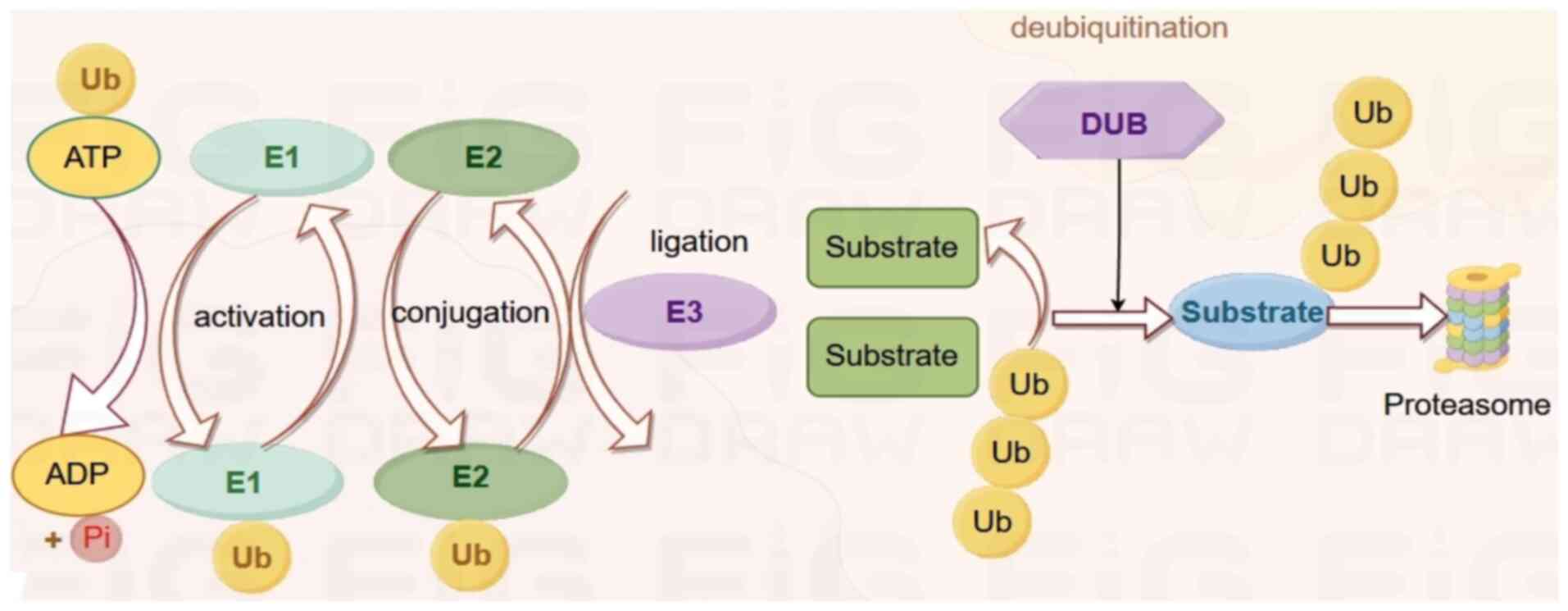
Ubiquitination is the sequential binding of
ubiquitin (Ub) molecules by three enzymes (E1 activator, E2
conjugating enzyme and E3 ligase) and the transfer of the small
modified protein Ub to lysine residues within the substrate
protein. Proteins can be modified with a single Ub at one or more
lysine residues or with a Ub chain formed through one of their
lysine residues or an N-terminal methionine residue (M1). In
humans, there are two Ub-specific E1 proteins, ~35 Ub-specific E2
binding enzymes and nearly 1,000 identified E3 ligases (1,2).
Ubiquitination is a general term for several different types of Ub
modifications, each having a different effect on the target
protein. Polyubiquitin chains allow proteins to be degraded by the
proteasome. Another form of ubiquitination is monoubiquitination,
which can alter subcellular localization. These different types of
Ub modifications represent the different effects of ubiquitination
on protein function (3). As a
post-translational modification (PTM), ubiquitination can affect
the stability, interaction, localization and/or activity of
thousands of proteins by forming conjugates of different
topologies, thereby controlling a large number of signaling events
in the cell, including proteasomal degradation, DNA damage repair
and cell cycle progression, amongst others (4,5).
Ubiquitination plays an important role in all aspects of cellular
physiology, including angiogenesis, which is the focus of this
review. The process of ubiquitination, on the other hand, is the
process by which ubiquitin molecules, in the presence of a series
of special enzymes, classify proteins in the cell and select from
them target protein molecules for specific modification (Fig. 1).
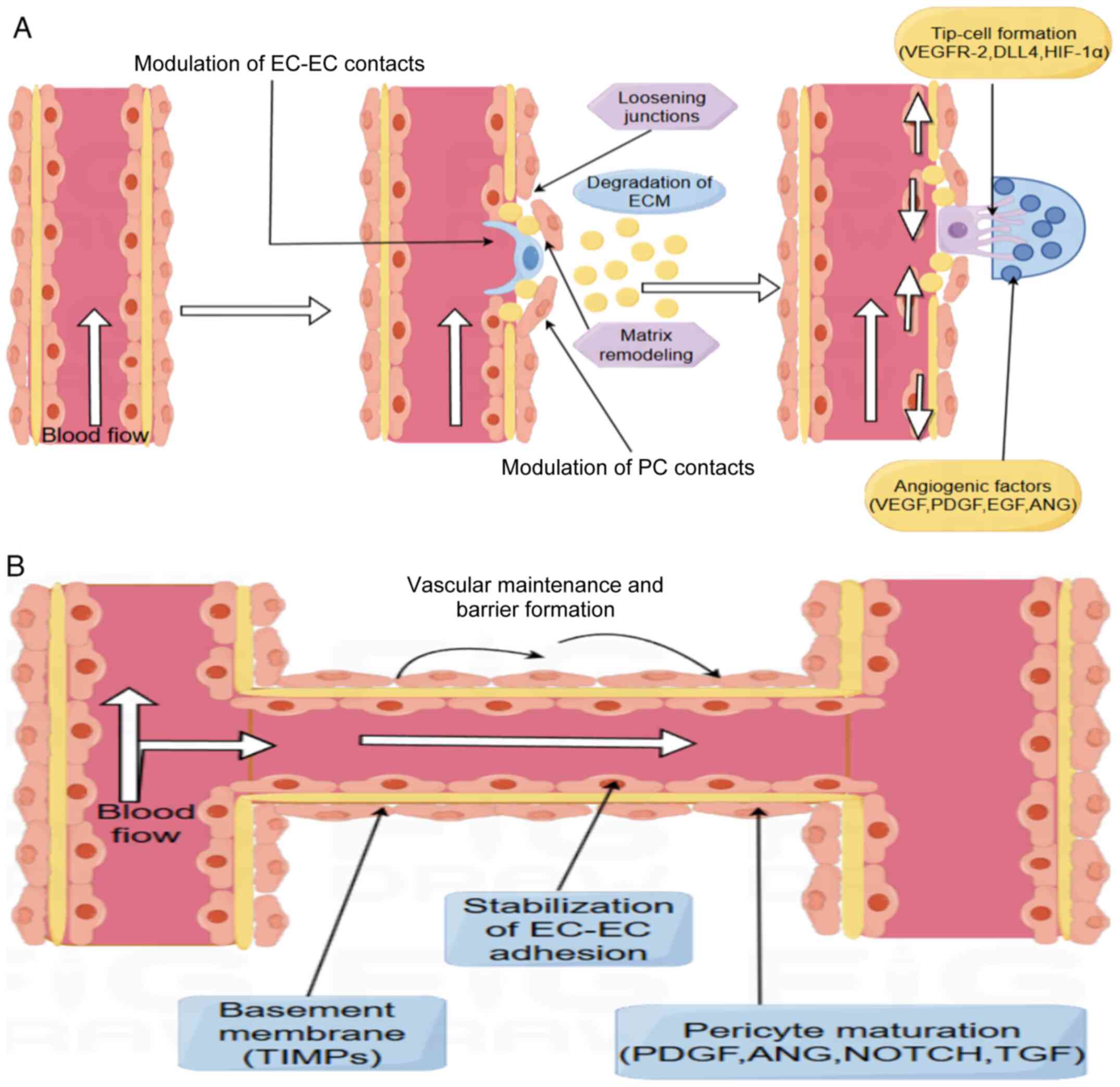
Angiogenesis is the process by which new capillaries
are formed from existing capillaries and occurs via two primary
mechanisms: Sprouting and non-sprouting. Angiogenesis aims to
respond to changes in the cellular machinery or metabolic
environment, and appears in the form of endothelial cell (EC)
sprouting and longitudinal division. The sprouting form is
considered the primary mechanism of angiogenesis during
physiological development and in cancer, where new capillaries
branch and protrude from existing ones, and this requires the
excessive proliferation of ECs (6). The non-budding process appears to
be more efficient compared to the budding approach. In the
non-budding approach to angiogenesis, the developed vasculature is
halved by longitudinal splitting within the capillaries, turning
one capillary into two. This reduces the need for EC proliferation
(7). Angiogenesis is a complex,
multi-step process involving the close interplay of cell
proliferation, differentiation, migration and signaling. Blood
vessels are composed of two main cell types, ECs and pericytes. ECs
constitute the main body of the blood vessel, forming a hollow tube
known as the lumen, which is lined with a specialized extracellular
matrix (ECM) known as the basement membrane. Pericytes are attached
to the lumen's surface and regulate the vessel's permeability and
stability as required by the environment, whilst also serving as
important support cells for the vessel (8). Angiogenesis begins when ECs receive
pro-angiogenic signals in the form of growth factors such as
vascular endothelial growth factor (VEGF), platelet-derived growth
factor (PDGF) and epidermal growth factor (EGF). Fibroblast growth
factors (FGFs) promote migration and proliferation in angiogenesis
by stimulating VEGF-mediated integrin levels and activating
proteolytic activity through tight binding to their respective
receptors (FGFRs) (9). Activated
ECs then invade the basement membrane and ECM, disrupting
integrin-mediated interactions. When ECs are activated, MMPs are
secreted from the tip cells to degrade the ECM and break down the
basement membrane in the region where they are located. By
digesting the basement membrane, ECs migrate and proliferate.
Angiopoietin (Ang) is then activated to initiate lumen formation
(10). In addition, cell
adhesion molecules (CAMs) play a central role in angiogenesis. ECs
use CAMs to achieve homogeneous and heterogeneous adhesion and to
adhere to and migrate through the ECM, a critical step in
angiogenesis. Increased expression of activated CAMs can also lead
to their detachment or release from ECs. Several soluble forms of
CAMs have been identified, such as intercellular adhesion
molecule-1 (ICAM-1), vascular CAM 1 (VCAM-1) and E-selectin
(11). Then, when the ECs
converge, they begin to interconnect and form tubular structures,
termed luminalization. Intercellular interactions, adhesion and
alignment of cells are particularly critical at this stage
(11). The EphB/ephrinB system
is involved in regulating the process of lumen formation, while the
involvement of pericytes and smooth muscle cells is required to
stabilize the newly formed vessels (12). Finally, the process of vascular
maturation requires the involvement of multiple cells and signals.
The interaction between ECs, SMCs and pericytes is particularly
important in this process. The encapsulation of SMCs and pericytes
not only enhances the structural strength of the vessel but also
regulates blood flow and vascular reactivity. Reconstruction of the
ECM and modulation of biosignals ensures that the vessel is
sufficiently resilient and functional to cope with the various
changes in pressure in the body. In addition, the neovasculature
needs to interface with the existing vascular network to form a
stable circulatory network (11,12). In this process, cell-to-cell
communication and microenvironmental regulation determine whether
the blood vessel can be truly 'generated' and function effectively
(11,12). There is also a process in
angiogenesis called arteriogenesis. This is the enlargement of an
artery's diameter and wall thickness. It occurs primarily in large
arteries and small arterioles. This process requires the
proliferation of SMCs and ECs. The most important stimulus involved
in the process of arteriogenesis is the hemodynamic stimulus of
shear stress (13). Acute
changes in fluid shear stress induce ECs to express chemokines and
CAMs, and chemokines trigger the recruitment of circulating
monocytes that adhere to activated endothelium-expressing CAMs.
Monocytes then migrate through the endothelium into the
subendothelial space, where they transform into macrophages and
produce inflammatory cytokines and growth factors that diffuse into
the intima-vascular layer and ultimately mediate the transformation
of the SMC phenotype by modulating SMC signaling pathways (Fig. 2A and B) (14).
There is increasing evidence that several diseases
are dependent on angiogenesis, including atherosclerosis, pulmonary
hypertension, inflammatory diseases, neurodegenerative diseases,
aneurysms and cancer (15).
Although these diseases have different etiologies and pathogenesis
in several respects, they all have dysregulated angiogenesis in
common. The most extensive research in the field of angiogenesis is
currently focused on tumors. Angiogenesis is one of the hallmarks
of cancer and angiogenesis begins during the early stages of tumor
development. For tumors to grow, they must acquire angiogenic
capabilities, which are usually achieved through hypoxia-induced
expression of angiogenesis-inducing molecules. To support expanding
tumor growth, the angiogenic switch is flipped, allowing the
proliferation and germination of ECs that are normally quiescent
(16). Angiogenesis is therefore
an important component of tumor growth and metastasis. Once the
angiogenic switch is activated, a series of responses are
triggered, including activation of various proteases by activated
ECs, leading to degradation of the basement membrane surrounding
existing vessels, migration and proliferation of ECs, formation of
vascular sprouting lumens, generation of new basement membranes by
recruiting pericytes and finally generation of new fusion vessels.
Under certain circumstances, when cells within the tissue respond
to hypoxia, certain oncogenes together with other hypoxia-inducible
genes induce the expression of VEGF, thereby inducing tumor
angiogenesis (17,18). However, ubiquitination occurs
throughout almost all steps of tumor angiogenesis. For instance, in
the hypoxic microenvironment of tumor cells, hypoxia-inducible
factor (HIF) induces angiogenesis to increase oxygen delivery by
controlling the transcription of multiple genes (e.g. VEGF). Tumor
cells produce angiogenic factors to stimulate the development of
new blood vessels. Several transcription factors (such as HIF) and
signaling pathways (such as Notch and Wnt) are involved in
angiogenesis (19). Among them,
VEGF and its associated receptors (VEGFR) are important players.
