Introduction
Myocardial hypertrophy represents a fundamental
adaptive response of the heart to diverse stressors, frequently
characterized by the enlargement of myocardial cells and increased
fibrosis. Initially, this adaptation functions as a compensatory
process aimed at preserving cardiac function. However, over time,
sustained hypertrophy disrupts myocardial performance,
precipitating heart failure (1).
Myocardial hypertrophy is widely recognized as an independent
predictor of cardiovascular-related morbidity and mortality, with
prolonged hypertrophy driving structural remodeling, functional
decline, heart failure progression and, in severe cases, sudden
cardiac death (2).
Epidemiological data demonstrate that myocardial hypertrophy is a
leading cause of morbidity and mortality in heart failure cases
(3). Cardiac hypertrophy
typically manifests with cardiomyocyte enlargement, fibrosis,
myofibrillar disarray, cell death, and increased protein synthesis
(4). Underlying mechanisms often
include mitochondrial dysfunction, cardiomyocyte fibrosis,
apoptosis and the overproduction of reactive oxygen species (ROS)
(5). Investigating
pharmacological interventions that target these pathways may offer
potential therapeutic strategies for controlling this pathological
condition (6).
Thymoquinone (TQ), the major bioactive compound in
Nigella sativa seeds, plays a significant role in mediating various
biological activities of the plant (7). Studies have identified the
therapeutic benefits of TQ across multiple pathological conditions,
including tumorigenesis, autoimmune disorders, diabetes and
neurodegenerative diseases, primarily through pathways involving
immune modulation, suppression of apoptosis, attenuation of
oxidative stress and neutralization of free radicals (8,9).
However, the precise mechanism through which TQ exerts its
protective effects on cardiac hypertrophy remains elusive.
Autophagy is a process that entails the engulfment
of cytoplasmic proteins or organelles, their encapsulation into
vesicles and subsequent fusion with lysosomes to form autolysosomes
for degradation. In cardiomyocytes, adaptive autophagy helps
eliminate damaged organelles, ensuring the preservation of cardiac
function. However, both excessive activation and marked suppression
of autophagy can compromise cardiac structure and function
(10). Accumulating evidence
indicates a key involvement of autophagy in cardiac hypertrophy.
Xue et al (11)
demonstrated that Sestrin 1 mitigated phenylephrine-induced cardiac
hypertrophy by modulating the AMP-activated protein kinase
(AMPK)/mTORC1 autophagy pathway. Furthermore, the deletion of
ATPase inhibitory factor 1 enhances AMPK activity, boosts autophagy
and alleviates transverse aortic constriction (TAC)-induced cardiac
hypertrophy (12). Thus,
targeting autophagy regulation offers potential therapeutic value
for cardiac hypertrophy.
Peroxisome proliferator-activated receptor-γ
(PPAR-γ) belongs to a family of ligand-induced transcription
factors within the nuclear receptor superfamily (13). Activation of PPAR-γ can reduce
inflammation, inhibit oxidative stress and enhance cardiomyocyte
energy metabolism, thus inhibiting myocardial remodeling and
mitigating myocardial hypertrophy (14). Research has demonstrated that
thiazolidinediones exhibit efficacy in enhancing insulin
sensitivity and mitigating hyperglycemia through their targeting of
PPAR-γ, and they have gained widespread application in the
treatment of type 2 diabetes (15). Since PPAR-γ targeted drugs have
been poorly studied in cardiac hypertrophy, it is of clinical
interest to investigate natural products that can effectively
activate PPAR-γ to alleviate cardiac hypertrophy.
The objective of the present study was to validate
the protective effect of TQ on cardiac hypertrophy both in
vitro and in vivo, as well as to delve into the
underlying mechanisms and provide key insights into the therapeutic
potential of TQ in both preventing and managing cardiac
hypertrophy.
Materials and methods
Reagents and chemicals
TQ (purity ≥99.59%), 3-Methyladenine (3-MA; purity
≥99.91%) and GW9662 (PPAR-γ antagonist; purity ≥99.87%) were
sourced from MedChemExpress, while pAD/14-3-3γ-short hairpin
(sh)RNA was procured from Cyagen Biosciences, Inc. AngII was
acquired from GLPBIO Technology LLC. PPAR-γ (cat. no. YT3836)
antibody was supplied by ImmunoWay Biotechnology Company. Primary
antibodies for LC3 (cat. no. 381544), p62 (cat. no. 380612),
14-3-3γ (cat. no. R381405) and secondary antibodies (goat
anti-mouse, cat. no. 511103; goat anti-rabbit, cat. no. 511203)
were purchased from Chengdu Zen-Bioscience Co., Ltd. Proteintech
Group, Inc. provided antibodies against collagen I (cat. no.
14695-1-AP), atrial natriuretic peptide (ANP; cat. no. 27426-1-AP)
and β-actin (cat. no. 66009-1-Ig). ABclonal, Inc. provided
antibodies against brain natriuretic peptide (BNP; cat. no.
A2179).
Cell model of cardiac hypertrophy and
treatment
The rat H9C2 cell line, obtained from the Cell Bank
of Type Culture Collection of the Chinese Academy of Sciences, was
maintained in H-DMEM (HyClone; Cytiva), supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
The H9C2 cells were incubated at 37°C in a controlled environment
(95% humidity, 21% O2, and 5% CO2) (16). The H9C2 cells were allocated into
six distinct experimental groups: i) Control group, H9C2 cells were
maintained in H-DMEM for 48 h under standard conditions; ii) AngII
group: H9C2 cells were cultured in H-DMEM for 24 h, followed by
exposure to 1 µM AngII for an additional 24 h (17); iii) AngII + TQ group: Cells were
pretreated with 10 µM TQ for 24 h before subsequent exposure
to 1 µM AngII for another 24 h; iv) AngII + TQ + 3-MA group:
Cells were incubated with 10 µM TQ and 5 mM 3-MA (18) for 24 h, followed by 1 µM
AngII treatment for 24 h; v) AngII + TQ + GW9662 group: Cells were
pretreated with 10 µM TQ and 10 µM GW9662 (19) for 24 h, then exposed to 1
µM AngII for an additional 24 h; and vi) AngII + TQ +
pAD/14-3-3γ-shRNA group: H9C2 cardiomyocytes were transfected with
pAd/14-3-3γ-shRNA at 37°C for 48 h, incubated with 10 µM TQ
for 24 h and subsequently treated with 1 µM AngII for
another 24 h.
Adenovirus transfection
H9C2 cardiomyocytes were pre-cultured in fresh
H-DMEM supplemented with 10% FBS and then transfected with either
pAd/14-3-3γ-shRNA or pAd/negative control-shRNA (Cyagen
Biosciences, Inc.; concentration not measured) using HighGene plus
Transfection reagent (cat. no. RM09014; ABclonal Biotech Co.,
Ltd.). The transfection efficiency was ~85% after 48 h at 37°C,
when the subsequent experiments were carried out. The shRNA
sequences are provided in Table
I.
 | Table ISequences of the shRNA used in the
study. |
Table I
Sequences of the shRNA used in the
study.
| Name | Sequence (5′ to
3′) |
|---|
| 14-3-3γshRNA |
CGGCGAAGGCAACAACTAACTCGAGTTAGTTGTTGCCTTCGCCG |
| pAD/NC-shRNA |
CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG |
Cell viability assay
Cell viability was determined via the Cell Counting
Kit-8 (CCK-8; GLPBIO Technology LLC). H9C2 cells were cultured in
96-well plates at a density of 2×104 cells/well and
exposed to TQ at concentrations of 1, 5, 10 and 20 µM for 24
h. Subsequently, 10 µl CCK-8 reagent was introduced to each
well for 1 h at 37°C, and the absorbance at 450 nm was recorded
using a microplate reader (Thermo Fisher Scientific, Inc.).
Determination of oxidative stress and
lysosome detection
ROS production levels were assessed via staining
with dichlorofluorescein diacetate (DCFH-DA; Beyotime Institute of
Biotechnology). H9C2 cells were exposed to 10 µM DCFH-DA for
20 min at 37°C in the dark, followed by three washes with H-DMEM
lacking 10% FBS. Intracellular ROS was subsequently visualized
using a fluorescence microscope (Olympus Corporation). Lyso-Tracker
Red (Beyotime Institute of Biotechnology), a lysosomal red
fluorescent probe, enables lysosome-specific staining in live cells
by penetrating cell membranes. H9C2 cells were incubated with the
Lyso-Tracker Red working solution for 30 min at 37°C in the dark,
and fluorescence microscopy (Olympus Corporation) was employed for
observation.
Transmission electron microscopy
(TEM)
Cells were harvested and fixed in 2% glutaraldehyde
at 25°C for 2 h, followed by sequential washing with PBS,
dehydration, embedding, sectioning and staining. The sections were
60-80 nm thick and were embedded at 37°C for 5-8 h using EPON 812.
Staining was performed using 2% uranyl acetate and 2.6% lead
citrate at 37°C for 8 min. Autophagosome structures in H9C2 cells
were analyzed via TEM (Hitachi 7800; Hitachi, Ltd.).
Western blot analysis
Total proteins were isolated from H9C2 cells and
mouse myocardial tissue using RIPA lysis buffer (Beijing Solarbio
Science & Technology Co., Ltd.). Protein concentrations were
determined via the BCA Assay Kit (GLPBIO Technology LLC).
Equivalent amounts of protein (30-40 µg) were resolved via
10-12.5% gradient SDS-PAGE and subsequently transferred to
polyvinylidene fluoride membranes (Pall Corporation). Membranes
were blocked with 5% skimmed milk at room temperature for 2 h,
followed by overnight incubation at 4°C with primary antibodies
against LC3 (1:1,000), p62 (1:1,000), 14-3-3γ (1:1,000), β-actin
(1:1,000), collagen I (1:500) and PPAR-γ (1:500) on a shaker. After
washing three times using TBST (0.05% Tween 20), the membranes were
incubated with HRP-conjugated secondary antibodies (1:5,000) for 2
h at room temperature. β-actin served as a loading control.
Finally, protein bands were visualized using the Ultra High
Sensitivity ECL kit (cat. no. GK10008; GLPBIO Technology LLC) and
protein band densities were semi-quantified using ImageJ software
(National Institutes of Health; v1.8.0.345).
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
RT-qPCR analysis was performed to quantify the mRNA
expression levels of ANP, BNP, NADPH oxidase 4 (NOX4), superoxide
dismutase 2 (SOD2) and collagen I. Total RNA was isolated with
TRIzol reagent (Beijing Solarbio Science & Technology Co.,
Ltd.), followed by RT using the PrimeScript RT reagent kit (Monad
Biotech Co., Ltd.) according to the manufacturer's instructions.
qPCR was performed using the SYBR Green qPCR Master Mix (cat. no.
