Cardiovascular disease (CVD) is a leading cause of
mortality worldwide, accounting for >50% of deaths related to
noncom-municable diseases, according to the World Economic Forum
(1). It has been estimated that
by 2030, CVD will be responsible for >22.2 million deaths
annually worldwide. Furthermore, the American Heart Association
estimates that 1.1 trillion dollars will be devoted to managing CVD
by 2035 (2). As a major public
health challenge, CVD is characterized by high mortality rates and
escalating healthcare expenditures, underscoring the importance of
understanding its underlying mechanisms and identifying therapeutic
targets. Regardless of the specific pathways involved,
cardiomyocyte damage in CVD ultimately results in cell death.
CVD is associated with several cell death
mechanisms, including ferroptosis, pyroptosis and autophagy
(3,4). Ferroptosis, first proposed by Dixon
et al (5), represents a
unique form of cell death, distinct from apoptosis, autophagy and
necroptosis, with defining morphological, biochemical and
immunological characteristics governed by specific regulatory genes
or proteins. Morphologically, it is marked by mitochondrial
shrinkage, increased density of the double-layered membranes and
the loss of mitochondrial crests. Biochemically, the depletion of
glutathione reduces the activity of glutathione peroxidase 4
(GPX4), impairing the breakdown of lipid peroxides. Consequently,
divalent iron converts lipids into reactive oxygen species (ROS),
triggering ferroptosis. In immunological contexts,
damage-associated molecular patterns (DAMPs) release
pro-inflammatory mediators such as HMGB1 (high mobility group box
1). Genetic research has focused on mutations that regulate iron
metabolism and lipid degradation (5,6).
Excess iron accumulation leads to irreversible tissue damage and
organ failure (7). Ferroptosis
is primarily characterized by oxidative damage caused by
mitochondrial dysfunction, driven by iron-dependent lipid
peroxidation (8). The mechanisms
of ferroptosis are closely linked to physiological processes such
as iron, amino acid and lipid metabolism.
Several signaling molecules and metabolic pathways
are involved in the regulation of ferroptosis. The present review
highlighted the key aspects of iron, amino acid and lipid
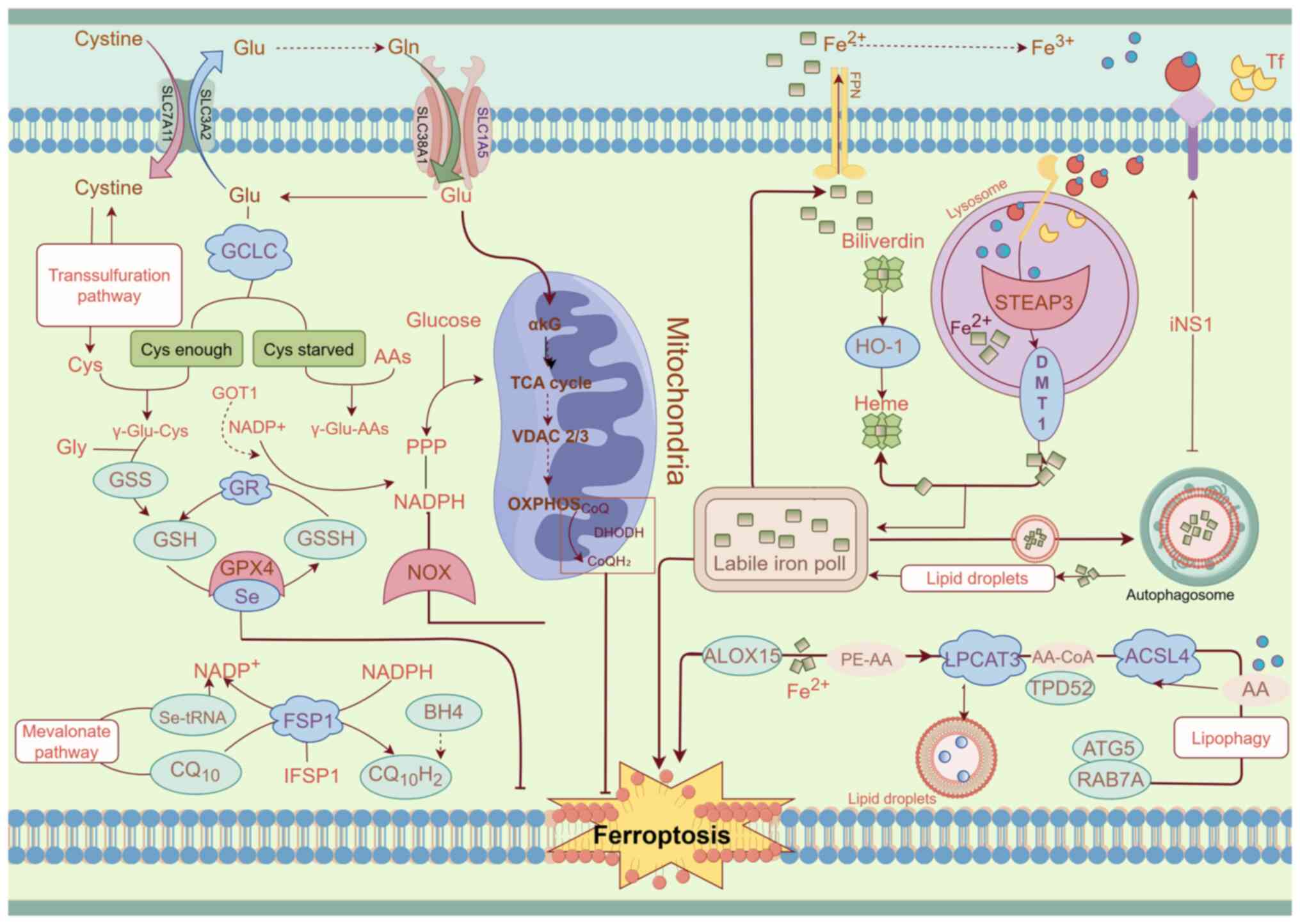
metabolism that drive the progression of ferroptosis (Fig. 1).
Arachidonic acid (AA) and adrenic acid (ADA), two
PUFAs present in membrane lipids, are precursors of PUFA-PE
(16,17). Lysophosphatidylcholine
acyltransferase 3 (LPCAT3) and acyl-CoA synthetase long-chain
family member 4 (ACSL4) are key enzymes involved in phospholipid
biosynthesis (18). ACSL4
acylates PUFAs to produce PUFA-acyl-coenzyme A (PUFA-CoA), which
LPCAT3 then re-esterifies to form PUFA-PEs through their reaction
with phosphatidylethanolamines (PEs). LOXs further oxidize PUFA-PEs
into harmful lipid hydroperoxides, such as PE-AA-OOH and
PE-ADA-OOH. Peroxidation predominantly occurs in plasma membranes
and the endoplasmic reticulum (ER), where ACSL4 is most active
(17). ACSL4, a critical
regulator of PUFA-CoA production, serves as a marker for
ferroptosis susceptibility (18). Knockdown of ACSL4 reduces PUFA-PE
synthesis and prevents RSL3-induced ferroptosis. Additionally,
thiazolidinediones, such as Rosiglitazone and Troglitazone, may
inhibit ACSL4, protecting cells from ferroptosis-associated damage
(19). LOXs, as non-heme
iron-dependent dioxygenases, contribute to ferroptosis by
oxygenating membrane PUFAs (20).
Iron is a vital nutrient for humans, is primarily
involved in synthesizing hemoglobin and myoglobin and serves a key
role in various physiological processes, including mitochondrial
respiration, intracellular enzymatic activities and oxygen
transport and storage (23).
Imbalances in iron metabolism can disrupt these processes, leading
to pathological conditions. A strong connection exists between
ferroptosis and iron-dependent lipid peroxidation. The body sources
most of its iron from an individual's dietary intake or through the
natural breakdown of red blood cells during the aging process.
Transferrin (TF) binds extracellular Fe3+ to form
TF-Fe3+, which is then absorbed by cells through
transferrin receptor 1 (TFR1). Inside the cell, Fe3+ is
released from endosomes by divalent metal transporter 1 (DMT1)
after being reduced by six-transmembrane epithelial antigen of
prostate 3 and enters the cytosol (24,25). Ferroportin 1 facilitates the
release of Fe2+ from ferritin molecules. When the
balance between iron absorption, utilization and recycling is
disrupted, ferroptosis is triggered. This occurs as free
Fe2+ reacts with hydrogen peroxide, generating highly
reactive lipid peroxides that drive the process of ferroptosis
(25). Several proteins and
regulatory factors involved in iron metabolism contribute to
ferroptosis. TFR1, which serves a critical role in iron transport,
increases cellular iron uptake and sensitivity to ferroptosis when
upregulated by hypoxia-inducible factor-1 and iron regulatory
protein (IRP). Limiting iron intake can prevent ferroptosis
(26). In cases where TFR1 is
absent, the metal cation transporter solute carrier family 39
member 14 can also facilitate iron entry into cells, leading to
ferroptosis (27). Additionally,
autophagy and ferroptosis are closely interconnected. Nuclear
receptor coactivator 4 (NCOA4) serves a key role in breaking down
ferritin through a process known as ferritinophagy. This mechanism
helps maintain intracellular iron homeostasis by regulating the
degradation of ferritin (28).
However, when NCOA4 mediates ferritin autophagy, cells become more
prone to ferroptosis due to the increased release of free iron
(29). Mitochondrial iron
transport is regulated by CDGSH iron sulfur domain 1 (CISD1) and
CISD2. Suppression of CISD1 enhances erastin-induced ferroptosis
and iron-dependent mitochondrial lipid peroxidation (30). Similarly, inhibition of CISD2
increases mitochondrial Fe2+ levels and lipid ROS,
further accelerating sulfasalazine-induced ferroptosis (31). These findings underscore the
pivotal role of iron metabolism in regulating ferroptosis.
L-g-glutamyl-L-cysteinylglycine, commonly known as
glutathione (GSH), is a tripeptide found in nearly all cells of the
body (32). The synthesis of GSH
is catalyzed by γ-glutamylcysteine synthetase, which converts
glutamate and cysteine into γ-glutamylcysteine. GSH serves a
critical role in protecting cells from ferroptosis by converting
potentially harmful lipid hydroperoxides, generated during cellular
metabolism, into less toxic lipid alcohols (33,34). GSH exists in two forms: Reduced
glutathione and oxidized glutathione disulfide (32). There are several pathways for GSH
synthesis. Of these pathways, one method involves the enzyme
glutathione reductase, which uses NADPH to reduce glutathione
disulfide to its active form, NADH. Another method involves
synthesizing GSH from intracellular cysteine, glycine and glutamate
with the help of specific enzymes (35,36). Cysteine is essential for GSH
synthesis and can be acquired through methionine production via the
transsulfuration pathway or through exogenous cysteine uptake. The
Xc complex (also known as System Xc−), a
glutamate-cystine antiporter composed of solute carrier family 3
member 2 and solute carrier family 7 member 11 (SLC7A11), is
responsible for importing cystine into cells, which is then reduced
to cysteine. Glutaminase 2 converts glutamine to glutamate, while
transporters, such as solute carrier family 38 member 1 and solute
carrier family 1 member 5, import glutamine into cells. The primary
role of GSH in cells is to neutralize potentially toxic lipid
hydrogen peroxides by converting them into less toxic lipid
alcohols, thereby preventing oxidative damage (37).
Research into the amino acid metabolic pathways
regulating ferroptosis has typically focused on key components such
as GPX4 and cysteine. The availability of cysteine is regulated by
multiple pathways, with significant attention given to the role of
SLC7A11 (37). In HT-1080
fibrosarcoma cells, overexpression of SLC7A11 was found to increase
resistance to ferroptosis, while its knockdown made cells more
susceptible to ferroptosis (38). The tumor suppressor gene p53 also
serves a critical role in regulating ferroptosis, metabolism and
neurodegeneration, with its effects depending on tissue type and
genotoxic stress levels (39-41). By downregulating SLC7A11, p53 can
inhibit cystine uptake through the system Xc−
transporter, reducing cellular antioxidant capacity, increasing
lipid ROS and inducing ferroptosis. Another key regulator of lipid
peroxidation and ferroptosis is nuclear factor erythroid 2-related
factor 2 (Nrf2), which controls genes involved in iron metabolism
and oxidative stress, such as heme oxygenase-1 (HO-1) and quinone
oxidoreductase 1 (NQO1). The Nrf2-HO-1 axis enhances cellular
resistance to ferroptosis by upregulating system Xc−
expression. Additionally, p62 prevents Nrf2 degradation, allowing
Nrf2 to accumulate and further protect against ferroptosis.
Knockdown of p62, NQO1, HO-1 and ferritin heavy chain 1 through RNA
interference exacerbates erastin-induced ferroptosis (42,43). The p53/p21 axis also delays
ferroptosis by reducing GSH consumption, although more research is
needed to clarify how p21 promotes GSH production (44). Erastin impairs cystine uptake
through system Xc−, leading to decreased GSH levels and
reduced GPX4 activity, which weakens cellular antioxidant defenses,
increases lipid ROS and induces ferroptosis. Furthermore, Ras
synthetic lethal 3 (RSL3) directly inhibits GPX4 enzymatic
activity, exacerbating ferroptosis.
VDACs are ion channels formed by porin proteins
located on the outer mitochondrial membrane (45). These channels serve a pivotal
role in regulating the exchange of ions and metabolites, such as
respiratory substrates, ADP and phosphate, between the cytosol and
mitochondria (46). In the open
state, VDACs permit the entry of these critical molecules into the
mitochondria, facilitating cellular respiration. However, when
VDACs are closed, this transport is inhibited. Microtubulin can
regulate mitochondrial metabolism and ion transport by obstructing
VDACs, leading to reduced mitochondrial function and a subsequent
decrease in ROS production. Paradoxically, this mechanism enhances
mitochondrial metabolism, reduces glycolysis and increases ROS
levels (47-49). Further research has shown that
reducing the expression of VDAC2 or VDAC3 via RNA interference
confers resistance to erastin and alters cell membrane permeability
(50). Erastin-induced
ferroptosis is linked to the opening of VDACs, which results in
diminished glycolysis and elevated ROS generation, causing
mitochondrial oxidative stress (51).
In eukaryotic cells, the ER serves a pivotal role in
protein synthesis, folding, assembly and transport, as well as
maintaining calcium homeostasis and cholesterol synthesis (52). Misfolded or unfolded proteins can
accumulate within the ER under stress conditions, such as
inflammation, hypoxia, glucose deprivation and oxidative stress,
triggering the unfolded protein response (UPR) and inducing ER
stress. The UPR is a protective mechanism aimed at restoring normal
protein folding or initiating cell death if stress persists. A
previous study showed that erastin and Sorafenib, in addition to
their effects on ferroptosis in HT1080 cells, also activated the ER
stress response (53). A
critical factor linking ER stress to ferroptosis is activating
transcription factor 4 (ATF4), which regulates genes involved in
amino acid metabolism and antioxidant defenses and is expressed
across various human tissues (54-57).
Recent studies have identified ferroptosis
suppressor protein 1 (FSP1) as a potent anti-ferroptosis factor,
independent of the glutathione pathway (58,59). Following myristoylation, FSP1
localizes to the plasma membrane, where it functions as an
oxidoreductase, converting NADPH to coenzyme Q10, which acts as a
lipid-soluble antioxidant (58,59). Coenzyme Q10 helps inhibit
ferroptosis by neutralizing lipid peroxidation in cell membranes.
Fin56, a type 3 ferroptosis inducer, promotes ferroptosis partly by
depleting coenzyme Q10, which serves as an endogenous ferroptosis
inhibitor (60). Additionally,
it has been reported that doxorubicin (DOX) may trigger FSP1
translocation in the heart by inducing lipid peroxidation products
(61). However, the specific
role of FSP1 in cardiac tissue remains poorly understood.
Conditional deletion of Fsp1 in mice could provide further insights
into the underlying mechanisms of its function in the heart.
