Introduction
Pendrin (SLC26A4) is an electroneutral exchanger for
monovalent anions that is expressed on the apical membrane of
distinct epithelial cells in several tissues (1-4).
In the inner ear, the physiological functions of pendrin are
complex and include the modification of the ion composition, pH and
volume of the endolymph. Specifically, Cl− reabsorption
linked to a pendrin-dependent
Cl−/HCO3− exchange residing on the
mitochondria-rich cells permits fluid reabsorption in the
endolymphatic sac, thereby controlling the volume of the endolymph
(5). In the cochlea,
HCO3− secretion from pendrin-expressing
non-sensory cells prevents acidification of the endolymph and is
fundamental in the maintenance of endolymphatic ion homeostasis
(6,7). In the thyroid, pendrin contributes
to the transport of iodide into the colloidal lumen of thyroid
follicles, most likely via a Cl−/I− exchange
(8). In the kidney, pendrin is
part of the molecular machinery of beta-intercalated cells that, in
the distal nephron, reacts to metabolic alkalosis by activating
HCO3− secretion into the pre-urine.
Simultaneously, pendrin drives a Cl− reabsorption that
permits salt and fluid reabsorption and contributes to controlling
systemic blood pressure (9-11).
Genetically determined loss of pendrin function in
humans leads to isolated autosomal recessive deafness B4 (DFNB4,
OMIM ID_600791) and Pendred syndrome, which is defined as the
association between hearing loss and thyroid dysfunction due to a
defective iodide organification (OMIM ID_274600) (12,13). The clinical course of hearing
loss varies among subjects with Pendred syndrome/DFNB4. Some
patients lose hearing during their early childhood, which severely
compromises language onset and discrimination. However, other
patients manifesting fluctuating and progressive or sudden hearing
loss lose hearing later in life (14). Hearing loss in Pendred
syndrome/DFNB4 is consistently associated with a malformation of
the temporal bone called enlarged vestibular aqueduct (EVA), while
a cochlear incomplete partition type II (Mondini cochlea) may or
may not be present. The findings of ears with EVA and normal
hearing indicate that EVA does not directly cause hearing loss but
can be considered a radiologic marker of the genetic etiology of
this condition (14,15).
Ablation of the Slc26a4 gene in the mouse was
instrumental in clarifying the pathological mechanism of
pendrin-related hearing loss. The most striking findings in pendrin
knockout mice are profound deafness and prominent enlargement of
all inner ear compartments, including the scala media of the
cochlea and the endolymphatic sac and duct (16,17). Hearing loss in these mice appears
to be the consequence of sequential and probably causally connected
perturbations of the ion and volume homeostasis of the endolymph,
starting with acidification of the cochlear endolymph due to lack
of pendrin-dependent bicarbonate secretion and expansion of the
endolymphatic compartment due to lack of pendrin-driven salt and
fluid reabsorption in the endolymphatic sac. The cochlear fluid
expansion was suggested to lead to metabolic stress in the stria
vascularis due to an increased rate of K+ secretion,
with loss of expression of the oxidative stress-sensitive
K+ channel KCNJ10/Kir4.1 and consequential loss of
endo-cochlear potential (7).
Both the loss of the endo-cochlear potential, which is positive in
the lumen of the scala media, and inhibition of the
pH-sensitive Ca++ channels TRPV5/TRPV6, would cause an
increase in the endolymphatic Ca2+ concentration,
ultimately leading to the degeneration of the sensory hair cells in
the organ of Corti (17,18). These derangements point to the
dysregulation of ion transport as a key pathogenic factor in
Pendred syndrome/DFNB4. Therefore, a potential therapeutic approach
should aim to recover the ion transport function of pathogenic
pendrin variants.
Studies in transgenic mice where pendrin expression
could be induced and terminated at defined time points revealed
that the time window between embryonic day 16.5 and post-natal (P)
day 2 is critical for hearing acquisition (15). Importantly, induction of pendrin
expression at P6 stabilized hearing in a mouse model of fluctuating
and progressive hearing loss with EVA (19). These findings indicate that
interventions to recover pendrin expression during the pre-natal or
early post-natal phase may have therapeutic potential in Pendred
syndrome/DFNB4 (17).
Interestingly, while pendrin knockout mice invariably exhibit
profound deafness, in our pendrin p.L236P knock-in mouse the degree
of hearing loss varies from mild to profound, more closely
resembling the human phenotype (20). Together, these considerations
support the existence of a temporal window of possible intervention
to prevent hearing loss or protect and stabilize residual hearing
in patients with Pendred syndrome/DFNB4. However, no mechanistic
approaches toward these therapeutic goals have been developed.
The great majority of inherited alterations of the
SLC26A4 gene lead to the production of pathogenic protein
variants with a single amino acid substitution. Seminal studies of
these pendrin variants in heterologous expression systems helped
reveal the pathological mechanism of disease. Pathogenic pendrin
variants showed a partial or total impairment of ion transport
activity, which was initially associated with misfolding of the
protein, retention within intracellular compartments, and
consequential failure in reaching the cell surface (21,22). Later, studies revealed that
several pendrin variants, although compromised concerning their ion
transport ability, are correctly expressed at the plasma membrane
(23-27). Importantly, it was found out that
reduction of total protein expression, rather than
mis-localization, appears to be a key feature of all functionally
impaired pendrin variants (26,27).
Very little is known about the mechanisms
controlling the protein levels of wild-type pendrin and its
variants. Misfolded cellular proteins undergo ubiquitination and
ubiquitin-proteasome system (UPS)-mediated degradation. It was
previously shown that the fusion protein wild-type pendrin-GFP is a
slow-folding protein that accumulates in the endoplasmic reticulum
(ER) and perinuclear aggregates and is ubiquitinated (28), but the ubiquitination sites
remained unknown. The ER membrane-associated ubiquitin ligase Rma1
was identified as a regulator of the cellular levels of wild-type
and p.H723R pendrin, but not those of p.L236P and other variants
(29). Interestingly, in
conditions of blockade of the ER-to-Golgi transit or ER stress, the
misfolded pendrin p.H723R can bypass the Golgi, reach the plasma
membrane, and exhibit significant ion transport function. This
phenomenon depends on the heat shock cognate protein 70 (Hsc70) and
the HSP70 co-chaperone DNAJC14 (30). Ubiquitination not only determines
protein degradation but also controls subcellular localization. The
E3 HECT ubiquitin ligase Nedd4-2 controls the trafficking of
wild-type pendrin at the apical membrane of beta-intercalated cells
in the mouse cortical collecting duct and connecting tubule, with
no major influence on the total protein abundance (31).
In the present study, it was demonstrated that the
cellular expression levels of wild-type pendrin and its pathogenic
protein variants are controlled by the UPS, and UPS inhibitors can
rescue pendrin protein expression and function.
Materials and methods
Mammalian expression vectors
The pTARGET (Promega Corporation) vector coding for
human SLC26A4 (NCBI Sequence ID: NM_000441.2) with or without a
hexa-histidine tag at the C-terminus was used for transfecting
cells for functional testing and some of the western blotting,
respectively. For functional testing, the enhanced yellow
fluorescent protein (EYFP) pEYFPN1 p.H148Q;I152L plasmid was also
used. This vector encodes for the EYFP variant p.H148Q;I152L, which
is sensitive to the intracellular iodide concentration (32). The pFLAG-CMV-4 (Sigma-Aldrich;
Merck KGaA) vector coding for SLC26A4 with a Met-FLAG tag at the
N-terminus was used for transfecting cells for immunoprecipitation,
half-life measurements, and western blotting.
For the determination of pendrin expression levels
via quantitative imaging, cells were transfected with the pEYFPN1
vector (Takara Bio USA, Inc.) to produce pendrin with the EYFP
fused to its C-terminus (SLC26A4-EYFP). In experiments where the
plasma membrane expression was evaluated, cells were co-transfected
with equimolar amounts of the SLC26A4-EYFP vector and a vector
coding for the enhanced cyan fluorescent protein (ECFP) (pECFPC1
vector; Takara Bio USA, Inc.).
Sequence alterations in the pendrin coding sequence
were performed using the QuikChange® site-directed
mutagenesis kit (Agilent Technologies, Inc.) according to the
manufacturer's protocol using the following primers: p.L236P
forward, 5′-GCT GGT CTC ACA GCC AAA GAT TGT CCT CAA TG-3′ and
reverse, 5′-CAT TGA GGA CAA TCT TTG GCT GTG AGA CCA GC-3′; and p.
R409H forward, 5′-ACT GCT CTT TCC CAC ACG GCC GTC CAG GA-3′ and
reverse, 5′-TCC TGG ACG GCC GTG TGG GAA AGA GCA GT-3′. The plasmid
vectors coding the other pendrin variants have already been
described (26). Sequential
C-terminal SLC26A4 truncations were obtained by inserting a STOP
codon at the appropriate nucleotide position in the SLC26A4
sequence into the pFLAG-CMV-4 vector. The integrity of all coding
sequences was verified by Sanger sequencing (Microsynth AG).
Animals
Homozygous mutant mice bearing the Slc26a4 variant
p.L236P were generated using the CRISPR/Cas9 genome editing
technology and have been previously described (20). The animals were kept in a
standard environment with a temperature of 22±1°C and a relative
humidity of 50-60%, and they were allowed free access to food and
water. The age of the mice used in the present study was 2-3
months-old and weighed >20 g. Mice were euthanized by cervical
dislocation. Each group of experiments involved 3 male mice. The
treatment of all the animals was in strict accordance with the
requirements of animal experiments at Shandong University (Jinan,
China), which were approved by the Ethics Committee of the School
of Life Science, Shandong University (approval no.
SYDWLL-2021-76).
Reverse-transcription quantitative PCR
(RT-qPCR)
For RT-qPCR, total RNA was extracted from mouse
kidneys with the TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Reverse transcription was conducted with
SPARKscript II All-in-one RT SuperMix for qPCR with gDNA Eraser
following the manufacturer's protocol (Shandong Sparkjade
Scientific Instruments Co., Ltd.), and qPCR was made with 2X SYBR
Green qPCR Mix with ROX (Shandong Sparkjade Scientific Instruments
Co., Ltd.). For qPCR, the initial denaturation temperature was
95°C, followed by 30 cycles of denaturation (95°C, 30 sec),
annealing (60°C, 30 sec) and extension (72°C, 30 sec). The Bio-Rad
Sequence Detection System (Bio-Rad Laboratories Inc.) was used to
detect the expression levels of Slc26a4. The primers used
were as follows: Slc26a4 forward, 5′-AA GAG AGC CTT TGG TGT
GGT A-3′ and reverse, 5′-CAG GGC ATA AGC CAT CCC TTG-3′; and
ACTB forward, 5′-GGC TGT ATT CCC CTC CAT CG-3′ and reverse,
5′-CCA GTT GGT AAC AAT GCC ATG T-3′. Relative gene expression
values were quantified using the comparative Ct method (33).