B21203; Selleck Chemicals) on the BIO-RAD CFX Connect Real-Time PCR
Detection System from Bio-Rad Laboratories, Inc. The qPCR protocol
comprised an initial denaturation step at 95°C for 10 min, followed
by 40 cycles of denaturation at 95°C for 15 sec and annealing plus
extension at 60°C for 1 min. β-actin served as the internal
reference and the relative mRNA expression levels of the target
genes were calculated employing the 2−ΔΔCq method
(20). The specific primers for
each gene are detailed in Table
II.
| Table IIPrimers used for of reverse
transcription-quantitative PCR. |
Table II
Primers used for of reverse
transcription-quantitative PCR.
| Gene | Species | Forward primer (5′
to 3′) | Reverse primer (5′
to 3′) |
|---|
| ANP | Rat |
ATCTGATGGATTTCAAGAACC |
CTCTGAGACGGGTTGACTTC |
| BNP | Rat |
ACAATCCACGATGCAGAAGCT |
GGGCCTTGGTCCTTTGAGA |
| ANP | Mouse |
CTCCGATAGATCTGCCCTCTTGAA |
GGTACCGGAAGCTGTTGCAGCCTA |
| BNP | Mouse |
GCTCTTGAAGGACCAAGGCCTCAC |
GATCCGATCCGGTCTATCTTGTGC |
| Collagen I | Rat |
GAGAGAGCATGACCGATGGATT |
TGGACATTAGGCGCAGGAA |
| Collagen I | Mouse |
AGGCTTCAGTGGTTTGGATG |
CACCAACAGCACCATCGTTA |
| NOX4 | Rat |
CTGACAGGTGTCTGCATGGT |
ACTTCAACAAGCCACCCGAA |
| NOX4 | Mouse |
CGGGATTTGCTACTGCCTCCAT |
GTGACTCCTCAAATGGGCTTCC |
| SOD2 | Rat |
GTGTCTGTGGGAGTCCAAGG |
TGCTCCCACACATCAATCCC |
| SOD2 | Mouse |
TAACGCGCAGATCATGCAGCTG |
AGGCTGAAGAGCGACCTGAGTT |
| ACTB | Rat |
AGATGACCCAGATCATGTTTGAGA |
CGCTCGGTCAGGATCTTCAT |
| ACTB | Mouse |
GGCTGTATTCCCCTCCATCG |
CCAGTTGGTAACAATGCCATGT |
Immunofluorescence staining
LC3 expression was evaluated using
immunofluorescence staining. After washing the cell samples with
PBS fixation was carried out with 4% paraformaldehyde (Biosharp
Life Sciences) at room temperature for 10 min, then
permeabilization for 10 min at room temperature using 0.2% Triton
X-100 (Beijing Solarbio Science & Technology Co., Ltd.),
followed by blocking with 2% BSA (cat. no. 9048-46-8; Shanghai
Yuanye Biotechnology Co., Ltd.) at room temperature for 30 min. The
cells were subsequently incubated overnight at 4°C with rabbit
anti-LC3 primary antibody (1:500). Following three washes with TBST
(0.05% Tween 20), fluorescent secondary goat anti-rabbit antibody
(1:200; cat.no. A32732; Thermo Fisher Scientific, Inc.) was applied
for 1 h at room temperature. DAPI counterstaining was performed at
room temperature for 5 min, and fluorescence microscopy was used
for observation.
Luciferase activity assay
Luciferase activity was assessed using a
dual-luciferase reporter assay system (Promega Corporation; cat.
no. E1910 Transfection of pcDNA3.1-PPAR-γ and psiCheck2-14-3-3γ
(Hunan Fenghui Biotechnology Co., Ltd.) constructs into cells was
carried out with LipoFiter (cat. no. HB-LF-1000; Hanbio
Biotechnology Co., Ltd.) for 6 h at 37°C. Subsequently, 100
µl each of firefly and Renilla luciferase detection
reagents were applied, followed by the measurement of relative
light units. The luciferase activity ratio between firefly and
Renilla was then calculated.
Molecular docking
The three-dimensional structures of TQ and PPAR-γ
were obtained from PubChem (Compound CID: 10281) and the Protein
Data Bank (PDB) (PDB ID: 3PRG), respectively. The CB-Dock2
(https://cadd.labshare.cn/cb-dock2/php/index.php)
server was utilized to carry out molecular docking.
Animal model of cardiac hypertrophy and
treatment
The experimental procedures followed the guidelines
established by the National Institutes of Health and were approved
by the Animal Experimentation Ethics Committee of The First
Affiliated Hospital, Jiangxi Medical College, Nanchang University
(Nanchang, China; approval no. CDYFY-IACUC-202407QR115). Male
C57BL/6 mice (22-24 g, 8 weeks old) were sourced from Changzhou
Cavens Laboratory Animal Ltd. The mice were maintained in
controlled environmental conditions, with a temperature of 23±1°C,
a humidity of 40-50%, a 12-h light/dark cycle to simulate day and
night, and ad libitum access to food and water to ensure
their well-being and experimental accuracy.
The mice were randomly allocated into four groups:
i) Sham operation, ii) TAC operation, iii) TAC operation with TQ
treatment, and iv) TAC operation with TQ treatment alongside PPAR-γ
antagonist (GW9662). Each group comprised 6 mice. The mouse model
of pressure overload-induced cardiac hypertrophy was generated
through TAC surgery, following the protocol outlined in a previous
study (21). For the operation,
animals were anesthetized in a chamber with isoflurane (induced
with 2% and maintained with 1.5%), supplemented during surgery with
0.1 mg/kg buprenorphine HCl via subcutaneous injection.
Post-surgery, the health and behavior of the mice were monitored
twice daily. In the TQ treatment group, TQ (50 mg/kg, dissolved in
corn oil) was administered via gavage for 6 weeks post-TAC
(22). The PPAR-γ antagonist
group received intraperitoneal injections of GW9662 (1 mg/kg) after
TAC surgery, administered every 3 days for 6 weeks.
The animal was euthanized if predefined humane
endpoints were met, including: i) Rapid weight loss of 15-20% of
the original body weight (BW); ii) the inability to eat, drink or
stand for up to 24 h; and iii) depression and hypothermia
(<37°C) without anesthesia or sedation. No mice exhibited signs
of reaching these endpoints during the experiment. Euthanasia was
induced by an overdose of pentobarbital sodium (200 mg/kg). Animals
were observed for 5-10 min post-injection to confirm death,
assessed by respiratory and cardiac arrest, and loss of corneal
reflex.
Histological analysis
Mouse heart weight (HW) and BW were recorded. Heart
tissue samples were preserved in formalin for 12 h at room
temperature and embedded in paraffin following established
protocols (23).
Paraffin-embedded specimens were sectioned at 5 µm thickness
and subjected to hematoxylin and eosin staining for 5 min at room
temperature as well as Masson's trichrome staining for 5 min at
room temperature, using standard procedures (24,25). The myocardial cell size was
observed from ventricular slices stained with fluorescein
isothiocyanate-labeled wheat germ agglutinin (WGA) at 37°C in the
dark for 2 h. Finally, the sections were examined under an inverted
fluorescence microscope (Nikon Eclipse Ti-SR), and images were
subsequently captured.
Echocardiography
After anesthetizing the mice with 1.5% isoflurane,
echocardiography was performed. Cardiac function was assessed using
a small animal ultrasound imaging system (VEVO2100; FUJIFILM
VisualSonics, Inc.; FUJIFILM Sonosite, Inc.) featuring a 30-MHz
probe.
Statistical analysis
Data analysis was conducted using GraphPad Prism 9.0
software (Dotmatics). For two-group comparisons, unpaired Student's
t-test was applied, while one-way ANOVA followed by the Tukey's
post hoc test was employed for multiple group comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
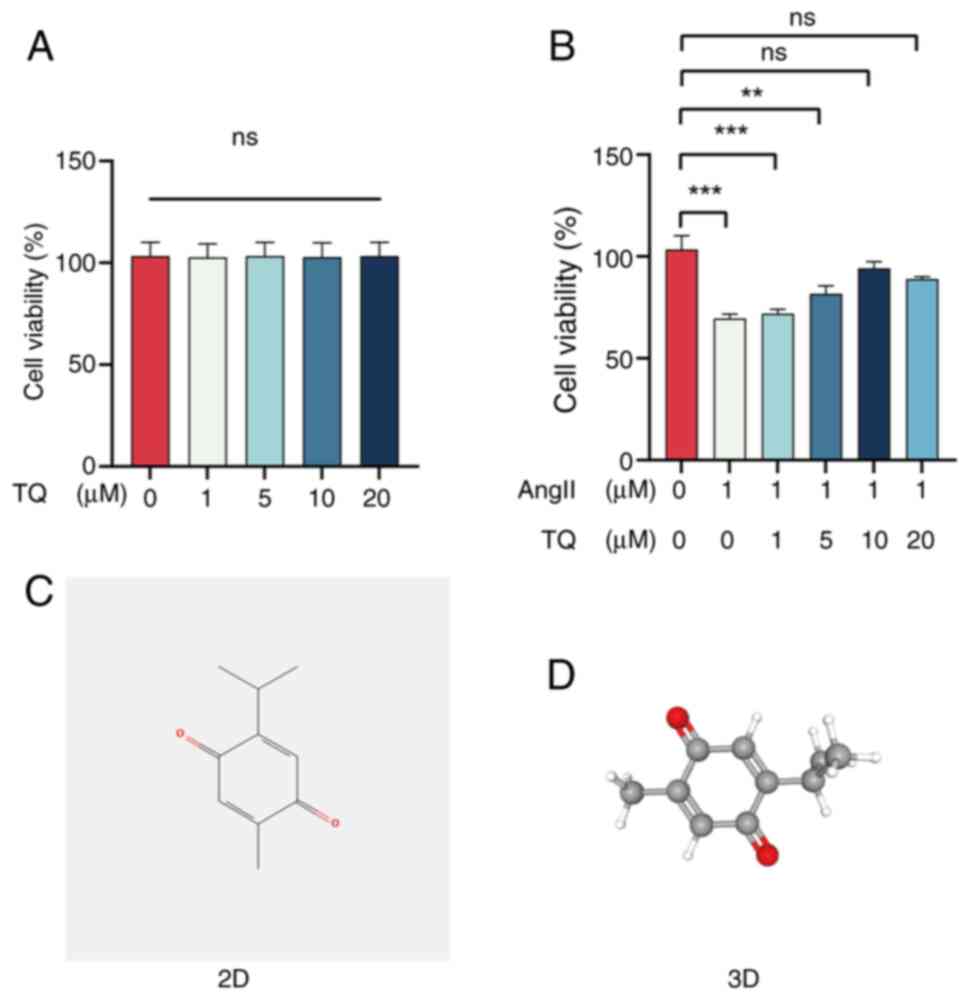
Effects of TQ on the viability of H9C2
cells
H9C2 cell viability under varying concentrations of
TQ (0, 1, 5, 10 and 20 µM) and AngII (1 µM) was
evaluated using the CCK-8 assay. Treatment with TQ at different
concentrations did not significantly alter cell viability compared
with the control (0 µM) group (Fig. 1A). By contrast, AngII (1
µM) significantly reduced cell viability, an effect that was
attenuated by TQ pretreatment (Fig.
1B). As the 10 µM concentration demonstrated the most
notable protective effect, it was selected for subsequent
experiments. The chemical structure of TQ is illustrated in
Fig. 1C and D.