Deubiquitinating enzymes regulate the stability of VEGFR, affecting
EC responses and vascular physiology (20). In addition, ubiquitination is
involved in basement membrane degradation, EC migration and
proliferation, vascular sprouting lumen formation, pericyte
recruitment and other angiogenic processes in tumors and other
diseases (discussed further below). Thus, angiogenesis is important
in tumorigenesis and other human diseases, and tubulogenesis is
considered a promising target for the treatment of related
diseases. Thus, angiogenesis is considered a promising target for
the management of related diseases (Fig. 3).
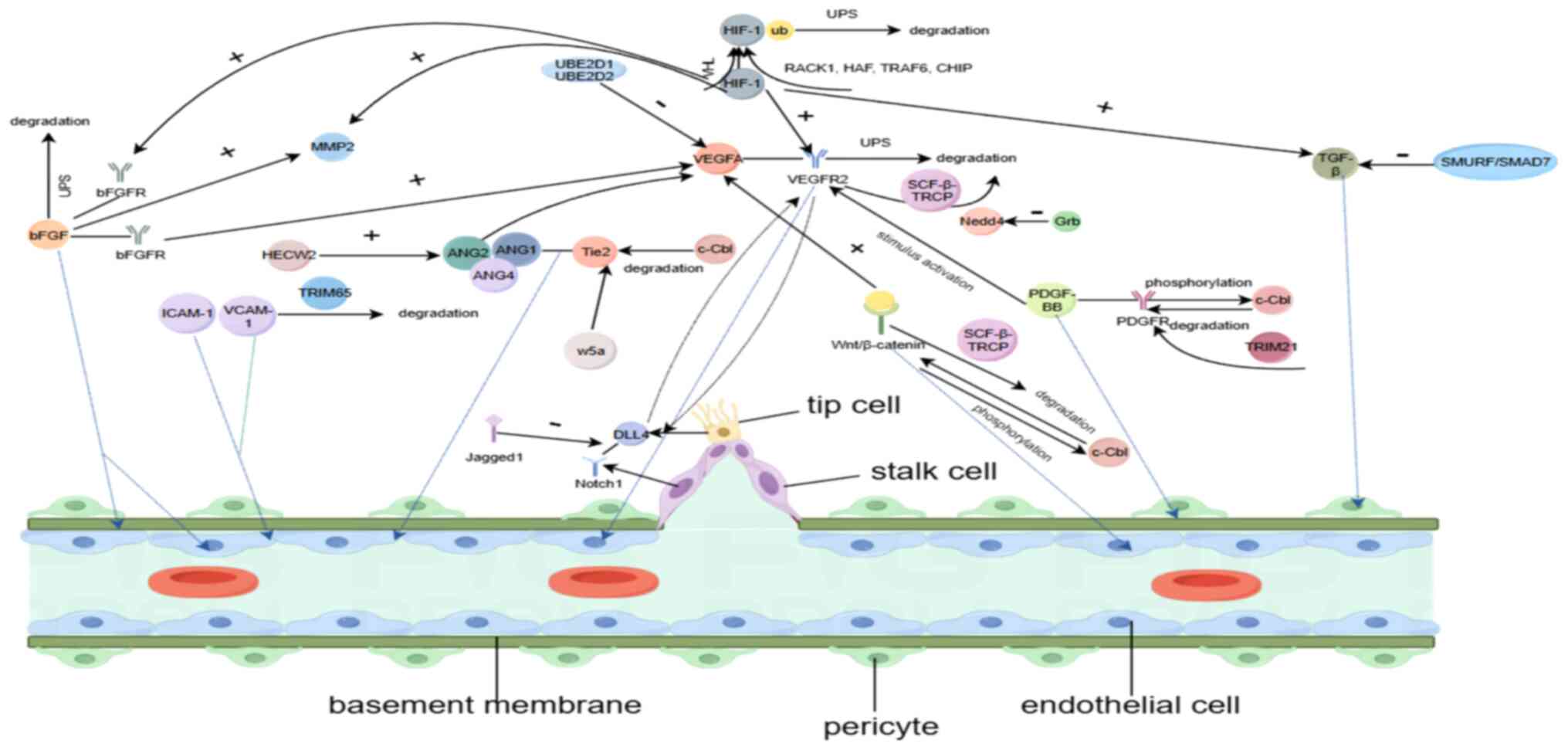
In angiogenesis, growth factors (GFs) enable blood
vessel formation by promoting EC proliferation and differentiation,
and recruitment of SMCs and fibroblasts (21). To date, several angiogenic GFs
have been identified, the most important of which are VEGF, PDGF,
basic FGF (bFGF/FGF2) and Ang. Ubiquitination regulates a wide
range of cellular functions, including protein processing and
transcriptional regulation, and these core pro-angiogenic proteins,
including angiogenic signaling proteins, as well as other non-VEGF
proteins, are an important basis for angiogenesis, and
ubiquitination plays a central role in regulating the function of
these signaling proteins and pathways.
Circulating angiogenesis-stimulating factors are
mediators of the angiogenic switch and VEGF is one of the most
important. At least five ligands associated with VEGF have been
identified: VEGF-A, -B, -C, -D and -E. VEGF-A is considered the
most important in stimulating angiogenesis (22). The effects of VEGF are mediated
by binding to three closely related receptor tyrosine kinases
(RTKs): VEGFR-1, -2 and -3 (23). VEGFR-2 belongs to the family of
RTKs, which play key roles in physiological and pathological
angiogenesis (24).
Structurally, the extracellular region of VEGFR-2 consists of seven
N-glycosylation-modified immunoglobulin-like regions, and the
intracellular region is affected by a variety of PTMs such as Tyr
and Ser/Thr phosphorylation, Arg and Lys methylation, acetylation
and ubiquitination (25). The
Ub-proteasome system (UPS) is the major pathway for protein
degradation in eukaryotic cells. This system controls a wide range
of cellular regulatory proteins, including transcription factors,
as well as cell cycle regulators, and has a major impact on the
onset of angiogenic responses through signaling via the VEGFR-2
pathway (26). The UPS consists
of two major components: Substrate-recruiting enzymes (E1, E2 and
E3) and substrate-degrading enzymes. E1 activates the peptide Ub in
an ATP-dependent manner, allowing it to be translocated to the
Ub-carrying enzyme E2 (27).
Inhibition of the proteasome significantly suppressed VEGFR-2 mRNA
accumulation. In addition, treatment with a proteasome inhibitor
significantly reduced the transcriptional activity of the VEGFR-2
promoter gene with a deletion at the 5′ end. Proteasome
inhibition-mediated repression was controlled by a GC-rich region
containing a consensus Sp1 binding site. Proteasome inhibition
reduces structural Sp1-dependent DNA binding (28). In addition, the presence of the
PEST structural domain in the carboxylated structural domain of
VEGFR-2 and the EST sequence, which is hypothesized to be an
unstructured region in certain protein sequences, may act as a
phospho-degrading agent that recruits the F-box-containing Ub E3
ligase, leading to ubiquitination and degradation, which may be the
key role of this carboxylated structural domain in the degradation
of VEGFR-2 (29). Of note,
ubiquitination can also affect cell signaling by targeting
activated proteins in a protein degradation-independent manner
(30). VEGF-A, which can be
secreted by several cell types, exerts its pro-angiogenic effects
primarily through interaction with VEGFR-2 (31). The levels of VEGFR-2 in the
plasma membrane significantly modulate VEGF-A-mediated signaling.
Activation of the MAPK (32),
phospholipase C (PLC)γ1 (33)
and AKT (34) signaling pathways
was also significantly increased by VEGF-A stimulation. E2
Ub-conjugating enzymes (UBE2D1 and UBE2D2) form a complex with
VEGFR-2, downregulating VEGFR-2 expression in ECs (35). A net pool of cellular regulatory
VEGFR-2 in the plasma membrane controls RTK-mediated signaling and
cellular responses to extracellular VEGFR-2. This
ligand-independent regulatory pathway mediates the availability of
VEGFR-2 at the plasma membrane for VEGF-A-stimulated signaling
functions. At the same time, the E1 enzyme UBA1 can regulate
basement membrane VEGFR-2 levels and influence VEGF-A-stimulated
activation of the PLC-ERK1 and γ/2 signaling pathways (36). Phosphorylation of RTKs such as
VEGFR or VEGFR-2 is followed by ubiquitination and regulated
intracellular trafficking. This endocytotic process depends on the
interaction between the ubiquitinated receptor and carrier proteins
with Ub-interacting motifs such as epsin, EGFR pathway substrate 15
and hepatocyte growth factor-regulated tyrosine kinase substrate
(37). In addition, the Ub E3
ligase stem cell factor (SCF) -β-transducin repeat containing E3
ubiquitin protein ligase pseudogene 1 (β-TRCP) can promote
ubiquitination and degradation of VEGFR-2 in a casein kinase I
(CKI)-dependent manner. Knockdown of the β-TRCP gene or inhibition
of CKI resulted in the accumulation of VEGFR-2, which led to an
increase in VEGFR-2 downstream signaling. By contrast,
β-TRCP-deficient ECs exhibited enhanced angiogenesis in
vitro (38). Hect E3 Ub
ligases are a class of E3 Ub ligases characterized by the HECT
structural domain (homologous to the C-terminus of E6AP), a
conserved C-terminal catalytic structural domain through which
ubiquitination of substrate proteins is achieved. NEDD4 E3
ubiquitin protein ligase (Nedd4) belongs to the E3 proteins
containing the Hect domain and the NEDD4 family all contain three
structural domains: A single C2 domain at the N-terminal end for
membrane binding, 1-4 WW domains in the center for specific
recognition of PY motif substrate proteins and the C-terminal HECT
domain for Ub-protein attachment. This feature allows the transfer
of Nedd4 Ub from E2 to the HECT domain of E3 and then to the
substrate (39,40). Nedd4 expression induces
intracellular degradation of VEGFR-2. While GF receptor-bound
protein 10 (Grb10) is a positive regulator of the VEGF signaling
pathway, Grb10 can stimulate vascular VEGFR-2 expression by
inhibiting Nedd4-mediated degradation of VEGFR-2 (40). Thus, ubiquitination can target
VEGFR-2 in a proteasome catabolism-dependent manner (the UPS body
pathway) and indirectly regulate VEGF by modulating gene expression
and/or spatial localization of VEGFR-2.
PDGFs belong to the family of GFs and five isoforms
have been identified to date; PDGF-AA, -AB, -BB, -CC and -DD. These
isoforms play a critical role in stimulating cell growth and
directing changes in cell shape and motility (21). Regarding angiogenesis, they are
critical for pericyte recruitment and differentiation (21). In the tumor microenvironment,
PDGF-BB is widely expressed in ECs and perivascular-like cells, and
there is a strong association between the expression of
angiogenesis-related genes, the infiltration of all
vascular-associated cells including pericytes and the enrichment of
angiogenesis-associated gene sets that play an important role in
tumor angiogenesis (41).
PDGF-BB binds to all PDGF receptors (PDGFR), leading to PDGFR
autophosphorylation, pathway activation and internalization
(42). PDGF-BB not only improves
angiogenesis in bone marrow endothelial progenitor cells (EPCs),
but also effectively promotes human umbilical vein EC (HUVEC)
angiogenesis (43). The Cbl
family is a group of E3 ligases that share the same structural
domain. These molecules consist of an N-terminal tyrosine kinase
binding domain, a linker region, a RING finger domain for
recruitment of the E2 enzyme, a proline-rich unfolded region for
binding to proteins containing the SH3 domain and a C-terminal
Ub-associated domain (44). The
RING finger domain has been implicated in the ubiquitination of
RTKs, including PDGFRs, and stimulation of PDGFR promotes
phosphorylation of Cbl proto-oncogene C (c-Cbl) and their
interactions. In turn, overexpression of c-Cbl accelerates
ligand-induced ubiquitination and subsequent degradation of PDGFR-A
and PDGFR-B, and inhibits PDGF pro-proliferative and
survival-dependent, negative regulation of PDGF-BB-induced
chemotaxis (45,46). The PDGF isoforms also bind to α-
and β-RTKs (PDGFRα and PDGFRβ, respectively) and promote receptor
dimerization, which leads to receptor autophosphorylation and
subsequent ubiquitination and internalization from the cell
membrane (47). PDGFRα and
PDGFRβ signaling play equally important roles in angiogenesis
(48). Inhibition of PDGFRα
phosphorylation inhibits angiogenesis in HUVECs (49), whereas overexpression of PDGFRβ
not only promotes angiogenesis in HUVECs in vitro, but also
regulates tumor angiogenesis (50). The Cbl family plays a key role in
the Ub-mediated internalization of PDGFRβ and PDGFRα. Depletion of
Cbl significantly reduces PDGFRβ ubiquitination (49) and overexpression of Cbl increases
PDGFRα ubiquitination (51).
Most tripartite motif (TRIM) family proteins are a type of E3 Ub
ligase and TRIM proteins are characterized by their N-terminal
TRIM-containing RING finger structural domain, one or two
zinc-finger structural domains named box B (box B1 and box B2), and
the associated coiled-coil regions. The multiple heterogeneous
heterodimeric forms of TRIM proteins may contribute to the
recognition of different substrates and play a role in enzyme
regulation through molecular interactions or dominant negative
effects in enzyme regulation (52). In addition, TRIM21 (an E3 ligase)
also promotes PDGFRβ ubiquitination (53). Thus, the PDGF family members are
primarily associated with pericyte recruitment during angiogenesis,
and the degradation of PDGF requires ubiquitination to promote, so
targeting the ubiquitination degradation process of PDGF may affect
its role in angiogenesis.