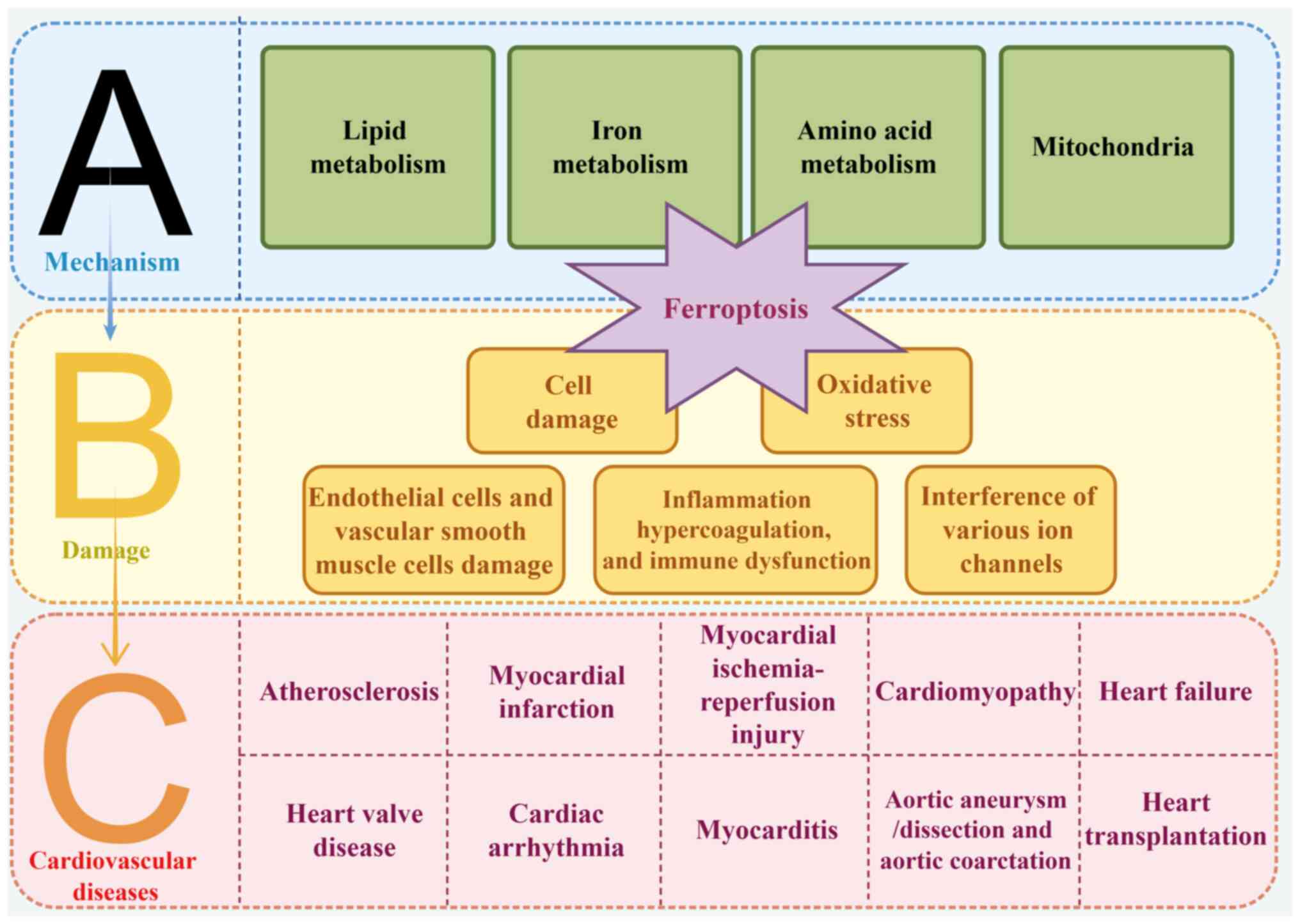
A concise overview of the link between ferroptosis
and various CVDs was summarized (Fig. 2).
Ferroptosis has been implicated in several CVDs,
including AS, a lipid metabolism disorder characterized by
inflammation, smooth muscle proliferation, endothelial dysfunction
and the formation of foam cells and lipid plaques. Pro-inflammatory
processes in AS are closely tied to ferroptosis, which is also
associated with altered blood iron levels. During ferroptosis,
excess iron catalyzes the Fenton reaction, leading to the
production of lipid ROS. These ROS oxidize low-density lipoprotein
(LDL), contributing to lipid deposition, foam cell formation and
endothelial dysfunction. This cascade ultimately weakens
endothelial function, enhances white blood cell adhesion and
increases the vulnerability of plaques, promoting new blood vessel
growth within the plaques and even causing intraplaque hemorrhaging
(62). Iron overload is a key
factor in the early development of AS, damaging endothelial
mitochondria and altering macrophage phenotypes. Iron-catalyzed
free radical reactions oxidize LDL in endothelial and smooth muscle
cells, exacerbating the progression of AS. Furthermore, iron excess
and foam cell formation are commonly seen as central to AS
pathology. According to Xiao et al (63), iron ions accelerate AS by
generating free radicals, which induce endothelial cell apoptosis
and oxidize LDL, promoting macrophage uptake.
MI is a leading cause of death among individuals
with CVDs. In a mouse model of MI, proteomic analysis revealed a
significant reduction in GPX4 protein levels, a change strongly
associated with increased lipid peroxidation and ROS accumulation.
These processes are central to the onset of ferroptosis, which
primarily occurs during the early and middle phases of MI. To
counteract this, the Nrf2 pathway is activated, serving as a
defense mechanism to inhibit ferroptosis (67). Cardiomyocytes from mice with mTOR
knockdown displayed elevated ROS production and increased cell
death. Conversely, cardiomyocytes with enhanced mTOR expression
showed resistance to ferroptosis when exposed to erastin or RSL3.
The upregulation of TfR1 and ferroportin in mice with mTOR
overexpression indicates that ferroptosis is mitigated by
maintaining intracellular iron balance, suggesting that targeting
this pathway could offer a novel therapeutic approach for MI
(68). Furthermore, exosomes
derived from human umbilical cord blood mesenchymal stem cells were
administered to mice following MI, effectively preventing
cardiomyocyte death from ferroptosis and reducing heart damage.
Detailed analysis showed that these exosomes targeted DMT1 and
delivered microRNA (miR)-23a-3p, which inhibited ferroptosis,
offering a promising potential treatment for MI (9).
Apoptosis, necrosis and autophagy-related cell death
have typically been considered the primary contributors to the
pathophysiology of ischemia/reperfusion (I/R) injury (69). However, previous studies suggest
that processes occurring during I/R, such as the release of ROS
leading to lipid peroxidation (70) and the accumulation of
intracellular iron (71), are
closely linked to ferroptosis. Researchers have observed elevated
levels of ACSL4, a key marker of ferroptosis, in the hearts of rats
after I/R or hypoxia-reoxygenation. Furthermore, a study reported
that ferroptosis can be inhibited by treatments such as iron
chelation with desferrioxamine or by blocking glutamine metabolism
(72). A previous study
confirmed that ferroptosis serves a significant role in cardiac I/R
injury (73), emphasizing its
importance in this context.
Several studies have focused on uncovering the exact
mechanisms by which ferroptosis contributes to I/R injury. First,
ferroptosis occurs at specific phases of the I/R process. A
previous study reported that ferroptosis is most prominent during
reperfusion, as interventions targeting ferroptosis primarily
reduce reperfusion-related damage but do not significantly affect
ischemia-related injury (74).
The distinct characteristics of the ischemia and reperfusion phases
explain this difference in cell death mechanisms. While
ischemia-induced cell death follows a certain pattern, reperfusion
of ischemic tissue triggers a significant surge in ROS production,
which is referred to as the oxidative burst. This phase of
reperfusion is more strongly linked to ferroptosis and is
characterized by lipid peroxidation, as the oxidative burst can
exacerbate I/R injury (75).
Thus, the reperfusion phase is more closely associated with
ferroptosis compared with the ischemic phase. Ferroptosis also
contributes to I/R injury due to its role in inducing ER stress
(ERS). ERS triggers apoptosis through mechanisms involving p53
upregulated modulator of apoptosis and C/EBP homologous protein
(CHOP) binding and its activation has been linked to ferroptosis
(76). The interaction between
ERS and ferroptosis can be explained in three phases. First, ERS
frequently occurs alongside ferroptosis. Ferroptosis inducers can
trigger the UPR, which activates the eukaryotic translation
initiation factor 2 α kinase 3 (PERK)/eukaryotic translation
initiation factor 2α/ATF4/CHOP pathway, leading to ERS. Upon
activation, PERK dimerizes in the cytoplasm, and the downstream
CHOP molecules, upregulated by ATF4 translation, promote apoptosis
and cellular damage (77).
Second, ferroptosis may exacerbate ERS by affecting system
Xc−, a critical transporter involved in cystine import
and glutathione synthesis, thus aggravating oxidative stress and
ERS (53). Finally, ROS,
produced during ferroptosis can initiate the ERS response (78). The generation of phospholipid
oxidation products during ROS accumulation, as observed in
intrarenal injury models, further supports this connection
(60). In rats, CHOP-mediated
ERS has been shown to contribute to I/R damage (53), indicating that
ferroptosis-induced ERS may serve as a key link between ferroptosis
and the exacerbation of I/R injury.
Cardiomyopathy is a progressive cardiac disorder
with multiple causes, leading to a heightened risk of mortality.
Recent advances have enhanced the understanding and classification
of cardiomyopathy (79,80). According to the European Society
of Cardiology, cardiomyopathy is defined as a condition in which
the heart muscle exhibits structural and functional abnormalities
that cannot be attributed to coronary artery disease, hypertension,
valvular disease or congenital heart defects (79). The cardiomyopathies discussed in
the present review were selected for their clinical significance
and the wealth of currently published data. All forms of
cardiomyopathy involve the loss of cardiomyocytes, with damaged
tissue being replaced by non-contractile fibrotic tissue (80). This loss of cardiomyocytes leads
to abnormal ventricular remodeling and, ultimately, heart
failure.
Chemotherapy-related cardiomyopathy. Anthracyclines,
including DOX, daunorubicin, epothilone and idarubicin, are widely
used in the treatment of cancers, such as breast cancer and
leukemia (81). However, their
clinical use is often limited by severe cardiotoxic side effects,
including irreversible cardiomyopathy and heart failure (82). Ferroptosis, a regulated form of
cell death, has been identified as a key mechanism of cardiomyocyte
loss in models of DOX-induced cardiotoxicity. It has been shown
that inhibiting ferroptosis significantly reduces DOX-induced
cardiomyocyte death. Sequencing analysis showed that DOX increases
free iron release and promotes cardiomyocyte heme degradation via
the NRF2/heme oxygenase 1 (HMOX1) pathway, triggering ferroptosis
(73). Mice pre-treated with
iron chelators and then exposed to DOX-induced myocardial damage
exhibit improved cardiac function compared with untreated controls
(73). Additionally, the
mitochondria-targeted antioxidant MitoTEMPO was found to
effectively reduce DOX-induced cardiomyopathy (DIC) by preventing
oxidative damage to mitochondria and inhibiting ferroptosis-induced
cardiomyocyte injury (73).
These observations suggest that mitochondria-specific
Fe2+ chelators could offer a novel approach to treating
DIC. Further studies highlight the role of mitochondrial GSH
depletion in DOX cardiomyopathy. The outer mitochondrial membrane
protein FUN14 domain containing 2 binds to and stabilizes the
mitochondrial GSH transporter protein solute carrier family 25
member 11, regulating its stability and preventing ferroptosis
(83). Melatonin, a potent
mitochondrial antioxidant, has also been shown to protect against
DOX-induced cardiotoxicity by modulating Yes-associated protein
expression and suppressing ferroptosis (84). Protein arginine methyltransferase
4 (PRMT4), accelerates ferroptosis by inhibiting the NRF2/GPX4
pathway, thereby exacerbating DIC (85). In neonatal rat ventricular
cardiomyocytes treated with DOX, researchers reported an elevated
expression of methyltransferase 14, N6-adenosine-methyltransferase
non-catalytic subunit (METTL14), a factor that induces ferroptosis
by increasing TFR expression and suppressing miR-7-5p levels.
Furthermore, the inhibition of miR-7-5p forms a positive feedback
loop that enhances METTL14 expression, thereby promoting
ferroptosis (86). These
findings provide a deeper understanding of the molecular mechanisms
underlying DOX-induced cardiotoxicity and suggest potential
therapeutic targets to mitigate its effects.
Radiation-induced cardiomyopathy. Radiation therapy
(RT) is commonly used to treat solid tumors and hematologic
malignancies (87), but it can
lead to cardiovascular complications, particularly
radiation-induced heart disease (RIHD) (88). A prevalent form of RIHD is
radiation-induced cardiomyopathy, characterized by endothelial
damage, myocardial fibrosis and long-term cardiac dysfunction
(87). Previous studies reported
that radiation can trigger various forms of cell death, including
necrosis, autophagy and apoptosis (89,90). Subsequent research has identified
vascular injury and endothelial dysfunction as key contributors to
the development of radiation-induced cardiomyopathy (91,92), primarily due to the heightened
sensitivity of endothelial cells to radiation (92).
Mechanistically, ROS are likely the main drivers of
endothelial dysfunction and cell death following radiation exposure
(93-95). Radiation is reported to enhance
ROS production by increasing the activity of NADPH oxidase 2 (NOX2)
and NOX4 (87,96). Additionally, it has been reported
that ionizing radiation leads to increased expression of ACSL4, a
key enzyme that contributes to the initiation of ferroptosis
(97). This upregulation
heightens the susceptibility of PUFAs to oxidation, thereby
promoting ferroptosis (98).
Specifically, a dose of 2 Gy of radiation therapy has been shown to
activate the cyclic GMP-AMP synthase-stimulator of interferon genes
protein pathway in HCECest2 cells, which line the coronary arteries
(99). Although patients may
remain asymptomatic for nearly a decade following radiation
treatment, radiation-induced cardiac fibrosis can still develop and
be detected later (100).
Septic cardiomyopathy (SCM). SCM, a form of
myocardial dysfunction resulting from septic shock, can lead to
severe outcomes such as heart failure or death. Recent studies
suggest that ferroptosis exacerbates heart damage in patients with
sepsis (101). Gong et
al (101) used
bioinformatics to identify cyclin dependent kinase inhibitor 1A,
NFE-2 like BZIP transcription factor 2, RELA proto-oncogene, NF-κB
subunit, prostaglandin-endoperoxide synthase 2 (Ptgs2) and Vim5 as
key genes interacting with ferroptosis. Islet cell autoantigen 69
(ICA69), a major regulator of the inflammatory response, is also
elevated in the hearts of lipopolysaccharide (LPS)-treated mice,
along with markers of ROS and ferroptosis. Knockdown of ICA69
significantly reduced the expression of ferroptosis markers,
suggesting ICA69 as a potential therapeutic target for septic
cardiomyopathy (102).
Coronavirus disease 2019 (COVID-19)-related
cardiomyopathy. The global outbreak of COVID-19, caused by severe
acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has been
associated with numerous cardiovascular complications, including
chest pain, myocarditis, palpitations, stress cardiomyopathy,
postural tachycardia, arrhythmias and MI (103-107). SARS-CoV-2 can inhibit the
production of certain selenoprotein mRNAs and reduce the activity
of GPX4 in patients with COVID-19, thereby exacerbating oxidative
stress (108). Additionally,
the viral protein ORF3a recruits Kelch-like ECH associated protein
1 (Keap1) to degrade NRF2, diminishing the cell's oxidative stress
defense and promoting ferroptosis (109). The virus can also activate
ACSL4, further inducing ferroptosis, and inhibiting ACSL4 has been
shown to reduce viral replication (110). This highlights ferroptosis as a
potential therapeutic target for managing COVID-19. Notably,
SARS-CoV-2 infection can trigger ferroptosis in sinoatrial node
cells, leading to their dysfunction. Antiviral agents like iron
chelators and tyrosine kinase inhibitors have demonstrated
protective effects by reducing viral infection and
ferroptosis-related damage (111,112).