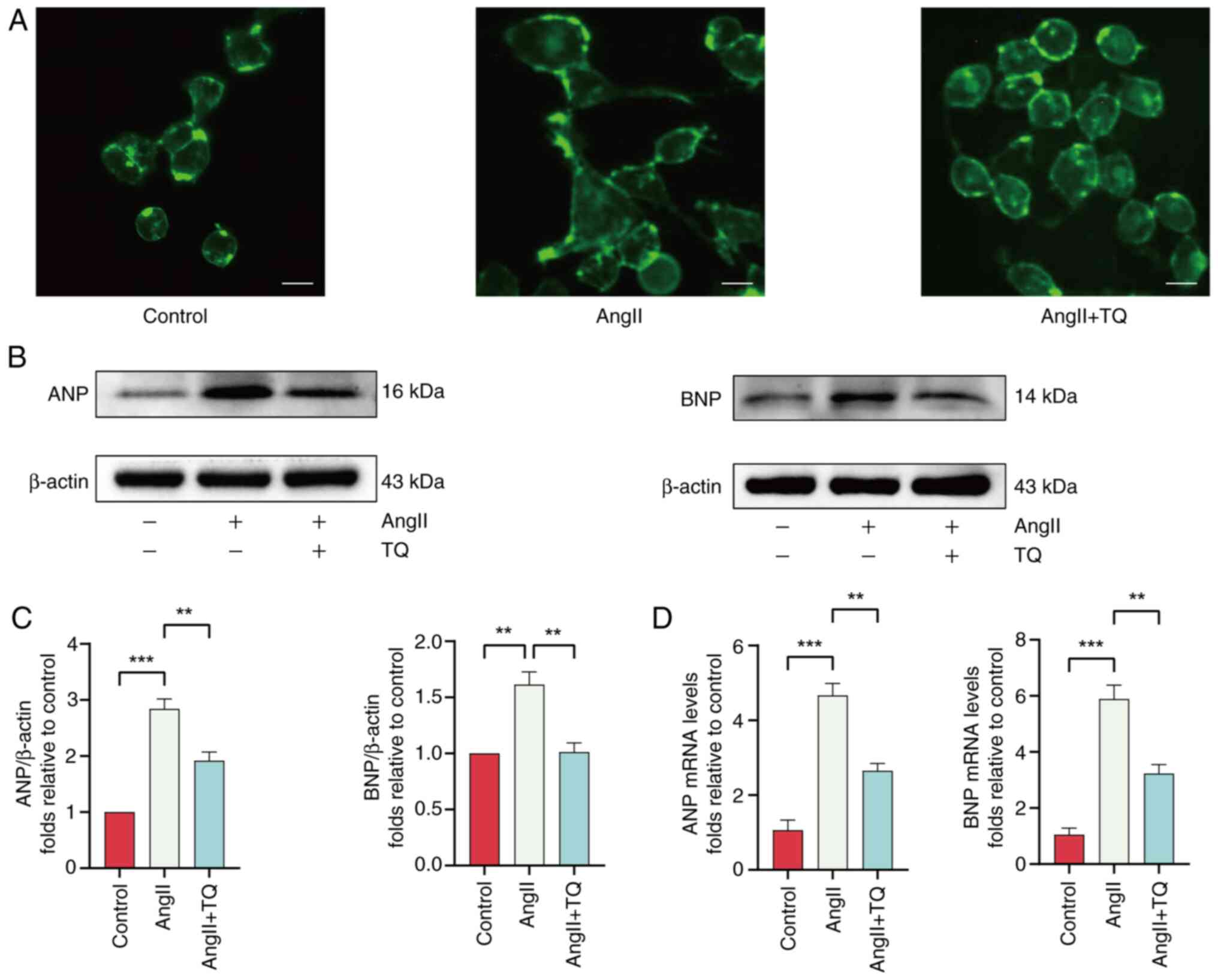
TQ attenuates cardiomyocyte hypertrophy
in vitro and in vivo
To assess the potential protective effects of TQ
against AngII-induced hypertrophy in H9C2 cells, cells were
pretreated with AngII (1 µM) for 24 h to induce hypertrophy,
followed by evaluation of the cell surface area through WGA
immunostaining. Pretreatment with TQ significantly decreased the
cell surface area compared with the AngII group (Fig. 2A). In addition, analysis of the
protein and mRNA expression levels of key hypertrophic markers, ANP
and BNP, indicated that TQ significantly downregulated the
expression of these markers (Fig.
2B-D). These results collectively indicated that TQ mitigated
the hypertrophic response triggered by AngII in H9C2 cells.
To assess the cardioprotective effects of TQ on
cardiac hypertrophy, a pressure overload hypertrophy mouse model
was induced via TAC surgery. Cardiac function was analyzed through
echocardiography (Fig. 3A). Mice
in the TAC group exhibited a decline in left ventricular ejection
fraction and fractional shortening, alongside an increase in left
ventricular internal dimension diastole and posterior wall
dimension, compared with the sham group (Fig. 3B-E). However, post-TAC
administration of TQ improved cardiac function and attenuated
hypertrophy in mice (Fig. 3A-E).
Furthermore, pretreatment with TQ alleviated TAC-induced
hypertrophy, as indicated by a reduction in cardiac size (Fig. 3G). Additionally, a marked
increase in heart size and myofibril degeneration was observed in
the TAC group, while the TQ-treated group exhibited reduced heart
size and improved myofibril integrity, as shown by hematoxylin and
eosin staining (Fig. 3H). The
HW/BW ratio was significantly lower in the TAC + TQ group compared
with the TAC group (Fig. 3F).
Additionally, the protein and mRNA expression levels of ANP and BNP
were notably down-regulated in the TAC + TQ group relative to the
TAC group (Fig. 3I-L). These
findings indicated that TQ attenuated pressure overload-induced
cardiac hypertrophy.
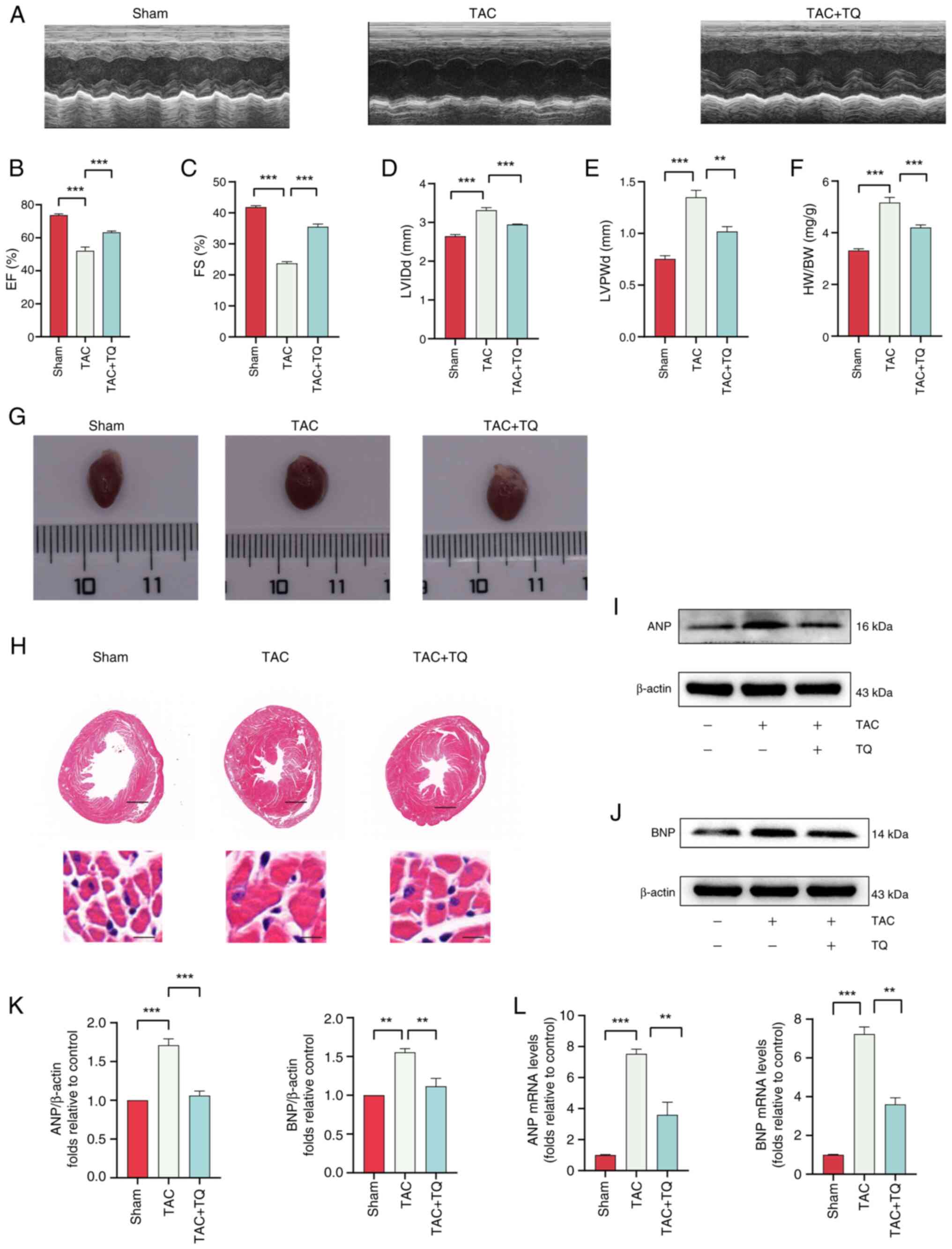 | Figure 3TQ attenuates cardiomyocyte
hypertrophy in vivo. (A) Echocardiography was conducted to
assess heart function. (B) EF, (C) FS, (D) LVPWd, (E) LVIDd and (F)
HW/BW ratios were measured. (G) Representative images of the whole
hearts. (H) Hematoxylin and eosin-stained cardiac cross-sections
(scale bars, 2.5 mm or 100 µm). (I and J) Immunoblotting
analysis of ANP and BNP protein expression levels in vivo,
(K) alongside the semi-quantification of the results. β-actin was
used as an internal control (n=3). (L) The effects of TQ on the
mRNA expression of ANP and BNP. Data are presented as the mean ±
SD, n=6 mice per group. **P<0.01,
***P<0.001. TQ, thymoquinone; EF, ejection fraction;
FS, fractional shortening (of the left ventricular diameter);
LVPWd, diastolic left ventricular posterior wall thickness; LVIDd,
diastolic left ventricular internal diameter; HW, heart weight; BW,
body weight; TAC, transverse aortic constriction; ANP, atrial