Among members of the FGF family, bFGF is the most
well-studied and common pro-angiogenic factor. The expression of
bFGF is increased at sites of chronic inflammation, after tissue
injury and in several types of cancer (54). bFGF exerts its pro-angiogenic
effects through the activation of FGFRs, including FGFR1, -2, -3
and -4 (55). It binds tightly
to the ECM of the vascular basement membrane under physiological
conditions and promotes bFGF production under certain conditions of
the angiogenic process. Certain conditions stimulate the production
of bFGF. For instance, during wound healing, bFGF is released from
the ECM via heparan sulfate-mediated enzymatic degradation. Under
hypoxic conditions, bFGF can exert a remodeling effect on the
perivascular ECM via MMPs (16).
bFGF induces angiogenesis through its effect on SMCs and ECs, and
also induces chemotaxis and proliferation of fibroblasts and ECs
(56). Conversely, bFGF can
indirectly promote angiogenesis at wounds by increasing the
expression levels of VEGF, MMP2 and MMP9 (57). Angiogenesis is associated with
inflammation in a variety of physiological and pathological
conditions, including cancer. bFGF is expressed by inflammatory
cells, and inflammatory mediators activate the synthesis and
release of bFGF from the vascular endothelium, stimulating
angiogenesis by autocrine secretion (54). For instance, the long non-coding
RNA taurine upregulated 1 is ubiquitylated following bFGF
ubiquitination, downregulating bFGF expression, and this ultimately
promotes osteogenic differentiation of tendon stem/progenitor cells
(58). The UPS degradation
pathway is important in controlling bFGF expression in angiogenesis
in the tumor microenvironment. By promoting ubiquitination of bFGF,
bFGF expression is downregulated, ultimately inhibiting
angiogenesis (55,59). However, there are fewer studies
on the role of ubiquitination in bFGF-induced angiogenesis and the
exact mechanism remains to be fully elucidated (60). Studies have confirmed that bFGF
plays an important role in angiogenesis under pathological and
physiological conditions, and the study of the ubiquitination
process of bFGF served as a major breakthrough point for targeting
bFGF to counteract angiogenesis (60,61).
Ang1-4 are important GFs whose activity is mediated
by the RTKs tyrosine kinase with immunoglobulin-like and EGF-like
domains 1 (Tie1) and Tie2. The best-characterized members are Ang1
and Ang2 (62). Ang1 is a potent
angiogenic factor that is essential for angiogenesis in vivo
and functions differently from VEGF (63). Ang1, secreted by perivascular
cells, has been identified as a major ligand for Tie2, and binding
of Ang1 to Tie2 induces several downstream signaling pathways,
primarily regulated by Akt, which induces a pro-angiogenic response
(64). Activation of Ang1-Tie2
signaling promotes interactions between ECs and peri-EC supporting
cells, which stabilizes the vasculature. In addition, Ang1
stimulates EC migration and promotes EC sprouting and tube
formation, which are key events in angiogenesis (65). Ang2 is another key regulator of
angiogenesis and its effect on vascular EC production is relevant
to the local environment. Ang2 binds to Tie2 and acts as a negative
regulator of Ang1-Tie2 signaling during angiogenesis, which in turn
controls the response of ECs to exogenous cytokines. The binding of
Ang2 to VEGF induces vascular sprouting, whereas in the absence of
VEGF, Ang-2 induces EC apoptosis (66). It has been suggested that Ang2
binds to integrins in Tie2 EC and subsequently induces
phosphorylation of the integrin adaptor protein focal adhesion
kinase in a Tie2-independent manner, leading to angiogenesis
(67). Furthermore, in cancer,
the angiogenic process is significantly accelerated when the
Ang2/Ang1 ratio is downregulated (68). During angiogenesis, Ang4
expression in ECs is induced by hypoxia, and Ang4 is required to
maintain the metabolic balance necessary for EC proliferation and
migration. Conditional Ang4 deficiency in vascular ECs in
vivo reduces vascular EC proliferation, thereby reducing
pathological angiogenesis (69).
Therefore, studying the role of Ang regulation during angiogenesis
is of particular importance. Prior to receptor-mediated
endocytosis, RTKs are often modified by the addition of Ub. The
addition of a single Ub molecule to RTKs is sufficient for
receptor-targeted internalization and degradation (70). The E3 Ub ligases known to be
involved in the ubiquitination of most RTKs are members of the Cbl
protein family (71).
Stimulation of Tie2-expressing cells with Ang1 resulted in their
ubiquitination, suggesting that this may be a necessary signal for
receptor conversion. c-Cbl interacts with the Tie2 signaling
complex in a stimulus-dependent manner and this interaction is
required for Tie2 ubiquitination, internalization and degradation
(72). Attenuation of Tie2
trafficking by eliminating Tie2 ubiquitination may result in the
maintenance of the signaling cascade for this receptor on the cell
surface, increasing vascular stability and integrity, and
potentially inhibiting angiogenesis (73). In addition, HECW2 is an EC Ub E3
ligase that plays a key role in stabilizing inter-EC junctions by
regulating the stability of angiomotor protein, and depletion of
HECW2 stimulates Yes-associated protein (YAP) translocation to the
nucleus, thereby promoting EC outgrowth during angiogenesis by
increasing ANG-2 expression (74). In summary, several GFs, including
VEGF, PDGF, FGF and ANG, play key roles in angiogenesis in
vivo. It is clear that ubiquitination plays an important role
in regulating the signaling and trafficking of several GFs in
angiogenesis, and that ubiquitination is an important pathway for
their internalized degradation. The study of the ubiquitination
process is important in the identification of novel targets of GFs
in angiogenesis.
Vascular physiology relies on the synergistic
movements of multiple cell types, including pericytes, ECs and
vascular smooth muscle cells (VSMCs). This interaction between cell
types is inherently dynamic (75). Pericytes are wall cells that
support vascular development, remodeling and homeostasis, and are
implicated in a variety of pathological conditions, including
cancer (76). ECs are the
primary cells responsible for the expansion of the new vasculature.
During angiogenesis, ECs proliferate, migrate against the blood
flow and differentiate into neovascular tips, capillaries and
arterial ECs (77). In addition,
during arteriogenesis, EC channels are covered by pericytes or
VSMCs, providing stability and controlling perfusion (78).
When resting vessels sense angiogenic signals (e.g.
VEGF, VEGF-C, ANG-2, FGF or chemokines) released following hypoxia,
inflammation or by tumor cells, pericytes first detach from the
vessel wall and are released from the basement membrane by
proteolytic degradation (78,79). The currently known effects of
ubiquitination in pericytes are primarily focused on the nervous
system. Brain pericytes surround the ECs of cerebral capillaries
and form the neurovascular unit responsible for maintaining the
integrity of the blood-brain barrier and controlling blood flow
(80). When pericytes are
exposed to Ub-proteasome inhibitors, severe cytotoxicity occurs due
to the production of reactive oxygen species leading to apoptosis
(81). Dynamic interactions
between pericytes and ECs are the basis of angiogenesis. Pericytes
are recruited to developing capillary buds by proliferation,
migrate independently of ECs and can proliferate on growing
capillaries (75). It has been
shown that Japanese encephalitis virus-infected pericytes release
biologically active molecules (such as IL-6) that activate the
Ub-proteasome. While expression of the E3 Ub ligase Ubr1 is
dependent on STAT activity induced by the IL-6/gp130 pathway,
proteasomal degradation of Ubr1 zona occludens 1-expressing
proteasomes is stimulated by IL-6 and leads to disruption of
endothelial barrier integrity (82). NEDD8 is a Ub-like protein and
MLN4924, a small-molecule inhibitor of NEDD8-activating enzymes
that can be activated by inhibiting cullin RING E3 ligase,
preserves chronic hypoperfusion-mediated loss of pericyte coverage
in callosal capillaries (83).
In addition, seven in absentia homolog 1 (Siah1) (an E3
ubiquitination ligase) may be involved in protein ubiquitination
and protease-mediated degradation (84). As pericytes are perivascular
cells closely associated with retinal capillaries, Siah1
association with GAPDH nuclear translocation may partially mediate
pericyte death secondary to high glucose exposure (85). In addition, the effect of
ubiquitination on pericytes has been demonstrated in the tumor
microenvironment. TGF-β induces the transformation of quiescent
pericytes into myofibroblasts, which promote tumor growth and
metastasis, whereas the E3 Ub ligase-SMAD ubiquitination regulatory
factor/recombinant mothers against decapentaplegic homolog 7
(SMURF/SMAD7) complex inhibits tumor growth by antagonizing TGF-β
signaling by promoting ubiquitination of the TGF-β receptor complex
and inhibiting pericyte transformation (86). Furthermore, in non-small cell
lung cancer, glioma-associated oncogene 1 (Gli1) can promote
pericyte and EC motility required for angiogenesis by promoting
bFGF expression, whereas inhibition of the Gli1-bFGF axis markedly
reduces pericyte motility for tumor angiogenesis (55). This shows that the ubiquitination
process has a wide range of effects on pericytes, but the effects
on pericytes during angiogenesis need to be further
investigated.
ECs play an important role in angiogenesis. First,
tightly coordinated activation/inactivation of Rho GTPase
signaling, a key molecule controlling actin dynamics, is essential
for the stabilization, disruption and reconstitution of endothelial
junctions (87). While
localization of Rho GTPases is critical for their activation and
downstream signaling, they are regulated by a variety of PTMs,
including ubiquitination (88).
Signaling by Rac family small GTPase 1 (rac1), a Rho GTPase, is
critical for the regulation of EC diffusion, and there is a strong
positive correlation between rac1 activity and its ubiquitination
level (89). In studies of the
mechanisms of EPC-mediated repair of damaged endothelium, Cbl was
found to increase the ubiquitination of Janus tyrosine kinase 2
(JAK2) and decrease the expression of JAK2 and STAT4, which
increases the expression of Runt-associated transcription factor 3
(Runx3) by regulating the level of histone H3 lysine 4
trimethylation. JAK2 and overexpression of STAT4 or Runx3 increased
apoptosis of HUVECs and abrogated the effect of Cbl on endothelial
function (90). In addition,
ubiquitination modifications play an important role in response to
oxidative stress in ECs, which are likely to be dominated by
certain E3 ligase family members. The E3 Ub ligase Smurf2 regulates
the stability of poly(ADP-ribose) polymerase 1, a highly conserved
protein associated with vascular EC injury, to attenuate oxidative
stress-induced HUVEC injury (91). In addition, E3 Ub protein ligase
2 (WWP2), which contains the WW structural domain, can regulate the
ubiquitination and stability of specific substrate proteins in the
following ways (92). WWP2 has
been shown to regulate angiotensin II-induced oxidative stress in
ECs by inducing ubiquitination and proteasome-mediated degradation
of Septin 4 (93). In addition,
WWP2 attenuates endothelial injury by targeting K63-linked
polyubiquitination and proteasomal degradation (94). It was found that TRIM47E3 (a Ub
ligase), a novel EC activator, can also induce inflammatory
responses in ECs by enhancing K63-linked ubiquitination of tumor
necrosis factor receptor-associated factor (TRAF)2, which in turn
activates NF-κB and MAPK signaling pathways (95). In addition to studies showing the
involvement of ubiquitination in oxidative stress (96) and inflammation (97), it also plays an important role in
cellular senescence. It has been indicated that the E3 Ub ligase
STIP1 homology and U-box containing protein 1 promotes
ubiquitination of basic helix-loop-helix ARNT-like 1, thereby
delaying cellular senescence (98). Inhibition of USP7 (a
deubiquitinating enzyme) via inhibits glycosylation end
product-induced cell cycle arrest and cellular senescence in HUVECs
by promoting p53 ubiquitination (99). In addition, YAP/tafazzin,
phospholipid-lysophospholipid transacylase (TAZ) regulates
sprouting angiogenesis during development and tumor growth
(100). Deletion of endothelial
FAT atypical cadherin 1 (FAT1) interferes with the degradation of
the Hippo signaling pathway effector protein YAP/TAZ, leading to
increased YAP/TAZ protein expression levels and expression of
canonical YAP/TAZ target genes, which, in turn, leads to increased
EC proliferation. By contrast, the E3 Ub ligase mind bomb-(Mib)2, a
FAT1-interacting protein, mediates FAT1-induced ubiquitination and
degradation of YAP/TAZ, leading to this effect (101,102). A study of gallocatechin gallate
(EGCG)-induced changes in the human EC proteome found a significant
increase in Ub-proteasome activity in EGCG-treated cells.
Interestingly, studies on angiogenic properties showed that EGCG
primarily reduced EC proliferation, migration/invasion, endothelial
tube formation, barrier function, transendothelial resistance and
the expression of VEGF-2 (103).
In angiogenesis, the effects of ubiquitination
modifications on VSMCs are primarily observed during
arteriogenesis. During arteriogenesis, collateral vascular function
requires the maintenance of a contractile state of VSMCs to
regulate vascular tone and blood flow, while synthesized VSMCs grow
and remodel in the middle layer of collateral branches to form more
stable ductal arteries (14).
Timely VSMC phenotypic switching requires the coordinated action of
several molecules and cellular mediators, resulting in dilatory
remodeling of the side branches to efficiently restore blood flow
to downstream ischemic tissues (14). Not only does arteriogenesis
require timely VSMC phenotypic switching, but maintenance of normal
VSMC numbers and function is also critical. TRIM is the most
abundant subfamily of the E3 Ub ligase family, and TRIM proteins
are involved in EC injury, inflammation, angiogenesis, oxidative
stress, and SMC proliferation and migration under conditions
associated with vascular disease (104). TRIM65 and TRIM32 are more
recently discovered E3 ligases that interact with and ubiquitinate
a variety of substrates. Overexpression of TRIM65 and TRIM32 was
found to activate PI3K/Akt signaling, leading to loss of the
contractile phenotype of VSMCs, as well as cell proliferation and
migration (105,106). In addition, high expression of
TRIM59 (other TRAM family members) promotes the proliferation of
pulmonary artery SMCs (PASMCs) in pulmonary hypertension (PH), and
this effect can be enhanced by YAP1/TEAD4 (107). In addition, YAP expression was
upregulated and activated in a rat model of MCT-induced PH,
accompanied by PASMC proliferation. Siah2 Ub ligase inhibited
excessive PASMC proliferation by promoting proteasomal degradation,
which, in turn, upregulated YAP expression, reduced its
phosphorylation and promoted its nuclear translocation (108). USPs cleave Ub chains from
specific protein substrates, thereby inhibiting the degradation of
the targeted substrate or modulating its subcellular localization
(109). USP7 was found to
upregulate MDM2, an E3 Ub ligase, to promote ubiquitination of
forkhead box O4 and subsequently increase cyclin D1 expression to
mediate PDGF-induced proliferation of PASMCs (110). In addition to the findings on
PASMCs, the ubiquitination process affects atherosclerotic VSMCs.