Diabetic cardiomyopathy. Diabetic cardiomyopathy,
characterized by myocardial fibrosis, hypertrophy and diastolic
dysfunction, is primarily caused by insulin resistance, type 2
diabetes and hyperinsulinemia (113). Oxidative stress serves a
significant role in the progression of diabetic cardiomyopathy
(114). In diabetic
retinopathy, ferroptosis has been identified as the primary cause
of death in pigment epithelial cells, while autophagy provides a
protective mechanism (115).
Moreover, ferroptosis inhibitors have shown efficacy in preventing
cardiomyocyte dysfunction induced by high glucose levels,
indicating ferroptosis is a key driver in the development of
diabetic cardiomyopathy (116).
Curcumin, a natural antioxidant, has been found to mitigate heart
damage caused by ferroptosis and improve cardiac function by
promoting NRF2 nuclear translocation and increasing GPX4 and HO-1
levels (117). However, HO-1
activation, which involves the breakdown of heme to release ferrous
iron, can elevate mitochondrial iron levels and enhance ferroptosis
(118,119). Curcumin may also regulate
ferroptosis by increasing ferritin and SLC7A11 levels and
activating NRF2 to inhibit ferroptosis (120).
Hypertensive cardiomyopathy. Hypertensive heart
disease, which arises from prolonged, untreated hypertension, leads
to structural and functional heart alterations and is a major
contributor to heart failure. The apelin receptor, abundantly
present in cardiac microvascular endothelial cells (CMVECs),
interacts with its ligand, elabela. Angiotensin II (Ang II)
significantly reduces elabela levels in rats. Elabela and
ferrostatin-1 have been shown to mitigate myocardial remodeling and
ultrastructural damage in Ang II-induced hypertension by enhancing
xCT/GPX4 signaling and inhibiting IL-6/STAT3 signaling, which
reduces iron content and lipid peroxidation in the heart. These
reports suggest that elabela may slow the progression of
hypertensive cardiomyopathy by blocking Ang II-induced ventricular
remodeling and CMVEC ferroptosis (121).
Sickle cell disease (SCD)-induced cardiomyopathy.
SCD-induced cardiomyopathy is a complication of SCD characterized
by excessive hemolysis (122).
Ferroptosis serves a key role in the pathogenesis of this
cardiomyopathy. The breakdown of heme by HMOX1 releases iron into
the bloodstream, promoting ferroptosis. In an SCD mouse model,
rising heme levels were correlated with increased ferroptosis
markers and worsening cardiomyopathy. Notably, treatment with
ferroptosis inhibitors improved cardiac function and previous
research suggests that activating or inhibiting Hmox1 can
respectively enhance or reduce cardiac ferroptosis (122).
Hypertrophic cardiomyopathy. Hypertrophic
cardiomyopathy is another common cardiac complication that can
progress to heart failure. In individuals with inherited
hemochromatosis, the most frequent cause of death is heart damage
resulting from iron overload, which is associated with cardiac
hypertrophy and decreased left ventricular ejection fraction
(123). In a hemochromatosis
mouse model, researchers found that cardiac iron levels increased
with age, suggesting iron deposition and oxidative stress (124). Ferroptosis is reported to
contribute to heart enlargement in certain patients. A diet high in
iron leads to reduced GSH levels and increased lipid peroxidation,
causing significant heart damage and the development of
hypertrophic cardiomyopathy. Treatment with ferrostatin-1 reversed
these changes, suggesting the role of ferroptosis in ventricular
hypertrophy (125). In
cardiomyocytes exposed to Ang II, hypertrophy was induced by the
downregulation of xCT mRNA and protein expression levels.
Impairment of xCT function exacerbates the progression of heart
hypertrophy and dysfunction. Compounds such as ferrostatin-1 and
elabela have shown potential in reducing cardiac remodeling by
inhibiting ferroptosis (121,126). Cardiac hypertrophy is also
linked to dysfunction of the small blood vessels within the heart
(127).
Interferon regulatory factor 3 (IRF3) inhibition
promotes ferroptosis by downregulating SLC7A11 transcription.
Conversely, docosahexaenoic acid enhances IRF3 expression,
protecting endothelial cells from pressure overload. This suggests
that increasing IRF3 levels may offer new therapeutic strategies
for treating heart failure and ventricular hypertrophy (128). In both human and animal models
of hypertrophic heart disease, apelin-13 injections increased
mitochondrial iron deposition in the heart and upregulated the
expression of NCOA4 and sideroflexin 1 (SFXN1). Apelin-13 was found
to reduce cardiac hypertrophy and reverse mitochondrial iron
overload by decreasing SFXN1 and NCOA4 expression. Furthermore,
NCOA4 mediates autophagy and ferroptosis, which are crucial in the
development of cardiac hypertrophy, and functions upstream of SFXN1
(129,130). Mice supplemented with iron
exhibited increased iron accumulation in lung tissue, elevated
pulmonary artery resistance and right ventricular hypertrophy.
Losartan, an Ang II-1 receptor blocker, effectively prevented
vascular remodeling, pulmonary hypertension and right ventricular
hypertrophy caused by iron overload (131).
Dilated cardiomyopathy. Dilated cardiomyopathy (DCM)
is the most prevalent form of nonischemic cardiomyopathy, affecting
~1/2,500 individuals (132).
Heart failure is a major symptom of this disease, and for those
with severe DCM, heart transplantation often becomes the only
viable treatment option (132,133). The causes of DCM can be
genetic, with ~20-30% of cases linked to hereditary factors, while
the majority arise from other forms of cardiomyopathy (134,135). Both genetic and non-genetic
forms of DCM share pathways with hypertrophic cardiomyopathy,
particularly in terms of mechanisms leading to ferroptosis
(136).
Post-transplant cardiomyopathy. Heart
transplantation offers a treatment option for severe heart failure,
but transplant recipients face significant risks of both immediate
and long-term complications, particularly graft degradation and
failure. I/R injury is recognized as a primary contributor to graft
dysfunction (GD) in the early post-transplant period (137). Sterile inflammation, driven by
neutrophil-mediated tissue damage, is a key mechanism of GD
(138,139). Neutrophil recruitment is
triggered by the release of DAMPs from dead cells, which bind to
Toll-like receptors (TLRs) on innate immune cells (139,140). Ferroptosis is thought to serve
a pivotal role in this process, as it induces cardiomyocyte death
and promotes the release of DAMPs (141). Research by Li et al
(139) showed that the
ferroptosis inhibitor Fer-1 reduced levels of
hydroperoxy-arachidonoyl-PE and decreased both ferroptosis and
cardiomyocyte death in a mouse heart transplantation model,
suggesting that ROS are a key factor in cardiomyocyte death.
Endothelial dysfunction, a defining feature of GD, is closely
associated with cardiac allograft vasculopathy (CAV), a condition
that affects the entire cardiac vasculature (142). Ferroptosis has been linked to
endothelial dysfunction in CAV (143,144). A previous study reported that
natural killer cells contribute to the development of CAV through a
CD8+ T-cell-mediated process (145). CD8+ T-cells secrete
IFN-γ, which can reduce SLC7A11 expression, thereby promoting
ferroptosis (146).
Post-transplant cardiomyopathy is a complex condition involving
multiple pathways (147). While
it is evident that ferroptosis contributes to the immune responses
in post-transplant cardiomyopathy, further research is needed to
fully understand the various mechanisms involved and to develop
targeted interventions.
Iron overload cardiomyopathy. Iron overload
cardiomyopathy arises when excess iron accumulates in the heart
muscle, leading to systolic or diastolic dysfunction (148,149). This condition is particularly
common in patients with thalassemia and primary hemochromatosis.
Early stages of iron overload cardiomyopathy typically show
preserved systolic function; however, as the disease progresses,
systolic dysfunction worsens and ventricular dilation occurs
(149,150). Excess free iron in
cardiomyocytes catalyzes the production of ROS, causing damage to
intracellular lipids, proteins and DNA (151). Mitochondrial damage and
oxidative stress occur as the mitochondria struggle to clear the
excess iron effectively (152).
Iron overload cardiomyopathy can develop when non-transferrin-bound
iron enters cardiomyocytes through type I Ca2+ channels,
competing with calcium ions. The increasing Fe2+
concentration further disrupts Ca2+ flow, leading to
reduced contractile activity (153,154). Male and female mice exhibit
differing risks for iron overload cardiomyopathy as males were less
affected by iron overload and females benefitted more from the iron
injection treatments (155).
This finding could have considerable consequences in clinical
practice. First, it highlights the importance of considering sex as
a biological variable when assessing patient risk for iron overload
cardiomyopathy. Female patients may inherently be at lower risk for
developing this condition, even under conditions of iron overload,
which could influence diagnostic algorithms and preventive
strategies. Conversely, male patients may require more vigilant
monitoring and earlier intervention to mitigate the development of
cardiomyopathy. Secondly, the observed sex-specific differences in
response to iron injection treatments suggest that therapeutic
approaches may need to be tailored to the patient's sex. In female
patients, who appear to be protected from iron-overload
cardiomyopathy, treatments aimed at reducing iron levels might need
to be less aggressive to avoid potential side effects without
conferring significant additional benefit. By contrast, male
patients, who are more susceptible, may require more robust
iron-reducing therapies to effectively manage their risk of
developing cardiomyopathy. Finally, the identification of estrogen
as a potential protective factor against iron-overload
cardiomyopathy opens new avenues for therapeutic intervention.
Estrogen replacement therapies or estrogen-mimetic drugs could be
explored as potential treatment options for male patients or for
postmenopausal female patients who may no longer benefit from their
estrogen levels. Additionally, understanding the molecular
mechanisms underlying the protective effects of estrogen could lead
to the development of novel therapies that target these pathways,
providing new strategies to prevent and treat iron overload
cardiomyopathy in both sexes (156).
Heart failure (HF) represents the final stage of
many CVDs, often linked to disturbances in iron metabolism, whether
it be deficiency or overload. Iron homeostasis in the heart
requires ferritin H, and its absence disrupts this balance.
Conversely, overexpression of Slc7a11, which increases GSH levels,
protects against cardiac ferroptosis in ferritin H-deficient
animals, suggesting a critical role for ferritin H in preventing HF
(125,157). Additionally, research has
identified that adipose tissue macrophages release miR-140-5p in
their extracellular vesicles, which may contribute to obesity
associated with a high-fat diet. miR-140-5p targets SLC7A11,
reducing GSH production and promoting ferroptosis in cardiomyocytes
(158). Further studies by Chen
et al (130) using
bioinformatics analysis revealed a connection between ferroptosis,
autophagy, TLR4 and NOX4. TLR4 binding to NOX4 triggers the
production of superoxide anions, hydrogen peroxide and
cardiomyocyte ferroptosis. In heart failure models, knockdown of
TLR4 and NOX4 expression using small interfering RNA lentivirus
reduced cardiomyocyte death and improved ventricular remodeling by
delaying the onset of autophagy and ferroptosis. This suggests that
the TLR4-NOX4 axis is a potential therapeutic target for treating
heart failure (130). The
primary mechanisms leading to heart failure following an I/R injury
are ventricular remodeling and the eventual development of cardiac
fibrosis, both of which are closely related to ferroptosis and
autophagy. These findings underscore the importance of targeting
pathways involved in oxidative stress and ferroptosis to prevent or
treat heart failure.
The heart valves serve a critical role in protecting
the electrical conduction system from the laminar shear stress
caused by blood flow. However, hemodynamic abnormalities, such as
those caused by hypertension or extreme stress, can lead to
extracellular matrix injury and the rupture of the basement
membrane. These issues, along with reduced laminar stress and
increased mechanical strain, can contribute to valve damage. A
common consequence of valve injury is lipid infiltration at the
damaged basement membrane, leading to the formation of dispersed
lipid deposits beneath the endothelium. These lesions are often
accompanied by high levels of oxidized LDL and the presence of
activated inflammatory cells (159). Pathological calcification of
the valve is frequently associated with the accumulation of ROS
(159). Iron likely contributes
to oxidative stress, which is linked to calcification in both the
valves and blood vessels. Ferroptosis, a type of cell death driven
by iron-dependent lipid peroxidation, may be involved in
calcification-associated valve degeneration, particularly in cases
involving intraleaflet hemorrhage (ILH), abnormalities in iron
metabolism and ROS production. ILH and interstitial iron deposition
can lead to inflammation in valve tissue and promote the
development of osteogenic cells, which contribute to calcium
accumulation in the valve. In degenerative aortic stenosis, ILH is
a recognized risk factor for valve calcification (160) and a previous study has reported
that the extent of calcification correlates with iron accumulation
(161). Heme iron has been
shown to catalyze oxidative damage to DNA, proteins and lipids
through Fenton and Haber-Weiss reactions, which generate ROS
(162). Moreover, the Nrf2/HO-1
axis serves a regulatory role in ferroptosis during heart valve
calcification (163). Nrf2
regulates several oxidative processes, and under normal conditions,
it is ubiquitinated and degraded upon binding to the Keap1 protein.
However, changes in the redox state stabilize Nrf2, allowing it to
enter the nucleus and regulate gene transcription related to
oxidative stress (164).
Atrial fibrillation (AF) is a prevalent arrhythmia
that has been linked to a significant number of clinical deaths.
The incidence of AF has doubled over the past 50 years (165). Several pathogenic factors
contribute to AF, including the potential activation of
ferroptosis. For example, heavy and frequent alcohol consumption
has been linked to increased AF incidence, partly by triggering
ferroptosis. However, ferroptosis inhibitors have shown the
potential to reduce AF risk (166), indicating that ferroptosis
serves a role in AF progression. Suppression of ferroptosis has
been found to reverse AF-related changes in various models, whereas
activation of ferroptosis significantly increased susceptibility to
AF in endotoxemia models, fast atrial pacing canines and mice with
chronic iron overload (167-169). Additionally, patients with beta
thalassemia, who experience cardiac iron deposition, have a higher
incidence of AF (170). In
these individuals, iron chelators may help prevent AF. Overall,
ferroptosis inhibitors represent promising therapeutic targets for
the prevention and treatment of AF across a range of health
conditions.
Sepsis-induced myocarditis is closely linked to
ferroptosis, with LPS commonly used to model toxemia. LPS triggers
inflammatory pathways in cardiomyocytes, leading to oxidative
stress and apoptosis, which ultimately result in cardiac damage
(171). LPS elevates NCOA4
expression and increases intracellular Fe2+ levels. This
rise in cytoplasmic Fe2+ further enhances the presence
of SFXN1 on the mitochondrial membrane, facilitating
Fe2+ transfer into mitochondria, promoting ROS
production and leading to mitochondrial iron accumulation (24). Additionally, LPS stimulates Ptgs2
production and raises lipid ROS levels, processes inhibited by
Fer-1 and dexrazoxane (172).
While Fer-1 alleviates LPS-induced cardiomyocyte damage, erastin
and sorafenib exacerbate it. Excessive iron accumulation, linked to
immune dysregulation, inflammation and hypercoagulability, is also
reported to serve a significant role in the pathogenesis of
COVID-19 (173). Evidence of
myocardial lipid peroxidation in a patient with COVID-19 suggests
ferroptosis may contribute to the cardiac damage associated with
the virus (174). These
findings suggest that targeting ferroptosis could represent a
promising therapeutic approach for preventing and managing
myocarditis.
Abdominal aortic aneurysms (AAAs) are characterized
by progressive segmental aortic dilation, thinning of the aortic
wall, and eventual rupture (175). As the diameter of the artery
increases, the risk of rupture escalates (176). Age is another key factor, with
the likelihood of vasodilation, AAA formation and rupture
increasing over time and rupture leading to death in up to 90% of
cases (177). A critical
pathological feature of AAA is the loss of vascular smooth muscle
cells (VSMCs) in the medial artery wall (178). VSMCs are particularly
vulnerable to smoking-induced cell death, which is exacerbated by
lipid peroxidation and depletion of GSH. Ferroptosis, a form of
regulated cell death, has been identified as a potential
therapeutic target for preventing AAA, as inhibitors of ferroptosis
can protect VSMCs from smoking-induced damage (179). Neutrophils contribute to AAA
development through the release of neutrophil extracellular traps
(NETs), which carry bactericidal proteins. Elevated levels of NET
markers have been detected in the bloodstream of patients with AAA.