natriuretic peptide; BNP, brain natriuretic peptide. |
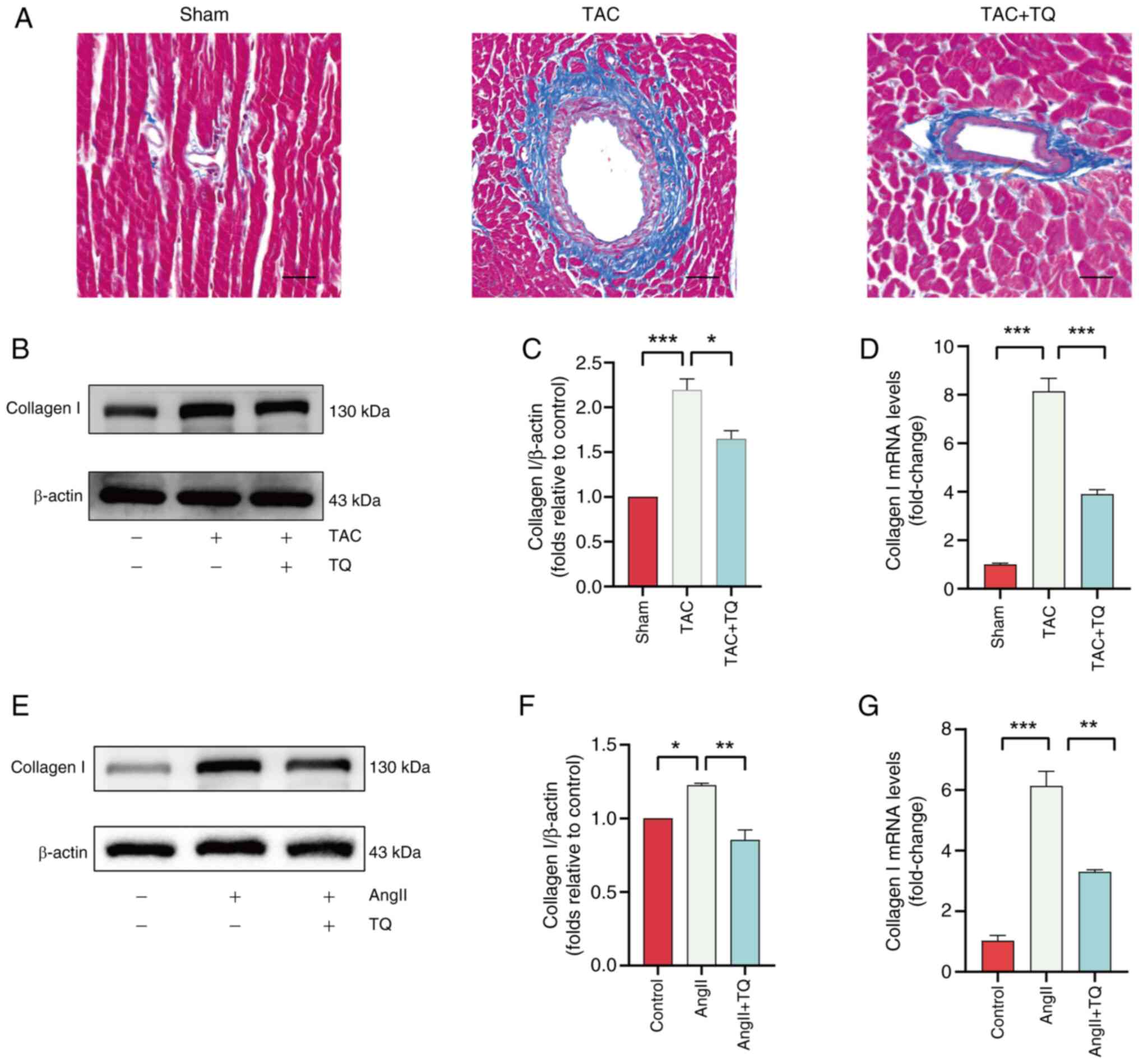
TQ inhibits cardiac fibrosis in vivo and
in vitro
The inter-relation between cardiac fibrosis and
hypertrophy has been well-established, with fibrosis progression
impairing myocardial contractility, ultimately contributing to
heart failure and potential mortality (26,27). Masson staining, immunoblotting
and RT-qPCR were employed to assess the effects of TQ on cardiac
fibrosis. Masson staining showed a marked increase in fibrosis in
mice subjected to TAC surgery, which was markedly attenuated by TQ
treatment (Fig. 4A). The protein
and mRNA levels of type I collagen were significantly upregulated
in the TAC group compared with the control but were reduced
following TQ administration (Fig.
4B-D). Similarly, in vitro experiments using H9C2 cells
revealed elevated expression of type I collagen in the AngII group,
which was mitigated by TQ, aligning with the in vivo
findings (Fig. 4E-G).
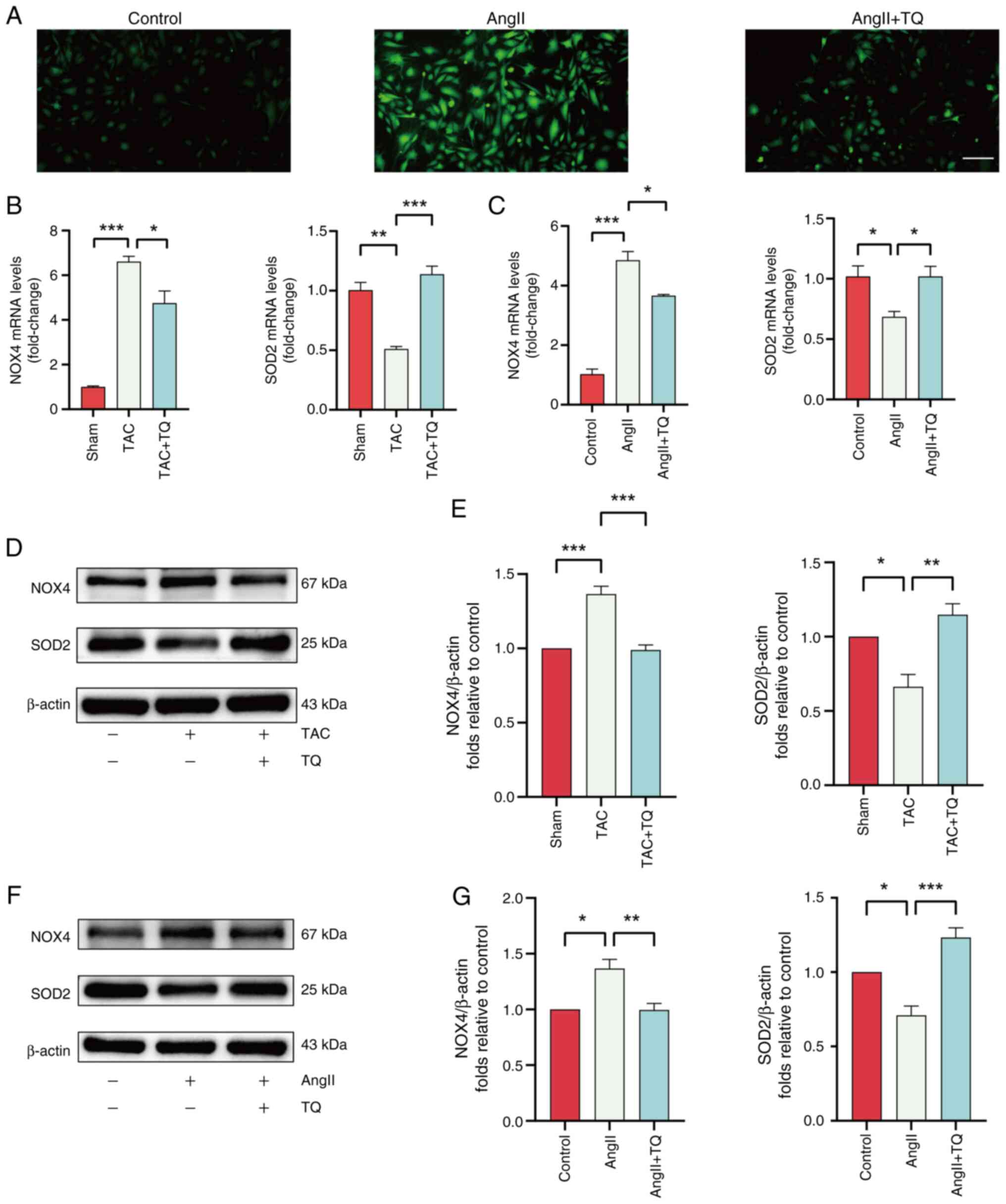
TQ attenuates oxidative stress levels in
cardiac hypertrophy cells
Excessive generation of ROS is recognized as a key
mechanism contributing to the progression of cardiac hypertrophy
(5). Elevated ROS accumulation
in cardiomyocytes has been shown to aggravate cardiac hypertrophy
and myocardial fibrosis, ultimately leading to heart failure
(28). To explore the potential
antioxidative effect of TQ in cardiac hypertrophy, ROS levels were
measured in treated cells. A notable increase in ROS levels were
observed in the AngII group, while TQ treatment markedly reduced
ROS levels (Fig. 5A).
Additionally, RT-qPCR and western blot analysis indicated that TQ
significantly downregulated the mRNA and protein expression of the
oxidation gene, NOX4, and significantly upregulated the mRNA and
protein expression of antioxidation gene, SOD2, in both the
TAC-induced and AngII-induced groups (Fig. 5B-G).
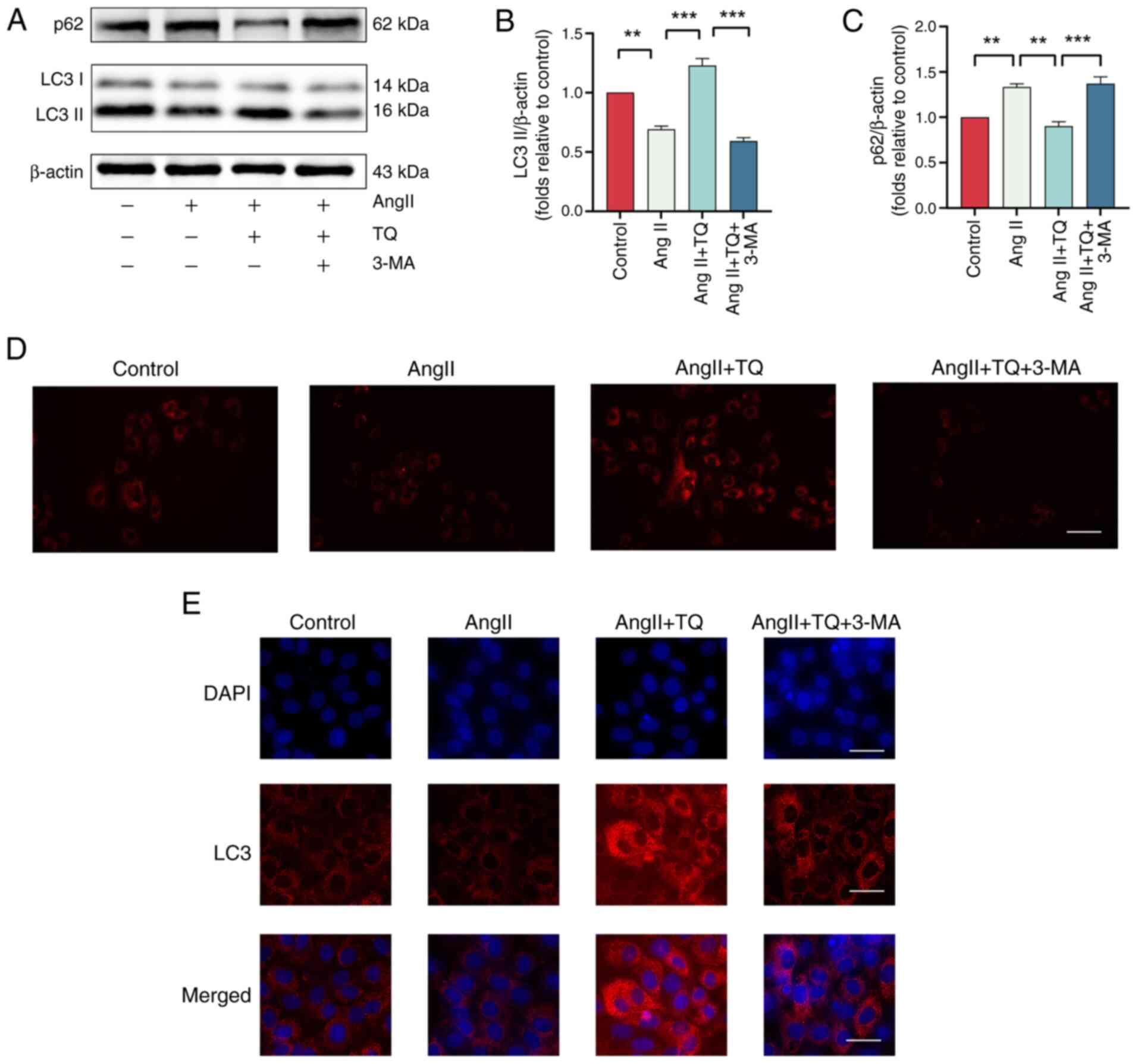
TQ activates adaptive autophagy in H9C2
hypertrophy cardiomyocytes
Autophagy plays a central role in the pathogenesis
of cardiac hypertrophy, with its dysregulation contributing to the
worsening of the condition (29,30). To evaluate the effect of TQ
pretreatment on adaptive autophagy in H9C2 cells, the expression
levels of autophagy markers, LC3II and p62, as well as lysosome
counts, were analyzed. As shown in Fig. 6A-C, TQ + AngII treatment
significantly increased LC3II expression while reducing p62 levels
compared with AngII alone. This modulation was attenuated by the
autophagy inhibitor, 3-MA. These results suggested that autophagy
was downregulated during the onset of hypertrophy in cardiac cells
and that TQ pretreatment restored autophagic activity under these
conditions. Furthermore, LysoTracker Red staining revealed a
reduction in lysosome numbers following AngII-induced treatment of
H9C2 cells, which was reversed upon TQ administration. Notably, the
addition of 3-MA markedly reduced fluorescence intensity in H9C2
cells (Fig. 6D).