For instance, carboxyl terminus of the Hsp70 interacting protein
(CHIP) is a co-chaperone protein that interacts with the C-terminus
of chaperone proteins such as heat shock proteins 70/90, and exerts
E3 ligase activity on its substrate proteins. CHIP interacts with
troponin, reduces troponin stability, inhibits troponin-dependent
SMC differentiation and inhibits troponin-independent SMC
differentiation by promoting Ub-mediated degradation of troponin in
SMC differentiation (111,112). In addition, nuclear receptor 6
subfamily A group member 1 (NR6A1) is involved in promoting the
apoptotic process in VSMCs, and the linear Ub chain assembly
complex inhibits VSMC apoptosis by inhibiting its expression by
promoting linear ubiquitination of NR6A1, leading to
dephosphorylation of amino/threonine protein kinase 3 (113). In addition, by stimulating
arterial SMCs, overexpression of TRIM37 reduced the expression of
B-connexin, c-Myc and cell cycle protein cyclin D1. In addition,
TRM37 inhibits arterial SMC proliferation and invasion by
inhibiting the Wnt/β-catenin signaling pathway (114). TRAF6 maintains VSMC viability
by stimulating cytoskeletal-associated smooth muscle protein Sm22α
ubiquitination, possibly by increasing glucose-6-phosphate
dehydrogenase activity and production (115). However, the above findings in
different VSMCs are not sufficient to address the true regulatory
role of ubiquitination on SMCs during angiogenesis. Therefore, the
regulatory role of ubiquitination on VSMC function in angiogenesis
remains to be validated by further studies.
The vascular ECM consists of the vascular basement
membrane and the interstitium, and is considered an essential
component of the vasculature (116). Interactions between the
cellular matrix and cells to control and influence angiogenesis and
the balanced activity between specific angiogenic molecules that
initiate the process and specific inhibitory molecules that block
the process are hypothesized to be critical for an optimal
angiogenic response. The ECM orchestrates complex signaling
cascades within the cell and influences several fundamental aspects
of its biology, including proliferation, migration, cytoskeletal
organization, cell shape, survival and ultimately vascular
stabilization (116,117). The ECM plays a critical role in
the regulation of vascular processes, which are driven by a variety
of positive and negative regulators. Degradation of the ECM results
in the degradation or partial modification of matrix molecules, the
release of soluble factors and the exposure of cryptic sites with
pro- and/or anti-angiogenic activity. ECM molecules and fragments
produced by proteolytic hydrolysis can also act directly as
inflammatory stimuli, thus exacerbating angiogenesis (117,118). The MMPs are a group of metal
(calcium and zinc) -dependent protein hydrolases that are capable
of degrading the ECM. MMPs are secreted intracellularly and
extracellularly as zymogen pro-MMP, which is activated by a series
of protease cascades in order to become hydrolytically competent.
MMP-2 has been implicated in angiogenesis; it proteolytically
hydrolyses ECM components, causing EC budding, fragmentation and
release of matrix-bound angiogenic factors, and interacts with
integrin αvβ3 (119). JWA is a
multifunctional microtubule-binding protein that is important in
regulating tumor metastasis by inhibiting MMP-2. JWA was found to
inhibit Sp1 activity through a Ub proteasome-dependent mechanism
and downregulate the expression of pro-angiogenic MMP-2, providing
new evidence for the inhibition of tumor angiogenesis in gastric
cancer (120). In addition,
inhibitor of growth family member 4 (ING4), a potential tumor
suppressor, has also been implicated in angiogenesis. ING4 was
found to inhibit the expression and transcriptional activity of Sp1
through Ub degradation and to downregulate the expression of the
pro-angiogenic gene MMP-2 downstream of Sp1, potently inhibiting
angiogenesis in colorectal cancer (121). TRIM25 can also inhibit
angiogenesis in gastric cancer by promoting the ubiquitination of
SP1 at K610, which further inhibits the expression of MMP-2
(122). In addition,
anti-angiotensin small inhibitory RNA reduced endogenous
angiotensin expression in HUVECs, which reduced MMP-2 activity and
expression. Downregulation of angiotensin and its analogs may be
achieved by inhibiting the degradation of the Ub-proteasome pathway
(123). Although, to the best
of our knowledge, to date, no study has directly demonstrated the
involvement of ubiquitination processes in the effects on
angiogenesis, it is clear from the available evidence that
ubiquitination plays a key role in the regulation of the ECM in
angiogenesis under both physiological and pathophysiological
conditions, and this relationship needs to be confirmed by further
studies.
Angiogenesis is a multifactorial process initially
influenced by the disassembly of endothelial junctions, then by the
detachment, proliferation and migration of ECs, and finally by the
re-establishment of cell-cell and cell-matrix contacts (124). Endothelial junctional CAMs,
such as platelet EC adhesion molecule (PECAM) (125) and junctional adhesion molecule
A (126), promote EC-cell
contact through homophilic binding and are involved in the
regulation of EC homeostasis and angiogenesis through various
mechanisms. VCAM-1 plays a role in promoting angiogenesis during
oxidative stress-induced neovascularization. In retinal neovascular
disease, there are interactions between adhesion molecules (VCAM-1
and ICAM-1) and pro-angiogenic factors (e.g. VEGF and PDGF) that
regulate angiogenesis (127).
Interestingly, ubiquitination of several CAMs associated with
angiogenesis has been identified. Membrane-associated ring-CH-type
finger IX, an E3 ligase, controls the expression of ICAM-1, a key
CAM (128). In addition, the E3
ligase TRIM65 selectively targets VCAM-1 and promotes its
ubiquitination and degradation (129). PECAM-1 may mediate the response
to vascular remodeling and collateral vessel formation during
angiogenesis by sensing shear stress (130). The cell cyclosome (APC) can
catalyze the ubiquitination of key molecules to regulate cell cycle
progression. APC/CDh1 (chromodomain helicase DNA binding protein,
one of the co-activators of APC) has been shown to promote
K48-linked polyubiquitination of PECAM-1, which maintains EC
function by degrading PECAM-1 under pulsatile shear stress
(131). In addition,
calcineurin 4, a major adhesion molecule in adherens junctions, can
induce angiogenesis in papillary thyroid carcinoma by inhibiting
the E3 Ub ligase β-TrCP-dependent ubiquitination of β-catenin, an
important structural component of the calcineurin adherens junction
(132). Thus, it is evident
that different types of CAMs affect the angiogenic process in a
similar manner, primarily through their effects on the endothelium,
and the role and mechanism of ubiquitination in this process need
to be verified by further studies.
Angiogenesis is a multi-step process that requires
the involvement of several biological signals and stimuli under
both physiological and pathological conditions. For example,
vascular injury, occlusion and reduced blood flow can lead to a
reduction in oxygen supply, a state known as hypoxia. Hypoxia is a
potent angiogenic trigger that stimulates the activity of
pro-angiogenic factors (133).
HIF is an important transcription factor of the cellular response
to changes in oxygen concentration. Under hypoxic conditions, HIF
is activated, inducing the expression of numerous genes necessary
for adaptation to hypoxia (134). HIF signaling mediates the
response to hypoxia by cell-autonomous mechanisms, with
non-autonomous effects on angiogenesis. The expression of
HIF-stimulated pro-angiogenic factors (e.g. VEGF, VEGFR, MMPs and
IL-8) is critical for angiogenesis in physiological and
pathophysiological settings (135,136). In eukaryotic cells, HIF-1 is a
major transcriptional mediator of the hypoxic response and a key
regulator of oxygen homeostasis. Under normoxic conditions,
hydroxylation of two proline residues and acetylation of one lysine
residue on the oxygen-dependent degradation structural domain of
HIF-1 triggers its binding to the von Hippel-Lindau (VHL) E3 ligase
complex, leading to degradation of HIF-1 via the Ub-proteasome
pathway. Under hypoxic conditions, the HIF-1 subunit is stabilized
and interacts with coactivators to regulate target gene expression
(134,137). Due to the rapid degradation of
the HIF-1α subunit via the proteasome pathway, its stability is
markedly reduced under normoxic conditions. In addition to hypoxia,
ubiquitination can also regulate the stability and transcriptional
activity of HIF-1α. In addition to the oxygen-dependent Ub E3
ligase VHL, which is involved in the oxygen-dependent regulation of
HIF degradation, there are also E3 Ub ligases involved in the
oxygen-dependent regulation of HIF-1α degradation, such as receptor
for activated C kinase 1, hypoxia-associated factor, TRAF6 and CHIP
(138). Specifically,
ubiquitination modifications are involved in multiple processes
that are affected by hypoxia during angiogenesis. For instance, the
transcriptional co-activator YAP1 is an important oncogenic
component of the Hippo signaling pathway that contributes to the
development and progression of several tumors (139). Induction of tumor angiogenesis
is primarily mediated by HIF, particularly HIF-1α (140). There may be a novel means by
which YAP1 is regulated under normal hypoxic conditions; for
example, it may interact with prolyl hydroxylase 2 (PHD2). This
leads to the hydroxylation of specific proline residues, which in
turn leads to its interaction with VHL and degradation by the
proteasome. Under hypoxic conditions, the interaction of YAP1 with
PHD2 and VHL is lost, and YAP1 interacts with HIF-1α and/or E2F1,
inducing a variety of genes involved in the angiogenic process,
including VEGF, as well as MMPs (141). In addition, leucine-rich repeat
protein 16 in breast cancer (142), ETS transcription factor ELK3 in
glioma (143), homeodomain
interacting protein kinase 2 in hepatocellular carcinoma (144) and histone deacetylase 1 in
colorectal cancer (145) can
directly bind to HIF-1α and induce its ubiquitination and
degradation, thereby controlling tumor angiogenesis. The inhibitory
member of ASPP of p53 family can bind directly to the β-structural
domain of VHL, which is also involved in the binding of HIF-1α, and
thus block the binding of VHL and therefore the degradation of the
HIF-1α protein in the normoxic state (146). In summary, the effect of HIF-1α
on angiogenesis in tumor cells may be influenced by the presence of
VEGF and regulated by the ubiquitination process. In addition to
the above findings in tumor cells, in a model of retinopathy of
prematurity, the circular RNA of phosphodiesterase 4B can inhibit
VEGF-A expression and ultimately pathological angiogenesis by
promoting VHL-mediated Ub degradation of HIF-1α (147). Taken together, these findings
lead to the conclusion that HIF orchestrates the angiogenic process
by participating in each step of angiogenesis and that
ubiquitination modifications play an important role in this
process. In addition, the interaction between HIF-1 and
proangiogenic factors is fundamental in the angiogenic process
under hypoxic conditions.
The signaling molecules Wnt and Notch are important
in angiogenesis. Wnt signaling can regulate angiogenesis through
cell proliferation and migration, and Notch signaling can regulate
the transcription of a series of genes involved in angiogenesis.
Notch signaling also plays a crucial role in the recruitment and
tight interaction of vascular ECs with pericytes and VSMCs. In
addition, Wnt and Notch are the most important signaling pathways
and downstream transcription factors determining EC arteriovenous
differentiation, driving endothelial progenitor cell
differentiation to arterial or venous EC through the coordinated
action of different family members (148,149). More importantly, tip cell
migration and stem cell proliferation require the involvement of
Notch and Wnt signaling. Cells neighboring the tip cells occupy
auxiliary positions as stem cells, which divide to lengthen the
stem (stimulated by Notch, Wnt and other signals) and establish the
lumen. Tip cells are equipped with filamentous pseudopods to sense
environmental guidance cues, while stem cells convey spatial
information about the position of their neighbors, thereby allowing
stem elongation (150).
Therefore, PTMs of receptors and ligands ensure precise signaling
regulation. From receptor synthesis to maturation, ubiquitination
plays a role in a series of processes to ensure the correct outcome
of signal transduction (151).
Wnt signaling controls a wide range of cellular
functions, including cell proliferation and migration. Wnt
signaling can be divided into classical/β-catenin-dependent
pathways and non-classical pathways (including Wnt signaling
through Ca2+, planar cell polarity and other signaling
mechanisms that do not involve β-catenin), which both regulate and
control angiogenesis (152,153). Wnt ligands trigger these
pathways by binding to appropriate receptors. These signaling
pathways are triggered by Wnt ligands by binding to appropriate
receptors, which belong to the family of seven-pass transmembrane
proteins called Frizzled. Activation of certain Wnt signaling
pathways requires a co-receptor, LDL receptor-related protein
(LRP)5/6, which is blocked when LRP5/6 binds to the secreted
inhibitory protein Dickkopf-1 (154). The binding of Wnt to these
receptors has now been shown to play a role in angiogenesis in
human ocular vascular disease (149). During tumor angiogenesis,
overactivation of the Wnt/β-catenin signaling pathway consistently
induces upregulation of pro-angiogenic factors (VEGF-A and VEGF-C)
(155). In addition to the
classical signaling pathways, non-classical signaling pathways also
play an important role in angiogenesis. Exogenous Wnt family member
5A (Wnt5a) expression in vascular EC can promote angiogenesis by
inducing the proliferation and survival of vascular ECs in
vitro. The underlying mechanism is related to regulation of
MMP-1 and Tie2 by the non-classical Wnt5a signaling pathway. In
pathological angiogenesis, macrophage-derived Wnt5a can stimulate
angiogenesis through inflammatory responses. Wnt5a released by
infiltrating monocytes is particularly important in driving
angiogenesis in inflammatory vessels (156). In addition to Wnt5a, Wnt1 and
Wnt3a have been shown to control EC migration and proliferation
(157). Secreted
frizzled-related protein can activate the Wnt/Ca2+
pathway to stimulate angiogenesis in tumors (158). Various signaling factors have
been shown to mediate the up- or downregulation of Wnt signaling
through PTMs. Among the numerous PTMs involved, most Wnt signaling
factors are regulated by ubiquitination and deubiquitination.