Cell transfection
293 Phoenix cells (34) (EcoPhoenix, https://ngvbcc.org/ReagentRepositoryView?prefillSearch=Cell%20Line),
a highly transfectable derivative of 293T cells (CVCL_0063), were a
kind gift from Prof. M. Baruscotti from the University of Milan
(Italy). HeLa cells (human cervical adenocarcinoma, CCL-2) were
directly obtained from the American Type Cell Culture Collection.
These cells were cultivated as previously described (26,27). 293 Phoenix cells were transfected
with the calcium phosphate co-precipitation method for 48 h, and
Hela cells were transfected with METAFECTENE PRO®
(Biontex Laboratories GmbH) for 72 h. The ratio µg DNA:
µl METAFECTENE PRO® was 1:2.
For western blotting, confocal imaging, half-life
measurements, and immunoprecipitation, cells were seeded into
six-well plates and transfected with 3 µg/well (293 Phoenix)
or 1.5 µg/well (HeLa) of the indicated plasmids. After 6-8
h, the transfection medium was replaced with a fresh medium.
For functional testing, 293 Phoenix cells were
seeded into black 96-well plates and co-transfected with 0.12
µg/well of the pTARGET plasmid encoding for wild-type
pendrin or its variants and 0.12 µg/well of the EYFP
p.H148Q; I152L plasmid. The endogenous iodide influx was determined
in cells co-transfected with 0.12 µg/well of the EYFP
p.H148Q; I152L vector and 0.12 µg/well of the empty pTARGET
vector. The background fluorescence was measured in cells
transfected with 0.24 µg/well of the pTARGET vector.
Pendrin functional test
The ion transport function of pendrin was measured
via a fluorometric method allowing for evaluation of iodide influx
in cells transfected with pendrin and the iodide-sensitive
fluorescent probe EYFP p.H148Q;I152L, as formerly described
(26,27,35-42). Additional details are provided in
Appendix S1.
Half-life measurements and western
blotting
For pendrin half-life measurements, 293 Phoenix
cells were seeded in 6-well plates and transfected with wild-type
FLAG-SLC26A4 or the p.L236P and p.R409H variants. After
transfection (24 h), the cells were treated with 25 µg/ml
cycloheximide (Sigma-Aldrich; Merck KGaA) to block the protein
synthesis and collected 0, 2, 4, 8 and 24 h after treatment. To
assess the effect of proteasome inhibition with MG132, N-terminally
flagged full-length pendrin and its C-terminal truncations were
expressed in 293 Phoenix cells for 48 h and incubated with 1-10
µM MG132 or the vehicle (0.01% DMSO) for 16 h. To assess the
effect of inhibition of the autophagosomal/lysosomal pathway,
N-terminally flagged wild-type pendrin and its p.R409H or p.L236P
variants were expressed in 293 Phoenix cells for 48 h and incubated
with 200 µM chloroquine or the vehicle (water) for 6 h. Each
cell pellet corresponding to one or two wells of a 6-well plate was
lysed by resuspension in 50-100 µl RIPA buffer
(Sigma-Aldrich; Merck KGaA) supplemented with 1X Halt Protease
Inhibitor Cocktail (Thermo Fisher Scientific, Inc.) to obtain total
proteins. Cell lysates were centrifuged at 900 x g for 10 min at
4°C, and the supernatant was submitted to western blotting. Where
indicated, whole cell lysates (20 µg total proteins) were
submitted to deglycosylation with 50,000 units/ml PNGase F (cat.
no. P0704; New England Biolabs) for 24 h at 37°C before western
blotting.
To assess the effect of proteasome inhibition with
MG132 on pendrin variants, 293 Phoenix cells were transfected for
48 h with wild-type pendrin or its variants and collected by
centrifugation. The total membrane protein fraction was obtained
with the Plasma Membrane Extraction kit (MBL International
Corporation) and subjected to western blotting.
Western blotting was performed with standard
methods. Additional details are given in Appendix S1. Pendrin
detection was done with a mouse monoclonal anti-FLAG® M2
antibody (cat. no. F3165; Sigma-Aldrich; Merck KGaA) diluted
1:1,000 (half-life measurements) or with a rabbit anti-pendrin
polyclonal antibody raised against amino acids 586-780 of human
pendrin (cat. no. sc-50346; Santa Cruz Biotechnology, Inc.) diluted
1:500. The housekeeping proteins alpha-tubulin or
calreticulin/calregulin were detected with mouse monoclonal
antibodies (1:2,000; cat. no. 05-829; Sigma-Aldrich; Merck KGaA; or
1:100; cat. no. sc-373863; Santa Cruz Biotechnology, Inc.) and
GAPDH was detected with a goat polyclonal antibody (1:1,000; cat.
no. PLA0302; Sigma-Aldrich; Merck KGaA). The secondary antibodies
(goat anti-rabbit, cat. no. 92632211; goat anti-mouse, cat. no.
926-32210; or donkey anti-goat, cat. no. 926-32214; conjugated to
IRDye-800 CW, all from LI-COR Biosciences) were diluted 1:20,000.
The signal of immunocomplexes was detected using the Odyssey
(LI-COR Biosciences) infrared imaging system. Densitometric
analysis was conducted using the ImageJ (1.53t) software (National
Institutes of Health).
Western blotting on total proteins of mouse kidney
was performed with a customized polyclonal antibody raised in
rabbit against two partially overlapping synthetic peptides
corresponding to the C-terminus of human pendrin (Davids
Biotechnologie GmbH). The antibody was diluted (1:1,000) and
membranes were blocked with 1% BSA (cat. no. 1.12018.0100;
Sigma-Aldrich; Merck KGaA) in TBST (0.1% Tween 20) at room
temperature for 2 h. β-actin was used as the housekeeping
protein.
Total protein expression
Quantitative imaging to determine total protein
expression was performed by confocal microscopy (Leica TCS SP5II
AOBS; Leica Microsystems GmbH) as formerly described (26,27). The expression levels of wild-type
or mutant SLC26A4-EYFP are given as the EYFP fluorescence intensity
normalized for the cell density. Additional details are reported in
Appendix S1.
Plasma membrane protein expression
The protein expression levels of wild-type or mutant
SLC26A4-EYFP in the plasma membrane region were determined by
confocal microscopy in live HeLa cells co-transfected with ECFP and
stained with the CellMask™ Deep Red plasma membrane stain (cat. no.
C10046; Molecular Probes; Thermo Fisher Scientific, Inc.) by using
a formerly optimized protocol (26). The ECFP signal allowed
normalization of the protein expression for the transfection
efficiency of the single cell. Additional information is provided
in Appendix S1.
Autophagosome staining and
co-localization
Hela cells seeded in 6-well plates were transfected
with 1.5 µg/well wild-type or mutant SLC26A4-EYFP plasmid
vector and 3 µl/well METAFECTENE PRO® (cat. no.
T040-2.0; Biontex Laboratories GmbH) at 37°C, transferred on glass
slides after 42 h, and incubated overnight with 100 µM
chloroquine or the vehicle. The total transfection time was 72 h.
After this, cells were fixed in 4% paraformaldehyde for 15 min,
permeabilized with 0.2% Triton X-100, blocked with 3% BSA for 1 h
at room temperature, incubated overnight with a rabbit polyclonal 1
µg/ml anti-LC3B antibody (cat. no. L10382;
InvitrogenTM; Thermo Fisher Scientific, Inc.; in 0.1%
BSA), thoroughly washed, and incubated for 1 h at room temperature
with a goat anti-rabbit Cy5®-conjugated IgG (cat. no.
ab6564; Abcam) diluted 1:2,500 in 0.1% BSA. Finally, nuclei were
counterstained with 0.1 µg/ml DAPI for 10 min, and cells
were kept and imaged in PBS. Imaging was performed by sequential
acquisition confocal microscopy. The imaging parameters were as
follows: DAPI, excitation wavelength 405 nm (diode laser), emission
range 450-490 nm. EYFP, excitation wavelength 514 nm (argon laser),
emission range 525-600 nm; Cy5, excitation wavelength 633 nm (HeNe
laser), emission range 650-750 nm. Co-localization between SLC26A4
(EYFP) and autophagosomes (Cy5) was quantified and expressed as
Pearson's correlation coefficient by using the colocalization
plugin of the LAS AF software (Leica Microsystems GmbH).
Cell viability tests
Cell proliferation was determined with the CellTiter
96® AQueous One Solution Cell Proliferation Assay system
(Promega Corporation). Cells were seeded in 96-well plates, treated
with proteasome inhibitors or their vehicle (DMSO) for 6-16 h, and
incubated with 10 µl CellTiter 96® AQueous One
Solution Reagent for 1.5 h in a humidified 5% CO2
atmosphere. The formation of formazan was determined by reading the
absorbance at 490 nm (VICTOR™ X3 Multilabel Plate Reader; Perkin
Elmer, Inc.). All readings were corrected for the absorbance of the
background.
To measure the cell density, cells were seeded in
24-well plates, treated with proteasome inhibitors or their
vehicle, fixed in 4% paraformaldehyde for 30 min, stained with 0.1
µg/ml DAPI for 10 min, thoroughly washed, and kept in HBSS.
The fluorescence of DAPI was read following excitation at 355 nm
(VICTOR™ X3 Multilabel Plate Reader) and subtracted for the
background fluorescence.