Immunofluorescence analysis indicated an elevation in LC3
expression following TQ pretreatment, which was markedly reversed
by 3-MA (Fig. 6E). Thus, we
found that TQ activated adaptive autophagy in H9C2 hypertrophy
cardiomyocyte.
TQ alleviates hypertrophy in cardiac
cells by upregulating 14-3-3γ expression to activate adaptive
autophagy
The 14-3-3 proteins, a group of highly conserved
acidic proteins found in all eukaryotic cells, comprise seven
isoforms (31). Research has
established their role in regulating autophagy and their protective
function in cardiomyocytes (16,32). To investigate whether TQ
conferred protection via autophagy regulation mediated by 14-3-3γ,
the expression of 14-3-3γ was knocked down in H9C2 cells, the
success of which was verified by western blot analysis (Fig. S1). As shown in Fig. 7A and B, TQ pretreatment led to a
significant upregulation of 14-3-3γ expression, an effect that was
reversed by pAD/14-3-3γ shRNA. In addition, knockdown of 14-3-3γ
significantly influenced autophagic activity (Fig. 7A, C and D). Specifically,
pAD/14-3-3γ shRNA reduced LC3II expression while simultaneously
increasing p62 expression compared with the TQ group. TEM further
confirmed a reduction in autophagic vesicles and therefore a
suppression of autophagy in H9C2 cells treated with pAD/14-3-3γ
shRNA (Fig. 7E). Additionally,
pAD/14-3-3γ shRNA upregulated the mRNA and protein expression
levels of ANP and BNP compared with the AngII + TQ group (Fig. 7F-H). These experiments confirmed
that TQ alleviated cardiac hypertrophy by upregulating 14-3-3γ.
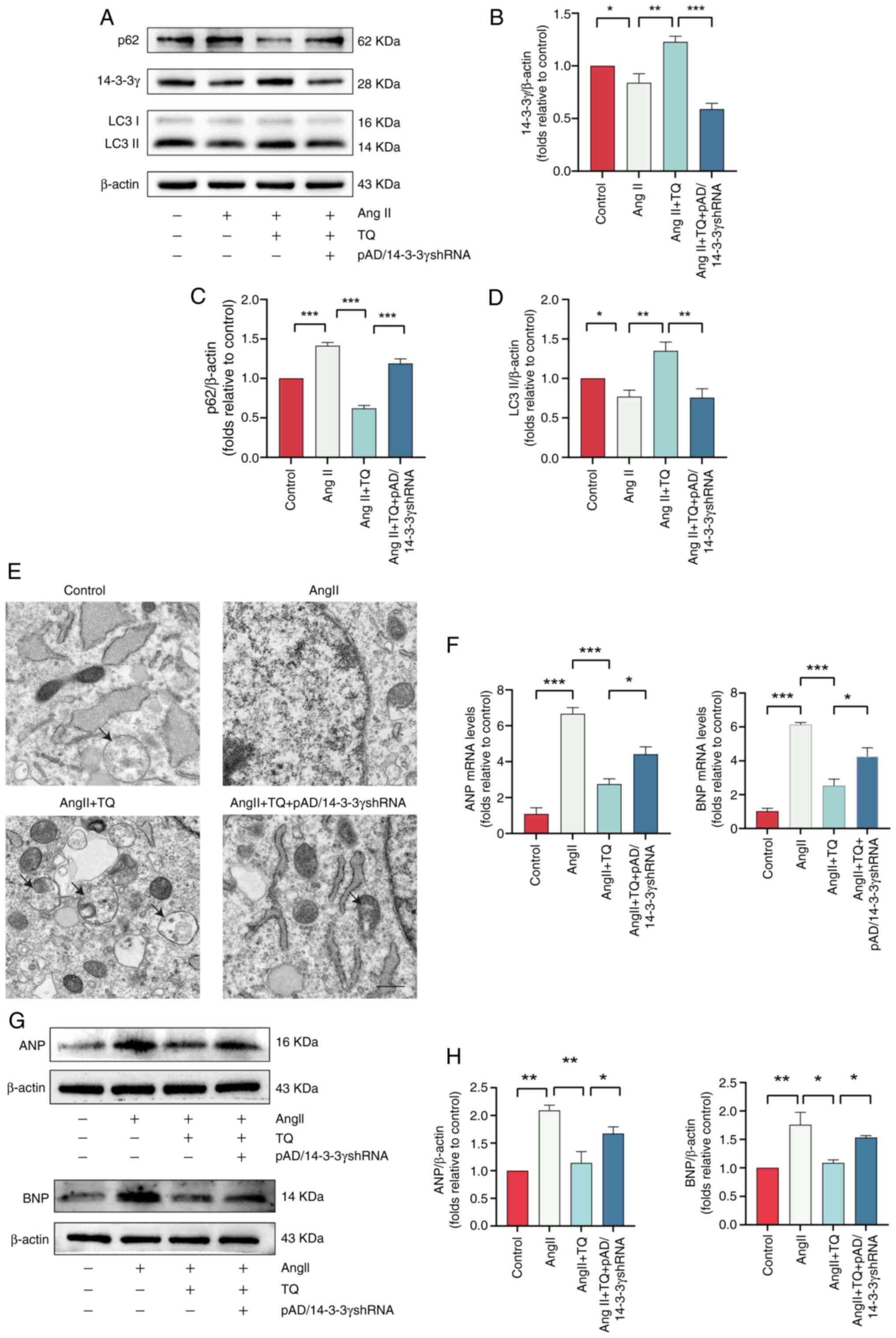 | Figure 7TQ alleviates cardiac hypertrophy by
upregulating 14-3-3γ expression to activate adaptive autophagy. (A)
Immunoblotting analysis of LC3, p62 and 14-3-3γ protein expression
levels in AngII-induced hypertrophic H9C2 cells, (B-D) alongside
the semi-quantification of the results. β-actin was used as an
internal control (n=3). TQ inhibited the protein expression levels
of p62 and upregulated the protein expression levels of LC3II,
effects that were abolished by the simultaneous knockdown of 14-3-3
γ. (E) Transmission electron microscopy images of H9C2 cells
(magnification, ×6,000; scale bar: 2 µm). (F) Expression
levels of the hypertrophic genes, ANP and BNP, in H9C2 cells. (G)
Immunoblotting analysis of the ANP and BNP protein expression
levels in H9C2 hypertrophic cardiomyocytes, (H) alongside the
semi-quantification of the results. β-actin was used as an internal
control (n=3). TQ inhibited the mRNA and protein expression levels
of ANP and BNP, effects that were abolished by the simultaneous
knockdown of 14-3-3γ. Data are presented as the mean ± SD.
*P<0.05, **P<0.01,
***P<0.001. TQ, thymoquinone; AngII, angiotensin II;
shRNA, short hairpin RNA; ANP, atrial natriuretic peptide; BNP,
brain natriuretic peptide. |
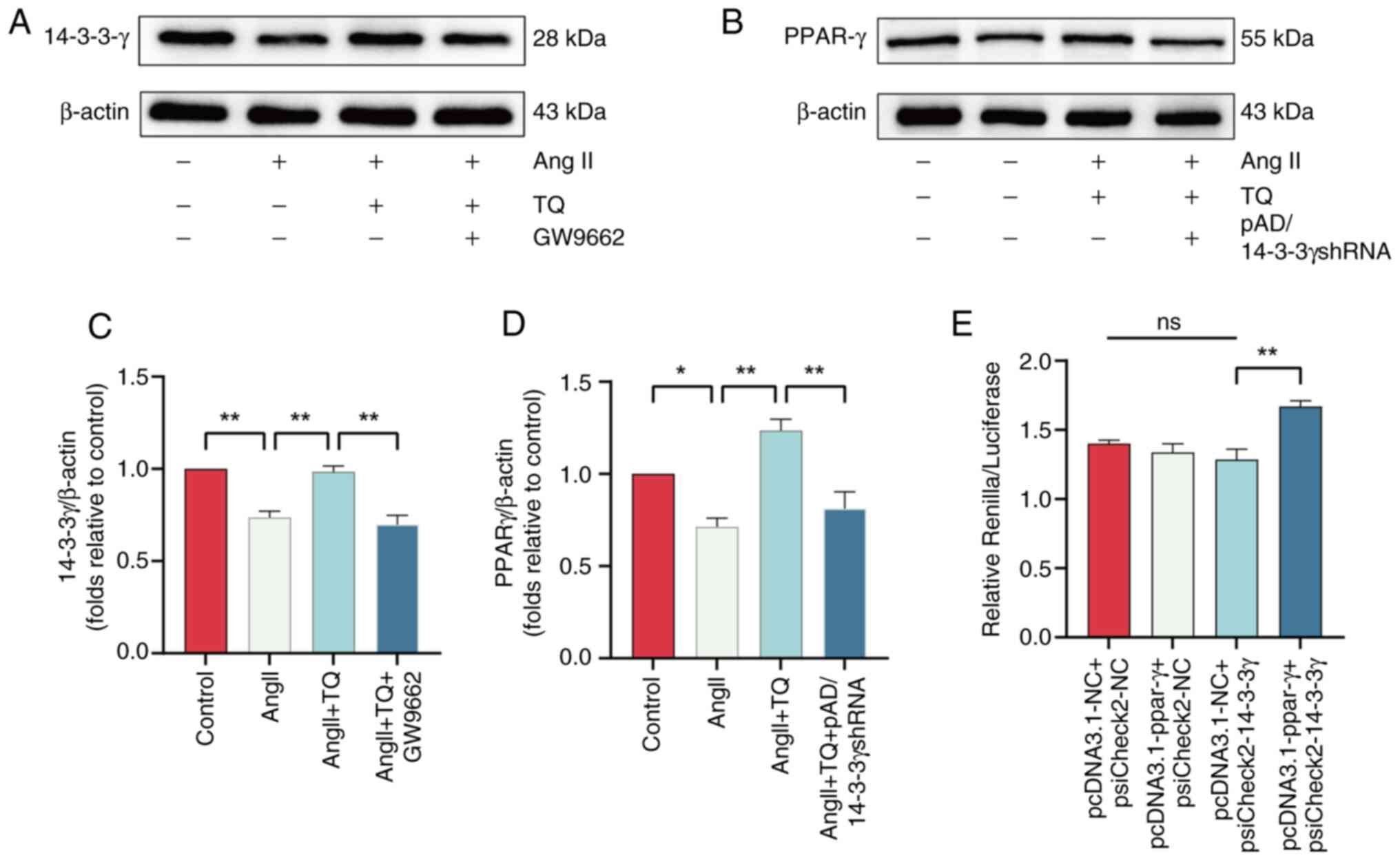
TQ targets PPAR-γ and attenuates cardiac
hypertrophy through activating adaptive autophagy
PPAR-γ, a protective regulator, is essential for
maintaining cardiac homeostasis and preventing heart failure
(33-35). We hypothesized that TQ could
specifically target PPAR-γ and activate adaptive autophagy, thereby
alleviating cardiac hypertrophy. To test this conjecture, the
protein expression level of PPAR-γ was detected using western blot
analysis. The results demonstrated that PPAR-γ expression in
hypertrophic cardiac tissue revealed was significantly
downregulated compared with the control group. However, TQ
treatment effectively restored PPAR-γ levels. Notably, the
co-administration of a PPAR-γ inhibitor (GW9662) reversed the
TQ-mediated upregulation of PPAR-γ (Fig. 8A-D). Molecular docking techniques
were employed to explore the regulatory interaction between TQ and
PPAR-γ, simulating their binding. This method is widely used to
evaluate compound interactions and activity (36). The analysis demonstrated that TQ
formed hydrogen bonds with several key sites on PPAR-γ (Fig. 8F), suggesting that PPAR-γ
modulation may be a potential mechanism of TQ's effects.