Research indicates that NETs promote AAA progression by inducing
ferroptosis in VSMCs through suppression of the PI3K/AKT pathway.
Notably, extracellular vesicles (EVs) derived from mesenchymal stem
cells (MSCs) can inhibit the release of NETs, making MSC-EVs a
promising therapeutic target for AAA treatment (180). Proprotein convertase
subtilisin/kexin type 9 (PCSK9) inhibitors, widely used to lower
cholesterol, may also influence AAA progression. Bioinformatics
analysis of human AAA tissue revealed an upregulation of PCSK9
expression, negatively correlated with GPX4, suggesting that PCSK9
could promote ferroptosis by regulating lipid metabolism (181).
Aortic dissection (AD), a severe and often fatal
vascular condition, is characterized by degeneration of the medial
aortic wall, including the death of smooth muscle cells (SMCs) and
rupture of elastic fibers (182). AD occurs when the intima and
lining of the aorta are damaged, leading to the formation of true
and false lumens. Studies have shown decreased expression of
SLC7A11, GPX4 and FSP1 in the aortic tissues of individuals with
Stanford type A AD, reinforcing the association between AD and
ferroptosis (182). Methylation
of m6A RNAs, regulated by the enzyme methyltransferase-like 3
(METTL3) (183), has been
linked to many CVDs. In human aortic smooth muscle cells, METTL3
promotes ferroptosis by suppressing SLC7A11 and FSP1 expression. In
mice with METTL3 knockdown, aortic degeneration and Alzheimer's
disease progression are slowed (182). BRD4770 (a histone
methyltransferase G9a inhibitor), when administered to a mouse
model of Alzheimer's disease, reduced AD progression by decreasing
inflammatory cell infiltration and preventing ferroptosis in SMCs
(184).
The outcomes for individuals with late-stage HF
have significantly improved with the global rise in heart
transplantation (HT). However, new challenges have emerged,
particularly I/R injury, which causes aseptic inflammation and
leads to complications. Primary graft dysfunction, resulting from
I/R injury, accounts for difficulties and mortality in up to 28% of
patients who underwent a heart transplant (185). It has been reported that using
ferroptosis inhibitors may enhance post-transplant prognosis by
mitigating I/R injury. Studies confirm that this process is
distinct from necrosis, with Fer-1 shown to improve outcomes
following HT (139). Similar
protective effects were observed in animal models of myocardial
I/R, where inhibiting ferroptosis improved recovery after coronary
artery ligation. In a multicenter clinical study involving 103,299
heart transplants from 1982 to 2011, cardiac allograft vasculopathy
(CAV) was identified as the leading cause of graft failure and
mortality, affecting between 20-65% of transplant recipients
(186). CAV primarily stems
from endothelial dysfunction due to allograft damage. Ferroptosis,
a key contributor to endothelial cell dysfunction, is linked to
damage across the cardiac vascular system, including intimal
thickening and plaque formation (142,187). Consequently, using ferroptosis
inhibitors during heart transplantation may reduce inflammation and
improve patient outcomes.
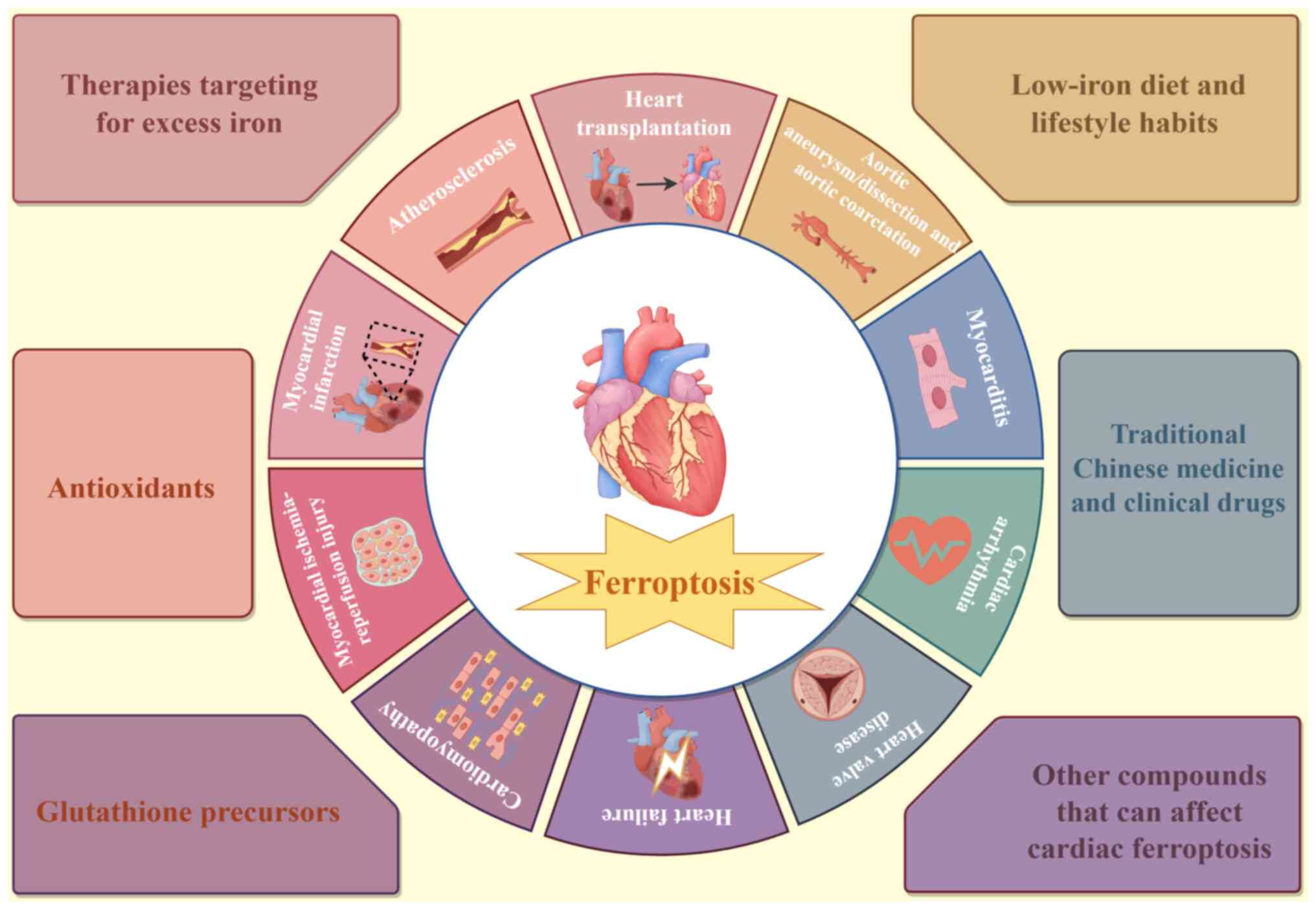
Ferroptosis, characterized by iron accumulation,
lipid peroxidation and inflammation, serves a key role in various
CVDs. Therefore, inhibiting ferroptosis represents an effective
therapeutic strategy for treating cardiac disorders associated with
this form of cell death. Various substances and treatments that
target ferroptosis were summarized (Fig. 3).
Iron homeostasis and quantity within cells are
regulated by mRNA-binding proteins IRP1 and IRP2. Patients with
conditions such as hereditary hemochromatosis often undergo
chelation therapy to manage excess iron. The Food and Drug
Administration (FDA) has approved three iron chelators for removing
iron from the heart: deferasirox, deferiprone and deferoxamine
(DFO) (188). DFO, a non-toxic
chelator, is commonly used to treat beta thalassemia and other iron
overload disorders. Studies have shown that DFO-treated mice
exhibit reduced ROS in cardiomyocytes, while patients with coronary
heart disease experience improved endothelium-dependent
vasodilation (189-191). In patients recovering from
thalassemia and undergoing bypass surgery, Paraskevaidis et
al (192) reported enhanced
heart function following DFO treatment. In another previous study
aimed at preventing I/R injury in perfused hearts, Jiang et
al (72) combined
L-glutamine, a glutaminase inhibitor, with DFO, demonstrating the
protective effects of chelators and ferroptosis inhibitors by
lowering iron levels. Additionally, the FDA-approved iron chelator
dexrazoxane protects patients with cancers from DOX-induced
cardiotoxicity, primarily by inhibiting ferroptosis (73). A novel iron chelator,
MitoFerroGreen, has shown promising cardioprotective effects in
mice treated with DOX, further highlighting the potential of iron
chelation strategies to mitigate ferroptosis-related heart damage
(193). These findings
underscore the therapeutic potential of iron chelators and
ferroptosis inhibitors in protecting against heart injury in
various clinical contexts.
Ferroptosis, a form of cell death driven by the
oxidation of phospholipids, can be mitigated by introducing
compounds that inhibit this oxidation process. Liproxstatin-1, a
member of the lipophilic radical-trapping antioxidants, effectively
halts the spread of lipid peroxyl radicals and reduces myocardial
infarction and I/R injury. This compound has demonstrated
significant efficacy in ferroptotic models (194). Another approach to chemically
counteract lipid peroxidation involves deuterium substitution at
the bis-allylic carbon of PUFAs. Deuterated PUFAs have been shown
to suppress ferroptosis effectively, while monounsaturated fatty
acids have also been identified as potent inhibitors, suggesting a
promising therapeutic strategy (195). Omega-3 fatty acids, known for
their cardioprotective properties, have been linked to a 25%
reduction in cardiovascular events, further supporting their
potential in improving cardiovascular outcomes (196).
Statins, such as fluvastatin, lovastatin and
simvastatin, are widely used to lower blood cholesterol by
inhibiting the enzyme HMG-CoA reductase, which regulates
cholesterol synthesis. In addition to lowering cholesterol, statins
also block the production of isopentyl pyrophosphate in the
mevalonate pathway, leading to reduced synthesis of selenoproteins
such as GPX4 and coenzyme Q10. This disruption promotes ferroptosis
in MSCs, indicating that statins may indirectly influence
ferroptosis-related cardiovascular conditions (197,198). Nucleotide pyrophosphatase 2, a
lipid kinase, has been shown to reduce the generation of ROS and
protect cardiomyocytes from erastin-induced ferroptosis (199). GSH, a key antioxidant, is
essential for preventing ferroptosis. A previous clinical study has
suggested that GSH supplementation, along with thiol compounds,
improves endothelial dysfunction by enhancing nitric oxide (NO)
activity (200).
Antioxidant-rich natural compounds, including vitamins, have been
found to inhibit ferroptosis and exert cardioprotective effects
(201). Specifically, vitamin E
may protect against atherosclerosis by preventing LDL oxidation,
highlighting its role in reducing ferroptosis-related damage in
CVDs (202-205).
Cystine, a key precursor for glutathione synthesis,
is imported into cells by the system xc− in its reduced
form. In vitro studies have demonstrated that supplementing
cell cultures with cystine or cysteine inhibits ferroptosis
(72). To enhance cysteine
bioavailability, N-acetyl cysteine (NAC) was developed, which has
shown positive effects on heart function (206,207). Research on ferroptosis induced
by cysteine depletion or system xc− inhibition has
demonstrated that NAC exhibits anti-ferroptotic properties
(5,208). Another study found that NAC
treatment reduced myocardial ischemia-reperfusion injury in
diabetic rats, suggesting its potential for treating human cardiac
conditions (209).
Fer-1, a potent ferroptosis inhibitor, reduces cell
damage by suppressing ROS and has shown promise in the treatment of
cardiovascular disorders (29).
Fer-1 protects against myocardium damage in conditions such as
septic cardiomyopathy and DIC (73,210). Additionally, Fer-1 has been
linked to reduced total creatine kinase release and neutrophil
recruitment following heart transplantation, further supporting its
cardioprotective role (139).
Another ferroptosis inhibitor, liproxstatin-1 (LIP-1), has
demonstrated potential cardioprotective effects by reducing the
expression of voltage-dependent anion channel 1, thereby preserving
mitochondrial integrity and mitigating myocardial infarction
severity (211). MitoTEMPO, a
mitochondria-targeted superoxide scavenger, alleviates heart
dysfunction and mitochondrial damage by reducing lipid peroxides,
further highlighting the critical role of antioxidants in managing
CVD (73). While these studies
underscore the therapeutic potential of antioxidants like NAC,
Fer-1, LIP-1 and MitoTEMPO in treating cardiovascular conditions,
further research is necessary to clarify their full therapeutic
applications and optimize their use.
TCM has a history spanning nearly 2,000 years,
emphasizes holistic balance and healing using a range of natural
substances, including herbs, animal products, medicinal minerals
and mineral extracts (212).
TCM's multifaceted approach to disease treatment, particularly
through the regulation of ferroptosis, has garnered interest in CVD
research (213,214). The natural substances in TCM
are reported to have a wide range of therapeutic targets and
minimal side effects, making them potentially valuable for the
study of future cardiovascular treatments (215).
TCM compounds can act as natural antioxidants and
regulate ferroptosis. Notable examples include artemisinin
(216), curculigoside (217), curcumin (117) and glycyrrhiza (218). These substances have been
explored for their cardioprotective effects, given their ability to
modulate ferroptosis pathways. For instance, baicalin has been
reported to enhance cell resistance to ferroptosis by inhibiting
erastin-induced GPX4 degradation and suppressing ACSL4 expression
(219). Other TCM compounds,
such as betulinic acid and ginsenoside Rd, protect against I/R
injury by reducing oxidative stress through the NRF2/HO-1 signaling
pathway (220,221). Resveratrol has also
demonstrated cardioprotective effects by upregulating GPX4 and
ferritin heavy chain (FTH) (222).
Although progress has been made, further research
is needed to understand how TCM regulates ferroptosis and other
cardiovascular conditions. For example, the
NAD+-dependent class III histone deacetylase SIRT1 has
shown potential in managing cardiovascular disorders (227,228). Fisetin, through the SIRT1/NRF2
pathway, has been shown to reduce DIC (229). Additionally, 6-Gingerol, a
polyphenol from ginger extract, possesses antioxidant,
anti-inflammatory and anti-apoptotic properties, and protects
against DCM by blocking ferroptosis and reducing ROS via the
NRF2/HO-1 pathway (230).
Puerarin, commonly used in the treatment of CVDs, exhibits notable
antioxidant properties (231).
Additionally, it inhibits ferroptosis and decelerates the
progression of heart failure by elevating the levels of FTH1 and
GPX4, while simultaneously lowering LIP and ROS levels (232).
The average human body contains 3-5 g of iron under
normal physiological conditions. Deviations from this range,
whether due to iron deficiency or excess, can lead to adverse
health outcomes (233).
Notably, a review of cohort studies has indicated that the risk of
death from CVDs rises with increased haem iron consumption,
suggesting that reducing haem iron intake could help lower the risk
of premature mortality from CVDs (234,235). Consequently, following a
low-iron diet may be an effective preventive strategy against
ferroptosis-related cardiovascular conditions. There is a link
between frequent heavy alcohol consumption and an elevated risk of
AF, compared with infrequent heavy drinking. Given that ferroptosis
serves a role in alcohol-induced AF, inhibiting ferroptosis could
potentially reduce AF vulnerability in these cases (166). Additionally, research has shown
that Beclin1 haploinsufficiency improves contractile dysfunction
and cardiac remodeling induced by acute ethanol exposure through a
ferroptosis-mediated mechanism (236). In rat VSMCs, cigarette smoke
extract (CSE) has been found to trigger key features of
ferroptosis, such as lipid peroxidation, depletion of intracellular
GSH and increased PTGS2 mRNA expression. A previous study reported
that the use of iron chelators and specific ferroptosis inhibitors
can completely prevent CSE-induced cell death in these cells
(179). Moreover, cigarette tar
accelerates atherosclerosis by promoting macrophage ferroptosis
through the NF-κB-activated hepcidin/FPN/SLC7A11 pathway (237). Natural ferroptosis inhibitors,
such as vitamin E, cyanidin-3-glucoside and baicalin, are found in
a variety of fruits and vegetables (219,238). Therefore, lifestyle changes,
including quitting smoking and excessive alcohol consumption, along
with increased intake of fruits and vegetables, may help mitigate
the risk of CVDs linked to ferroptosis.