Immunoprecipitation and ubiquitination
assay
293 Phoenix cells seeded in 6-well plates were
transfected for 48 h with wild-type or mutant FLAG-SLC26A4 or left
untreated and collected by centrifugation. Each cell pellet
corresponding to a whole 6-well plate was lysed in 300 µl
Pierce™ IP Lysis Buffer (Thermo Fisher Scientific, Inc.)
supplemented with 1X Halt Protease Inhibitor Cocktail (Thermo
Fisher Scientific, Inc.). Cell lysates were centrifuged at 900 x g
for 10 min at 4°C, the supernatant was saved and the protein
content was quantified. Total protein (1 mg) was incubated
overnight at 4°C with 10 µg of a mouse monoclonal
anti-FLAG® M2 antibody (cat. no. F3165; Sigma-Aldrich;
Merck KGaA) in 400 µl IP Lysis Buffer on a tube rotator.
Subsequently, this sample was incubated with 50 µl
Dynabeads™ Protein A (Invitrogen™; Thermo Fisher Scientific, Inc.)
for 3 h at room temperature on a tube rotator. Beads were washed 3
times with 0.2% Tween-20 in PBS and eluted in 20 µl sample
buffer containing 100 mM dithiothreitol and 8% SDS. The eluates
were boiled at 95°C for 5 min and subjected to western blotting
with anti-FLAG antibody (1:1,000 in 5% non-fat dry milk in TBST) to
confirm the presence of pendrin in the immunoprecipitated samples.
GAPDH (goat anti-GAPDH; 1:1,000; cat. no. PLA0302; Sigma-Aldrich;
Merck KGaA) was detected to ensure the absence of unrelated
cellular proteins in the immunoprecipitated samples. To detect
ubiquitination, membranes were stripped and probed with a mouse
monoclonal anti-ubiquitin antibody (P4D1; 1:1,000; cat. no.
sc-8017; Santa Cruz Biotechnology, Inc.). In a subset of
experiments, the immunoprecipitated proteins were subjected to
deglycosylation by incubating the beads with 50,000 units/ml PNGase
F (New England Biolabs) for 24 h at 37°C.
Mass spectrometry (MS)
For ubiquitination analysis by MS, 293 Phoenix cells
were transfected with the pFLAG vector coding for wild-type pendrin
or the pendrin variant p.R409H and incubated overnight with the
proteasome inhibitor MG132 (1 µM; cat. no. C2211;
Sigma-Aldrich; Merck KGaA) or its vehicle (0.01% DMSO). Protein
extracts from cell lysates obtained in RIPA buffer were
immunoprecipitated with an anti-FLAG antibody as described above,
separated on a 9% acrylamide gel, and the band corresponding to
pendrin (90-120 kDa) was cut from the gel. Trypsin digestion for
the liquid chromatography-tandem mass spectrometry (LC-MS/MS)
ubiquitination analysis was performed overnight at 37°C directly on
the gel slice. Peptides were extracted from the gel slice,
lyophilized, and resuspended in 0.1% formic acid before the
analysis. Peptides were separated in an Ultimate™ 3000 RSLCnano
UHPLC system (Thermo Fisher Scientific, Inc.) with a flow rate of
250 nl/min. Additional details are given in Appendix S1. Raw MS
output files were analyzed and searched against the human pendrin
reference sequence (NP_000432.1) using Maxquant (1.6.2.6) in order
to identify GlyGly-modified peptides. The analysis was performed in
duplicate for both wild-type pendrin and the p.R409H variant, with
and without MG132 treatment.
Salt and chemicals
Bortezomib (cat. no. PS-341), carfilzomib (cat. no.
PR-171), delanzomib (cat. no. CEP-18770), ataluren (cat. no.
PTC124), elexacaftor (cat. no. VX-445), lumacaftor (cat. no.
VX-809) and tezacaftor (cat. no. VX-661) were obtained from Selleck
Chemicals. MG132, tenidap (cat. no. PZ0196) and rapamycin (cat. no.
553211) were from Sigma-Aldrich; Merck KGaA. To prepare stock
solutions, compounds were dissolved in dimethyl-sulfoxide (DMSO)
(Sigma-Aldrich; Merck KGaA) and stored at −80°C.
Correlation analysis and statistics
For the correlation analysis shown in Fig. 1, panels D-F, raw data for ion
transport function, total protein abundance, and plasma membrane
protein abundance of pendrin variants are obtained either from a
previous study by the authors (26) (p.P142L, p.G149R, p.T193I,
p.C282Y, p.Q413R, p.L445W and p.R776C) or from the present study
(p.L236P and p.R409H). Ion transport function values have been
subtracted for the endogenous iodide influx determined in cells
transfected with the empty vector to obtain the net iodide influx
of each variant. All data are expressed as a fraction of the
wild-type (0-1). A positive correlation between data sets was
tested by linear regression and expressed as Pearson's r
coefficient.
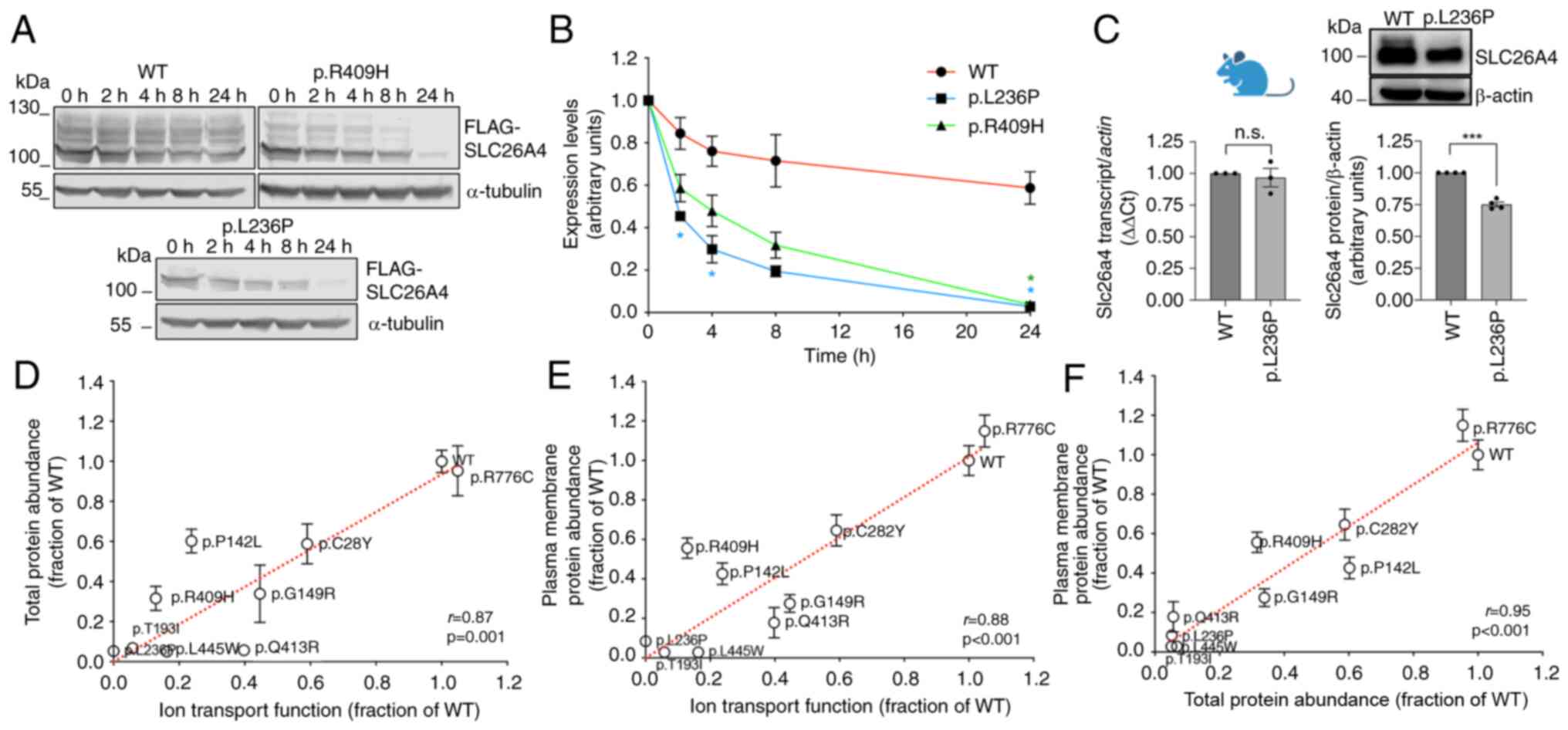 | Figure 1The protein half-life of pathogenic
pendrin variants is significantly reduced compared with the
wild-type, and reduced protein levels correlate to reduced
function. (A) Original western blots and (B) half-life measurements
of wild-type pendrin or pendrin variants p.L236P and p.R409H in 293
Phoenix cells transfected for 24 h with the pFLAG vector and
incubated with 25 µg/ml cycloheximide for 0-24 h.
*P<0.05, vs. wild-type, two-way ANOVA with Tukey's
multiple comparison post-test. 3<n<4; n refers to the number
of independent experiments. For each time point, data have been
normalized for the expression levels of the respective variant at
time 0. (C) Slc26a4 transcript levels (n=3) and original western
blot and densitometry (n=4) of pendrin expression in the crude
lysates of whole kidneys from SLC26A4 p.L236P mice and their
wild-type littermates. n refers to the number of independent
samples. ***P<0.001, one-sample Student's t-test.
n.s., not significant. (D-F) Ion transport function (the iodide
influx of each individual variant was subtracted for the endogenous
iodide influx measured in control cells; 42<n<72 independent
measurements from at least 3 independent experiments), total
protein abundance (n=16 independent measurements from at least 3
independent experiments), and plasma membrane protein abundance
(n=16 independent measurements from at least 3 independent
experiments) of wild-type pendrin and seven pendrin variants [raw
data received from de Moraes et al (26)] plus p.L236P and p.R409H (raw data
taken from Figs. 4D, 3B and 4B respectively, white bars) were
expressed as fraction of the wild-type and positive correlation
between data sets was tested by linear regression. Pearson's r and
two-tailed P-values are indicated. |
Data are expressed as arithmetic mean ± standard
error of the mean (S.E.M.). GraphPad Prism (version 10.2.3 for Mac
OS, GraphPad Software, Inc.; Dotmatics) and Excel (Microsoft
Corporation) software were used for statistical analysis and
graphics generation. Significant differences between two or more
data sets were determined by the one-sample or two-tailed unpaired
Student's t-test or one-way or two-way ANOVA with Bonferroni's,
Dunnet's, or Tukey's post hoc test, as appropriate. P<0.05 was
considered to indicate a statistically significant difference; (n)
corresponds to the number of independent samples.