Furthermore, similar results in AngII-pretreated H9C2 cells as
those obtained from in vivo experiments were observed. The
results indicated that the autophagy levels were suppressed as
LC3II expression decreased and p62 expression increased in the
AngII + TQ + GW9662 group treated compared with the AngII + TQ
group (Fig. 8A-D). TEM further
confirmed the reduction of autophagic vesicles in the PPAR-γ
inhibitor-treated group, consistent with the western blot results
(Fig. 8E). Additionally, PPAR-γ
inhibition reversed the protective effects of TQ on cardiac
hypertrophy, resulting in an enlarged cardiomyocyte surface area
(Fig. 8G) and elevated the
protein and mRNA expression of ANP and BNP (Fig. 8H-J).
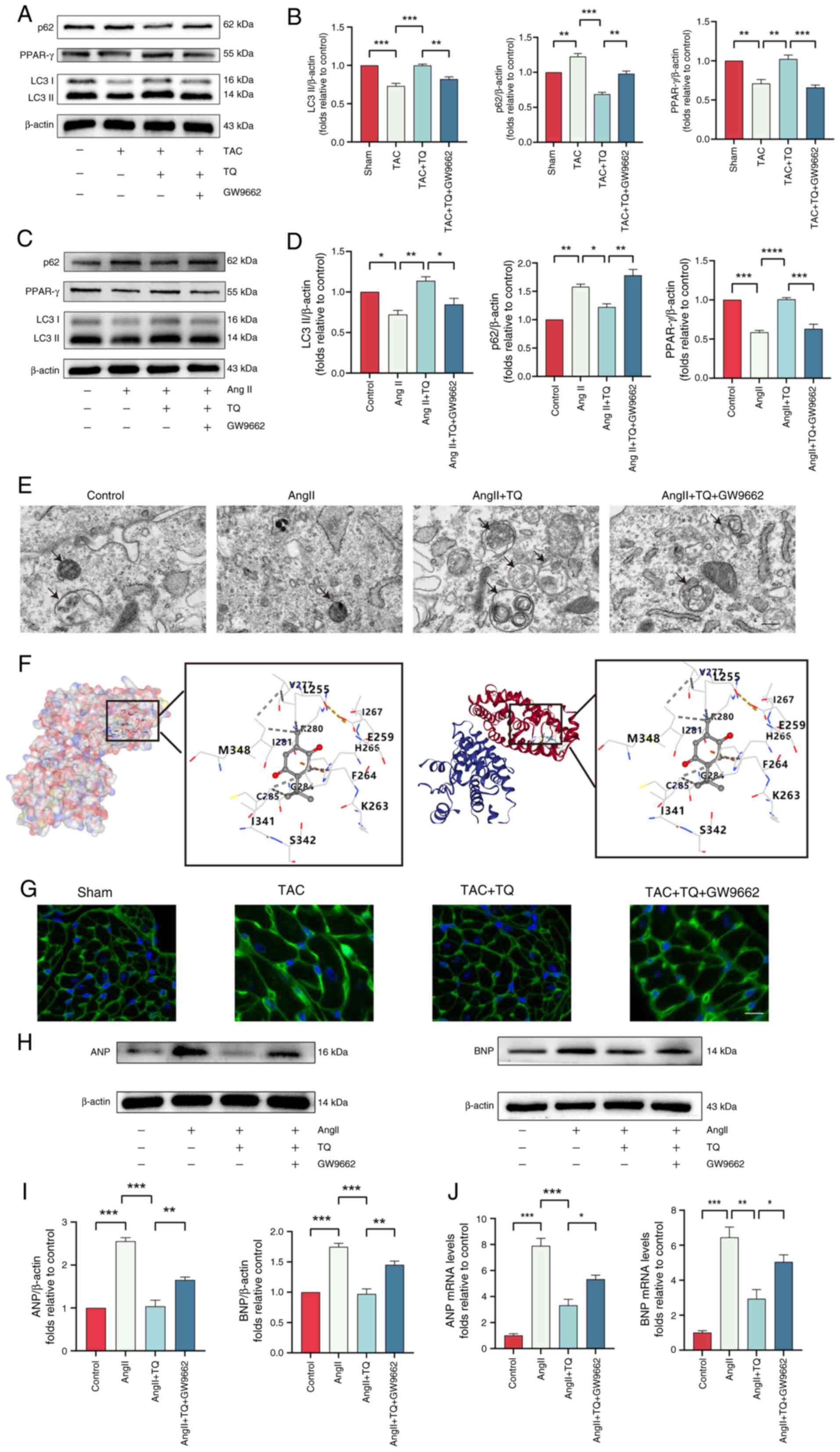 | Figure 8TQ targets PPAR-γ to attenuate
cardiac hypertrophy through activating adaptive autophagy.
Immunoblotting analysis of LC3, p62 and PPAR-γ protein expression
levels (A) in vivo and (C) in vitro after
pretreatment with TQ and GW9662, (B and D) alongside the
semi-quantification of the results. β-actin was used as an internal
control (n=3). (E) Transmission electron microscopy images of H9C2
cells (magnification, ×6,000; scale bar, 2 µm). (F)
Molecular docking images illustrate the binding positions of TQ
within PPAR-γ, highlighting the interactions between the ligand and
the residues. The docking mode employed was structure-based blind
docking. The potential binding sites of the queried ligands were
ranked according to the AutoDock Vina score (kcal/mol), and those
with the lowest binding energy are selected for display. (G)
Representative images of fluorescein isothiocyanate--conjugated
wheat germ agglutinin stained transverse sections. (H)
Immunoblotting analysis of the ANP and BNP protein expression
levels in H9C2 hypertrophic cardiomyocytes after pretreatment with
TQ and GW9662, (I) alongside the semi-quantification of the
results. β-actin was used as an internal control (n=3). (J) The
expression levels of the hypertrophic genes, BNP and ANP in H9C2
hypertrophic cardiomyocytes after pretreatment with TQ and GW9662.
Data are presented as the mean ± SD. *P<0.05,
**P<0.01, ***P<0.001. TQ, thymoquinone;
AngII, angiotensin II; TAC, transverse aortic constriction; PPAR-γ,
peroxisome proliferator-activated receptor-γ; ANP, atrial
natriuretic peptide; BNP, brain natriuretic peptide. |
TQ attenuates cardiac hypertrophy by
activating adaptive autophagy through the PPAR-γ/14-3-3γ
pathway
Inhibition of 14-3-3γ expression was observed upon
treatment with the PPAR-γ inhibitor, GW9662. The suppression of
PPAR-γ resulted in a corresponding reduction in 14-3-3γ levels
(Fig. 9A and C). Similarly,
knockdown of 14-3-3γ via pAD/14-3-3γ shRNA also diminished PPAR-γ
expression (Fig. 9B and D).
Moreover, the dual-luciferase assay demonstrated that PPAR-γ
upregulated 14-3-3γ promoter activity (Fig. 9E). These results suggest that TQ
mitigates cardiac hypertrophy by promoting adaptive autophagy
through the PPAR-γ/14-3-3γ signaling axis (Fig. S2).
Discussion
Myocardial hypertrophy constitutes a significant
risk factor for numerous cardiac pathologies. Prolonged hypertrophy
often leads to a diminished left ventricular ejection fraction and
progressive cardiac dysfunction, ultimately resulting in heart
failure and increased mortality rates (37). At present, no specific
pharmacological intervention exists for the effective treatment of
cardiac hypertrophy in clinical practice. Thus, investigating the
molecular mechanisms underlying the development of cardiac
hypertrophy is essential for the discovery and advancement of novel
therapeutic agents. In the present study, TQ treatment demonstrated
a notable reduction in cardiac hypertrophy both in vitro,
using H9C2 cells, and in vivo, utilizing a TAC-induced
cardiac hypertrophy mouse model. TQ was found to markedly lower ROS
levels, mitigate myocardial fibrosis, enhance autophagic activity
in the context of cardiac hypertrophy and provide protection
against hypertrophy-induced cardiac dysfunction. Additionally, TQ
activated the PPAR-γ and 14-3-3γ signaling pathways, leading to
enhanced autophagy and suppression of pathological cardiac
hypertrophy. These results suggest that TQ holds considerable
promise as a potential therapeutic agent for the treatment of
cardiac hypertrophy and heart failure, with strong prospects for
clinical application.
