
|
|
31
|
Sawashita Y, Hirata N, Yoshikawa Y, Terada
H, Tokinaga Y and Yamakage M: Remote ischemic preconditioning
reduces myocardial ischemia-reperfusion injury through unacylated
ghrelin-induced activation of the JAK/STAT pathway. Basic Res
Cardiol. 115:502020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu X, Liu L, Zheng Q, Ye H, Yang H, Hao H
and Li P: Dihydrotanshinone I preconditions myocardium against
ischemic injury via PKM2 glutathionylation sensitive to ROS. Acta
Pharm Sin B. 13:113–127. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu LM, Dong X, Xue XD, Zhang J, Li Z, Wu
HJ, Yang ZL, Yang Y and Wang HS: Naringenin improves mitochondrial
function and reduces cardiac damage following ischemia-reperfusion
injury: the role of the AMPK-SIRT3 signaling pathway. Food Funct.
10:2752–2765. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wan J, Zhang Z, Wu C, Tian S, Zang Y, Jin
G, Sun Q, Wang P, Luan X, Yang Y, et al: Astragaloside IV
derivative HHQ16 ameliorates infarction-induced hypertrophy and
heart failure through degradation of lncRNA4012/9456. Signal
Transduct Target Ther. 8:4142023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang C, Shi Q, Yang J, Ren H, Zhang L,
Chen S, Si J, Liu Y, Sha D, Xu B and Ni J: Ceria nanozyme
coordination with curcumin for treatment of sepsis-induced cardiac
injury by inhibiting ferroptosis and inflammation. J Adv Res.
63:159–170. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ruan Y, Xiong Y, Fang W, Yu Q, Mai Y, Cao
Z, Wang K, Lei M, Xu J, Liu Y, et al: Highly sensitive
curcumin-conjugated nanotheranostic platform for detecting
amyloid-beta plaques by magnetic resonance imaging and reversing
cognitive deficits of Alzheimer's disease via NLRP3-inhibition. J
Nanobiotechnology. 20:3222022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X, Zhang Y, Yang Y, Zhang W, Luo L,
Han F, Guan H, Tao K and Hu D: Curcumin pretreatment protects
against hypoxia/reoxgenation injury via improvement of
mitochondrial function, destabilization of HIF-1α and activation of
Epac1-Akt pathway in rat bone marrow mesenchymal stem cells. Biomed
Pharmacother. 109:1268–1275. 2019. View Article : Google Scholar
|
|
38
|
Kim YS, Kwon JS, Cho YK, Jeong MH, Cho JG,
Park JC, Kang JC and Ahn Y: Curcumin reduces the cardiac
ischemia-reperfusion injury: Involvement of the toll-like receptor
2 in cardiomyocytes. J Nutr Biochem. 23:1514–1523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bo H and Feng X: Post-treatment curcumin
reduced ischemia-reperfusion-induced pulmonary injury via the
Notch2/Hes-1 pathway. J Int Med Res. 48:3000605198924322020.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang M, Yu LM, Zhao H, Zhou XX, Yang Q,
Song F, Yan L, Zhai ME, Li BY, Zhang B, et al:
2,3,5,4′-Tetrahydroxystilbe ne-2-O-β-D-glucoside protects murine
hearts against ischemia/reperfusion injury by activating
Notch1/Hes1 signaling and attenuating endoplasmic reticulum stress.
Acta Pharmacol Sin. 38:317–330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou XL, Wan L, Xu QR, Zhao Y and Liu JC:
Notch signaling activation contributes to cardioprotection provided
by ischemic preconditioning and postconditioning. J Transl Med.