Results
Pathogenic variants of the pendrin
protein undergo rapid cellular degradation
As aforementioned, it has been previously shown that
reduction in protein expression is a common feature of pathogenic
pendrin variants (26,27), but the underlying mechanisms
remained unknown. To test whether reduced expression stems from
accelerated protein degradation, the half-life of ectopically
expressed wild-type pendrin and pendrin variants p.L236P and
p.R409H, which are two of the most common pathogenic pendrin
variants in Caucasians, was measured (43,44). The results clearly revealed that,
after the block of the protein synthesis (time 0), pendrin protein
abundance was significantly influenced by time (P<0.0001,
two-way ANOVA) and variant type (P<0.0001). In addition, there
was a significant interaction of variant type and time on pendrin
abundance (P<0.0001), that is, the pendrin half-life differed
depending on the individual variant. Specifically, pendrin protein
variants p.L236P and p.R409H were degraded more rapidly compared
with the wild-type (Fig. 1A and
B). For each variant, data have been normalized for the
corresponding expression levels at time 0 and therefore show
expression level 1 at this time point (Fig. 1B), but absolute expression levels
of p.L236P and p.R409H variants are indeed reduced compared with
the wild-type (Fig. 1A, 0
h).
The transcript and protein expression of pendrin
were also tested in a knock-in mouse model of Pendred
syndrome/DFNB4 bearing the pendrin variant p.L236P. The mouse
exhibits mild to profound hearing loss and vestibular dysfunction
(20). The protein expression
levels of p.L236P variant were significantly reduced in the kidneys
of homozygous mutant mice compared with their wild-type littermates
with no reduction in transcript levels, ruling out the possibility
that the reduced pendrin protein abundance in p.L236P mice is due
to reduced transcription or mRNA instability and indicating that an
accelerated degradation of the pathogenic pendrin variant also
occurs in native tissue (Fig.
1C). The ion transport function determined as iodide influx in
transfected cells, the total protein abundance, and the plasma
membrane protein abundance of wild-type pendrin and seven pendrin
variants characterized in our previous study (26), as well as pendrin variants
p.L236P and p.R409H, are shown in Fig. 1D-F. Total protein abundance
(Fig. 1D), as well as plasma
membrane protein abundance (Fig.
1E), correlated positively with ion transport function. These
observations support the hypothesis that a reduction in protein
function derives from a reduction in protein expression. Total
protein abundance and plasma membrane protein abundance were also
positively correlated (Fig. 1F).
As the plasma membrane fraction is the functional form of the anion
exchanger, these data indicate that increasing the total protein
abundance might rescue plasma membrane protein abundance and,
consequently, protein function.
Wild-type pendrin and its pathogenic
variants are ubiquitinated
293 Phoenix cells were transfected with N-terminally
flagged wild-type pendrin and pendrin variants p.R409H and p.L236P
and treated with a specific inhibitor of 26S proteasome (1
µM MG132 for 16 h) to induce the accumulation of
ubiquitinated proteins. Pendrin was immunoprecipitated with an
anti-FLAG antibody from whole-cell lysates and the
immunoprecipitated proteins were probed with an anti-ubiquitin
antibody. A clear band corresponding to the MW of pendrin denoted
that wild-type pendrin and both variants are abundantly
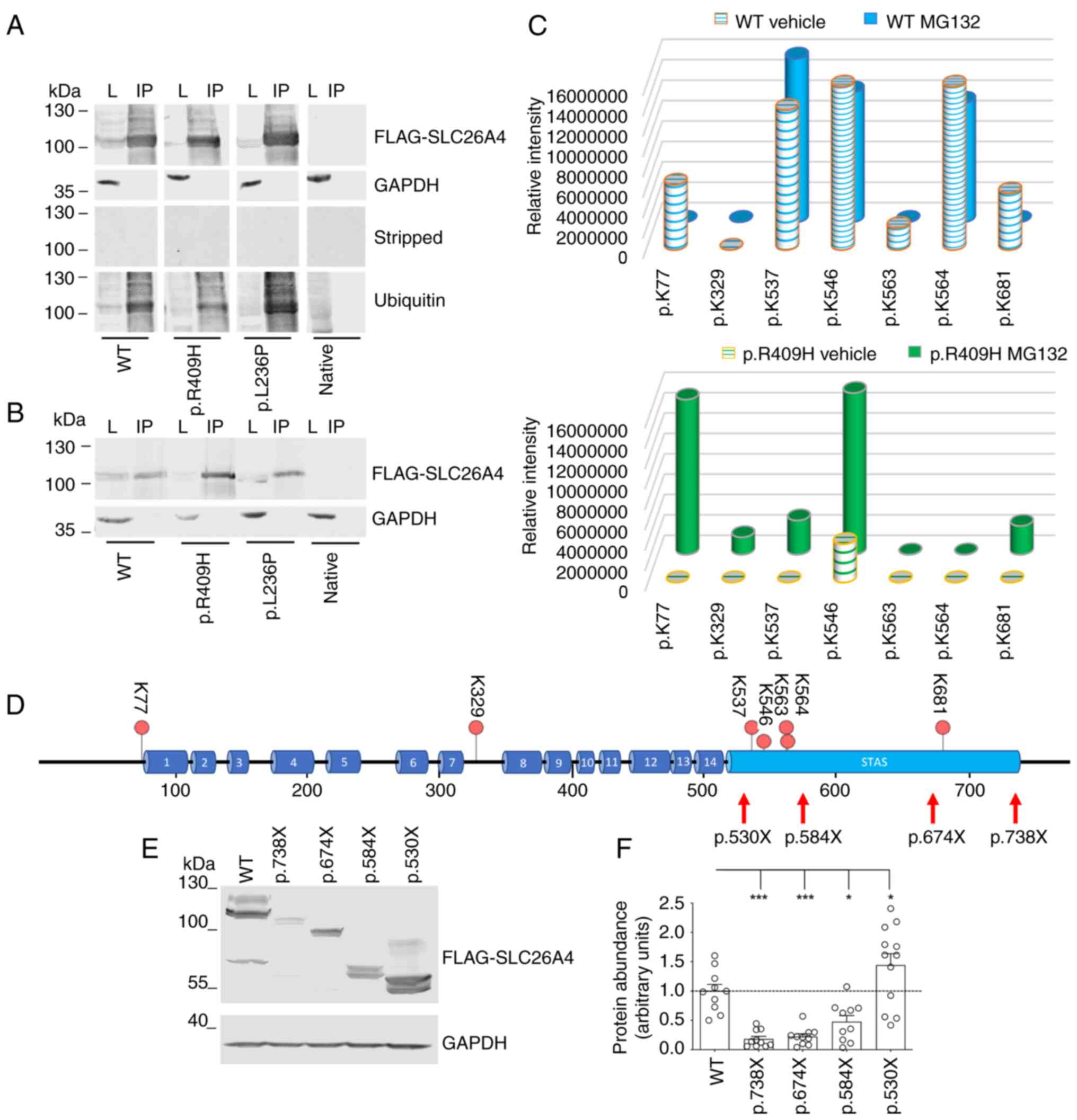
ubiquitinated (Fig. 2A and B).
These results indicated that the reduction of expression of
pathogenic pendrin variants may derive from their ubiquitination
and consequent accelerated degradation via the UPS.
To confirm the ubiquitination of the pendrin protein
and to experimentally identify sites of ubiquitination within the
polypeptide, wild-type pendrin and pendrin variant p.R409H were
immunoprecipitated from 293 Phoenix cells treated with MG132 or the
vehicle and submitted to LC-MS/MS analysis (Fig. 2C). After trypsin enzymatic
digestion, 29 peptides were produced for the wild-type and 28 for
the p.R409H variant (Table SI).
From the MS/MS analysis, 6 GlyGly-modified peptides were identified
in the MG132-treated and vehicle-treated wild-type pendrin samples
and 5 in the MG132-treated and vehicle-treated pendrin p.R409H
samples. The lysine residue at position p.77 was found to be
modified in the vehicle-treated wild-type samples, albeit with low
intensity, reflecting a lower abundance of the corresponding
modified peptide, and in the MG132-treated p.R409H samples at
greater intensity. Lysine 329 was only modified in MG132-treated
p.R409H samples. Lysine at position 537 was found modified in both
MG132-treated and vehicle-treated wild-type samples and the
MG132-treated p.R409H samples. Lysine 546 was found modified in all
samples but with lower intensity in the vehicle-treated p.R409H
samples. Lysine 563 and 564 were found to be modified only in the
wild-type samples. Lysine 681 was found modified in the
vehicle-treated wild-type samples and the MG132-treated p.R409H
samples (Fig. 2C and D).
To verify which of these ubiquitination sites
control pendrin degradation, sequential C-terminal SLC26A4
truncations were designed (Fig.
2D), and the stability of the corresponding polypeptides was
verified by western blotting (Fig.
2E and F). Expression of these constructs in cells gives rise
to glycosylated polypeptides (Fig.
S1A, left panel), as confirmed by deglycosylation of protein
lysates prior to western blotting (Fig. S1A, right panel). As expected for
pendrin variants, deletion of the C-terminus of pendrin leads to
accelerated degradation of the p.738X, p.674X and p.584X protein
products, as reflected by a reduction of protein abundance compared
with full-length pendrin (Fig.
2F). However, removal of the four ubiquitination sites Lys537,
Lys546, Lys563 and Lys564 led to stabilization of the p.530X
fragment compared with full-length pendrin and the p.738X, p.674X
and p.584X fragments, denoting that these ubiquitination sites play
an important role in the degradation of wild-type pendrin (Fig. 2F).
Exposure of cells to MG132 led to a significant
increase in protein expression of full-length pendrin (109%
increase) and p.738X (156% increase), p.674X (233% increase) and
p.584X (497% increase) fragments, but only a modest increase (37%)
of p.530X fragment, denoting that the amino acid sequence 530-584,
containing Lys537, Lys546, Lys563 and Lys564, is essential for
responsiveness to proteasome inhibitors and supporting the
important role of these ubiquitination sites in pendrin degradation
(Fig. S1B).
Wild-type pendrin and its pathogenic
variants are degraded by the UPS
It was investigated whether ubiquitination and
increased degradation by the UPS might explain the reduction in the
expression of naturally occurring pathogenic pendrin variants.