Cardiac ferroptosis can be inhibited by several
compounds that target mechanisms beyond those typically discussed.
In mice, zinc protoporphyrin IX, a competitive inhibitor of HO1,
reduces ferroptosis and DOX-induced iron accumulation in the heart
by preventing haem breakdown and the release of free iron (73). Additionally, compound 968, an
inhibitor of glutaminolysis, has been shown to reduce cardiac I/R
injury in vitro by limiting glutamine availability, which is
essential for cysteine deprivation-induced ferroptosis (72). Another protective agent, P22077,
inhibits ubiquitin-specific protease 7, activates p53 and
subsequently lowers TFR1 levels, thereby suppressing ferroptosis
and protecting the heart from I/R damage (239). Dexmedetomidine, an
α2-adrenergic receptor agonist used in clinical sedation, has been
found to reduce sepsis-induced cardiac damage by upregulating the
ferroptosis suppressor GPX4 (240). Puerarin, an isoflavone from
kudzu root, has also been shown to mitigate pressure-induced heart
failure in rats and protect cardiomyocytes from ferroptosis induced
by erastin and isoprenaline (232). Moreover, atorvastatin has
demonstrated potential in preventing hypertrophic cardiomyopathy by
improving isoprenaline-induced ventricular dysfunction and
remodeling through inhibition of ferritinophagy-mediated
ferroptosis (241).
Additionally, some commonly prescribed heart medications may
possess previously unrecognized anti-ferroptotic effects.
Carvedilol, widely used for treating hypertension and heart
failure, has been found to inhibit ferroptosis (242,243), likely due to its capacity to
scavenge lipid peroxides and bind iron (244).
CVD represents a significant threat to human health
and longevity. Maintaining cardiac function and preventing heart
failure depends largely on reducing cardiomyocyte death. Over the
past decade, substantial experimental evidence has emerged
indicating that ferroptosis, a regulated form of cell death driven
by iron, serves a critical role in the progression of CVDs.
Characterized by lipid oxidation, ferroptosis has been associated
with various cardiovascular conditions and is increasingly viewed
as a potential therapeutic target. Unlike other forms of cell
death, ferroptosis is marked by the accumulation of intracellular
iron, lipid peroxides and mitochondrial changes. However, the
precise mechanisms underlying many CVDs remain only partially
understood, and current knowledge of ferroptosis is insufficient to
develop comprehensive treatment strategies. Furthermore, multiple
pathways of regulated cell death, including ferroptosis, are often
interconnected rather than isolated. Greater clarity is required
regarding the relationship between ferroptosis and other cell death
mechanisms that have recently gained attention. Investigating these
interactions could provide deeper insights into the progression of
biological processes and lead to more effective therapeutic
interventions. Empirical evidence has highlighted the potential of
inhibiting ferroptosis as a novel therapeutic approach for a range
of CVDs. Iron chelation therapy, in particular, shows promise for
patients with iron overload-related cardiomyopathy. Encouraging
preclinical results suggest that ferroptosis-specific antagonists
could be developed for various clinical applications (72,73,193,240). Despite this potential,
significant challenges remain in addressing both CVDs and
ferroptosis concurrently. Research must prioritize uncovering the
precise pathways linking ferroptosis to CVDs to identify new
treatment targets. Moreover, the current body of clinical research
involving human participants is limited, with most studies on
ferroptosis and CVDs focusing on cellular or animal models.
Additional clinical trials are essential to confirm the efficacy of
ferroptosis inhibitors in treating CVDs. Furthermore, it is
essential to explore the potential adverse effects of these
inhibitors on other organs, as their long-term impact remains
unclear. The relationship between ferroptosis and inflammation also
requires further investigation. While ferroptosis can exacerbate
inflammation and ROS production, these processes are not always
harmful; in certain contexts, they may be benign or even
beneficial. It is crucial to assess whether ferroptosis activators
could be used to modulate inflammation or ROS levels in specific
cases. Given the widespread use of anti-inflammatory drugs in CVD
treatment, large-scale, multicenter clinical trials could establish
a stronger link between ferroptosis modulation and inflammation
management in cardiovascular therapy.
Not applicable.
SQ and CZ reviewed the literature and wrote the
manuscript. CC and ZS collected and analyzed the existing data from
the previously published research articles. YC reviewed and
critically revised the manuscript. Data authentication is not
applicable. All authors read and approved the final version of the
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
No funding was received.
|
1
|
Virani SS, Alonso A, Benjamin EJ,
Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR,
Cheng S, Delling FN, et al: Heart Disease and Stroke
Statistics-2020 Update: A Report From the American Heart
Association. Circulation. 141:e139–e596. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roth GA, Mensah GA, Johnson CO, Addolorato
G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ, Benjamin EJ,
Benziger CP, et al: Global Burden of Cardiovascular Diseases and
Risk Factors, 1990-2019: Update From the GBD 2019 Study. J Am Coll
Cardiol. 76:2982–3021. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental Mechanisms of Regulated Cell Death and
Implications for Heart Disease. Physiol Rev. 99:1765–1817. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reis AH: From Assessing Risk Factors to
Understanding, Preventing, and Treating Cardiovascular Diseases: An
urgent journey. Discov Med. 34:199–204. 2022.
|
|
5
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell
Death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao M, Yi J, Zhu J, Minikes AM, Monian P,
Thompson CB and Jiang X: Role of Mitochondria in Ferroptosis. Mol
Cell. 73:354–363.e3. 2019. View Article : Google Scholar :
|
|
7
|
Powell LW, Seckington RC and Deugnier Y:
Haemochromatosis. Lancet. 388:706–716. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park E and Chung SW: ROS-mediated
autophagy increases intracellular iron levels and ferroptosis by
ferritin and transferrin receptor regulation. Cell Death Dis.
10:8222019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Song Y, Wang B, Zhu X, Hu J, Sun J, Xuan J
and Ge Z: Human umbilical cord blood-derived MSCs exosome attenuate
myocardial injury by inhibiting ferroptosis in acute myocardial
infarction mice. Cell Biol Toxicol. 37:51–64. 2021. View Article : Google Scholar
|
|
10
|
Li W, Li W, Leng Y, Xiong Y and Xia Z:
Ferroptosis is involved in diabetes myocardial ischemia/reperfusion
injury through endoplasmic reticulum stress. DNA Cell Biol.
39:210–225. 2020. View Article : Google Scholar
|
|
11
|
Gaschler MM and Stockwell BR: Lipid
peroxidation in cell death. Biochem Biophys Res Commun.
482:419–425. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A Regulated Cell Death Nexus Linking
Metabolism, Redox Biology, and Disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stoyanovsky DA, Tyurina YY, Shrivastava I,
Bahar I, Tyurin VA, Protchenko O, Jadhav S, Bolevich SB, Kozlov AV,
Vladimirov YA, et al: Iron catalysis of lipid peroxidation in
ferroptosis: Regulated enzymatic or random free radical reaction?
Free Radic Biol Med. 133:153–161. 2019. View Article : Google Scholar :
|
|
14
|
Lin Z, Liu J, Kang R, Yang M and Tang D:
Lipid Metabolism in Ferroptosis. Adv Biol (Weinh). 5:e21003962021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Conrad M and Pratt DA: The chemical basis
of ferroptosis. Nat Chem Biol. 15:1137–1147. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Naoe S, Tsugawa H, Takahashi M, Ikeda K
and Arita M: Characterization of lipid profiles after dietary
intake of polyunsaturated fatty acids using integrated untargeted
and targeted lipidomics. Metabolites. 9:2412019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar
|
|
18
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar :
|
|
19
|
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian
D, Liu D, Zhang F, Ning S, Yao J and Tian X: Ischemia-induced ACSL4
activation contributes to ferroptosis-mediated tissue injury in
intestinal ischemia/reperfusion. Cell Death Differ. 26:2284–2299.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Orafaie A, Mousavian M, Orafai H and
Sadeghian H: An overview of lipoxygenase inhibitors with approach
of in vivo studies. Prostaglandins Other Lipid Mediat.
148:1064112020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Smith LM, Aitken HM and Coote ML: The Fate
of the Peroxyl Radical in Autoxidation: How does polymer
degradation really occur? Acc Chem Res. 51:2006–2013. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ilari S, Giancotti LA, Lauro F, Gliozzi M,
Malafoglia V, Palma E, Tafani M, Russo MA, Tomino C, Fini M, et al:
Natural antioxidant control of neuropathic pain-exploring the role
of mitochondrial SIRT3 pathway. Antioxidants (Basel). 9:11032020.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fan X, Li A, Yan Z, Geng X, Lian L, Lv H,
Gao D and Zhang J: From iron metabolism to ferroptosis: Pathologic
changes in coronary heart disease. Oxid Med Cell Longev.
2022:62918892022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu H, Chen Y, Jing L, Zhai C and Shen L:
The Link Between Ferroptosis and Cardiovascular Diseases: A novel
target for treatment. Front Cardiovasc Med. 8:7109632021.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen Z, Yan Y, Qi C, Liu J, Li L and Wang
J: The Role of Ferroptosis in Cardiovascular Disease and Its
Therapeutic Significance. Front Cardiovasc Med. 8:7332292021.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Xin L, Xiang M, Shang C, Wang Y,
Wang Y, Cui X and Lu Y: The molecular mechanisms of ferroptosis and
its role in cardiovascular disease. Biomed Pharmacother.
145:1124232022. View Article : Google Scholar
|
|
27
|
Yu Y, Jiang L, Wang H, Shen Z, Cheng Q,
Zhang P, Wang J, Wu Q, Fang X, Duan L, et al: Hepatic transferrin
plays a role in systemic iron homeostasis and liver ferroptosis.
Blood. 136:726–739. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mancias JD, Wang X, Gygi SP, Harper JW and
Kimmelman AC: Quantitative proteomics identifies NCOA4 as the cargo
receptor mediating ferritinophagy. Nature. 509:105–109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fang X, Ardehali H, Min J and Wang F: The
molecular and metabolic landscape of iron and ferroptosis in
cardiovascular disease. Nat Rev Cardiol. 20:7–23. 2023. View Article : Google Scholar
|
|
30
|
Yuan H, Li X, Zhang X, Kang R and Tang D:
CISD1 inhibits ferroptosis by protection against mitochondrial
lipid peroxidation. Biochem Biophys Res Commun. 478:838–844. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim EH, Shin D, Lee J, Jung AR and Roh JL:
CISD2 inhibition overcomes resistance to sulfasalazine-induced
ferroptotic cell death in head and neck cancer. Cancer Lett.
432:180–190. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sies H: Glutathione and its role in
cellular functions. Free Radic Biol Med. 27:916–921. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu G, Fang YZ, Yang S, Lupton JR and
Turner ND: Glutathione Metabolism and Its Implications for Health.
J Nutr. 134:489–492. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pompella A, Visvikis A, Paolicchi A, Tata
VD and Casini AF: The changing faces of glutathione, a cellular
protagonist. Biochem Pharmacol. 66:1499–1503. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Paul BD, Sbodio JI and Snyder SH: Cysteine
Metabolism in Neuronal Redox Homeostasis. Trends Pharmacol Sci.
39:513–524. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic
Biol Med. 152:175–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Desideri E, Ciccarone F and Ciriolo MR:
Targeting glutathione metabolism: Partner in crime in anticancer
therapy. Nutrients. 11:19262019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Y, Zhao Y, Wang H, Zhang C, Wang M,
Yang Y, Xu X and Hu Z: Histone demethylase KDM3B protects against
ferroptosis by upregulating SLC7A11. FEBS Open Bio. 10:637–643.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu J, Zhang C, Hu W and Feng Z: Tumor
suppressor p53 and metabolism. J Mol Cell Biol. 11:284–292. 2019.
View Article : Google Scholar :
|
|
40
|
Kastenhuber ER and Lowe SW: Putting p53 in
Context. Cell. 170:1062–1078. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zille M, Karuppagounder SS, Chen Y, Gough
PJ, Bertin J, Finger J, Milner TA, Jonas EA and Ratan RR: Neuronal
death after hemorrhagic stroke in vitro and in vivo shares features
of ferroptosis and necroptosis. Stroke. 48:1033–1043. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016. View Article : Google Scholar
|
|
43
|
Dodson M, Castro-Portuguez R and Zhang DD:
NRF2 plays a critical role in mitigating lipid peroxidation and
ferroptosis. Redox Biol. 23:1011072019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kang R, Kroemer G and Tang D: The tumor
suppressor protein p53 and the ferroptosis network. Free Radic Biol
Med. 133:162–168. 2019. View Article : Google Scholar
|
|
45
|
Lipper CH, Stofleth JT, Bai F, Sohn YS,
Roy S, Mittler R, Nechushtai R, Onuchic JN and Jennings PA:
Redox-dependent gating of VDAC by mitoNEET. Proc Natl Acad Sci USA.
116:19924–19929. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rostovtseva TK, Tan W and Colombini M: On
the Role of VDAC in Apoptosis: Fact and Fiction. J Bioenerg
Biomembr. 37:129–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fang D and Maldonado EN: VDAC regulation:
A mitochondrial target to stop cell proliferation. Adv Cancer Res.
138:41–69. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mazure NM: VDAC in cancer. Biochim Biophys
Acta Bioenerg. 1858:665–673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lemasters JJ: Evolution of
Voltage-Dependent Anion Channel Function: From molecular sieve to
governator to actuator of ferroptosis. Front Oncol. 7:3032017.
View Article : Google Scholar
|
|
50
|
Yagoda N, von Rechenberg M, Zaganjor E,
Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM,
Boniface JJ, et al: RAS-RAF-MEK-dependent oxidative cell death
involving voltage-dependent anion channels. Nature. 447:865–868.
2007. View Article : Google Scholar
|
|
51
|
DeHart DN, Fang D, Heslop K, Li L,
Lemasters JJ and Maldonado EN: Opening of voltage dependent anion
channels promotes reactive oxygen species generation, mitochondrial
dysfunction and cell death in cancer cells. Biochem Pharmacol.
148:155–162. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Iurlaro R and Muñoz-Pinedo C: Cell death
induced by endoplasmic reticulum stress. FEBS J. 283:2640–2652.
2016. View Article : Google Scholar
|
|
53
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e025232014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang
L, Wang D, Xing J, Hou B, Li H, et al: Dihydroartemisinin-induced
unfolded protein response feedback attenuates ferroptosis via
PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res.
38:4022019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Luo Y, Guo Q, Zhang L, Zhuan Q, Meng L, Fu
X and Hou Y: Dihydroartemisinin exposure impairs porcine ovarian
granulosa cells by activating PERK-eIF2α-ATF4 through endoplasmic
reticulum stress. Toxicol Appl Pharmacol. 403:1151592020.