11:2512013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou XL, Zhao Y, Fang YH, Xu QR and Liu
JC: Hes1 is upregulated by ischemic postconditioning and
contributes to cardioprotection. Cell Biochem Funct. 32:730–736.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang L, Lai S, Zou H, Zhou X, Wan Q, Luo
Y, Wu Q, Wan L, Liu J and Huang H: Ischemic
preconditioning/ischemic post-conditioning alleviates
anoxia/reoxygenation injury via the Notch1/Hes1/VDAC1 axis. J
Biochem Mol Toxicol. 36:e231992022. View Article : Google Scholar
|
|
44
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang X, Chen X, Zhou W, Men H, Bao T, Sun
Y, Wang Q, Tan Y, Keller BB, Tong Q, et al: Ferroptosis is
essential for diabetic cardiomyopathy and is prevented by
sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 12:708–722.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Galy B, Conrad M and Muckenthaler M:
Mechanisms controlling cellular and systemic iron homeostasis. Nat
Rev Mol Cell Biol. 25:133–155. 2024. View Article : Google Scholar
|
|
47
|
Jankowska EA, Kasztura M, Sokolski M,
Bronisz M, Nawrocka S, Oleśkowska-Florek W, Zymliński R, Biegus J,
Siwołowski P, Banasiak W, et al: Iron deficiency defined as
depleted iron stores accompanied by unmet cellular iron
requirements identifies patients at the highest risk of death after
an episode of acute heart failure. Eur Heart J. 35:2468–2476. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fang X, Cai Z, Wang H, Han D, Cheng Q,
Zhang P, Gao F, Yu Y, Song Z, Wu Q, et al: Loss of cardiac ferritin
H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ
Res. 127:486–501. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fleming RE and Ponka P: Iron overload in
human disease. N Engl J Med. 366:348–359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li D, Zhang G, Wang Z, Guo J, Liu Y, Lu Y,
Qin Z, Xu Y, Cao C, Wang B, et al: Idebenone attenuates ferroptosis
by inhibiting excessive autophagy via the ROS-AMPK-mTOR pathway to
preserve cardiac function after myocardial infarction. Eur J
Pharmacol. 943:1755692023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ye Y, Chen A, Li L, Liang Q, Wang S, Dong
Q, Fu M, Lan Z, Li Y, Liu X, et al: Repression of the antiporter
SLC7A11/glutathione/glutathione peroxidase 4 axis drives
ferroptosis of vascular smooth muscle cells to facilitate vascular
calcification. Kidney Int. 102:1259–1275. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ikeda S, Zablocki D and Sadoshima J: The
role of autophagy in death of cardiomyocytes. J Mol Cell Cardiol.
165:1–8. 2022. View Article : Google Scholar :
|
|
53
|
Liu W, Chen C, Gu X, Zhang L, Mao X, Chen
Z and Tao L: AM1241 alleviates myocardial ischemia-reperfusion
injury in rats by enhancing Pink1/Parkin-mediated autophagy. Life
Sci. 272:1192282021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li Y, Liang P, Jiang B, Tang Y, Liu X, Liu
M, Sun H, Chen C, Hao H, Liu Z, et al: CARD9 promotes autophagy in
cardiomyocytes in myocardial ischemia/reperfusion injury via
interacting with Rubicon directly. Basic Res Cardiol. 115:292020.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wen L, Cheng X, Fan Q, Chen Z, Luo Z, Xu
T, He M and He H: TanshinoneⅡA inhibits excessive autophagy and
protects myocardium against ischemia/reperfusion injury via
14-3-3η/Akt/Beclin1 pathway. Eur J Pharmacol. 954:1758652023.
View Article : Google Scholar
|
|
56
|
Fan G, Yu J, Asare PF, Wang L, Zhang H,
Zhang B, Zhu Y and Gao X: Danshensu alleviates cardiac
ischaemia/reperfusion injury by inhibiting autophagy and apoptosis
via activation of mTOR signalling. J Cell Mol Med. 20:1908–1919.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mohamed DZ, El-Sisi A, Sokar SS, Shebl AM
and Abu-Risha SE: Targeting autophagy to modulate hepatic
ischemia/reperfusion injury: A comparative study between octreotide
and melatonin as autophagy modulators through
AMPK/PI3K/AKT/mTOR/ULK1 and Keap1/Nrf2 signaling pathways in rats.
Eur J Pharmacol. 897:1739202021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mancardi D, Pagliaro P, Ridnour LA,
Tocchetti CG, Miranda K, Juhaszova M, Sollott SJ, Wink DA and
Paolocci N: HNO protects the myocardium against reperfusion injury,
inhibiting the mPTP opening via PKCε activation. Antioxidants
(Basel). 11:3822022. View Article : Google Scholar
|
|
59
|
Pan P, Zhang H, Su L, Wang X and Liu D:
Melatonin balance the autophagy and apoptosis by regulating UCP2 in
the LPS-induced cardiomyopathy. Molecules. 23:6752018. View Article : Google Scholar : PubMed/NCBI
|