Ubiquitination by E3 ligases attaches Ub to target proteins and
usually induces proteasomal degradation of Wnt signaling factors
such as β-catenin, Axin and glycogen synthase kinase (GSK)3
(159-162). In the Wnt/β-catenin signaling
pathway, the Wnt ligand is the initiator, while β-catenin is the
key mediator. In the absence of the Wnt ligand, cytoplasmic
β-catenin is continuously phosphorylated by the destruction
complex. Phosphorylated β-catenin is recognized by the E3 Ub
ligase, β-TrCP, and then degraded by the proteasomal complex to
reduce nuclear translocation of β-catenin (163). Axin is recognized by four
proteasomal ligands, Siah-1, ring finger protein (RNF)146, Smurf1
and Smurf2, and four other E3 ligases. Ubiquitination of Siah-1 is
achieved by binding to the VxP motif of Axin, which is a positive
regulator of the Wnt signaling pathway. RNF146 induces degradation
and facilitates K48-linked ubiquitination of Axin. Smurf1
ubiquitination occurs at residue K505 of Axin, leading to its
degradation, while Smurf1 ubiquitination does not lead to Axin
degradation, but interferes with the interaction between Axin and
LRP6, acting as a negative regulator of the Wnt pathway (159,160). GSK3κ is a negative regulator of
the Wnt/κ-c-catenin pathway and activates the Wnt/κ-c-catenin
pathway by promoting the ubiquitinated degradation of GSK3κ,
thereby stimulating angiogenesis in tumors (164). In the absence of Wnt ligands
(Wnt-off phase), regulation of transcriptionally active nuclear
catenin is essential to ensure controlled induction of Wnt target
genes (165). In ECs, active
catenin is ubiquitinated by c-Cbl, and Wnt activation promotes
c-Cbl phosphorylation at tyrosine 731 (Y731), which increases c-Cbl
dimerization and binding of Y731 to β-catenin. Y731 phosphorylation
and dimerization mediates the nuclear translocation of c-Cbl and
leads to the degradation of nuclear-active β-catenin in the Wnt-on
phase. In the Wnt-off phase, termination of dimerization then
disrupts the binding of 731 to β-catenin (166). The activity of c-Cbl also
inhibits the pro-angiogenic Wnt targets IL-8 and VEGF levels and
inhibits angiogenesis independently of β-catenin (167). In colorectal cancer, regulation
of nuclear β-catenin by c-Cbl requires phosphorylation of c-Cbl at
Tyr371 and the Y371H mutant interacts with nuclear β-catenin but
does not ubiquitinate it. The nuclear localization of c-Cbl Y371H
mutants contributes to their dominant negative effect on nuclear
β-catenin (168). LRP5 is an
essential coreceptor in the Wnt/β-catenin signaling pathway.
sp90ab1 exerts these functions by interacting with LRP5 and
inhibiting Ub-mediated degradation of LRP5 (169). In addition, YAP is essential
for ubiquitination and proteasomal degradation of β-catenin. In the
cytoplasm, YAP directly interacts with β-catenin and restricts its
nuclear translocation. YAP is also critical for the recruitment of
β-TrCP to the destruction complex, promoting the degradation of
β-catenin (170). Given the
potent angiogenic activity of Wnt signaling, components of the Wnt
pathway have long been recognized as important targets in the
control of angiogenesis. Further studies on the regulation of Wnt
by ubiquitination may help to identify drug targets that are not
limited to angiogenesis in tumor pathology.
The Notch signaling pathway is a highly
evolutionarily conserved signaling mechanism that controls cell
fate specification, tissue morphology, cell proliferation, survival
and differentiation in a wide range of cell types in all
multicellular organisms (171).
Notch ligands can be structurally classified into two groups:
δ-like ligands (δ-like 1, 3 and 4) and serrate-like ligands
(Jagged-1 and -2). Both classes of ligands are transmembrane
proteins with numerous tandem EGF-like repeats in their
extracellular structural domains and unique δ-, Serrate- and
Jagged-2-binding structural domains at the amino terminus, which
are required for receptor interaction (172). Classical Notch signaling
requires the interaction of Notch ligands on the membrane of the
cell sending the signal with Notch receptors on the cell receiving
the signal, which induces Notch receptor protein hydrolysis
(173). Notch signaling also
plays an important and complex role in angiogenesis. It has been
suggested that Notch signaling may regulate angiogenesis directly
or indirectly through VEGFR (174). There appears to be a feedback
loop between Notch signaling and VEGF, in which Notch signaling is
downstream of VEGF, and when Notch signaling is activated, it
downregulates the expression of VEGFR2. Increased δ-like 4/Notch
signaling results in transcriptional repression of VEGFR2 and its
co-receptor neuropilin 1 (175). δ-like 4 is expressed in ECs and
is associated with tip and stem-cell differentiation during
angiogenesis. δ-like 4 acts as a negative regulator to inhibit EC
growth, proliferation and metastasis, and δ-like 4 upregulation
inhibits excessive vessel sprouting and branching (176). In addition, the ligand Jagged
1, a potent pro-angiogenic regulator, antagonizes δ-like 4-mediated
Notch signaling in angiogenesis (177). The effect of Notch on
angiogenesis depends on the interplay between multiple angiogenic
pathways. The Ang-1/Tie-2 pathway is an important angiogenic
signaling pathway that enhances Notch signaling through
Akt-mediated β-catenin activation. β-catenin activation enhances
Notch signaling to regulate vasoreactivity (178). The magnitude and duration of
the Notch response depend on the PTMs of the activated Notch
receptor-Notch intracellular structural domain (NICD). Factor
inhibiting HIF hydroxylates NICD1 under normoxic conditions,
leading to USP10 recruitment and subsequent deubiquitination and
stabilization of NICD1. Under hypoxic conditions, this regulatory
loop is disrupted, resulting in an attenuated Notch response
(179). In addition, the cell
fate determinant NUMB endocytic adaptor protein reduces Notch
signaling by inhibiting the ubiquitination of the intracellular
structural domain of Notch1. The E3 ligase Mdm2 can also bind to
Notch1 through the RAM and ANK repeat regions of Notch1 and
ubiquitinate Notch1 to promote its stability and signaling
activity. It can also ubiquitinate NUMB, a negative regulator of
Notch, for proteasomal degradation (151). The mature Notch receptor on the
cell membrane is partly bound to ligands via the classical pathway
and partly endocytosed, degraded in lysosomes or activated in
nuclear endosomes (ligand-independent) to participate in
non-classical pathways (180).
By contrast, the E3 Ub ligases Deltex (Itch-associated protein) and
suppressor of dx (mammalian Itch) ubiquitinate the intracellular
structural domains of Notch, facilitating the process of
endocytosis and thereby activating the non-classical pathway of
Notch signaling (181). Typical
δ/Serrate/LAG-2 (DSL) ligands are the most frequent activators
accounting for the major inducer of Notch signaling. Two distinct
families of RING-containing E3 ligases, Neuralised (Neur1 and -2)
and Mib1 and -2 in mammals, directly promote mono-ubiquitination of
DSL ligands and are also required for DSL ligand endocytosis
(182,183). In addition, F-box and WD repeat
domain containing 7 acts as a substrate recognition element for
S-phase kinase-associated protein 1, cullin (CUL)1, protein F
(SCF)-type E3 Ub ligases and is a potent positive regulator of
angiogenesis, limiting Notch activity in the endothelium of growing
vessels (184).
BCL-6-associated zinc finger protein has been found to bind to the
VEGF-A-induced Notch signaling factor CBF1 and promote degradation
of CBF1 through polyubiquitination in the CBF1-cullin3 (CUL3) E3
ligase complex to downregulate Notch signaling (185). The mechanisms of Notch
regulation in angiogenesis are complex and ubiquitination as a
substrate recognition element is a potent positive regulator of
Notch activity in the growing vascular endothelium. The regulatory
mechanisms of Notch in angiogenesis are also complex, and
ubiquitination acts at multiple levels of the Notch pathway and
plays a critical role in maintaining Notch, its ligands, and the
signaling activity. It is therefore particularly important to
understand how ubiquitination leads to angiogenesis by affecting
the Notch signaling pathway (Fig.
4, Table I).
Angiogenesis is an important physiological process
involved in development and wound healing, as well as in
pathological states, such as tumor growth, diabetes and ischemic
heart disease. Studying the mechanisms and factors involved in this
process is a major challenge, as angiogenesis is not limited to the
imbalance between pro- and anti-angiogenic factors, but also
involves the complex interplay of several signaling pathways that
regulate these factors in different environments. As our
understanding of cellular differentiation and tissue structure
continues to expand, the study of in vitro models of
angiogenesis has become more widespread. Through the use of in
vivo and in vitro models, it has been established that
several interrelated molecular pathways regulate angiogenesis,
including angiogenic signaling, vascular cell stabilization and
homeostasis, the cellular matrix and intercellular interactions. In
addition, PTMs, including ubiquitination, tightly regulate these
mechanisms and play a key role in controlling this process.
Therefore, ubiquitination plays an important role in the regulation
of angiogenesis, and understanding its mechanism is important in
developing novel treatments for angiogenesis-related diseases.
However, ubiquitination remains to be fully elucidated and its
involvement in the regulation of angiogenesis remains largely
uncharacterized. Unraveling the role of this important PTM and its
contribution to angiogenesis has also become a major challenge.
Further efforts are required to overcome this challenge to gain a
more complete understanding.
Not applicable.
TC and KW contributed to the conception of the
article, wrote the manuscript and revised the manuscript. ZS
contributed to the conception of the article and revised the
manuscript. All authors contributed to the review and have read and
approved the submitted version. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
No funding was received.
|
1
|
Qin W, Steinek C, Kolobynina K, Forné I,
Imhof A, Cardoso MC and Leonhardt H: Probing protein ubiquitination
in live cells. Nucleic Acids Res. 50:e1252022.
|
|
2
|
Popovic D, Vucic D and Dikic I:
Ubiquitination in disease pathogenesis and treatment. Nat Med.
20:1242–1253. 2014.
|
|
3
|
Xu G and Jaffrey SR: The new landscape of
protein ubiquitination. Nat Biotechnol. 29:1098–1100. 2011.
|
|
4
|
Lacoursiere RE, Hadi D and Shaw GS:
Acetylation, phosphorylation, ubiquitination (Oh My!): Following
post-translational modifications on the ubiquitin road.
Biomolecules. 12:4672022.
|
|
5
|
Rape M: Ubiquitylation at the crossroads
of development and disease. Nat Rev Mol Cell Biol. 19:59–70.
2018.
|
|
6
|
Rieger J, Kaessmeyer S, Al Masri S,
Hünigen H and Plendl J: Endothelial cells and angiogenesis in the
horse in health and disease-A review. Anat Histol Embryol.
49:656–678. 2020.
|
|
7
|
Akbarian M, Bertassoni LE and Tayebi L:
Biological aspects in controlling angiogenesis: current progress.
Cell Mol Life Sci. 79:3492022.
|
|
8
|
Francescone R and Vendramini-Costa DB: In
vitro models to study angiogenesis and vasculature. Methods Mol
Biol. 2514:15–28. 2022.
|
|
9
|
Ahmad A and Nawaz MI: Molecular mechanism
of VEGF and its role in pathological angiogenesis. J Cell Biochem.
123:1938–1965. 2022.
|
|
10
|
Mezu-Ndubuisi OJ and Maheshwari A: The
role of integrins in inflammation and angiogenesis. Pediatr Res.
89:1619–1626. 2021.
|
|
11
|
Li W, Wen L, Rathod B, Gingras AC, Ley K
and Lee HS: Kindlin2 enables EphB/ephrinB bi-directional signaling
to support vascular development. Life Sci Alliance.
6:e2022018002022.
|
|
12
|
Rabquer BJ, Amin MA, Teegala N, Shaheen
MK, Tsou PS, Ruth JH, Lesch CA, Imhof BA and Koch AE: Junctional
adhesion molecule-C is a soluble mediator of angiogenesis. J
Immunol. 185:1777–1785. 2010.
|
|
13
|
Rizzi A, Benagiano V and Ribatti D:
Angiogenesis versus arteriogenesis. Rom J Morphol Embryol.
58:15–19. 2017.
|
|
14
|
Ashraf JV and Al Haj Zen A: Role of
vascular smooth muscle cell phenotype switching in arteriogenesis.
Int J Mol Sci. 22:105852021.
|
|
15
|
Liu Y, Yang Y, Wang Z, Fu X, Chu XM, Li Y,
Wang Q, He X, Li M, Wang K, et al: Insights into the regulatory
role of circRNA in angiogenesis and clinical implications.
Atherosclerosis. 298:14–26. 2020.
|
|
16
|
Lugano R, Ramachandran M and Dimberg A:
Tumor angiogenesis: Causes, consequences, challenges and
opportunities. Cell Mol Life Sci. 77:1745–1770. 2020.
|
|
17
|
Liu ZL, Chen HH, Zheng LL, Sun LP and Shi
L: Angiogenic signaling pathways and anti-angiogenic therapy for
cancer. Signal Transduct Target Ther. 8:1982023.
|
|
18
|
Anderson NM and Simon MC: The tumor
microenvironment. Curr Biol. 30:R921–R925. 2020.
|
|
19
|
Maxwell PH and Ratcliffe PJ: Oxygen
sensors and angiogenesis. Semin Cell Dev Biol. 13:29–37. 2002.
|
|
20
|
Bui QT, Hong JH, Kwak M, Lee JY and Lee
PC: Ubiquitin-conjugating enzymes in cancer. Cells.
10:13832021.
|
|
21
|
Omorphos NP, Gao C, Tan SS and Sangha MS:
Understanding angiogenesis and the role of angiogenic growth
factors in the vascularization of engineered tissues. Mol Biol Rep.
48:941–950. 2021.
|
|
22
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1027. 2005.
|
|
23
|
Ferrara N: Vascular endothelial growth
factor: Basic science and clinical progress. Endocr Rev.