View Article : Google Scholar
|
|
56
|
Rozpedek W, Pytel D, Mucha B, Leszczynska
H, Diehl JA and Majsterek I: The Role of the PERK/eIF2α/ATF4/CHOP
Signaling Pathway in Tumor Progression During Endoplasmic Reticulum
Stress. Curr Mol Med. 16:533–544. 2016. View Article : Google Scholar
|
|
57
|
Yan L, Luo H, Li X and Li Y: d-Pinitol
protects against endoplasmic reticulum stress and apoptosis in
hepatic ischemia-reperfusion injury via modulation of
AFT4-CHOP/GRP78 and caspase-3 signaling pathways. Int J
Immunopathol Pharmacol. 35:1–15. 2021. View Article : Google Scholar
|
|
58
|
Chen Y, Mi Y and Zhang X: The CoQ
oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. J
Cell Physiol. 575:688–692. 2019.
|
|
59
|
Doll S, Freitas FP, Shah R, Aldrovandi M,
da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius
E, Scheel CH, et al: FSP1 is a glutathione-independent ferroptosis
suppressor. Nature. 575:693–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shimada K, Skouta R, Kaplan A, Yang WS,
Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ and
Stockwell BR: Global survey of cell death mechanisms reveals
metabolic regulation of ferroptosis. Nat Chem Biol. 12:497–503.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Miriyala S, Thippakorn C, Chaiswing L, Xu
Y, Noel T, Tovmasyan A, Batinic-Haberle I, Vander Kooi CW, Chi W,
Latif AA, et al: Novel role of 4-hydroxy-2-nonenal in
AIFm2-mediated mitochondrial stress signaling. Free Radic Biol Med.
91:68–80. 2016. View Article : Google Scholar :
|
|
62
|
Martinet W, Coornaert I, Puylaert P and De
Meyer GRY: Macrophage death as a pharmacological target in
atherosclerosis. Front Pharmacol. 10:3062019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Xiao L, Luo G, Guo X, Jiang C, Zeng H,
Zhou F, Li Y, Yu J and Yao P: Macrophage iron retention aggravates
atherosclerosis: Evidence for the role of autocrine formation of
hepcidin in plaque macrophages. Biochim Biophys Acta Mol Cell Biol
Lipids. 1865:1585312020. View Article : Google Scholar
|
|
64
|
Kiechl S, Willeit J, Egger G, Poewe W and
Oberhollenzer F: Body Iron Stores and the Risk of Carotid
Atherosclerosis: Prospective results from the Bruneck study.
Circulation. 96:3300–3307. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hou JW: Iron-load exacerbates the severity
of atherosclerosis via inducing inflammation and enhancing the
glycolysis in macrophages. Nutrition. 38:329–334. 2002.
|
|
66
|
Luo EF, Li HX, Qin YH, Qiao Y, Yan GL, Yao
YY, Li LQ, Hou JT, Tang CC and Wang D: Role of ferroptosis in the
process of diabetes-induced endothelial dysfunction. World J
Diabetes. 12:124–137. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Park TJ, Park JH, Lee GS, Lee JY, Shin JH,
Kim MW, Kim YS, Kim JY, Oh KJ, Han BS, et al: Quantitative
proteomic analyses reveal that GPX4 downregulation during
myocardial infarction contributes to ferroptosis in cardiomyocytes.
Cell Death Dis. 10:8352019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Baba Y, Higa JK, Shimada BK, Horiuchi KM,
Suhara T, Kobayashi M, Woo JD, Aoyagi H, Marh KS, Kitaoka H, et al:
Protective effects of the mechanistic target of rapamycin against
excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart
Circ Physiol. 314:H659–H668. 2018. View Article : Google Scholar :
|
|
69
|
Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv
F, Liu Y, Zheng W, Shang H, Zhang J, et al: CaMKII is a RIP3
substrate mediating ischemia- and oxidative stress-induced
myocardial necroptosis. Nat Med. 22:175–182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Farmer EE and Mueller MJ: ROS-Mediated
Lipid Peroxidation and RES-Activated Signaling. Annu Rev Plant
Biol. 64:429–450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Scindia Y PhD, Leeds J MD and Swaminathan
S MD: Iron homeostasis in healthy kidney and its role in acute
kidney injury. Semin Nephrol. 39:76–84. 2019. View Article : Google Scholar
|
|
72
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tang LJ, Luo XJ, Tu H, Chen H, Xiong XM,
Li NS and Peng J: Ferroptosis occurs in phase of reperfusion but
not ischemia in rat heart following ischemia or
ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol.
394:401–410. 2021. View Article : Google Scholar
|
|
75
|
Hess ML and Manson NH: Molecular oxygen:
Friend and foe*The role of the oxygen free radical system in the
calcium paradox, the oxygen paradox and ischemia/reperfusion
injury. J Mol Cell Cardiol. 37:969–985. 1984. View Article : Google Scholar
|
|
76
|
Ghosh AP, Klocke BJ, Ballestas ME and Roth
KA: CHOP Potentially Co-Operates with FOXO3a in Neuronal Cells to
Regulate PUMA and BIM Expression in Response to ER Stress. PLoS
One. 7:e395862012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lee YS, Lee DH, Choudry HA, Bartlett DL
and Lee YJ: Ferroptosis-Induced Endoplasmic Reticulum Stress:
Cross-talk between Ferroptosis and Apoptosis. Mol Cancer Res.
16:1073–1076. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sampilvanjil A, Karasawa T and Yamada N:
Endoplasmic Reticulum Stress and oxidative stress in cell fate
decision and human disease. Proc Natl Acad Sci USA. 160:303–318.
2020.
|
|
79
|
Elliott P, Andersson B, Arbustini E,
Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B,
McKenna WJ, et al: Classification of the cardiomyopathies: A
position statement from the European Society Of Cardiology Working
Group On Myocardial And Pericardial Diseases. Eur Heart J.
29:270–276. 2008. View Article : Google Scholar
|
|
80
|
Hashimoto H, Olson EN and Bassel-Duby R:
Therapeutic approaches for cardiac regeneration and repair. Nat Rev
Cardiol. 15:585–600. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Young RC, Ozols RF and Myers CE: The
anthracycline antineoplastic drugs. N Engl J Med. 305:139–153.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Singal PK and Iliskovic N:
Doxorubicin-induced cardiomyopathy. N Engl J Med. 339:900–905.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Tadokoro T, Ikeda M and Ide T:
Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis
and contributes to doxorubicin-induced cardiomyopathy. Am J Physiol
Heart Circ Physiol. 119:e21173961192022.
|
|
84
|
Sun X, Sun P, Zhen D, Xu X, Yang L, Fu D,
Wei C, Niu X, Tian J and Li H: Melatonin alleviates
doxorubicin-induced mitochondrial oxidative damage and ferroptosis
in cardiomyocytes by regulating YAP expression. Toxicol Appl
Pharmacol. 437:1159022022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wang Y, Yan S, Liu X, Deng F, Wang P, Yang
L, Hu L, Huang K and He J: PRMT4 promotes ferroptosis to aggravate
doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4
pathway. Cell Death Differ. 29:1982–1995. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhuang S, Ma Y, Zeng Y, Lu C, Yang F,
Jiang N, Ge J, Ju H, Zhong C, Wang J, et al: METTL14 promotes
doxorubicin-induced cardiomyocyte ferroptosis by regulating the
KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biol Toxicol. 39:1015–1035. 2023.
View Article : Google Scholar
|
|
87
|
Wang B, Wang H, Zhang M, Ji R, Wei J, Xin
Y and Jiang X: Radiation-induced myocardial fibrosis: Mechanisms
underlying its pathogenesis and therapeutic strategies. J Cell Mol
Med. 24:7717–7729. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wang H, Wei J, Zheng Q, Meng L, Xin Y, Yin
X and Jiang X: Radiation-induced heart disease: A review of
classification, mechanism and prevention. Int J Biol Sci.
15:2128–2138. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kuwahara Y, Oikawa T, Ochiai Y, Roudkenar
MH and Fukumoto M, Shimura T, Ohtake Y, Ohkubo Y, Mori S, Uchiyama
Y and Fukumoto M: Enhancement of autophagy is a potential modality
for tumors refractory to radiotherapy. Cell Death Dis. 2:e1772011.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Jelonek K, Walaszczyk A, Gabryś D,
Pietrowska M, Kanthou C and Widłak P: Cardiac endothelial cells
isolated from mouse heart-a novel model for radiobiology. Acta
Biochim Pol. 58:397–404. 2011. View Article : Google Scholar
|
|
91
|
Taunk NK, Haffty BG, Kostis JB and Goyal
S: Radiation-Induced Heart Disease: Pathologic Abnormalities and
Putative Mechanisms. Front Oncol. 5:392015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Klein D, Steens J, Wiesemann A, Schulz F,
Kaschani F, Röck K, Yamaguchi M, Wirsdörfer F, Kaiser M, Fischer
JW, et al: Mesenchymal stem cell therapy protects lungs from
radiation-induced endothelial cell loss by restoring superoxide
dismutase 1 expression. Antioxid Redox Signal. 26:563–582. 2017.
View Article : Google Scholar :
|
|
93
|
Liao JK: Linking endothelial dysfunction
with endothelial cell activation. Front Oncol. 123:540–541.
2013.
|
|
94
|
Van Der Meeren A, Squiban C, Gourmelon P,
Lafont H and Gaugler MH: Differential regulation by IL-4 and IL-10
of radiation-induced IL-6 AND IL-8 production and ICAM-1 expression
by human endothelial cells. Cytokine. 11:831–838. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Baselet B, Belmans N, Coninx E, Lowe D,
Janssen A, Michaux A, Tabury K, Raj K, Quintens R, Benotmane MA, et
al: Functional gene analysis reveals cell cycle changes and
inflammation in endothelial cells irradiated with a single X-ray
dose. Front Pharmacol. 8:2132017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Mortezaee K, Goradel NH, Amini P, Shabeeb
D, Musa AE, Najafi M and Farhood B: NADPH oxidase as a target for
modulation of radiation response; Implications to carcinogenesis
and radiotherapy. Curr Mol Pharmacol. 12:50–60. 2019. View Article : Google Scholar
|
|
97
|
Lei G, Zhang Y, Koppula P, Liu X, Zhang J,
Lin SH, Ajani JA, Xiao Q, Liao Z, Wang H and Gan B: The role of
ferroptosis in ionizing radiation-induced cell death and tumor
suppression. Cell Res. 30:146–162. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Dixon SJ, Winter GE, Musavi LS, Lee ED,
Snijder B, Rebsamen M, Superti-Furga G and Stockwell BR: Human
haploid cell genetics reveals roles for lipid metabolism genes in
nonapoptotic cell death. ACS Chem Biol. 10:1604–1609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Philipp J, Le Gleut R, Toerne CV, Subedi
P, Azimzadeh O, Atkinson MJ and Tapio S: Radiation response of
human cardiac endothelial cells reveals a central role of the
cGAS-STING pathway in the development of inflammation. Proteomes.
8:302020. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Heidenreich PA, Hancock SL, Lee BK,
Mariscal CS and Schnittger I: Asymptomatic cardiac disease
following mediastinal irradiation. J Am Coll Cardiol. 42:743–749.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Gong CW, Yuan MM, Qiu BQ, Wang LJ, Zou HX,
Hu T, Lai SQ and Liu JC: Identification and Validation of
Ferroptosis-Related Biomarkers in Septic Cardiomyopathy via
Bioinformatics Analysis. Front Genet. 13:8275592022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Kong C, Ni X, Wang Y, Zhang A, Zhang Y,
Lin F, Li S, Lv Y, Zhu J, Yao X, et al: ICA69 aggravates
ferroptosis causing septic cardiac dysfunction via STING
trafficking. Cell Death Discov. 8:1872022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Raman B, Bluemke DA, Lüscher TF and
Neubauer S: Long COVID: Post-acute sequelae of COVID-19 with a
cardiovascular focus. Eur Heart J. 43:1157–1172. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Xie Y, Xu E, Bowe B and Al-Aly Z:
Long-term cardiovascular outcomes of COVID-19. Nat Med. 28:583–590.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Katsoularis I, Fonseca-Rodríguez O,
Farrington P, Lindmark K and Fors Connolly AM: Risk of acute
myocardial infarction and ischaemic stroke following COVID-19 in
Sweden: A self-controlled case series and matched cohort study.
Lancet. 398:599–607. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Xie Y, Bowe B, Maddukuri G and Al-Aly Z:
Comparative evaluation of clinical manifestations and risk of death
in patients admitted to hospital with covid-19 and seasonal
influenza: Cohort study. BMJ. 371:m46772020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Merad M, Blish CA, Sallusto F and Iwasaki
A: The immunology and immunopathology of COVID-19. Science.
375:1122–1127. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Wang Y, Huang J, Sun Y, Stubbs D, He J, Li
W, Wang F, Liu Z, Ruzicka JA, Taylor EW, et al: SARS-CoV-2
suppresses mRNA expression of selenoproteins associated with
ferroptosis, endoplasmic reticulum stress and DNA synthesis. Food
Chem Toxicol. 153:1122862021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Liu L, Du J, Yang S, Zheng B, Shen J,
Huang J, Cao L, Huang S, Liu X, Guo L, et al: SARS-CoV-2 ORF3a
sensitizes cells to ferroptosis via Keap1-NRF2 axis. Redox Biol.
63:1027522023. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Bai T, Li M and Liu Y: Acyl-coenzyme a
synthetase long-chain family member 4 is involved in viral
replication organelle formation and facilitates virus replication
via ferroptosis. Free Radic Biol Med. 63:194–209. 2020.
|
|
111
|
Han Y, Zhu J, Yang L, Nilsson-Payant BE,
Hurtado R, Lacko LA, Sun X, Gade AR, Higgins CA, Sisso WJ, et al:
SARS-CoV-2 infection induces ferroptosis of sinoatrial node
pacemaker cells. Circ Res. 130:963–977. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Habib HM, Ibrahim S, Zaim A and Ibrahim
WH: The role of iron in the pathogenesis of COVID-19 and possible
treatment with lactoferrin and other iron chelators. Biomed
Pharmacother. 136:1112282021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Jia G, DeMarco VG and Sowers JR: Insulin
resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat
Rev Endocrinol. 12:144–153. 2016. View Article : Google Scholar :
|
|
114
|
Ritchie RH and Abel ED: Basic mechanisms
of diabetic heart disease. Circ Res. 126:1501–1525. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Liu C, Sun W, Zhu T, Shi S, Zhang J, Wang
J, Gao F, Ou Q, Jin C, Li J, et al: Glia maturation factor-β
induces ferroptosis by impairing chaperone-mediated autophagic
degradation of ACSL4 in early diabetic retinopathy. Redox Biol.
52:1022922022. View Article : Google Scholar
|
|
116
|
Chen L, Yin Z, Qin X, Zhu X, Chen X, Ding
G, Sun D, Wu NN, Fei J, Bi Y, et al: CD74 ablation rescues type 2
diabetes mellitus-induced cardiac remodeling and contractile
dysfunction through pyroptosis-evoked regulation of ferroptosis.
Pharmacol Res. 176:1060862022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wei Z, Shaohuan Q, Pinfang K and Chao S:
Curcumin attenuates ferroptosis-induced myocardial injury in
diabetic cardiomyopathy through the Nrf2 pathway. Cardiovasc Ther.
2022:31597172022. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Kwon MY, Park E, Lee SJ and Chung SW: Heme
oxygenase-1 accelerates erastin-induced ferroptotic cell death.
Oncotarget. 6:24393–24403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chang LC, Chiang SK, Chen SE, Yu YL, Chou
RH and Chang WC: Heme oxygenase-1 mediates BAY 11-7085 induced
ferroptosis. Cancer Lett. 416:124–137. 2018. View Article : Google Scholar
|
|
120
|
Wang X, Chen X, Zhou W, Men H, Bao T, Sun
Y, Wang Q, Tan Y, Keller BB, Tong Q, et al: Ferroptosis is
essential for diabetic cardiomyopathy and is prevented by
sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 12:708–722.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhang Z, Tang J, Song J, Xie M, Liu Y,
Dong Z, Liu X, Li X, Zhang M, Chen Y, et al: Elabela alleviates
ferroptosis, myocardial remodeling, fibrosis and heart dysfunction
in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling.
Free Radic Biol Med. 181:130–142. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Menon AV, Liu J, Tsai HP, Zeng L, Yang S,
Asnani A and Kim J: Excess heme upregulates heme oxygenase 1 and
promotes cardiac ferroptosis in mice with sickle cell disease.