Chinese herbal medicines have gained prominence in
the treatment of cardiovascular diseases (38). Baicalein, for instance, enhances
catalase expression to eliminate ROS, binds to FOXO3a, promotes its
transcriptional activity and activates autophagy, thereby
alleviating cardiac hypertrophy (39). Similarly, Sophora effectively
mitigates TAC-induced cardiac hypertrophy, protects against
hypertrophy-related cardiac dysfunction, reduces myocardial
fibrosis and activates AMPK/mTORC1-mediated autophagy to counteract
hypertrophy (40). Berberine
administration also ameliorates TAC-induced cardiac hypertrophy,
reduces myocardial apoptosis, limits fibrosis and upregulates
autophagy, providing cardioprotective effects in hypertrophy models
(41). Despite these advances,
research on the effects of TQ in cardiac hypertrophy remains
limited. The present study highlights TQ's potential to attenuate
cardiac hypertrophy, presenting a novel therapeutic strategy for
managing the condition.
Oxidative stress arises from an imbalance between
antioxidant defenses and ROS production, with excessive ROS leading
to cellular damage. A major contributor to cardiac hypertrophy is
oxidative stress driven by elevated ROS levels in cardiomyocytes
(42). Persistent oxidative
stress has been implicated in the progression of myocardial
hypertrophy to heart failure (43). Evidence suggests that controlling
oxidative stress can mitigate cardiac hypertrophy and hinder its
transition to heart failure (44,45). In the present study, in the
AngII-induced H9C2 cell hypertrophy model, ROS levels were assessed
through DCFH-DA staining under fluorescence microscopy and the
expression of ROS-related genes via qPCR. AngII markedly elevated
ROS production in cells, while TQ pretreatment attenuated ROS
levels, attributed to its potent antioxidant properties. However, a
ROS inhibitor was not employed to further verify the impact of TQ
on cardiac hypertrophy.
Autophagy is a conserved cellular mechanism
responsible for degrading proteins and damaged organelles, playing
a vital role in maintaining survival, development and homeostasis,
and is therefore integral to human health and development (46). This homeostatic process ensures
the degradation and recycling of cellular components under both
physiological and stress conditions. Impaired autophagy results in
disrupted ubiquitination, ROS accumulation and compromised
mitochondrial function (47).
Autophagy activation has been shown to alleviate cardiac
hypertrophy and mitigate cardiac dysfunction in stress-induced
hypertrophy models (48). The
present study reinforces the hypothesis that enhancing autophagy
can reduce the progression of cardiac hypertrophy. The present
study utilized TEM and the Lyso-Tracker method to examine
autophagic lysosomes, while western blotting was used to quantify
the LC3 protein levels to evaluate autophagic activity. LC3, a
widely recognized autophagy marker, is commonly used to assess
autophagy, with LC3II levels closely linked to autophagosome
numbers, and the LC3II/LC3I ratio serving as an indicator of
autophagic flux (49). In the
cardiac hypertrophy model, the protein expression level of LC3II
was significantly reduced compared with the TQ-treated group,
indicating that TQ mitigates cardiac hypertrophy and improves
cardiac function by promoting autophagy. This observation is
consistent with a prior study (48). Notably, to the best of our
knowledge, the present study is the first to establish the role of
TQ in attenuating cardiac hypertrophy through autophagy
enhancement.
PPARs are part of the ligand-activated transcription
factor family within the nuclear receptor superfamily, comprising
three isoforms: PPAR-α, PPAR-δ and PPAR-γ (50). PPAR-γ, predominantly expressed in
adipose tissue, governs adipocyte differentiation and the
regulation of genes involved in lipid storage (13). PPAR-γ has been identified as a
crucial regulator of adipose development and systemic metabolism,
with therapeutic potential in enhancing insulin sensitivity in
diabetic patients (51).
Activation of PPAR-γ has been shown to prevent myocardial
hypertrophy and attenuate post-myocardial infarction remodeling by
reducing inflammation, oxidative stress, cell death and improving
cardiomyocyte energy metabolism (13,50,52,53). In the present study, AngII
exposure inhibited PPAR-γ expression in cultured H9C2 cells, while
TQ treatment restored its activation. Notably, the cardioprotective
effect of TQ disappeared upon administration of the PPAR-γ
inhibitor, GW9662. To the best of our knowledge, the present study
presents the first evidence that TQ mitigates cardiac hypertrophy
via PPAR-γ activation.
The 14-3-3 proteins are ubiquitously expressed
across animal and plant tissues, serving critical roles in cellular
biology and signal transduction pathways (54). Numerous studies have identified
14-3-3 proteins as endogenous cardioprotective agents, offering
protection in various heart injury models. For instance, curcumin
has been shown to mitigate doxorubicin (DOX)-induced cardiotoxicity
by upregulating 14-3-3γ, which in turn reduces serum lactate
dehydrogenase (LDH) activity, inhibits apoptosis and limits
mitochondrial damage (55).
Quercetin has been shown to enhance cardiomyocyte viability,
elevate SOD and catalase activity, and decrease LDH, ROS and
malondialdehyde levels through the upregulation of 14-3-3γ, thereby
mitigating DOX-induced cardiotoxicity. The protective effect is
diminished when 14-3-3γ expression is knocked down (56). However, the role of 14-3-3γ in
cardiac hypertrophy remains uncertain. In the present study,
14-3-3γ expression was reduced in the cardiac hypertrophy model but
increased in the TQ-treated group. Knockdown of 14-3-3γ using
pAD/14-3-3γ in the TQ group weakened the protective effects of TQ
and suppressed autophagy. To the best of our knowledge, the present
study is the first to demonstrate that TQ alleviates cardiac
hypertrophy by upregulating 14-3-3γ and promoting autophagy.
Research has demonstrated that PPAR-γ plays a
central role in regulating lipid metabolism and inflammation
(57). PPAR-γ is crucial for
maintaining cellular energy homeostasis, modulating inflammatory
responses and controlling fibrosis (58). As a transcription factor, PPAR-γ
regulates TGF-β1 expression and Smad2/3 phosphorylation, thereby
inhibiting hepatic stellate cell activation and mitigating liver
fibrosis (59). PPAR-γ
activation also suppresses platelet derived growth factor and
tissue inhibitory of metalloproteinase-2 expression in
non-alcoholic fatty liver disease, reducing inflammation and
fibrosis (60). We hypothesize
that PPAR-γ may bind to the 14-3-3γ promoter to alleviate cardiac
hypertrophy. Notably, in the present study, both 14-3-3γ and PPAR-γ
protein expression increased in the TQ group but declined following
PPAR-γ inhibitor (GW9662) treatment. A dual-luciferase reporter
assay confirmed that PPAR-γ enhanced 14-3-3γ promoter activity.
However, the absence of chromatin immunoprecipitation, Baf
A1-induced LC3-I/II conversion and mRFP-LC3 tandem fluorescence
assays in the present study limits its conclusiveness,
necessitating further investigation. Additionally, primary
cardiomyocytes, which were not used in the present study, may
better mimic the in vivo environment and produce more
convincing findings.
In conclusion, TQ appears to play a notable role in
preventing pressure overload-induced cardiac hypertrophy by
reducing oxidative stress and inhibiting fibrosis. TQ alleviates
cardiac hypertrophy by modulating 14-3-3γ expression and enhancing
autophagy through the activation of PPAR-γ transcriptional
activity. This mechanism offers a potential therapeutic strategy
for the clinical treatment of cardiac hypertrophy. Nonetheless, the
present study contains certain limitations. First, while TQ
exhibited a protective effect, its efficacy remains limited,
necessitating further investigation across additional cardiac
hypertrophy models. Second, the precise mechanism by which TQ
induces autophagy through the PPAR-γ/14-3-3γ pathway requires
further elucidation.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
RBQ conducted the cell experiments and analyzed and
mapped the experimental data. STZ provided the experimental design
and data for analysis. ZQX conducted animal experiments and
analyzed the results. RYZ, ZCQ, HZP and LFZ contributed to the cell
experiments. LJH enhanced the language of the article and analyzed
the data. YPC and LW designed the experiments and provided
financial support. RYZ, ZCQ and HZP confirm the authenticity of all
the raw data. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Animal experiments followed the guidelines of the
National Institutes of Health and were authorized by the Animal
Experimentation Ethics Committee of the First Affiliated Hospital,
Jiangxi Medical College, Nanchang University (Nanchang, China;
approval no. CDYFY-IACUC-202407QR115).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This study was funded by the Natural Science Foundation of
Jiangxi Province (grant no. 20212ACB206011) and the National
Natural Science Foundation of China (grant nos. 82460057, 81860082
and 82260059).
References
|
1
|
Frey N and Olson EN: Cardiac hypertrophy:
The good, the bad, and the ugly. Annu Rev Physiol. 65:45–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu M, Wan CX, Huang SH, Wang HB, Fan D, Wu
HM, Wu QQ, Ma ZG, Deng W and Tang QZ: Oridonin protects against
cardiac hypertrophy by promoting P21-related autophagy. Cell Death
Dis. 10:4032019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ritterhoff J and Tian R: Metabolic
mechanisms in physiological and pathological cardiac hypertrophy:
New paradigms and challenges. Nat Rev Cardiol. 20:812–829. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maillet M, Van Berlo JH and Molkentin JD:
Molecular basis of physiological heart growth: Fundamental concepts
and new players. Nat Rev Mol Cell Biol. 14:38–48. 2013. View Article : Google Scholar
|
|
7
|
Gali-Muhtasib H, Roessner A and
Schneider-Stock R: Thymoquinone: A promising anti-cancer drug from
natural sources. Int J Biochem Cell Biol. 38:1249–1253. 2006.
View Article : Google Scholar
|
|
8
|
Darakhshan S, Bidmeshki Pour A,
Hosseinzadeh Colagar A and Sisakhtnezhad S: Thymoquinone and its
therapeutic potentials. Pharmacol Res. 95-96:138–158. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Woo CC, Kumar AP, Sethi G and Tan KH:
Thymoquinone: Potential cure for inflammatory disorders and cancer.
Biochem Pharmacol. 83:443–451. 2012. View Article : Google Scholar
|
|
10
|
Mialet-Perez J and Vindis C: Autophagy in
health and disease: Focus on the cardiovascular system. Essays
Biochem. 61:721–732. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xue R, Zeng J, Chen Y, Chen C, Tan W, Zhao
J, Dong B, Sun Y, Dong Y and Liu C: Sestrin 1 ameliorates cardiac
hypertrophy via autophagy activation. J Cell Mol Med. 21:1193–1205.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang K, Long Q, Saja K, Huang F, Pogwizd
SM, Zhou L, Yoshida M and Yang Q: Knockout of the ATPase inhibitory
factor 1 protects the heart from pressure overload-induced cardiac
hypertrophy. Sci Rep. 7:105012017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kelly DP: PPARs of the heart: Three is a
crowd. Circ Res. 92:482–484. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Majdalawieh A and Ro HS: PPARgamma1 and
LXRalpha face a new regulator of macrophage cholesterol homeostasis
and inflammatory responsiveness, AEBP1. Nucl Recept Signal.
8:e0042010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kung J and Henry RR: Thiazolidinedione
safety. Expert Opin Drug Saf. 11:565–579. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peng Y, Wang L, Zhang Z, He X, Fan Q,
Cheng X, Qiao Y, Huang H, Lai S, Wan Q, et al: Puerarin activates
adaptive autophagy and protects the myocardium against
doxorubicin-induced cardiotoxicity via the 14-3-3γ/PKCε pathway.