25:581–611. 2004.
|
|
24
|
Shah AA, Kamal MA and Akhtar S: Tumor
Angiogenesis and VEGFR-2: Mechanism, pathways and current
biological therapeutic interventions. Curr Drug Metab. 22:50–59.
2021.
|
|
25
|
Rahimi N and Costello CE: Emerging roles
of post-translational modifications in signal transduction and
angiogenesis. Proteomics. 15:300–309. 2015.
|
|
26
|
Rahimi N: The ubiquitin-proteasome system
meets angiogenesis. Mol Cancer Ther. 11:538–548. 2012.
|
|
27
|
Han D, Wang L, Jiang S and Yang Q: The
ubiquitin-proteasome system in breast cancer. Trends Mol Med.
29:599–621. 2023.
|
|
28
|
Meissner M, Reichenbach G, Stein M,
Hrgovic I, Kaufmann R and Gille J: Down-regulation of vascular
endothelial growth factor receptor 2 is a major molecular
determinant of proteasome inhibitor-mediated antiangiogenic action
in endothelial cells. Cancer Res. 69:1976–1984. 2009.
|
|
29
|
Meyer RD, Srinivasan S, Singh AJ, Mahoney
JE, Gharahassanlou KR and Rahimi N: PEST motif serine and tyrosine
phosphorylation controls vascular endothelial growth factor
receptor 2 stability and downregulation. Mol Cell Biol.
31:2010–2025. 2011.
|
|
30
|
Xu D, Wu J, Dong L, Luo W, Li L, Tang D
and Liu J: Serpinc1 acts as a tumor suppressor in hepatocellular
carcinoma through inducing apoptosis and blocking macrophage
polarization in an ubiquitin-proteasome manner. Front Oncol.
11:7386072021.
|
|
31
|
Wiszniak S and Schwarz Q: Exploring the
intracrine functions of VEGF-A. Biomolecules. 11:1282021.
|
|
32
|
Wang Y and Yang C: Enhanced VEGF-A
expression and mediated angiogenic differentiation in human
gingival fibroblasts by stimulating with TNF-α in vitro. J Dent
Sci. 17:876–881. 2022.
|
|
33
|
Watari K, Shibata T, Fujita H, Shinoda A,
Murakami Y, Abe H, Kawahara A, Ito H, Akiba J, Yoshida S, et al:
NDRG1 activates VEGF-A-induced angiogenesis through PLCγ1/ERK
signaling in mouse vascular endothelial cells. Commun Biol.
3:1072020.
|
|
34
|
Husain A, Khadka A, Ehrlicher A,
Saint-Geniez M and Krishnan R: Substrate stiffening promotes VEGF-A
functions via the PI3K/Akt/mTOR pathway. Biochem Biophys Res
Commun. 586:27–33. 2022.
|
|
35
|
Critchley WR, Smith GA, Zachary IC,
Harrison MA and Ponnambalam S: The E2 ubiquitin-conjugating enzymes
UBE2D1 and UBE2D2 regulate VEGFR2 dynamics and endothelial
function. J Cell Sci. 136:jcs2606572023.
|
|
36
|
Smith GA, Fearnley GW, Abdul-Zani I,
Wheatcroft SB, Tomlinson DC, Harrison MA and Ponnambalam S:
Ubiquitination of basal VEGFR2 regulates signal transduction and
endothelial function. Biol Open. 6:1404–1415. 2017.
|
|
37
|
Murakami T, Felinski EA and Antonetti DA:
Occludin phosphorylation and ubiquitination regulate tight junction
trafficking and vascular endothelial growth factor-induced
permeability. J Biol Chem. 284:21036–21046. 2009.
|
|
38
|
Shaik S, Nucera C, Inuzuka H, Gao D,
Garnaas M, Frechette G, Harris L, Wan L, Fukushima H, Husain A, et
al: SCF(β-TRCP) suppresses angiogenesis and thyroid cancer cell
migration by promoting ubiquitination and destruction of VEGF
receptor 2. J Exp Med. 209:1289–1307. 2012.
|
|
39
|
Tian X, Chen Y, Peng Z, Lin Q and Sun A:
NEDD4 E3 ubiquitin ligases: Promising biomarkers and therapeutic
targets for cancer. Biochem Pharmacol. 214:1156412023.
|
|
40
|
Murdaca J, Treins C, Monthouël-Kartmann
MN, Pontier-Bres R, Kumar S, Van Obberghen E and Giorgetti-Peraldi
S: Grb10 prevents Nedd4-mediated vascular endothelial growth factor
receptor-2 degradation. J Biol Chem. 279:26754–26761. 2004.
|
|
41
|
Wu R, Gandhi S, Tokumaru Y, Asaoka M, Oshi
M, Yan L, Ishikawa T and Takabe K: Intratumoral PDGFB gene
predominantly expressed in endothelial cells is associated with
angiogenesis and lymphangiogenesis, but not with metastasis in
breast cancer. Breast Cancer Res Treat. 195:17–31. 2022.
|
|
42
|
Liu W, Guo S, Tang Z, Wei X, Gao P, Wang
N, Li X and Guo Z: Magnesium promotes bone formation and
angiogenesis by enhancing MC3T3-E1 secretion of PDGF-BB. Biochem
Biophys Res Commun. 528:664–670. 2020.
|
|
43
|
Kim DY, Park G, Hong HS, Kim S and Son Y:
Platelet-derived growth factor-BB priming enhances vasculogenic
capacity of bone marrow-derived endothelial precursor like cells.
Tissue Eng Regen Med. 20:695–704. 2023.
|
|
44
|
Pinilla-Macua I and Sorkin A: Cbl and
Cbl-b independently regulate EGFR through distinct receptor
interaction modes. Mol Biol Cell. 34:ar1342023.
|
|
45
|
Tang R, Langdon WY and Zhang J: Negative
regulation of receptor tyrosine kinases by ubiquitination: Key
roles of the Cbl family of E3 ubiquitin ligases. Front Endocrinol
(Lausanne). 13:9711622022.
|
|
46
|
Rorsman C, Tsioumpekou M, Heldin CH and
Lennartsson J: The ubiquitin ligases c-Cbl and Cbl-b negatively
regulate platelet-derived growth factor (PDGF) BB-induced
chemotaxis by affecting PDGF receptor β (PDGFRβ) internalization
and signaling. J Biol Chem. 291:11608–11618. 2016.
|
|
47
|
Wang K, Papadopoulos N, Hamidi A,
Lennartsson J and Heldin CH: SUMOylation of PDGF receptor α affects
signaling via PLCγ and STAT3, and cell proliferation. BMC Mol Cell
Biol. 24:192023.
|
|
48
|
Tsioumpekou M, Cunha SI, Ma H, Åhgren A,
Cedervall J, Olsson AK, Heldin CH and Lennartsson J: Specific
targeting of PDGFRβ in the stroma inhibits growth and angiogenesis
in tumors with high PDGF-BB expression. Theranostics. 10:1122–1135.
2020.
|
|
49
|
Lv F, Li X and Wang Y: Lycorine inhibits
angiogenesis by docking to PDGFRα. BMC Cancer. 22:8732022.
|
|
50
|
Sang BT, Wang CD, Liu X, Guo JQ, Lai JY
and Wu XM: PDGF-BB/PDGFRβ induces tumour angiogenesis via enhancing
PKM2 mediated by the PI3K/AKT pathway in Wilms' tumour. Med Oncol.
40:2402023.
|
|
51
|
Miyake S, Lupher ML Jr, Druker B and Band
H: The tyrosine kinase regulator Cbl enhances the ubiquitination
and degradation of the platelet-derived growth factor receptor
alpha. Proc Natl Acad Sci USA. 95:7927–7932. 1998.
|
|
52
|
Hatakeyama S: TRIM family proteins: Roles
in autophagy, immunity, and carcinogenesis. Trends Biochem Sci.
42:297–311. 2017.
|
|
53
|
Sarri N, Papadopoulos N, Lennartsson J and
Heldin CH: The E3 ubiquitin ligase TRIM21 regulates basal levels of
PDGFRβ. Int J Mol Sci. 24:77822023.
|
|
54
|
Zahra FT, Sajib MS and Mikelis CM: Role of
bFGF in acquired resistance upon anti-VEGF therapy in cancer.
Cancers (Basel). Cancer (Basel). 13:14222021.
|
|
55
|
Lei X, Li Z, Huang M, Huang L, Huang Y, Lv
S, Zhang W, Chen Z, Ke Y, Li S, et al: Gli1-mediated tumor
cell-derived bFGF promotes tumor angiogenesis and pericyte coverage
in non-small cell lung cancer. J Exp Clin Cancer Res.
43:832024.
|
|
56
|
Przybylski M: A review of the current
research on the role of bFGF and VEGF in angiogenesis. J Wound
Care. 18:516–519. 2009.
|
|
57
|
Li L, Ma Q, Mou J, Wang M, Ye J and Sun G:
Basic fibroblast growth factor gel preparation induces angiogenesis
during wound healing. Int J Artif Organs. 46:171–181. 2023.
|
|
58
|
Yu Y, Chen Y, Zheng YJ, Weng QH, Zhu SP
and Zhou DS: LncRNA TUG1 promoted osteogenic differentiation
through promoting bFGF ubiquitination. In Vitro Cell Dev Biol Anim.
56:42–48. 2020.
|
|
59
|
Sukhthankar M, Yamaguchi K, Lee SH,
McEntee MF, Eling TE, Hara Y and Baek SJ: A green tea component
suppresses post-translational expression of basic fibroblast growth
factor in colorectal cancer. Gastroenterology. 134:1972–1980.
2008.
|
|
60
|
Wang R, Ma Y, Zhan S, Zhang G, Cao L,
Zhang X, Shi T and Chen W: B7-H3 promotes colorectal cancer
angiogenesis through activating the NF-κB pathway to induce VEGFA
expression. Cell Death Dis. 11:552020.
|
|
61
|
Xiong Z, Xu X, Zhang Y, Ma C, Hou C, You
Z, Shu L, Ke Y and Liu Y: IFITM3 promotes glioblastoma stem
cell-mediated angiogenesis via regulating JAK/STAT3/bFGF signaling
pathway. Cell Death Dis. 15:452024.
|
|
62
|
Akwii RG, Sajib MS, Zahra FT and Mikelis
CM: Role of Angiopoietin-2 in vascular physiology and
pathophysiology. Cells. 8:4712019.
|
|
63
|
Skóra JP, Antkiewicz M, Kupczyńska D,
Kulikowska K, Strzelec B, Janczak D and Barć P: Local intramuscular
administration of ANG1 and VEGF genes using plasmid vectors
mobilizes CD34+ cells to peripheral tissues and promotes
angiogenesis in an animal model. Biomed Pharmacother.
143:1121862021.
|
|
64
|
Zhou H, Chen T, Li Y, You J, Deng X, Chen
N, Li T, Zheng Y, Li R, Luo M, et al: Glycation of Tie-2 inhibits
angiopoietin-1 signaling activation and angiopoietin-1-induced
angiogenesis. Int J Mol Sci. 23:71372022.
|
|
65
|
Pan L, Liu Z, Chen Y, Yang B and Cheng B:
Angiopoietin-1: Can be produced by endothelial cells and act in an
autocrine agonistic manner? Clin Hemorheol Microcirc. 74:341–345.
2020.
|
|
66
|
Scholz A, Plate KH and Reiss Y:
Angiopoietin-2: A multifaceted cytokine that functions in both
angiogenesis and inflammation. Ann N Y Acad Sci. 1347:45–51.
2015.
|
|
67
|
Felcht M, Luck R, Schering A, Seidel P,
Srivastava K, Hu J, Bartol A, Kienast Y, Vettel C, Loos EK, et al:
Angiopoietin-2 differentially regulates angiogenesis through TIE2
and integrin signaling. J Clin Invest. 122:1991–2005. 2012.
|
|
68
|
Vimalraj S: A concise review of VEGF,
PDGF, FGF, Notch, angiopoietin, and HGF signalling in tumor
angiogenesis with a focus on alternative approaches and future
directions. Int J Biol Macromol. 221:1428–1438. 2022.
|
|
69
|
Chaube B, Citrin KM, Sahraei M, Singh AK,
de Urturi DS, Ding W, Pierce RW, Raaisa R, Cardone R, Kibbey R, et
al: Suppression of angiopoietin-like 4 reprograms endothelial cell
metabolism and inhibits angiogenesis. Nat Commun. 14:82512023.
|
|
70
|
Thien CB and Langdon WY: Negative
regulation of PTK signalling by Cbl proteins. Growth Factors.
23:161–167. 2005.
|
|
71
|
Mohapatra B, Ahmad G, Nadeau S, Zutshi N,
An W, Scheffe S, Dong L, Feng D, Goetz B, Arya P, et al: Protein
tyrosine kinase regulation by ubiquitination: critical roles of
Cbl-family ubiquitin ligases. Biochim Biophys Acta. 1833:122–139.
2013.
|
|
72
|
Wehrle C, Van Slyke P and Dumont DJ:
Angiopoietin-1-induced ubiquitylation of Tie2 by c-Cbl is required
for internalization and degradation. Biochem J. 423:375–380.
2009.
|
|
73
|
Augustin HG, Koh GY, Thurston G and
Alitalo K: Control of vascular morphogenesis and homeostasis
through the angiopoietin-Tie system. Nat Rev Mol Cell Biol.
10:165–177. 2009.
|
|
74
|
Choi KS, Choi HJ, Lee JK, Im S, Zhang H,
Jeong Y, Park JA, Lee IK, Kim YM and Kwon YG: The endothelial E3
ligase HECW2 promotes endothelial cell junctions by increasing
AMOTL1 protein stability via K63-linked ubiquitination. Cell
Signal. 28:1642–1651. 2016.
|
|
75
|
Chiaverina G, di Blasio L, Monica V,
Accardo M, Palmiero M, Peracino B, Vara-Messler M, Puliafito A and
Primo L: Dynamic interplay between pericytes and endothelial cells
during sprouting angiogenesis. Cells. 8:11092019.
|
|
76
|
Armulik A, Genové G and Betsholtz C:
Pericytes: Developmental, physiological, and pathological
perspectives, problems, and promises. Dev Cell. 21:193–215.