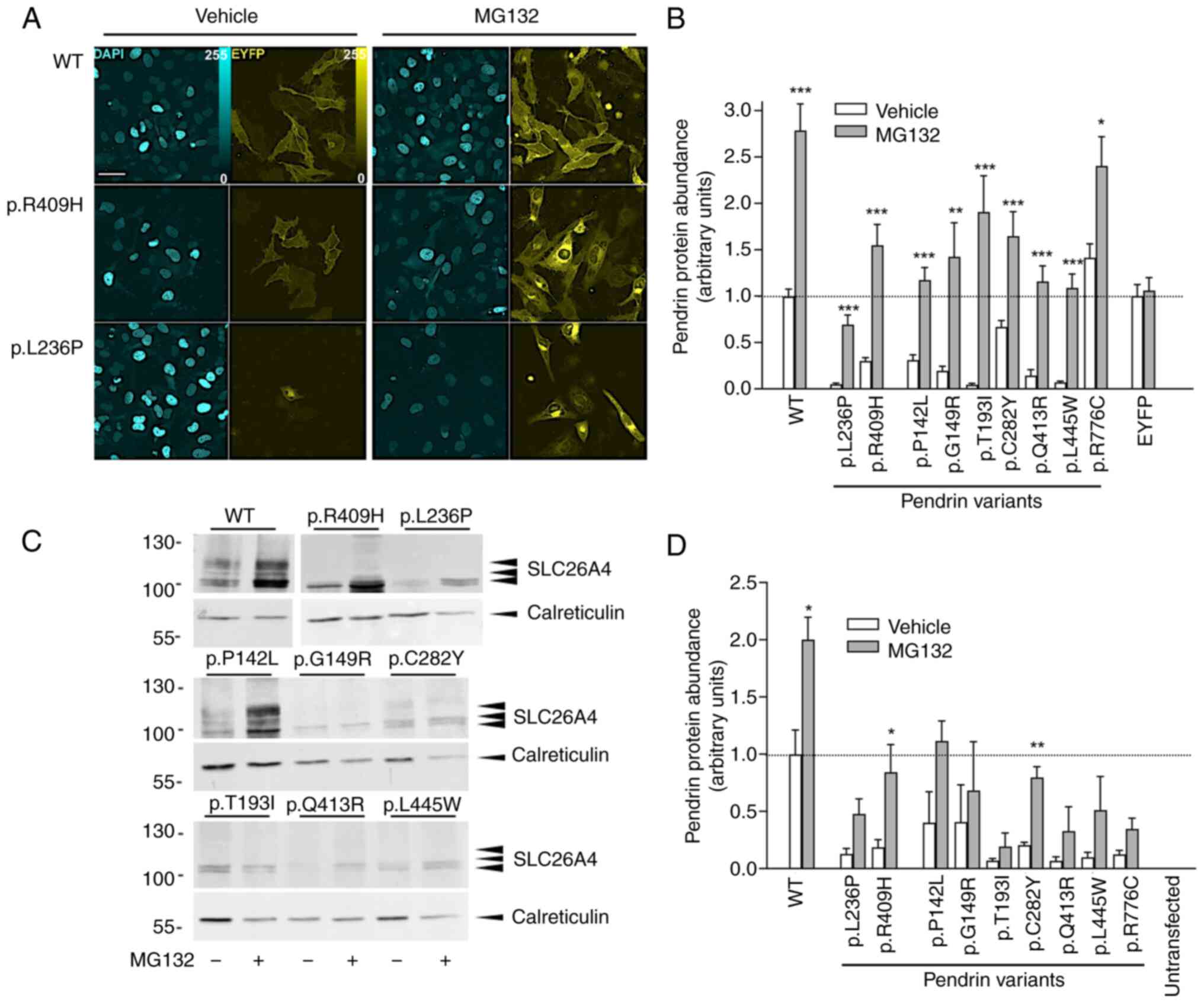
Pendrin variants were ectopically expressed, and their total
protein abundance was measured using quantitative imaging (Fig. 3A and B). The expression of all
variants, except for the fully functional variant p.R776C, was
significantly reduced compared with the wild-type (one-way ANOVA
with Dunnet's post-hoc test), which is in agreement with our
previous findings (26).
Exposure of cells to MG132 significantly increased the total
protein levels of all variants tested. Expression of the
transfection marker EYFP was unaffected. Testing of protein
expression via western blotting on total cellular membranes led to
similar results (Fig. 3C and
D).
Inhibitors of the 26S proteasome rescue
plasma membrane expression and function of pathogenic pendrin
variants
Pendrin variants were ectopically expressed and
their protein abundance in the plasma membrane region was measured
by quantitative imaging in the presence and absence of MG132. The
plasma membrane expression levels of all variants, except for the
fully functional variant p.R776C, were significantly reduced
compared with the wild-type (one-way ANOVA with Dunnet's post-hoc
test), consistent with previous results (26). The plasma membrane protein
expression significantly increased following treatment with MG132
for most variants, except the wild-type and the fully functional
variant p.R776C (Fig. 4A and
B).
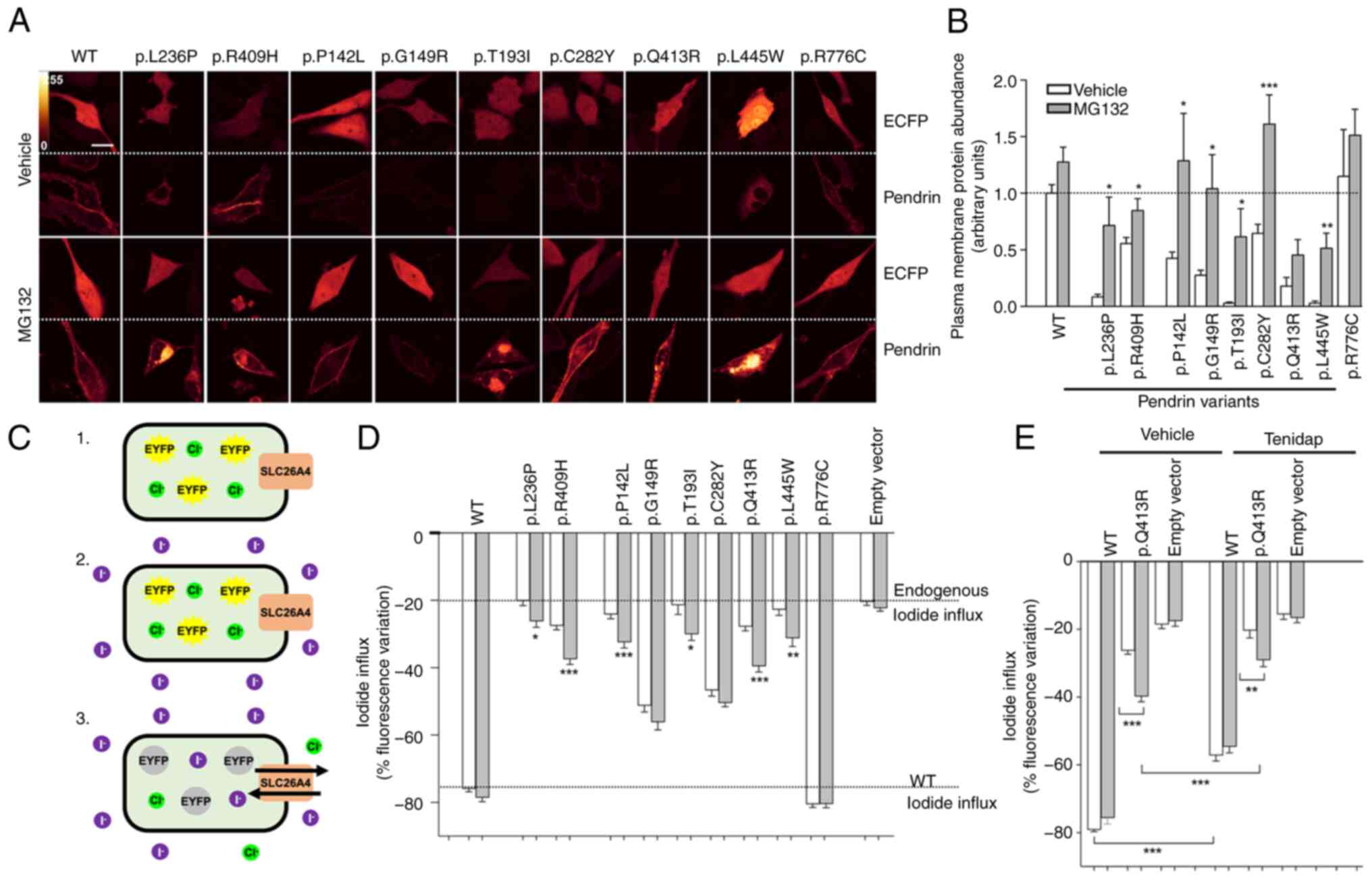 | Figure 4Inhibition of ubiquitin-proteasome
system increases plasma membrane protein levels and function of
pathogenic pendrin variants. (A) HeLa cells were transfected for 72
h with pEYFPN1 vectors encoding the wild-type or mutant
SLC26A4-EYFP and the transfection marker ECFP and incubated with
the proteasome inhibitor MG132 (10 µM) or the vehicle (0.1%
DMSO) for 16 h. Scale bar, 25 µm. (B) Plasma membrane levels
of pendrin variants were determined by quantitative imaging and
normalized for those of the wild-type. ***P<0.001,
**P<0.01 and *P<0.05 vs. vehicle,
two-tailed, unpaired Student's t-test. 11<n<21 from 3
independent experiments. n corresponds to the number of cells. (C)
Fluorometric method for evaluation of pendrin ion transport. Cells
transfected with wild-type or mutant pendrin (pTARGET vector) and
the iodide sensor EYFP H148Q:I152L for 48 h (1) are exposed to an iodide-rich
extracellular solution (2).
Iodide (magenta) enters the cell in exchange for chloride (green),
determining the quenching of EYFP H148Q:I152L fluorescence
(3). The % fluorescence decrease
is a measure of ion transport efficiency. (D and E) 293 Phoenix
cells were co-transfected for 48 h with the pTARGET vector encoding
the wild-type or mutant pendrin or an empty vector and the iodide
sensor EYFP H148Q:I152L and incubated with the proteasome inhibitor
MG132 (10 µM) or the vehicle (0.1% DMSO) for 6 h. Ion
transport activity was determined with the fluorometric method.