Biomed Pharmacother. 153:1134032022. View Article : Google Scholar
|
|
17
|
Cheng Y, Shen A, Wu X, Shen Z, Chen X, Li
J, Liu L, Lin X, Wu M, Chen Y, et al: Qingda granule attenuates
angiotensin II-induced cardiac hypertrophy and apoptosis and
modulates the PI3K/AKT pathway. Biomed Pharmacother.
133:1110222021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He H, Wang L, Qiao Y, Yang B, Yin D and He
M: Epigallocatechin-3-gallate pretreatment alleviates
doxorubicin-induced ferroptosis and cardiotoxicity by upregulating
AMPKα2 and activating adaptive autophagy. Redox Biol.
48:1021852021. View Article : Google Scholar
|
|
19
|
Lu Q, Hu S, Guo P, Zhu X, Ren Z, Wu Q and
Wang X: PPAR-γ with its anti-fibrotic action could serve as an
effective therapeutic target in T-2 toxin-induced cardiac fibrosis
of rats. Food Chem Toxicol. 152:1121832021. View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Rockman HA, Ross RS, Harris AN, Knowlton
KU, Steinhelper ME, Field LJ, Ross J Jr and Chien KR: Segregation
of atrial-specific and inducible expression of an atrial
natriuretic factor transgene in an in vivo murine model of cardiac
hypertrophy. Proc Natl Acad Sci USA. 88:8277–8281. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen H, Zhuo C, Zu A, Yuan S, Zhang H,
Zhao J and Zheng L: Thymoquinone ameliorates pressure
overload-induced cardiac hypertrophy by activating the AMPK
signalling pathway. J Cell Mol Med. 26:855–867. 2022. View Article : Google Scholar
|
|
23
|
Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng
KQ, Jiang X, Wang PX, Huang Z and Li H: The ubiquitin E3 ligase
TRAF6 exacerbates pathological cardiac hypertrophy via
TAK1-dependent signalling. Nat Commun. 7:112672016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li HH, Kedar V, Zhang C, McDonough H, Arya
R, Wang DZ and Patterson C: Atrogin-1/muscle atrophy F-box inhibits
calcineurin-dependent cardiac hypertrophy by participating in an
SCF ubiquitin ligase complex. J Clin Invest. 114:1058–1071. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xie X, Bi HL, Lai S, Zhang YL, Li N, Cao
HJ, Han L, Wang HX and Li HH: The immunoproteasome catalytic β5i
subunit regulates cardiac hypertrophy by targeting the autophagy
protein ATG5 for degradation. Sci Adv. 5:eaau04952019. View Article : Google Scholar
|
|
26
|
Gyöngyösi M, Winkler J, Ramos I, Do QT,
Firat H, McDonald K, González A, Thum T, Díez J, Jaisser F, et al:
Myocardial fibrosis: Biomedical research from bench to bedside. Eur
J Heart Fail. 19:177–191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bacmeister L, Schwarzl M, Warnke S,
Stoffers B, Blankenberg S, Westermann D and Lindner D: Inflammation
and fibrosis in murine models of heart failure. Basic Res Cardiol.
114:192019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Doroszko A, Dobrowolski P,
Radziwon-Balicka A and Skomro R: New insights into the role of
oxidative stress in onset of cardiovascular disease. Oxid Med Cell
Longev. 2018:95638312018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Z, Song Y, Liu L, Hou N, An X, Zhan D,
Li Y, Zhou L, Li P, Yu L, et al: miR-199a impairs autophagy and
induces cardiac hypertrophy through mTOR activation. Cell Death
Differ. 24:1205–1213. 2017. View Article : Google Scholar :
|
|
30
|
Simonson B, Subramanya V, Chan MC, Zhang
A, Franchino H, Ottaviano F, Mishra MK, Knight AC, Hunt D, Ghiran
I, et al: DDiT4L promotes autophagy and inhibits pathological
cardiac hypertrophy in response to stress. Sci Signal.
10:eaaf59672017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Obsil T and Obsilova V: Structural basis
of 14-3-3 protein functions. Semin Cell Dev Biol. 22:663–672. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu W, Yang B, Qiao Y, Zhou Q, He H and He
M: Kaempferol protects mitochondria and alleviates damages against
endotheliotoxicity induced by doxorubicin. Biomed Pharmacother.
126:1100402020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Diebold I, Hennigs JK, Miyagawa K, Li CG,
Nickel NP, Kaschwich M, Cao A, Wang L, Reddy S, Chen PI, et al:
BMPR2 preserves mitochondrial function and DNA during reoxygenation
to promote endothelial cell survival and reverse pulmonary
hypertension. Cell Metab. 21:596–608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Caglayan E, Stauber B, Collins AR, Lyon
CJ, Yin F, Liu J, Rosenkranz S, Erdmann E, Peterson LE, Ross RS, et
al: Differential roles of cardiomyocyte and macrophage peroxisome
proliferator-activated receptor gamma in cardiac fibrosis.
Diabetes. 57:2470–2479. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Duan SZ, Ivashchenko CY, Russell MW,
Milstone DS and Mortensen RM: Cardiomyocyte-specific knockout and
agonist of peroxisome proliferator-activated receptor-gamma both
induce cardiac hypertrophy in mice. Circ Res. 97:372–379. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li T, Guo R, Zong Q and Ling G:
Application of molecular docking in elaborating molecular
mechanisms and interactions of supramolecular cyclodextrin.
Carbohydr Polym. 276:1186442022. View Article : Google Scholar
|
|
37
|
Oka T, Akazawa H, Naito AT and Komuro I:
Angiogenesis and cardiac hypertrophy: Maintenance of cardiac
function and causative roles in heart failure. Circ Res.
114:565–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang C, Liu J, Pan H, Yang X and Bian K:
Mitochondrial dysfunction induced by excessive ROS/RNS-metabolic
cardiovascular disease and traditional Chinese medicines
intervention. Zhongguo Zhong Yao Za Zhi. 36:2423–2428. 2011.In
Chinese. PubMed/NCBI
|
|
39
|
Liu BY, Li L, Liu GL, Ding W, Chang WG, Xu
T, Ji XY, Zheng XX, Zhang J and Wang JX: Baicalein attenuates
cardiac hypertrophy in mice via suppressing oxidative stress and
activating autophagy in cardiomyocytes. Acta Pharmacol Sin.
42:701–714. 2021. View Article : Google Scholar :
|
|
40
|
Gao M, Hu F, Hu M, Hu Y, Shi H, Zhao GJ,
Jian C, Ji YX, Zhang XJ, She ZG, et al: Sophoricoside ameliorates
cardiac hypertrophy by activating AMPK/mTORC1-mediated autophagy.
Biosci Rep. 40:BSR202006612020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li MH, Zhang YJ, Yu YH, Yang SH, Iqbal J,
Mi QY, Li B, Wang ZM, Mao WX, Xie HG and Chen SL: Berberine
improves pressure overload-induced cardiac hypertrophy and
dysfunction through enhanced autophagy. Eur J Pharmacol. 728:67–76.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Togliatto G, Lombardo G and Brizzi MF: The
future challenge of reactive oxygen species (ROS) in hypertension:
From bench to bed side. Int J Mol Sci. 18:19882017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dhalla AK, Hill MF and Singal PK: Role of
oxidative stress in transition of hypertrophy to heart failure. J
Am Coll Cardiol. 28:506–514. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Qin F, Lennon-Edwards S, Lancel S, Biolo
A, Siwik DA, Pimentel DR, Dorn GW, Kang YJ and Colucci WS:
Cardiac-specific overexpression of catalase identifies hydrogen
peroxide-dependent and -independent phases of myocardial remodeling
and prevents the progression to overt heart failure in
G(alpha)q-overexpressing transgenic mice. Circ Heart Fail.
3:306–313. 2010. View Article : Google Scholar
|
|
45
|
Liu C, Wu QQ, Cai ZL, Xie SY, Duan MX, Xie
QW, Yuan Y, Deng W and Tang QZ: Zingerone attenuates aortic
banding-induced cardiac remodelling via activating the eNOS/Nrf2
pathway. J Cell Mol Med. 23:6466–6478. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hansen M, Rubinsztein DC and Walker DW:
Autophagy as a promoter of longevity: Insights from model
organisms. Nat Rev Mol Cell Biol. 19:579–593. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sun M, Ouzounian M, de Couto G, Chen M,
Yan R, Fukuoka M, Li G, Moon M, Liu Y, Gramolini A, et al:
Cathepsin-L ameliorates cardiac hypertrophy through activation of
the autophagy-lysosomal dependent protein processing pathways. J Am
Heart Assoc. 2:e0001912013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Drosatos K, Khan RS, Trent CM, Jiang H,
Son NH, Blaner WS, Homma S, Schulze PC and Goldberg IJ: Peroxisome
proliferator-activated receptor-γ activation prevents
sepsis-related cardiac dysfunction and mortality in mice. Circ
Heart Fail. 6:550–562. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Desvergne B and Wahli W: Peroxisome
proliferator-activated receptors: Nuclear control of metabolism.
Endocr Rev. 20:649–688. 1999.PubMed/NCBI
|
|
52
|
Herrmann JE, Heale J, Bieraugel M, Ramos
M, Fisher RL and Vickers AE: Isoproterenol effects evaluated in
heart slices of human and rat in comparison to rat heart in vivo.
Toxicol Appl Pharmacol. 274:302–312. 2014. View Article : Google Scholar
|
|
53
|
Yang J, Liu Y, Fan X, Li Z and Cheng Y: A
pathway and network review on beta-adrenoceptor signaling and beta
blockers in cardiac remodeling. Heart Fail Rev. 19:799–814. 2014.
View Article : Google Scholar
|
|
54
|
Yamaguchi O: Autophagy in the heart. Circ
J. 83:697–704. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
He H, Luo Y, Qiao Y, Zhang Z, Yin D, Yao
J, You J and He M: Curcumin attenuates doxorubicin-induced
cardiotoxicity via suppressing oxidative stress and preventing
mitochondrial dysfunction mediated by 14-3-3γ. Food Funct.
9:4404–4418. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen X, Peng X, Luo Y, You J, Yin D, Xu Q,
He H and He M: Quercetin protects cardiomyocytes against
doxorubicin-induced toxicity by suppressing oxidative stress and
improving mitochondrial function via 14-3-3γ. Toxicol Mech Methods.
29:344–354. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chen H, Tan H, Wan J, Zeng Y, Wang J, Wang
H and Lu X: PPAR-γ signaling in nonalcoholic fatty liver disease:
Pathogenesis and therapeutic targets. Pharmacol Ther.
245:1083912023. View Article : Google Scholar
|
|
58
|
Botta M, Audano M, Sahebkar A, Sirtori CR,
Mitro N and Ruscica M: PPAR agonists and metabolic syndrome: An
established role? Int J Mol Sci. 19:11972018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
NiX X, Li XY, Wang Q and Hua J: Regulation
of peroxisome proliferator-activated receptor-gamma activity
affects the hepatic stellate cell activation and the progression of
NASH via TGF-β1/Smad signaling pathway. J Physiol Biochem.
77:35–45. 2021. View Article : Google Scholar
|
|
60
|
Deng W, Meng Z, Sun A and Yang Z:
Pioglitazone suppresses inflammation and fibrosis in nonalcoholic
fatty liver disease by down-regulating PDGF and TIMP-2: Evidence
from in vitro study. Cancer Biomark. 20:411–415. 2017. View Article : Google Scholar
|