2011.
|
|
77
|
Lee HW, Xu Y, He L, Choi W, Gonzalez D,
Jin SW and Simons M: Role of venous endothelial cells in
developmental and pathologic angiogenesis. Circulation.
144:1308–1322. 2021.
|
|
78
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011.
|
|
79
|
van Splunder H, Villacampa P,
Martínez-Romero A and Graupera M: Pericytes in the disease
spotlight. Trends Cell Biol. 34:58–71. 2024.
|
|
80
|
Rustenhoven J, Jansson D, Smyth LC and
Dragunow M: Brain pericytes as mediators of neuroinflammation.
Trends Pharmacol Sci. 38:291–304. 2017.
|
|
81
|
Stevenson TJ, Johnson RH, Savistchenko J,
Rustenhoven J, Woolf Z, Smyth LCD, Murray HC, Faull RLM, Correia J,
Schweder P, et al: Pericytes take up and degrade α-synuclein but
succumb to apoptosis under cellular stress. Sci Rep.
12:173142022.
|
|
82
|
Chen CJ, Ou YC, Li JR, Chang CY, Pan HC,
Lai CY, Liao SL, Raung SL and Chang CJ: Infection of pericytes in
vitro by Japanese encephalitis virus disrupts the integrity of the
endothelial barrier. J Virol. 88:1150–1161. 2014.
|
|
83
|
Yang X, Chang L, Liu Z, Geng X, Wang R,
Yin X, Fan W and Zhao BQ: Neddylation in the chronically
hypoperfused corpus callosum: MLN4924 reduces blood-brain barrier
injury via ERK5/KLF2 signaling. Exp Neurol. 371:1145872024.
|
|
84
|
Huang F, Feng Y, Peterlin BM and Fujinaga
K: P-TEFb is degraded by Siah1/2 in quiescent cells. Nucleic Acids
Res. 50:5000–5013. 2022.
|
|
85
|
Suarez S, McCollum GW, Jayagopal A and
Penn JS: High glucose-induced retinal pericyte apoptosis depends on
association of GAPDH and Siah1. J Biol Chem. 290:28311–28320.
2015.
|
|
86
|
Liu C, Billadeau DD, Abdelhakim H, Leof E,
Kaibuchi K, Bernabeu C, Bloom GS, Yang L, Boardman L, Shah VH and
Kang N: IQGAP1 suppresses TβRII-mediated myofibroblastic activation
and metastatic growth in liver. J Clin Invest. 123:1138–1156.
2013.
|
|
87
|
Mosaddeghzadeh N and Ahmadian MR: The RHO
family GTPases: mechanisms of regulation and signaling. Cells.
10:18312021.
|
|
88
|
Majolée J, Kovačević I and Hordijk PL:
Ubiquitin-based modifications in endothelial cell-cell contact and
inflammation. J Cell Sci. 132:jcs2277282019.
|
|
89
|
Majolée J, Podieh F, Hordijk PL and
Kovačević I: The interplay of Rac1 activity, ubiquitination and GDI
binding and its consequences for endothelial cell spreading. PLoS
One. 16:e02543862021.
|
|
90
|
Jin Q, Lin L, Zhao T, Yao X, Teng Y, Zhang
D, Jin Y and Yang M: Overexpression of E3 ubiquitin ligase Cbl
attenuates endothelial dysfunction in diabetes mellitus by
inhibiting the JAK2/STAT4 signaling and Runx3-mediated H3K4me3. J
Transl Med. 19:4692021.
|
|
91
|
Qian H, Zhang N, Wu B, Wu S, You S, Zhang
Y and Sun Y: The E3 ubiquitin ligase Smurf2 regulates PARP1
stability to alleviate oxidative stress-induced injury in human
umbilical vein endothelial cells. J Cell Mol Med. 24:4600–4611.
2020.
|
|
92
|
Zou J, Zhou L, Le Y, Fang Z, Zhong M, Nie
F, Wei X, Zhang X, Chen Z, Cai L, et al: WWP2 drives the
progression of gastric cancer by facilitating the ubiquitination
and degradation of LATS1 protein. Cell Commun. Signal.
21:382023.
|
|
93
|
Zhang N, Zhang Y, Wu B, You S and Sun Y:
Role of WW domain E3 ubiquitin protein ligase 2 in modulating
ubiquitination and Degradation of Septin4 in oxidative stress
endothelial injury. Redox Biol. 30:1014192020.
|
|
94
|
You S, Xu J, Yin Z, Wu B, Wang P, Hao M,
Cheng C, Liu M, Zhao Y, Jia P, et al: Down-regulation of WWP2
aggravates type 2 diabetes mellitus-induced vascular endothelial
injury through modulating ubiquitination and degradation of DDX3X.
Cardiovasc Diabetol. 22:1072023.
|
|
95
|
Qian Y, Wang Z, Lin H, Lei T, Zhou Z,
Huang W, Wu X, Zuo L, Wu J, Liu Y, et al: TRIM47 is a novel
endothelial activation factor that aggravates
lipopolysaccharide-induced acute lung injury in mice via K63-linked
ubiquitination of TRAF2. Signal Transduct Target Ther.
7:1482022.
|
|
96
|
Liu J, Lu S, Zheng L, Guo Q, Cao L, Xiao
Y, Chen D, Zou Y, Liu X, Deng C, et al: ATM-CHK2-TRIM32 axis
regulates ATG7 ubiquitination to initiate autophagy under oxidative
stress. Cell Rep. 42:1134022023.
|
|
97
|
Cockram PE, Kist M, Prakash S, Chen SH,
Wertz IE and Vucic D: Ubiquitination in the regulation of
inflammatory cell death and cancer. Cell Death Differ. 28:591–605.
2021.
|
|
98
|
Ullah K, Chen S, Lu J, Wang X, Liu Q,
Zhang Y, Long Y, Hu Z and Xu G: The E3 ubiquitin ligase STUB1
attenuates cell senescence by promoting the ubiquitination and
degradation of the core circadian regulator BMAL1. J Biol Chem.
295:4696–4708. 2020.
|
|
99
|
Li X, Wang T, Tao Y, Wang X, Li L and Liu
J: Inhibition of USP7 suppresses advanced glycation end-induced
cell cycle arrest and senescence of human umbilical vein
endothelial cells through ubiquitination of p53. Acta Biochim
Biophys Sin (Shanghai). 54:311–320. 2022.
|
|
100
|
Mason DE, Collins JM, Dawahare JH, Nguyen
TD, Lin Y, Voytik-Harbin SL, Zorlutuna P, Yoder MC and Boerckel JD:
YAP and TAZ limit cytoskeletal and focal adhesion maturation to
enable persistent cell motility. J Cell Biol. 218:1369–1389.
2019.
|
|
101
|
Uematsu A, Kido K, Takahashi H, Takahashi
C, Yanagihara Y, Saeki N, Yoshida S, Maekawa M, Honda M, Kai T, et
al: The E3 ubiquitin ligase MIB2 enhances inflammation by degrading
the deubiquitinating enzyme CYLD. J Biol Chem. 294:14135–14148.
2019.
|
|
102
|
Li R, Shao J, Jin YJ, Kawase H, Ong YT,
Troidl K, Quan Q, Wang L, Bonnavion R, Wietelmann A, et al:
Endothelial FAT1 inhibits angiogenesis by controlling YAP/TAZ
protein degradation via E3 ligase MIB2. Nat Commun.
14:19802023.
|
|
103
|
Gallemit PEM, Yoodee S, Malaitad T and
Thongboonkerd V: Epigallocatechin-3-gallate plays more predominant
roles than caffeine for inducing actin-crosslinking,
ubiquitin/proteasome activity and glycolysis, and suppressing
angiogenesis features of human endothelial cells. Biomed
Pharmacother. 141:1118372021.
|
|
104
|
Goyani S, Roy M and Singh R: TRIM-NHL as
RNA binding ubiquitin E3 Ligase (RBUL): Implication in development
and disease pathogenesis. Biochim Biophys Acta Mol Basis Dis.
1867:1660662021.
|
|
105
|
Zhou ZX, Ma XF, Xiong WH, Ren Z, Jiang M,
Deng NH, Zhou BB, Liu HT, Zhou K, Hu HJ, et al: TRIM65 promotes
vascular smooth muscle cell phenotypic transformation by activating
PI3K/Akt/mTOR signaling during atherogenesis. Atherosclerosis.
390:1174302024.
|
|
106
|
Hu Z, Song Q, Ma H, Guo Y, Zhang T, Xie H
and Luo X: TRIM32 inhibits the proliferation and migration of
pulmonary artery smooth muscle cells through the inactivation of
PI3K/Akt pathway in pulmonary arterial hypertension. J Bioenerg
Biomembr. 53:309–320. 2021.
|
|
107
|
Liu Y, Zhu L, Ming Y, Wu Z, Zhang L, Chen
Q and Qi Y: A role of TRIM59 in pulmonary hypertension: Modulating
the protein ubiquitylation modification. J Transl Med.
21:8212023.
|
|
108
|
Wang Q, Shi W, Zhang Q, Feng W, Wang J,
Zhai C, Yan X and Li M: Inhibition of Siah2 ubiquitin ligase
ameliorates monocrotaline-induced pulmonary arterial remodeling
through inactivation of YAP. Life Sci. 242:1171592020.
|
|
109
|
Kitamura H: Ubiquitin-specific proteases
(USPs) and metabolic disorders. Int J Mol Sci. 24:32192023.
|
|
110
|
Zhu Y, Zhang Q, Yan X, Liu L, Zhai C, Wang
Q, Chai L and Li M: Ubiquitin-specific protease 7 mediates
platelet-derived growth factor-induced pulmonary arterial smooth
muscle cells proliferation. Pulm Circ.
11:204589402110461312021.
|
|
111
|
Zhou ZX, Ren Z, Yan BJ, Qu SL, Tang ZH,
Wei DH, Liu LS, Fu MG and Jiang ZS: The role of ubiquitin E3 ligase
in atherosclerosis. Curr Med Chem. 28:152–168. 2021.
|
|
112
|
Matsumura Y, Sakai J and Skach WR:
Endoplasmic reticulum protein quality control is determined by
cooperative interactions between Hsp/c70 protein and the CHIP E3
ligase. J Biol Chem. 288:31069–31079. 2013.
|
|
113
|
Cai Z, He X, Liu S, Bai Y, Pan B and Wu K:
Linear ubiquitination modification of NR6A1 by LUBAC inhibits RIPK3
kinase activity and attenuates apoptosis of vascular smooth muscle
cells. J Biochem Mol Toxicol. 36:e230912022.
|
|
114
|
Dai Y, Li Y, Cheng R, Gao J, Li Y and Lou
C: TRIM37 inhibits PDGF-BB-induced proliferation and migration of
airway smooth muscle cells. Biomed Pharmacother. 101:24–29.
2018.
|
|
115
|
Dong LH, Li L, Song Y, Duan ZL, Sun SG,
Lin YL, Miao SB, Yin YJ, Shu YN, Li H, et al: TRAF6-mediated SM22α
K21 ubiquitination promotes G6PD activation and NADPH production,
contributing to GSH homeostasis and VSMC survival in vitro and in
vivo. Circ Res. 117:684–694. 2015.
|
|
116
|
Marchand M, Monnot C, Muller L and Germain
S: Extracellular matrix scaffolding in angiogenesis and capillary
homeostasis. Semin Cell Dev Biol. 89:147–156. 2019.
|
|
117
|
Neve A, Cantatore FP, Maruotti N, Corrado
A and Ribatti D: Extracellular matrix modulates angiogenesis in
physiological and pathological conditions. Biomed Res Int.
2014:7560782014.
|
|
118
|
Mongiat M, Andreuzzi E, Tarticchio G and
Paulitti A: Extracellular matrix, a hard player in angiogenesis.
Int J Mol Sci. 17:18222016.
|
|
119
|
Pai FC, Huang HW, Tsai YL, Tsai WC, Cheng
YC, Chang HH and Chen Y: Inhibition of FABP6 reduces tumor cell
invasion and angiogenesis through the decrease in MMP-2 and VEGF in
human glioblastoma cells. Cells. 10:27822021.
|
|
120
|
Chen Y, Huang Y, Huang Y, Xia X, Zhang J,
Zhou Y, Tan Y, He S, Qiang F, Li A, et al: JWA suppresses tumor
angiogenesis via Sp1-activated matrix metalloproteinase-2 and its
prognostic significance in human gastric cancer. Carcinogenesis.
35:442–451. 2014.
|
|
121
|
Chen Y, Huang Y, Hou P, Zhang Z, Zhang Y,
Wang W, Sun G, Xu L, Zhou J, Bai J and Zheng J: ING4 suppresses
tumor angiogenesis and functions as a prognostic marker in human
colorectal cancer. Oncotarget. 7:79017–79031. 2016.
|
|
122
|
Chen JJ, Ren YL, Shu CJ, Zhang Y, Chen MJ,
Xu J, Li J, Li AP, Chen DY, He JD, et al: JP3, an antiangiogenic
peptide, inhibits growth and metastasis of gastric cancer through
TRIM25/SP1/MMP2 axis. J Exp Clin Cancer Res. 39:1182020.
|
|
123
|
Yang X, Rothman VL, L'Heureux DZ and
Tuszynski G: Reduction of angiocidin expression in human umbilical
vein endothelial cells via siRNA silencing inhibits angiogenesis.
Exp Mol Pathol. 81:108–114. 2006.
|
|
124
|
Huang MT, Mason JC, Birdsey GM, Amsellem
V, Gerwin N, Haskard DO, Ridley AJ and Randi AM: Endothelial
intercellular adhesion molecule (ICAM)-2 regulates angiogenesis.
Blood. 106:1636–1643. 2005.
|
|
125
|
Kitazume S, Imamaki R, Ogawa K and
Taniguchi N: Sweet role of platelet endothelial cell adhesion
molecule in understanding angiogenesis. Glycobiology. 24:1260–1264.