***P<0.001, **P<0.01 and
*P<0.05 compared with vehicle, two-tailed, unpaired
Student's t-test. (E) 100 µM tenidap or its vehicle (0.1%
DMSO) were added to the experimental solutions.
***P<0.001 and **P<0.01, one-way ANOVA
with Bonferroni's post-hoc test. Data are from at least 3
independent experiments with n=6 each. n corresponds to a well of a
96-well plate. WT, wild-type. |
The ion transport activity of all pendrin variants
was measured as iodide influx in transfected cells via a
fluorometric method in the presence and absence of MG132 (Fig. 4C). For this, cells were
co-transfected with the pTARGET and pEYFPN1 p.H148Q;I152L vectors.
The successful transfection of the pTARGET constructs into cells is
documented by western blotting (Fig.
3C and D; white bars). The activity of all variants, except for
the fully functional variant p.R776C, was significantly reduced
compared with the wild-type (one-way ANOVA with Dunnet's post-hoc
test), in agreement with our previous findings (26). Exposure of cells to MG132 led to
a statistically significant rescue of ion transport activity in 75%
(6/8) of the pathogenic variants tested. Specifically, the
partially functional pathogenic variant p.R409H exhibited a ~113%
recovery of ion transport function (n=30, P<0.001 vs. vehicle,
Fig. 4D). The basal iodide
influx measured in cells not expressing pendrin did not respond to
MG132, indicating that the MG132-increased iodide influx is
independent of endogenous ion transporters and/or channels.
It was previously reported that pendrin is resistant
to the classical inhibitor of anion exchangers
4,4′-diisothiocyano-2, 2′-stilbene-disulfonic acid (DIDS) but is
inhibited by the anti-inflammatory drug tenidap (41). Accordingly, the function of
pendrin variant p.Q413R measured in the presence of MG132 was
significantly inhibited by tenidap (Fig. 4E), further supporting the
conclusion that the MG132-induced iodide influx is
pendrin-dependent. Collectively, these findings indicated that the
UPS regulates the expression levels of wild-type pendrin and leads
to accelerated degradation of pathogenic pendrin variants,
therefore causing their loss of function. Inhibition of UPS can
lead to a partial recovery of the ion transport activity of some of
the functionally impaired pendrin variants.
The autophagosomal and lysosomal
degradation pathways are not involved in the regulation of
wild-type pendrin and pendrin variants
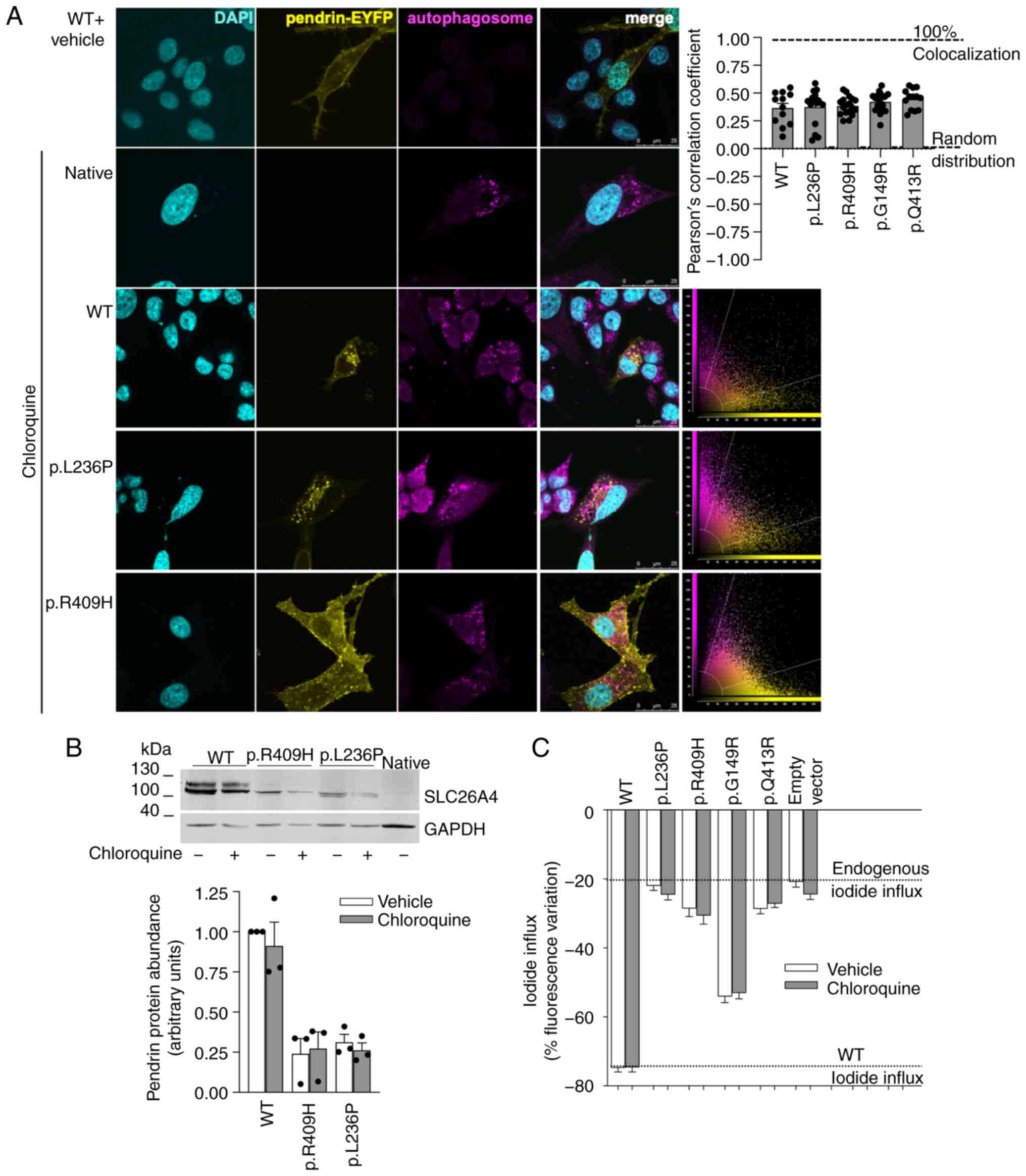
It was tested whether the autophagosomal and
lysosomal pathways might be responsible for pendrin degradation.
Pendrin variants were ectopically expressed and the accumulation of
autophagosomes was induced following exposure of cells to
chloroquine. Chloroquine mainly inhibits the autophagic flux by
impairing the fusion of autophagosomes with lysosomes (45). The possible presence of pendrin
and its variants within autophagosomes was verified by
co-localization with an autophagosomal marker. Results show a lack
of preferential localization of wild-type pendrin or pendrin
variants within autophagosomes (Fig.
5A). In addition, chloroquine failed to increase the expression
levels of pendrin variants p.L236P and p.R409H (Fig. 5B) and did not rescue the function
of four pendrin variants, three of which were responsive to MG132
(Fig. 5C). These findings
denoted that the autophagosomal and lysosomal degradation pathways
are likely not involved in the regulation of the expression levels
of pendrin and its variants and can unlikely be targeted to rescue
their expression and/or function.
Clinically approved UPS inhibitors rescue
plasma membrane expression and function of pathogenic pendrin
variants
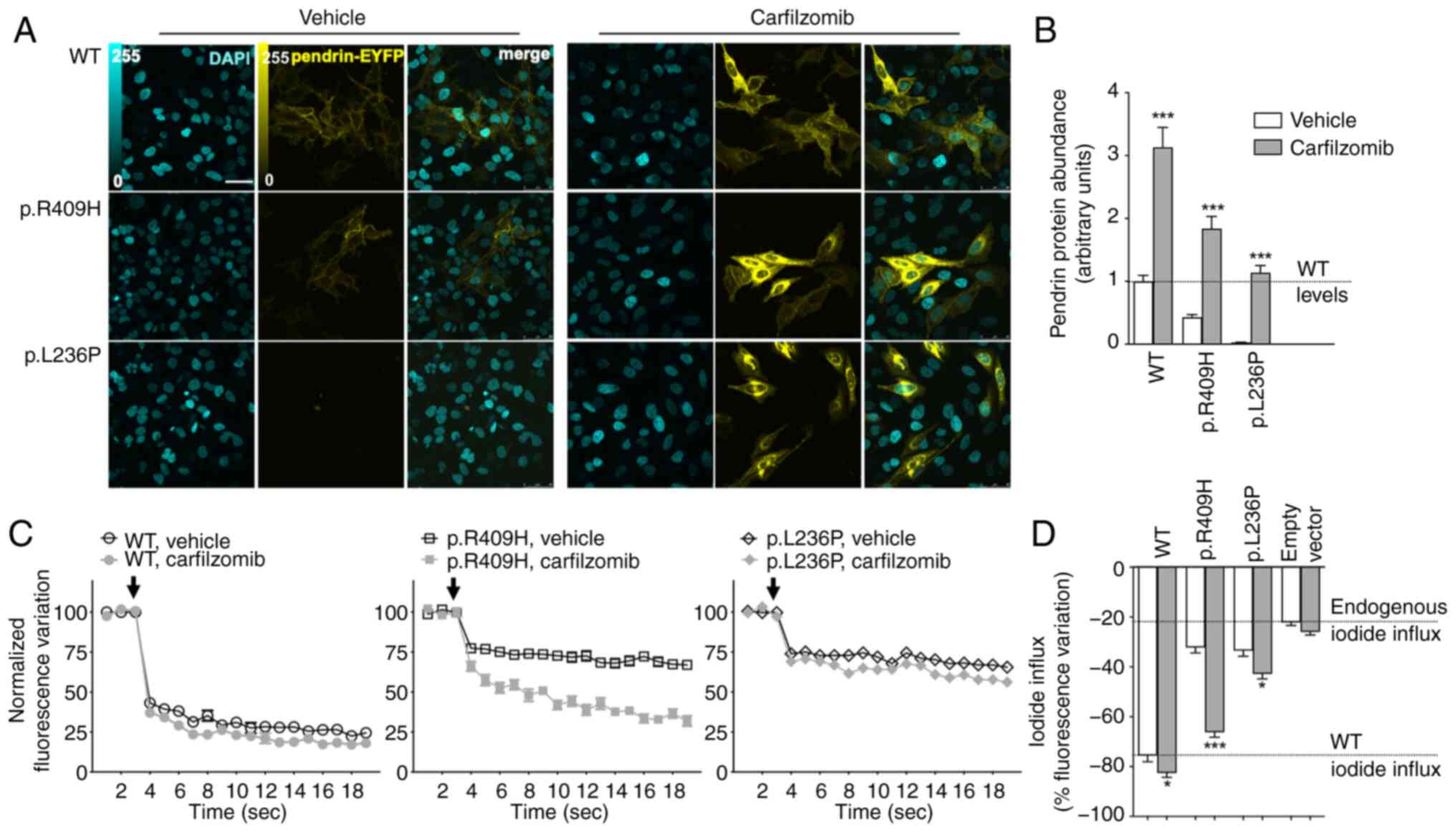
The FDA/EMA-approved proteasome inhibitors
bortezomib and carfilzomib and the investigational proteasome
inhibitor delanzomib were tested on wild-type, p.L236P and p.R409H
pendrin expression and function. Exposure of cells to 1 µM
bortezomib, carfilzomib, or delanzomib for 16 h significantly
increased the total protein abundance of wild-type pendrin and both
variants (Fig. S2). These
proteasome inhibitors (1-10 µM for 6-16 h) were also tested
on pendrin function, and a significant rescuing effect was observed
in most conditions (Figs.