Blood. 139:936–941. 2022. View Article : Google Scholar :
|
|
123
|
Rozwadowska K, Raczak G, Sikorska K,
Fijałkowski M, Kozłowski D and Daniłowicz-Szymanowicz L: Influence
of hereditary haemochromatosis on left ventricular wall thickness:
Does iron overload exacerbate cardiac hypertrophy? Folia Morphol
(Warsz). 78:746–753. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Sukumaran A, Chang J, Han M, Mintri S,
Khaw BA and Kim J: Iron overload exacerbates age-associated cardiac
hypertrophy in a mouse model of hemochromatosis. Sci Rep.
7:57562017. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Fang X, Cai Z, Wang H, Han D, Cheng Q,
Zhang P, Gao F, Yu Y, Song Z, Wu Q, et al: Loss of Cardiac Ferritin
H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ
Res. 127:486–501. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Zhang X, Zheng C, Gao Z, Chen H, Li K,
Wang L, Zheng Y, Li C, Zhang H, Gong M, et al: SLC7A11/xCT prevents
cardiac hypertrophy by inhibiting ferroptosis. Cardiovasc Drugs
Ther. 36:437–447. 2022. View Article : Google Scholar
|
|
127
|
Papayannopoulos V: Methylation-dependent
transcriptional repression of RUNX3 by KCNQ1OT1 regulates mouse
cardiac microvascular endothelial cell viability and inflammatory
response following myocardial infarction. Exp Mol Med. 53:401–426.
2019.
|
|
128
|
Shi P, Song C, Qi H, Ren J, Ren P, Wu J,
Xie Y, Zhang M, Sun H and Cao Y: Up-regulation of IRF3 is required
for docosahexaenoic acid suppressing ferroptosis of cardiac
microvascular endothelial cells in cardiac hypertrophy rat. J Nutr
Biochem. 104:1089722022. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Tang M, Huang Z, Luo X, Liu M, Wang L, Qi
Z, Huang S, Zhong J, Chen JX, Li L, et al: Ferritinophagy
activation and sideroflexin1-dependent mitochondria iron overload
is involved in apelin-13-induced cardiomyocytes hypertrophy. Free
Radical Biol Med. 134:445–457. 2019. View Article : Google Scholar
|
|
130
|
Chen X, Xu S, Zhao C and Liu B: Role of
TLR4/NADPH oxidase 4 pathway in promoting cell death through
autophagy and ferroptosis during heart failure. Biochem Biophys Res
Commun. 516:37–43. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Bertoli SR, Marques VB, Rossi EM, Krause
M, Carneiro MTWD, Simões MR and Dos Santos L: Chronic iron overload
induces vascular dysfunction in resistance pulmonary arteries
associated with right ventricular remodeling in rats. Toxicol Lett.
295:296–306. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Ciarambino T, Menna G, Sansone G and
Giordano M: Cardiomyopathies: An overview. Int J Mol Sci.
22:77222021. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Reichart D, Magnussen C, Zeller T and
Blankenberg S: Dilated cardiomyopathy: From epidemiologic to
genetic phenotypes. J Intern Med. 286:362–372. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Eldemire R, Mestroni L and Taylor MRG:
Genetics of Dilated Cardiomyopathy. Annu Rev Med. 75:417–426. 2024.
View Article : Google Scholar :
|
|
135
|
Kadhi A, Mohammed F and Nemer G: The
Genetic Pathways Underlying Immunotherapy in Dilated
Cardiomyopathy. Front Cardiovasc Med. 8:6132952022. View Article : Google Scholar
|
|
136
|
Li D, Pi W, Sun Z, Liu X and Jiang J:
Ferroptosis and its role in cardiomyopathy. Biomed Pharmacother.
153:1132792022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Al-Adhami A, Avtaar Singh SS, De SD, Singh
R, Panjrath G, Shah A, Dalzell JR, Schroder J and Al-Attar N:
Primary graft dysfunction after heart transplantation-unravelling
the enigma. Curr Probl Cardiol. 47:1009412022. View Article : Google Scholar
|
|
138
|
Gong CW, Yuan MM and Qiu BQ: Sterile
inflammation in thoracic transplantation. Molecular Cell.
78:581–601. 2021. View Article : Google Scholar
|
|
139
|
Li W, Feng G, Gauthier JM, Lokshina I,
Higashikubo R, Evans S, Liu X, Hassan A, Tanaka S, Cicka M, et al:
Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil
recruitment after heart transplantation. J Clin Invest.
129:2293–2304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Green DR: The coming decade of cell death
research: Five Riddles. Cell. 177:1094–1107. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Shi L, Liu Y, Li M and Luo Z: Emerging
roles of ferroptosis in the tumor immune landscape: from danger
signals to anti-tumor immunity. FEBS J. 289:3655–3665. 2022.
View Article : Google Scholar
|
|
142
|
Mallah SI, Atallah B, Moustafa F, Naguib
M, El Hajj S, Bader F and Mehra MR: Evidence-based pharmacotherapy
for prevention and management of cardiac allograft vasculopathy.
Prog Cardiovasc Dis. 63:194–209. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Jin JL, Zhang HW, Cao YX, Liu HH, Hua Q,
Li YF, Zhang Y, Wu NQ, Zhu CG, Xu RX, et al: Association of small
dense low-density lipoprotein with cardiovascular outcome in
patients with coronary artery disease and diabetes: A prospective,
observational cohort study. Cardiovasc Diabetol. 19:452020.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Li L, Wang H, Zhang J, Chen X, Zhang Z and
Li Q: Effect of endothelial progenitor cell-derived extracellular
vesicles on endothelial cell ferroptosis and atherosclerotic
vascular endothelial injury. Cell Death Discov. 7:2352021.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
McNerney ME, Lee KM, Zhou P, Molinero L,
Mashayekhi M, Guzior D, Sattar H, Kuppireddi S, Wang CR, Kumar V
and Alegre ML: Role of natural killer cell subsets in cardiac
allograft rejection. Am J Transplant. 6:505–513. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Wang W, Green M, Choi JE, Gijón M, Kennedy
PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al: CD8+ T
cells regulate tumour ferroptosis during cancer immunotherapy.
Nature. 569:270–274. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Kinsella A, Rao V, Fan CP, Manlhiot C,
Stehlik J, Ross H and Alba AC: Post-transplant survival in adult
congenital heart disease patients as compared to dilated and
ischemic cardiomyopathy patients; an analysis of the thoracic ISHLT
registry. Clin Transplant. 34:2020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Aldouri MA, Wonke B, Hoffbrand AV, Flynn
DM, Ward SE, Agnew JE and Hilson AJ: High incidence of
cardiomyopathy in beta-thalassaemia patients receiving regular
transfusion and iron chelation: Reversal by intensified chelation.
Acta Haematol. 84:113–117. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Liu P and Olivieri N: Iron overload
cardiomyopathies: New insights into an old disease. Cardiovasc Drug
Ther. 8:101–110. 1994. View Article : Google Scholar
|
|
150
|
Engle MA, M Erlandson M and Smith CH: Late
cardiac complications of chronic, severe, refractory anemia with
hemochromatosis. Circulation. 30:698–705. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Quinlan GJ, Evans TW and Gutteridge JM:
Iron and the redox status of the lungs. Free Radic Biol Med.
33:1306–1313. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Shvartsman M, Fibach E and Cabantchik ZI:
Transferrin-iron routing to the cytosol and mitochondria as studied
by live and real-time fluorescence. Biochem J. 429:185–193. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Oudit GY, Sun H, Trivieri MG, Koch SE,
Dawood F, Ackerley C, Yazdanpanah M, Wilson GJ, Schwartz A, Liu PP
and Backx PH: L-type Ca2+ channels provide a major pathway for iron
entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med.
9:1187–1194. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
154
|
Tsushima RG, Wickenden AD, Bouchard RA,
Oudit GY, Liu PP and Backx PH: Modulation of iron uptake in heart
by L-Type Ca2+ channel modifiers. Circ Res.
84:1302–1309. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Das SK, Patel VB, Basu R, Wang W,
DesAulniers J, Kassiri Z and Oudit GY: Females are protected from
iron-overload cardiomyopathy independent of iron metabolism: Key
role of oxidative stress. J Am Heart Assoc. 6:e0034562017.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Brewer C, Otto-Duessel M, Wood RI and Wood
JC: Sex differences and steroid modulation of cardiac iron in a
mouse model of iron overload. Transl Res. 163:151–159. 2014.
View Article : Google Scholar :
|
|
157
|
Zhang H, Zhabyeyev P, Wang S and Oudit GY:
Role of iron metabolism in heart failure: From iron deficiency to
iron overload. Biochim Biophys Acta Mol Basis Dis. 1865:1925–1937.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Zhao X, Si L, Bian J, Pan C, Guo W, Qin P,
Zhu W, Xia Y, Zhang Q and Wei K: Adipose tissue macrophage-derived
exosomes induce ferroptosis via glutathione synthesis inhibition by
targeting SLC7A11 in obesity-induced cardiac injury. Free Radic
Biol Med. 182:232–245. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Nguyen NT, Nguyen TT, Da Ly D, Xia JB, Qi
XF, Lee IK, Cha SK and Park KS: Oxidative stress by Ca2+
overload is critical for phosphate-induced vascular calcification.
Am J Physiol Heart Circ Physiol. 319:H1302–H1312. 2020. View Article : Google Scholar
|
|
160
|
Akahori H, Tsujino T, Naito Y, Matsumoto
M, Lee-Kawabata M, Ohyanagi M, Mitsuno M, Miyamoto Y, Daimon T, Hao
H, et al: Intraleaflet haemorrhage is associated with rapid
progression of degenerative aortic valve stenosis. Eur Heart J.
32:888–896. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Laguna-Fernandez A, Carracedo M, Jeanson
G, Nagy E, Eriksson P, Caligiuri G, Franco-Cereceda A and Bäck M:
Iron alters valvular interstitial cell function and is associated
with calcification in aortic stenosis. Eur Heart J. 37:3532–3535.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Morvan M, Arangalage D, Franck G, Perez F,
Cattan-Levy L, Codogno I, Jacob-Lenet MP, Deschildre C, Choqueux C,
Even G, et al: Relationship of iron deposition to calcium
deposition in human aortic valve leaflets. J Am Coll Cardiol.
73:1043–1054. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Xu Y, Lin H, Wang H, Pang J and Zhou Y:
Fraxetin attenuates ferroptosis in myocardial infarction via
AKT/Nrf2/HO-1 signaling. Am J Transl Res. 13:10315–10327.
2021.PubMed/NCBI
|
|
164
|
Liberman M, Bassi E, Martinatti MK, Lario
FC, Wosniak J Jr, Pomerantzeff PM and Laurindo FR: Oxidant
generation predominates around calcifying foci and enhances
progression of aortic valve calcification. Arterioscler Thromb Vasc
Biol. 28:463–470. 2008. View Article : Google Scholar
|
|
165
|
Schnabel RB, Yin X, Gona P, Larson MG,
Beiser AS, McManus DD, Newton-Cheh C, Lubitz SA, Magnani JW,
Ellinor PT, et al: 50 year trends in atrial fibrillation
prevalence, incidence, risk factors, and mortality in the
Framingham Heart Study: A cohort study. Lancet. 386:154–156. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Dai C, Kong B, Qin T, Xiao Z, Fang J, Gong
Y, Zhu J, Liu Q, Fu H, Meng H, et al: Inhibition of ferroptosis
reduces susceptibility to frequent excessive alcohol
consumption-induced atrial fibrillation. Toxicology.
465:1530552022. View Article : Google Scholar
|
|
167
|
Rose RA, Sellan M, Simpson JA,
Izaddoustdar F, Cifelli C, Panama BK, Davis M, Zhao D, Markhani M,
Murphy GG, et al: Iron overload decreases CaV1.3-dependent L-type
Ca2+ currents leading to bradycardia, altered electrical
conduction, and atrial fibrillation. Circ Arrhythm Electrophysiol.
4:733–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Fang J, Kong B, Shuai W, Xiao Z, Dai C,
Qin T, Gong Y, Zhu J, Liu Q and Huang H: Ferroportin-mediated
ferroptosis involved in new-onset atrial fibrillation with
LPS-induced endotoxemia. Front Pharmacol. 913:1746222021.
|
|
169
|
Liu D, Yang M, Yao Y, He S, Wang Y, Cao Z,
Chen H, Fu Y, Liu H and Zhao Q: Cardiac fibroblasts promote
ferroptosis in atrial fibrillation by secreting Exo-miR-23a-3p
Targeting SLC7A11. Oxid Med Cell Longev.
2022:39614952022.PubMed/NCBI
|
|
170
|
Russo V, Rago A, Pannone B, Di Meo F, Papa
AA, Mayer MC, Spasiano A, Russo MG, Golino P, Calabrò R and Nigro
G: Early electrocardiographic evaluation of atrial fibrillation
risk in beta-thalassemia major patients. Int J Hematol. 93:446–451.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Lee AS, Jung YJ, Thanh TN, Lee S, Kim W,
Kang KP and Park SK: Paricalcitol attenuates
lipopolysaccharide-induced myocardial inflammation by regulating
the NF-κB signaling pathway. Int J Mol Med. 37:1023–1029. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C,
Wu H, Deng W, Shen D and Tang Q: Ferritinophagy-mediated
ferroptosis is involved in sepsis-induced cardiac injury. Free
Radic Biol Med. 160:303–318. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Calne DB and Calne JS: Normality and
Disease. Can J Neurol Sci. 15:3–4. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Kung YA, Chiang HJ and Li ML: Fatal
lymphocytic cardiac damage in coronavirus disease 2019 (COVID-19):
Autopsy reveals a ferroptosis signature. Clin Transplant.
130:113–117. 1990.
|
|
175
|
Wang YD, Liu ZJ, Ren J and Xiang MX:
Pharmacological Therapy of Abdominal Aortic Aneurysm: An Update.
Curr Vasc Pharmacol. 16:114–124. 2018. View Article : Google Scholar
|
|
176
|
Wang K, Song Y, Li H, Song J and Wang S:
Identification of differentially expressed ferroptosis-related
genes in abdominal aortic aneurysm: Bioinformatics analysis. Front
Cardiovasc Med. 9:9916132022. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Golledge J: Abdominal aortic aneurysm:
Update on pathogenesis and medical treatments. Nat Rev Cardiol.
16:225–242. 2019. View Article : Google Scholar
|
|
178
|
Quintana RA and Taylor WR: Cellular
Mechanisms of Aortic Aneurysm Formation. Circ Res. 124:607–618.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Sampilvanjil A, Karasawa T, Yamada N,
Komada T, Higashi T, Baatarjav C, Watanabe S, Kamata R, Ohno N and
Takahashi M: Cigarette smoke extract induces ferroptosis in
vascular smooth muscle cells. Am J Physiol Heart Circ Physiol.
318:H508–H518. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Chen L, Liu Y, Wang Z, Zhang L, Xu Y, Li
Y, Zhang L, Wang G, Yang S and Xue G: Mesenchymal stem cell-derived
extracellular vesicles protect against abdominal aortic aneurysm
formation by inhibiting NET-induced ferroptosis. Exp Mol Med.
55:939–951. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Zhuang J, Zhu H, Cheng Z, Hu X, Yu X, Li
J, Liu H, Tang P, Zhang Y, Xiong X and Deng H: PCSK9, a novel
immune and ferroptosis related gene in abdominal aortic aneurysm
neck. Sci Rep. 13:60542023. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Li N, Yi X, He Y, Huo B, Chen Y, Zhang Z,
Wang Q, Li Y, Zhong X, Li R, et al: Targeting ferroptosis as a
novel approach to alleviate aortic dissection. Int J Biol.
18:4118–4134. 2022. View Article : Google Scholar
|
|
183
|
Shi H, Wei J and He C: Where, When, and
How: Context-Dependent Functions of RNA Methylation Writers,
Readers, and Erasers. Mol Cell. 74:640–650. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Chen Y, Yi X, Huo B, He Y, Guo X, Zhang Z,
Zhong X, Feng X, Fang ZM, Zhu XH, et al: BRD4770 functions as a
novel ferroptosis inhibitor to protect against aortic dissection.