2014.
|
|
126
|
Kummer D and Ebnet K: Junctional adhesion
molecules (JAMs): The JAM-integrin connection. Cells. 7:252018.
|
|
127
|
Kaur G, Sharma D, Bisen S, Mukhopadhyay
CS, Gurdziel K and Singh NK: Vascular cell-adhesion molecule 1
(VCAM-1) regulates JunB-mediated IL-8/CXCL1 expression and
pathological neovascularization. Commun Biol. 6:5162023.
|
|
128
|
Hoer S, Smith L and Lehner PJ: MARCH-IX
mediates ubiquitination and downregulation of ICAM-1. FEBS Lett.
581:45–51. 2007.
|
|
129
|
Li Y, Huang X, Guo F, Lei T, Li S,
Monaghan-Nichols P, Jiang Z, Xin HB and Fu M: TRIM65 E3 ligase
targets VCAM-1 degradation to limit LPS-induced lung inflammation.
J Mol Cell Biol. 12:190–201. 2020.
|
|
130
|
Park S, Sorenson CM and Sheibani N:
PECAM-1 isoforms, eNOS and endoglin axis in regulation of
angiogenesis. Clin Sci (Lond). 129:217–234. 2015.
|
|
131
|
Liu J, Yao Q, Xiao L, Li F, Ma W, Zhang Z,
Xie X, Yang C, Cui Q, Tian Y, et al: APC/Cdh1 targets PECAM-1 for
ubiquitination and degradation in endothelial cells. J Cell
Physiol. 235:2521–2531. 2020.
|
|
132
|
Wu L, Xiao J, Yi D, Ding H, Wang R, Duan
Z, Liu Z, Shi X, Shen M and Sang J: Cytosolic Cadherin 4 promotes
angiogenesis and metastasis in papillary thyroid cancer by
suppressing the ubiquitination/degradation of β-catenin. J Transl
Med. 22:2012024.
|
|
133
|
Zimna A and Kurpisz M: Hypoxia-Inducible
Factor-1 in physiological and pathophysiological angiogenesis:
Applications and therapies. Biomed Res Int. 2015:5494122015.
|
|
134
|
Tirpe AA, Gulei D, Ciortea SM, Crivii C
and Berindan-Neagoe I: Hypoxia: Overview on hypoxia-mediated
mechanisms with a focus on the role of HIF Genes. Int J Mol Sci.
20:61402019.
|
|
135
|
Chen L, Endler A and Shibasaki F: Hypoxia
and angiogenesis: Regulation of hypoxia-inducible factors via novel
binding factors. Exp Mol Med. 41:849–857. 2009.
|
|
136
|
Wicks EE and Semenza GL: Hypoxia-inducible
factors: Cancer progression and clinical translation. J Clin
Invest. 132:e1598392022.
|
|
137
|
Ke Q and Costa M: Hypoxia-inducible
factor-1 (HIF-1). Mol Pharmacol. 70:1469–1480. 2006.
|
|
138
|
Kubaichuk K and Kietzmann T: Involvement
of E3 ligases and deubiquitinases in the control of HIF-α subunit
abundance. Cells. 8:5982019.
|
|
139
|
Ajani JA, Xu Y, Huo L, Wang R, Li Y, Wang
Y, Pizzi MP, Scott A, Harada K, Ma L, et al: YAP1 mediates gastric
adenocarcinoma peritoneal metastases that are attenuated by YAP1
inhibition. Gut. 70:55–66. 2021.
|
|
140
|
Koyasu S, Kobayashi M, Goto Y, Hiraoka M
and Harada H: Regulatory mechanisms of hypoxia-inducible factor 1
activity: Two decades of knowledge. Cancer Sci. 109:560–571.
2018.
|
|
141
|
Bora-Singhal N, Saha B, Mohankumar D,
Padmanabhan J, Coppola D and Chellappan S: A novel
PHD2/VHL-mediated regulation of YAP1 contributes to VEGF expression
and angiogenesis. Cancer Res Commun. 2:624–638. 2022.
|
|
142
|
Kim YJ, Zhao Y, Myung JK, Yi JM, Kim MJ
and Lee SJ: Suppression of breast cancer progression by FBXL16 via
oxygen-independent regulation of HIF1α stability. Cell Rep.
37:1099962021.
|
|
143
|
Yueyang M, Yaqin H, Guolian X, Wenjian Z,
Yang J, Chen L, Haiyan C, Min C, Jianping D, Penggao D, et al:
Glioma angiogenesis is boosted by ELK3 activating the
HIF-1[Formula: See text]/VEGF-A signaling axis. BMC Cancer.
23:6622023.
|
|
144
|
Chen P, Duan X, Li X, Li J, Ba Q and Wang
H: HIPK2 suppresses tumor growth and progression of hepatocellular
carcinoma through promoting the degradation of HIF-1α. Oncogene.
39:2863–2876. 2012.
|
|
145
|
Chen C, Wei M, Wang C, Sun D, Liu P, Zhong
X, He Q and Yu W: The histone deacetylase HDAC1 activates
HIF1α/VEGFA signal pathway in colorectal cancer. Gene.
754:1448512020.
|
|
146
|
Hu L, Lv X, Li D, Zhang W, Ran G, Li Q and
Hu J: The anti-angiogenesis role of FBXW7 in diabetic retinopathy
by facilitating the ubiquitination degradation of c-Myc to
orchestrate the HDAC2. J Cell Mol Med. 25:2190–2202. 2021.
|
|
147
|
Deng Y, Li S, Li S, Yu C, Huang D, Chen H
and Yin X: CircPDE4B inhibits retinal pathological angiogenesis via
promoting degradation of HIF-1α though targeting miR-181c. IUBMB
Life. 72:1920–1929. 2020.
|
|
148
|
Pitulescu ME, Schmidt I, Giaimo BD,
Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D,
Rocha SF, et al: Dll4 and Notch signalling couples sprouting
angiogenesis and artery formation. Nat Cell Biol. 19:915–927.
2017.
|
|
149
|
Parmalee NL and Kitajewski J: Wnt
signaling in angiogenesis. Curr Drug Targets. 9:558–564. 2008.
|
|
150
|
Shaw P, Dwivedi SKD, Bhattacharya R,
Mukherjee P and Rao G: VEGF signaling: Role in angiogenesis and
beyond. Biochim Biophys Acta Rev Cancer. 1879:1890792024.
|
|
151
|
Dutta D, Sharma V, Mutsuddi M and
Mukherjee A: Regulation of notch signaling by E3 ubiquitin ligases.
FEBS J. 289:937–954. 2022.
|
|
152
|
Zhang B and Ma JX: Wnt pathway antagonists
and angiogenesis. Protein Cell. 1:898–906. 2010.
|
|
153
|
Choi HJ, Park H, Lee HW and Kwon YG: The
Wnt pathway and the roles for its antagonists, DKKS, in
angiogenesis. IUBMB Life. 64:724–731. 2012.
|
|
154
|
Zerlin M, Julius MA and Kitajewski J:
Wnt/Frizzled signaling in angiogenesis. Angiogenesis. 11:63–69.
2008.
|
|
155
|
Dejana E: The role of wnt signaling in
physiological and pathological angiogenesis. Circ Res. 107:943–952.
2010.
|
|
156
|
Shi YN, Zhu N, Liu C, Wu HT, Gui Y, Liao
DF and Qin L: Wnt5a and its signaling pathway in angiogenesis. Clin
Chim Acta. 471:263–269. 2017.
|
|
157
|
Mankuzhy P, Dharmarajan A, Perumalsamy LR,
Sharun K, Samji P and Dilley RJ: The role of Wnt signaling in
mesenchymal stromal cell-driven angiogenesis. Tissue Cell.
85:1022402023.
|
|
158
|
van Loon K, Huijbers EJM and Griffioen AW:
Secreted frizzled-related protein 2: A key player in noncanonical
Wnt signaling and tumor angiogenesis. Cancer Metastasis Rev.
40:191–203. 2021.
|
|
159
|
Park HB, Kim JW and Baek KH: Regulation of
Wnt signaling through ubiquitination and deubiquitination in
cancers. Int J Mol Sci. 21:39042020.
|
|
160
|
Kikuchi A: Modulation of Wnt signaling by
Axin and Axil. Cytokine Growth Factor Rev. 10:255–265. 1999.
|
|
161
|
Huang SM, Mishina YM, Liu S, Cheung A,
Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner
S, et al: Tankyrase inhibition stabilizes axin and antagonizes Wnt
signalling. Nature. 461:614–620. 2009.
|
|
162
|
Law SM and Zheng JJ: Premise and peril of
Wnt signaling activation through GSK-3β inhibition. iScience.
25:1041592022.
|
|
163
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009.
|
|
164
|
Li Q, Luo H, Dai FQ, Wang RT, Fan XQ, Luo
YY, Deng MS, Wang Y, Long T, Guo W, et al: SAMD9 promotes
postoperative recurrence of esophageal squamous cell carcinoma by
stimulating MYH9-Mediated GSK3β/β-Catenin signaling. Adv Sci
(Weinh). 10:e22035732023.
|
|
165
|
Lyle CL, Belghasem M and Chitalia VC:
c-Cbl: An important regulator and a target in angiogenesis and
tumorigenesis. Cells. 8:4982019.
|
|
166
|
Shivanna S, Harrold I, Shashar M, Meyer R,
Kiang C, Francis J, Zhao Q, Feng H, Edelman ER, Rahimi N and
Chitalia VC: The c-Cbl ubiquitin ligase regulates nuclear β-catenin
and angiogenesis by its tyrosine phosphorylation mediated through
the Wnt signaling pathway. J Biol Chem. 290:12537–12546. 2015.
|
|
167
|
Chitalia V, Shivanna S, Martorell J, Meyer
R, Edelman E and Rahimi N: c-Cbl, a ubiquitin E3 ligase that
targets active β-catenin: A novel layer of Wnt signaling
regulation. J Biol Chem. 288:23505–23517. 2013.
|
|
168
|
Kumaradevan S, Lee SY, Richards S, Lyle C,
Zhao Q, Tapan U, Jiangliu Y, Ghumman S, Walker J, Belghasem M, et
al: c-Cbl expression correlates with human colorectal cancer
survival and Its Wnt/β-catenin suppressor function is regulated by
Tyr371 phosphorylation. Am J Pathol. 188:1921–1933. 2018.
|
|
169
|
Wang H, Deng G, Ai M, Xu Z, Mou T, Yu J,
Liu H, Wang S and Li G: Hsp90ab1 stabilizes LRP5 to promote
epithelial-mesenchymal transition via activating of AKT and
Wnt/β-catenin signaling pathways in gastric cancer progression.
Oncogene. 38:1489–1507. 2019.
|
|
170
|
Chen C, Zhu D, Zhang H, Han C, Xue G, Zhu
T, Luo J and Kong L: YAP-dependent ubiquitination and degradation
of β-catenin mediates inhibition of Wnt signalling induced by
Physalin F in colorectal cancer. Cell Death Dis. 9:5912018.
|
|
171
|
Harper JA, Yuan JS, Tan JB, Visan I and
Guidos CJ: Notch signaling in development and disease. Clin Genet.
64:461–472. 2003.
|
|
172
|
Hasan SS and Fischer A: Notch signaling in
the vasculature: Angiogenesis and angiocrine functions. Cold Spring
Harb Perspect Med. 13:a0411662023.
|
|
173
|
Tetzlaff F and Fischer A: Control of blood
vessel formation by notch signaling. Adv Exp Med Biol.
1066:319–338. 2018.
|
|
174
|
Luo Z, Shang X, Zhang H, Wang G, Massey
PA, Barton SR, Kevil CG and Dong Y: Notch signaling in
osteogenesis, osteoclastogenesis, and angiogenesis. Am J Pathol.
189:1495–1500. 2019.
|
|
175
|
Sainson RC and Harris AL: Regulation of
angiogenesis by homotypic and heterotypic notch signalling in
endothelial cells and pericytes: From basic research to potential
therapies. Angiogenesis. 11:41–51. 2008.
|
|
176
|
Jiang N, Hu Y, Wang M, Zhao Z and Li M:
The notch signaling pathway contributes to angiogenesis and tumor
immunity in breast cancer. Breast Cancer (Dove Med Press).
14:291–309. 2022.
|
|
177
|
Benedito R, Roca C, Sörensen I, Adams S,
Gossler A, Fruttiger M and Adams RH: The notch ligands Dll4 and
Jagged1 have opposing effects on angiogenesis. Cell. 137:1124–1135.
2009.
|
|
178
|
Garcia A and Kandel JJ: Notch: A key
regulator of tumor angiogenesis and metastasis. Histol Histopathol.
27:151–156. 2012.
|
|
179
|
Ferrante F, Giaimo BD, Friedrich T, Sugino
T, Mertens D, Kugler S, Gahr BM, Just S, Pan L, Bartkuhn M, et al:
Hydroxylation of the NOTCH1 intracellular domain regulates Notch
signaling dynamics. Cell Death Dis. 13:6002022.
|
|
180
|
Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu
K and Chu Q: Notch signaling pathway: architecture, disease, and
therapeutics. Signal Transduct Target Ther. 7:952022.
|
|
181
|
Revici R, Hosseini-Alghaderi S, Haslam F,
Whiteford R and Baron M: E3 ubiquitin ligase regulators of notch
receptor endocytosis: From flies to humans. Biomolecules.
12:2242022.
|
|
182
|
Le Bras S, Loyer N and Le Borgne R: The
multiple facets of ubiquitination in the regulation of notch
signaling pathway. Traffic. 12:149–161. 2011.
|
|
183
|
Koo BK, Yoon KJ, Yoo KW, Lim HS, Song R,
So JH, Kim CH and Kong YY: Mind bomb-2 is an E3 ligase for Notch
ligand. J Biol Chem. 280:22335–22342. 2005.
|
|
184
|
Izumi N, Helker C, Ehling M, Behrens A,
Herzog W and Adams RH: Fbxw7 controls angiogenesis by regulating
endothelial Notch activity. PLoS One. 7:e411162012.
|
|
185
|
Ohnuki H, Inoue H, Takemori N, Nakayama H,
Sakaue T, Fukuda S, Miwa D, Nishiwaki E, Hatano M, Tokuhisa T, et
al: BAZF, a novel component of cullin3-based E3 ligase complex,
mediates VEGFR and Notch cross-signaling in angiogenesis. Blood.
119:2688–2698. 2012.
|