S3-S5). Again, the partially functional variant p.R409H proved
to be particularly responsive to proteasome inhibition, and its
activity was rescued to levels close to those of the wild-type (60%
of vehicle-treated wild-type activity subtracted for the
proteasome-treated endogenous iodide influx with 1 µM
bortezomib for 16 h; 75% with 10 µM carfilzomib for 16 h;
and 61% with 10 µM delanzomib for 16 h). Concerning variant
p.L236P, the best rescuing effect (31.5% of wild-type activity) was
obtained with 10 µM carfilzomib for 16 h. Although the
expression of the wild-type was increased following exposure to
proteasome inhibitors (Fig.
S2), the wild-type function and the endogenous iodide transport
were mostly unaffected (Figs.
S3-S5). Data of carfilzomib are summarized in Fig. 6. Although cell viability was
reduced following incubation with the proteasome inhibitors
according to the cell proliferation test (Fig. S6A-D), the number of cells was
not reduced (Fig. S6E),
indicating that the cell proliferation test revealed a metabolic
slowdown rather than cell death, which would have caused cell
loss.
Lumacaftor, tezacaftor, elexacaftor,
rapamycin, and ataluren failed to rescue the function of pathogenic
pendrin variants
Lumacaftor (VX-809) is a pharmaceutical chaperone
referred to as a cystic fibrosis transmembrane conductance
regulator (CFTR) corrector acting on the ER. It allows a fraction
of ΔF508 CFTR to adopt a properly folded form and mobilize from the
ER to the cell surface for normal functioning (46). Tezacaftor (VX-661) and
elexacaftor (VX-445) also act as CFTR correctors and are used in
combination with ivacaftor, which is a potentiator that increases
the ion channel function (47).
Ataluren is a potent non-sense-suppressing agent that selectively
induces ribosomal read-through of premature but not normal
termination codons and was proposed for class I mutations in cystic
fibrosis (48). Concentrations
of lumacaftor, tezacaftor, and elexacaftor well above their EC50
failed to rescue the function of pendrin p.L236P and p.R409H
variants in our experimental setting. Ataluren was ineffective in
increasing the function of pendrin p.335X (Fig. S7).
Rapamycin is an mTOR inhibitor and was shown to
relieve cell death of patient-derived iPSC cells (49). Rapamycin failed to rescue the
function of pendrin p.L236P and p.R409H variants in our
experimental setting (Fig.
S7).
Discussion
Several studies tested pathogenic pendrin variants
in heterologous expression systems and showed that only ~50% of
these variants remain trapped within the ER and manifest a severe
loss of function. In contrast, other pathogenic variants are
correctly localized at the plasma membrane and have a significant
residual function (26,27,36,37,39,40,42,50). It has been previously shown that
the total protein abundance and plasma membrane protein levels of
pathogenic pendrin variants were significantly reduced compared
with the wild-type when ectopically expressed, regardless of their
subcellular localization. Reduction in protein levels was not
associated with an alteration in the corresponding mRNA levels
(26,27). Half-life measurements showed that
decreased protein expression derives from accelerated protein
degradation of the pathogenic variants rather than decreased
protein production or cell loss (Fig. 1A and B). The protein expression
of the common variant p.L236P was reduced compared with that of the
wild-type in the kidneys of a knock-in mouse (20). A reduction in transcript levels
was not observed, leading to the exclusion of defective
transcription or mRNA instability as causative factors in this
context (Fig. 1C). Thus, the
reduction in protein levels, rather than ER trapping, constitutes
the main pathological mechanism of Pendred syndrome/DFNB4.
MS analysis confirmed the ubiquitination of
wild-type pendrin and showed the ubiquitination of pendrin variants
p.L236P and p.R409H, with multiple modification sites being
identified (Fig. 2A-C). A total
of 4 identical ubiquitination sites (p.K77, p.K537, p.K546 and
p.K681) were found in the wild-type as well as the p.R409H variant,
while p.K563 and p.K564 were only ubiquitinated in the wild-type
and p.K329 only in the p.R409H samples. Interestingly, the lysines
at positions 77, 329, 546 and 681 were also predicted as highly
probable ubiquitination sites by the online tool RUBI (51), with a probability score of
0.5112, 0.2972, 0.3236 and 0.3627, respectively. Assessing the
stability of pendrin following sequential deletions of the
C-terminus (Fig. 2D; the pendrin
structure is represented according to (52) allowed for determining which of
these ubiquitination sites controls the turnover of the wild-type
variant. Removal of the C-terminus of pendrin (p.738X) greatly
destabilizes the protein, and removal of Lys681 (p.674X construct)
does not improve protein stability, the marginal role of Lys681
being also confirmed by its scarce ubiquitination levels. By
contrast, removing the four ubiquitination sites between Lys537 and
Lys564 (p.530X construct) leads to the stabilization of the
polypeptide (Fig. 2E and F) and
greatly reduces responsiveness to proteasome inhibition (Fig. S1B), pointing to a fundamental
role of these residues in the degradation of wild-type pendrin.
For the wild-type samples, the ubiquitination
profile is similar before and after inhibition of UPS with MG132,
which did not increase the ubiquitination abundance. Interestingly,
the p.R409H variant showed a different pattern, with very scarce
ubiquitination levels and only one site being modified (p.K546) in
the untreated samples and five ubiquitinated lysine residues,
including p.K546, being detected upon treatment with MG132. Two of
these sites (p.K77 and p.K546) showed particularly high
ubiquitination levels, and one of these (p.K77) was not detected in
the MG132-treated wild-type samples. This points to an important
role of p.K77 in the degradation of the p.R409H variant.
MG132 treatment allows for the accumulation of those
protein species that would otherwise undergo rapid proteasomal
degradation. The low level of modified lysine residues in the
vehicle-treated p.R409H samples is consistent with the prompt
degradation of this pendrin variant by the UPS, whereas blocking
the proteasome with MG132 allows for ubiquitinated proteins to
accumulate in the cell, which permits their detection. Conversely,
the wild-type form is more stable than the variant, and the
ubiquitinated protein can be detected even in the absence of
proteasomal inhibition. This is consistent with the half-life
measurements showing a relatively longer half-life of the wild-type
polypeptide and rapid degradation of the p.R409H variant (Fig. 1A and B).
Membrane transporters and channels are large and
complex amphipathic multi-domain proteins, and their folding
process may be slow and inefficient. Therefore, the same pathway
may be involved in the degradation of the wild-type and mutant
proteins. The folding of both the wild-type and the cystic
fibrosis-associated mutant ΔF508 CFTR is inefficient, and mutant
proteins are degraded by a process that is kinetically and
pharmacologically indistinguishable from the process involved in
the degradation of the misfolded immature wild-type CFTR (53). Additional examples of wild-type
and pathogenic variants targeted to proteasomal degradation by the
same mechanism include the ClC1 channels (54), Kv10.1 K+ channels
(55) and long QT syndrome 2
HERG channels (56). As
wild-type pendrin is a slow-folding protein (28), it is unsurprising that the UPS
can control the expression levels of both the wild-type protein and
its pathogenic variants. However, differential ubiquitination to a
specific site might lead to preferential degradation of the
pathogenic variants. Comparing the wild-type and p.R409H variant
ubiquitination profile upon UPS inhibition, it appears that
ubiquitination of lysine residue p.K546 might participate in the
degradation of both the wild-type and p.R409H variant, while lysine
residue p.K77 can be responsible for the preferential degradation
of p.R409H variant, and possibly other pendrin variants (Fig. 2C).
It was tested whether UPS inhibition might rescue
the protein levels of pendrin and its variants. Inhibition of UPS
with MG132 effectively increased the total protein levels of all
pendrin variants tested, including the wild-type and a benign
variant (Fig. 3). These data
show that, following ubiquitination, wild-type pendrin and its
variants undergo UPS-mediated protein degradation, with minor
involvement of the lysosomal/autophagosomal pathway (Fig. 5) and provide proof of concept
that pharmacological inhibition of UPS rescues their
expression.
As total protein abundance, plasma membrane protein
abundance and ion transport function are positively correlated
(Fig. 1D-F), it was postulated
that increasing total protein abundance should increase plasma
membrane protein abundance and, hence, ion transport function. As
expected, the increase in pendrin total protein level following UPS
inhibition (Fig. 3) was
paralleled by a significant increase in protein levels of
pathogenic variants in the plasma membrane (Fig. 4A and B) and led to a modest,
although significant, increase in function for most of these
variants (Fig. 4D). This
indicates that misfolded pendrin variants might conserve at least
in part their ion transport ability. However, the different
variants variably responded to MG132, and the increase in
expression did not always lead to a significant increase in
function. The ion transport function of variants p.G149R and
p.C282Y did not respond to MG132 (Fig. 4D) despite the significant
increase in their plasma membrane levels (Fig. 4B). By contrast, the function of
variants p.R409H and p.Q413R showed a 112 and 135% increase in the
presence of MG132, respectively (the iodide influx of each variant
was corrected for the endogenous iodide influx measured in cells
transfected with the empty vector, P<0.001, unpaired, two-tailed
Student's t-test; Fig. 4D). The
reason for this inhomogeneous behavior amongst variants is
currently unclear and might reflect biophysical properties of each
individual variant, such as alterations in affinity for the
transported ion(s) and/or interference with the conformational
changes necessary to ion transport, that are not rescued by an
increase in protein levels. Thus, although all tested variants
responded to proteasomal inhibition concerning their expression
levels, it cannot be assumed that an increase in protein abundance
will increase the function of all pathogenic pendrin variants.