Pharmacol Res. 177:1061222022. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Kobashigawa J, Zuckermann A, Macdonald P,
Leprince P, Esmailian F, Luu M, Mancini D, Patel J, Razi R,
Reichenspurner H, et al: Report from a consensus conference on
primary graft dysfunction after cardiac transplantation. J Heart
Lung Transplant. 33:327–340. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Lund LH, Edwards LB, Kucheryavaya AY,
Dipchand AI, Benden C, Christie JD, Dobbels F, Kirk R, Rahmel AO,
Yusen RD, et al: The Registry of the International Society for
Heart and Lung Transplantation: Thirtieth Official Adult Heart
Transplant Report-2013; Focus Theme: Age. J Heart Lung Transplant.
32:951–964. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Bai T, Li M, Liu Y, Qiao Z and Wang Z:
Inhibition of ferroptosis alleviates atherosclerosis through
attenuating lipid peroxidation and endothelial dysfunction in mouse
aortic endothelial cell. Free Radic Biol Med. 160:92–102. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Neufeld EJ: Oral chelators deferasirox and
deferiprone for transfusional iron overload in thalassemia major:
New data, new questions. Blood. 107:3436–3441. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Philipp S, Cui L, Ludolph B, Kelm M,
Schulz R, Cohen MV and Downey JM: Desferoxamine and
ethyl-3,4-dihydroxybenzoate protect myocardium by activating NOS
and generating mitochondrial ROS. Am J Physiol Heart Circ Physiol.
290:H450–H457. 2006. View Article : Google Scholar
|
|
190
|
Duffy SJ, Biegelsen ES, Holbrook M,
Russell JD, Gokce N, Keaney JF Jr and Vita JA: Iron chelation
improves endothelial function in patients with coronary artery
disease. Circulation. 103:2799–2804. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Wong CAC and Leitch HA: Delayed time from
RBC transfusion dependence to first cardiac event in lower IPSS
risk MDS patients receiving iron chelation therapy. Leuk Res.
83:1061702019. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Paraskevaidis IA, Iliodromitis EK,
Vlahakos D, Tsiapras DP, Nikolaidis A, Marathias A, Michalis A and
Kremastinos DT: Deferoxamine infusion during coronary artery bypass
grafting ameliorates lipid peroxidation and protects the myocardium
against reperfusion injury: Immediate and long-term significance.
Eur Heart J. 26:263–270. 2005. View Article : Google Scholar
|
|
193
|
Tadokoro T, Ikeda M, Ide T, Deguchi H,
Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada KI,
et al: Mitochondria-dependent ferroptosis plays a pivotal role in
doxorubicin cardiotoxicity. JCI Insight. 5:e1327472020. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Zilka O, Shah R, Li B, Friedmann Angeli
JP, Griesser M, Conrad M and Pratt DA: On the Mechanism of
Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the role of
lipid peroxidation in ferroptotic cell death. ACS Cent Sci.
3:232–243. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Watanabe Y and Tatsuno I: Prevention of
cardiovascular events with omega-3 polyunsaturated fatty acids and
the mechanism involved. J Atheroscler Thromb. 27:183–198. 2020.
View Article : Google Scholar :
|
|
197
|
Bibli SI, Hu J, Leisegang MS, Wittig J,
Zukunft S, Kapasakalidi A, Fisslthaler B, Tsilimigras D, Zografos
G, Filis K, et al: Shear stress regulates cystathionine γ lyase
expression to preserve endothelial redox balance and reduce
membrane lipid peroxidation. Redox Biol. 28:1013792020. View Article : Google Scholar
|
|
198
|
Chakraborty A, Li Y, Zhang C, Li Y,
LeMaire SA and Shen YH: Simvastatin Ameliorates Diabetic
Cardiomyopathy by Attenuating Oxidative Stress and Inflammation in
Rats. Progress in Cardiovascular Diseases. 84:113–117. 1990.
|
|
199
|
Bai YT, Chang R, Wang H, Xiao FJ, Ge RL
and Wang LS: ENPP2 protects cardiomyocytes from erastin-induced
ferroptosis. Biochem Biophys Res Commun. 499:44–51. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Prasad A, Andrews NP, Padder FA, Husain M
and Quyyumi AA: Glutathione reverses endothelial dysfunction and
improves nitric oxide bioavailability. J Am Coll Cardiol.
34:507–514. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Imai H, Matsuoka M, Kumagai T, Sakamoto T
and Koumura T: Lipid peroxidation-dependent cell death regulated by
GPx4 and ferroptosis. Curr Top Microbiol Immunol. 403:143–170.
2017.PubMed/NCBI
|
|
202
|
Hinman A, Holst CR, Latham JC, Bruegger
JJ, Ulas G, McCusker KP, Amagata A, Davis D, Hoff KG, Kahn-Kirby
AH, et al: Vitamin E hydroquinone is an endogenous regulator of
ferroptosis via redox control of 15-lipoxygenase. PLoS One.
13:e02013692018. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Upston JM, Kritharides L and Stocker R:
The role of vitamin E in atherosclerosis. Prog Lipid Res.
42:405–422. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Steinberg D: Is there a potential
therapeutic role for vitamin E or other antioxidants in
atherosclerosis? Curr Opin Lipidol. 11:603–607. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Cogny A, Paul JL, Soni T, Atger V and
Moatti N: Vitamin E: Metabolism and role in atherosclerosis. Ann
Biol Clin (Paris). 52:515–522. 1994.
|
|
206
|
Dludla PV, Dias SC, Obonye N, Johnson R,
Louw J and Nkambule BB: A Systematic Review on the Protective
Effect of N-Acetyl Cysteine Against Diabetes-Associated
Cardiovascular Complications. Am J Cardiovasc Drugs. 18:283–298.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Bartekova M, Barancik M, Ferenczyova K and
Dhalla NS: Beneficial Effects of N-acetylcysteine and
N-mercaptopropionylglycine on ischemia reperfusion injury in the
heart. Curr Med Chem. 25:355–366. 2018. View Article : Google Scholar
|
|
208
|
Badgley MA, Kremer DM, Maurer HC,
DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J,
Firl CEM, et al: Cysteine depletion induces pancreatic tumor
ferroptosis in mice. Science. 368:85–89. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang
Z and Yao W: Diabetes aggravates myocardial ischaemia reperfusion
injury via activating Nox2-related programmed cell death in an
AMPK-dependent manner. J Cell Mol Med. 24:6670–6679. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Xiao Z, Kong B, Fang J, Qin T, Dai C,
Shuai W and Huang H: Ferrostatin-1 alleviates
lipopolysaccharide-induced cardiac dysfunction. Bioengineered.
12:9367–9376. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Feng Y, Madungwe NB, Imam Aliagan AD,
Tombo N and Bopassa JC: Liproxstatin-1 protects the mouse
myocardium against ischemia/reperfusion injury by decreasing VDAC1
levels and restoring GPX4 levels. Biochem Biophys Res Commun.
520:606–611. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Fang X, Wang H and Han D: Traditional
Chinese medicine for cardiovascular disease. Proc Natl Acad Sci
USA. 12:9367–9376. 2021.
|
|
213
|
Li W, Feng G and Gauthier JM: Translating
traditional herbal formulas into modern drugs: a network-based
analysis of Xiaoyao decoction. Cell Death Differ.
119:1183562020.
|
|
214
|
Ferrara PE, Codazza S, Cerulli S, Maccauro
G, Ferriero G and Ronconi G: Physical modalities for the
conservative treatment of wrist and hand's tenosynovitis: A
systematic review. Semin Arthritis Rheum. 50:1280–1290. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Wang R, Wang M, Zhou J, Wu D, Ye J, Sun G
and Sun X: Saponins in Chinese Herbal Medicine Exerts Protection in
Myocardial Ischemia-Reperfusion Injury: Possible mechanism and
target analysis. Front Pharmacol. 11:5708672021. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Chen GQ, Benthani FA, Wu J, Liang D, Bian
ZX and Jiang X: Artemisinin compounds sensitize cancer cells to
ferroptosis by regulating iron homeostasis. Cell Death Differ.
27:242–254. 2020. View Article : Google Scholar :
|
|
217
|
Wang S, Liu W, Wang J and Bai X:
Curculigoside inhibits ferroptosis in ulcerative colitis through
the induction of GPX4. Life Sci. 259:1183562020. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Zhu K, Zhu X, Liu S, Yu J, Wu S and Hei M:
Glycyrrhizin attenuates hypoxic-ischemic brain damage by inhibiting
ferroptosis and neuroinflammation in neonatal rats via the
HMGB1/GPX4 Pathway. Oxid Med Cell Longev. 2022:84385282022.
View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Fan Z, Cai L, Wang S, Wang J and Chen B:
Baicalin prevents myocardial ischemia/reperfusion injury through
inhibiting ACSL4 mediated ferroptosis. Front Pharmacol.
12:6289882021. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Zeng X, Li J and Li Z: Ginsenoside Rd
mitigates myocardial ischemia-reperfusion injury via Nrf2/HO-1
signaling pathway. Int J Clin Exp Med. 8:14497–14504.
2015.PubMed/NCBI
|
|
221
|
Wang D, Chen T and Liu F: Betulinic acid
alleviates myocardial hypoxia/reoxygenation injury via inducing
Nrf2/HO-1 and inhibiting p38 and JNK pathways. Eur J Pharmacol.
838:53–59. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Li T, Tan Y, Ouyang S, He J and Liu L:
Resveratrol protects against myocardial ischemia-reperfusion injury
via attenuating ferroptosis. Gene. 808:1459682022. View Article : Google Scholar
|
|
223
|
Cao H, Wang P, Li N, Liu D, Ma J, Fan R
and Zhou Z: Practice of Comparative Effectiveness Research to
Identify Treatment Characteristics of Similar Chinese Patent
Medicine for Angina Pectoris. Evid Based Complement Alternat Med.
2017:70627142017. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Wu L, Fan Z, Gu L, Liu J, Cui Z, Yu B, Kou
J and Li F: QiShenYiQi dripping pill alleviates myocardial
ischemia-induced ferroptosis via improving mitochondrial dynamical
homeostasis and biogenesis. J Ethnopharmacol. 308:1162822023.
View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Xu S, Wu B, Zhong B, Lin L, Ding Y, Jin X,
Huang Z, Lin M, Wu H and Xu D: Naringenin alleviates myocardial
ischemia/reperfusion injury by regulating the nuclear
factor-erythroid factor 2-related factor 2 (Nrf2)/System
xc-/glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis.
Bioengineered. 12:10924–10934. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Zhao L, Shi H, Zhang F, Xue H and Han Q:
Hederagenin protects against myocardial ischemia-reperfusion injury
via attenuating ALOX5-mediated ferroptosis. Naunyn-Schmiedeberg's
Arch Pharmacol. 397:3411–3424. 2024. View Article : Google Scholar
|
|
227
|
D'Onofrio N, Servillo L and Balestrieri
ML: SIRT1 and SIRT6 signaling pathways in cardiovascular disease
protection. Antioxid Redox Signal. 28:711–732. 2018. View Article : Google Scholar :
|
|
228
|
Kane AE and Sinclair DA: Sirtuins and
NAD(+)in the development and treatment of metabolic and
cardiovascular diseases. Circ Res. 123:868–885. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Li D, Liu X, Pi W, Zhang Y, Yu L, Xu C,
Sun Z and Jiang J: Fisetin Attenuates doxorubicin-induced
cardiomyopathy in vivo and in vitro by inhibiting ferroptosis
through SIRT1/Nrf2 signaling pathway activation. Front Pharmacol.
12:8084802022. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Wu S, Zhu J, Wu G, Hu Z, Ying P, Bao Z,
Ding Z and Tan X: 6-Gingerol alleviates ferroptosis and
inflammation of diabetic cardiomyopathy via the Nrf2/HO-1 pathway.
Oxid Med Cell Longev. Dec 31–2022.Epub ahead of print. View Article : Google Scholar
|
|
231
|
Zhou YX, Zhang H and Peng C: Puerarin: A
Review of Pharmacological Effects. Phytother Res. 28:961–975. 2014.
View Article : Google Scholar
|
|
232
|
Liu B, Zhao C, Li H, Chen X, Ding Y and Xu
S: Puerarin protects against heart failure induced by pressure
overload through mitigation of ferroptosis. Biochem Biophys Res
Commun. 497:233–240. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Zhang D, Zhang Y, Gao Y, Chai X, Pi R,
Chan G and Hu Y: Current understanding of iron homeostasis. Gene.
106:1559S–1566S. 2017.
|
|
234
|
Fang X, An P, Wang H, Wang X, Shen X, Li
X, Min J, Liu S and Wang F: Dietary intake of heme iron and risk of
cardiovascular disease: A dose-response meta-analysis of
prospective cohort studies. Nutr Metab Cardiovasc Dis. 25:24–35.
2015. View Article : Google Scholar
|
|
235
|
Han M, Guan L and Ren Y: Physical
modalities for the conservative treatment of wrist and hand's
tenosynovitis: A systematic review. Asia Pac J Clin Nutr.
29:309–321. 2020.
|
|
236
|
Zhao L, Shi H and Zhang F: Deficiency in
Beclin1 attenuates alcohol-induced cardiac dysfunction via
inhibition of ferroptosis. Front Pharmacol. 2021:1–15. 2021.
|
|
237
|
Bao X, Luo X, Bai X, Lv Y, Weng X, Zhang
S, Leng Y, Huang J, Dai X, Wang Y, et al: Cigarette tar mediates
macrophage ferroptosis in atherosclerosis through the
hepcidin/FPN/SLC7A11 signaling pathway. Free Radic Biol Med.
201:76–88. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Violi F, Nocella C, Loffredo L, Carnevale
R and Pignatelli P: Interventional study with vitamin E in
cardiovascular disease and meta-analysis. Free Radic Biol Med.
178:26–41. 2022. View Article : Google Scholar
|
|
239
|
Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang
JJ, Luo XJ and Peng J: Ubiquitin-specific protease 7 promotes
ferroptosis via activation of the p53/TfR1 pathway in the rat
hearts after ischemia/reperfusion. Free Radic Biol Med.
162:339–352. 2021. View Article : Google Scholar
|
|
240
|
Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang
CF, Li Y and Zhang Z: Dexmedetomidine alleviated sepsis-induced
myocardial ferroptosis and septic heart injury. Mol Med Rep.
22:175–184. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Ning D, Yang X, Wang T, Jiang Q, Yu J and
Wang D: Atorvastatin treatment ameliorates cardiac function and
remodeling induced by isoproterenol attack through mitigation of
ferroptosis. Biochem Biophys Res Commun. 574:39–47. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Mishima E, Sato E, Ito J, Yamada KI,
Suzuki C, Oikawa Y, Matsuhashi T, Kikuchi K, Toyohara T, Suzuki T,
et al: Drugs repurposed as antiferroptosis agents suppress organ
damage, including AKI, by functioning as lipid peroxyl radical
scavengers. J Am Soc Nephrol. 31:280–296. 2020. View Article : Google Scholar :
|
|
243
|
Conlon M, Poltorack CD, Forcina GC,
Armenta DA, Mallais M, Perez MA, Wells A, Kahanu A, Magtanong L,
Watts JL, et al: A compendium of kinetic modulatory profiles
identifies ferroptosis regulators. Nat Chem Biol. 17:665–674. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Oettl K, Greilberger J, Zangger K,
Haslinger E, Reibnegger G and Jürgens G: Radical-scavenging and
iron-chelating properties of carvedilol, an antihypertensive drug
with antioxidative activity. Biochem Pharmacol. 62:241–248. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
245
|
Hong M, Rong J, Tao X and Xu Y: The
emerging role of ferroptosis in cardiovascular diseases. Front
Pharmacol. 13:8220832022. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Fang W, Xie S and Deng W: Ferroptosis
mechanisms and regulations in cardiovascular diseases in the past,
present, and future. Cell Biol Toxicol. 40:172024. View Article : Google Scholar : PubMed/NCBI
|