Systematic functional testing of variants will be required to
identify or predict those responding to proteasomal inhibition.
It also has to be noted that some variants deviate
from the linear relationship between protein levels and function.
For example, p.Q413R variant exhibits significant function (40% of
the wild-type) despite almost undetectable protein levels (Fig. 1D). It is tempting to hypothesize
that the amino acid substitution of glutamine (uncharged and polar)
to arginine (positively charged at physiological pH) may lead to
protein misfolding, degradation and reduced cellular abundance
while simultaneously conferring an increased affinity for the
transported ions, thereby increasing the transport efficiency
(26). Accordingly, the function
of this variant responded well to the inhibition of proteasomal
degradation (Fig. 4D) despite a
modest increase in protein levels in the plasma membrane (Fig. 4B).
Variants p.R409H and p.L236P were selected for
further studies in that they are relatively common in the Caucasian
population and are representative of pendrin variants that remain
trapped in the ER with minimal residual function (p.L236P) and
variants with correct targeting to the plasma membrane with
significant residual function (p.R409H). Inhibition of UPS with
bortezomib, carfilzomib and delanzomib effectively increased
protein levels and ion transport function of pendrin p.R409H and
p.L236P (Figs. 6 and S2-S5). Carfilzomib, which irreversibly
inhibits the chymotrypsin-like activity of the 20S proteasome via a
covalent bond, gave the best results and rescued the function of
pendrin p.R409H to wild-type levels (Fig. 6). These findings indicate that,
among pendrin variants, those targeting the plasma membrane and
conserving residual function can represent candidates for molecular
rescue.
Bortezomib, delanzomib and carfilzomib are in
clinical use or being tested in clinical trials for the treatment
of some forms of cancer including multiple myeloma (57) and, among these, bortezomib is a
favorable candidate for drug repositioning in kidney fibrosis and
systemic lupus erythematosus (58,59). At the concentration and
incubation time tested, the toxicity of these compounds was mostly
due to a decrease in cell metabolic activity rather than cell death
in our experimental setting (Fig.
S6).
While UPS was initially considered to act as an
unspecific degradation mechanism of misfolded proteins, it is now
widely recognized as a regulatory pathway involved in the
fine-tuning of the cellular abundance and, hence, control of the
activity of specific targets by selective enzymatic reactions and
discriminatory receptors (60).
The mechanism of degradation by UPS requires polyubiquitination of
the target protein via the sequential activity of E1
(ubiquitin-activating), E2 (ubiquitin-conjugating) and E3
(ubiquitin ligases) enzymes (61). Skp1, Cullins, F-box (SCF)-type E3
ligases act in a macromolecular complex that includes Cullins 1-7
and various classes of adaptor and receptor proteins to recruit a
specific target for poly-ubiquitination and proteasomal degradation
(62,63), thereby offering numerous
opportunities for pharmacological intervention and drug development
(61,64,65). Multiple plasma membrane ion and
water transport proteins, including the chloride channel CFTR, the
large conductance Ca2+ and voltage-activated
K+ (BK) channels, as well as ClC1 chloride channels, are
regulated by the UPS (66,67,54).
The precise understanding of the mechanism leading
to the degradation of pathogenic variants of proteins has proved to
be crucial for the identification of novel drug targets and
mechanistic therapeutic strategies. Studies on the misfolding and
subsequent proteasomal degradation of CFTR led to the development
of novel drugs for the treatment of cystic fibrosis, including
lumacaftor/ivacaftor and triple therapy with the next-generation
corrector elexacaftor plus tezacaftor and ivacaftor (46,47,68). Genetic ablation and
pharmacological inhibition of the E3 ligase Rma1 (RNF5) ameliorated
the pathological phenotype in a mouse model of cystic fibrosis and
rescued the activity of ΔF508 CFTR in primary bronchial epithelial
cells derived from patients (69,70). These studies underscore that the
different molecular players of the UPS may represent promising drug
targets. Further studies are needed to elucidate the composition of
the molecular complex leading to the recruitment of pendrin to
proteasomal degradation.
To conclude, these findings show that i) the
cellular levels of wild-type pendrin are regulated by the UPS
following ubiquitination of Lys residues between amino acid
positions 537 and 564; ii) pathogenic pendrin variants undergo
accelerated degradation via the UPS following abundant and
preferential ubiquitination of Lys77, which reduces expression
levels and leads to loss of function; iii) UPS inhibitors can
rescue expression of pathogenic pendrin variants at the cell
surface and ion transport function. Although inhibitors of the
end-activity of the proteasome can unlikely be used in the clinical
setting of hearing loss, these experiments suggest that targeting
specific molecular players within the degradation pathway of
pendrin might represent a novel concept for the treatment of
Pendred syndrome/DFNB4, at least concerning the common p.R409H
variant and some selected variants.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
EB and SD conceptualized the study, curated data
and developed methodology. EB, RJ, AM, FH, HN, SK, HZ, JG and SD
conducted formal analysis, investigation and visualization, and
wrote, reviewed and edited the manuscript. SD prepared the original
of the manuscript and acquired funding. EB and SD confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved (approval no.
SYDWLL-2021-76) by the Ethics Committee of the School of Life
Science, Shandong University (Jinan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
CFTR
|
cystic fibrosis transmembrane
conductance regulator
|
|
DFNB4
|
deafness autosomal recessive 4
|
|
ECFP
|
enhanced cyan fluorescent protein
|
|
EVA
|
enlarged vestibular aqueduct
|
|
ER
|
endoplasmic reticulum
|
|
EYFP
|
enhanced yellow fluorescent
protein
|
|
MS
|
mass spectrometry
|
|
LC-MS/MS
|
liquid chromatography-tandem MS
|
|
UPS
|
ubiquitin-proteasome system
|
Acknowledgements
The authors acknowledge Mrs Elisabeth Mooslechner,
Institute of Pharmacology and Toxicology, Paracelsus Medical
University, Salzburg, Austria, for her expert secretarial
assistance.
Funding
The present study was supported in part by the Research and
Innovation Fund of Paracelsus Medical University (grant nos.
2022-Iif-004-DOSSENA and FIZ RM&NT Talent Pool Senior
Researcher 042023).
References
|
1
|
Everett LA, Morsli H, Wu DK and Green ED:
Expression pattern of the mouse ortholog of the Pendred's syndrome
gene (Pds) suggests a key role for pendrin in the inner ear. Proc
Natl Acad Sci USA. 96:9727–9732. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Royaux IE, Wall SM, Karniski LP, Everett
LA, Suzuki K, Knepper MA and Green ED: Pendrin, encoded by the
Pendred syndrome gene, resides in the apical region of renal
intercalated cells and mediates bicarbonate secretion. Proc Natl
Acad Sci USA. 98:4221–4226. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Royaux IE, Suzuki K, Mori A, Katoh R,
Everett LA, Kohn LD and Green ED: Pendrin, the protein encoded by
the Pendred syndrome gene (PDS), is an apical porter of iodide in
the thyroid and is regulated by thyroglobulin in FRTL-5 cells.
Endocrinology. 141:839–845. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dossena S and Paulmichl M: The role of
Pendrin in health and disease. Springer International Publishing;
Switzerland: 2017, View Article : Google Scholar
|
|
5
|
Honda K, Kim SH, Kelly MC, Burns JC,
Constance L, Li X, Zhou F, Hoa M, Kelley MW, Wangemann P, et al:
Molecular architecture underlying fluid absorption by the
developing inner ear. Elife. 6:e268512017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim HM and Wangemann P: Epithelial cell
stretching and luminal acidification lead to a retarded development
of stria vascularis and deafness in mice lacking pendrin. PLoS One.
6:e179492011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wangemann P, Itza EM, Albrecht B, Wu T,
Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, et
al: Loss of KCNJ10 protein expression abolishes endocochlear
potential and causes deafness in Pendred syndrome mouse model. BMC
Med. 2:302004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fugazzola L, Cerutti N, Mannavola D,
Vannucchi G and Beck-Peccoz P: The role of pendrin in iodide
regulation. Exp Clin Endocrinol Diabetes. 109:18–22. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soleimani M: The multiple roles of pendrin
in the kidney. Nephrol Dial Transplant. 30:1257–1266. 2015.
View Article : Google Scholar :
|
|
10
|
Brazier F, Corniere N, Picard N, Chambrey
R and Eladari D: Pendrin: Linking acid base to blood pressure.
Pflugers Arch. 476:533–543. 2024. View Article : Google Scholar
|
|
11
|
Wall SM: Regulation of blood pressure and
salt balance by Pendrin-Positive intercalated cells: Donald seldin
lecture 2020. Hypertension. 79:706–716. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smith RJH: Pendred Syndrome/Nonsyndromic
Enlarged Vestibular Aqueduct. GeneReviews®. Adam MP, Ardinger HH,
Pagon RA, et al: Seattle, WA: 1993
|
|
13
|
Fraser GR: Association of congenital
deafness with goitre (Pendred's Syndrome) a study of 207 families.
Ann Hum Genet. 28:201–249. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Griffith AJ and Wangemann P: Hearing loss
associated with enlargement of the vestibular aqueduct: Mechanistic
insights from clinical phenotypes, genotypes, and mouse models.
Hear Res. 281:11–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi BY, Kim HM, Ito T, Lee KY, Li X,
Monahan K, Wen Y, Wilson E, Kurima K, Saunders TL, et al: Mouse
model of enlarged vestibular aqueducts defines temporal requirement
of Slc26a4 expression for hearing acquisition. J Clin Invest.
121:4516–4525. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wangemann P: Mouse models for
pendrin-associated loss of cochlear and vestibular function. Cell
Physiol Biochem. 32:157–165. 2013. View Article : Google Scholar
|
|
17
|
Wangemann P and Griffith AJ: Mouse models
reveal the role of pendrin in the inner ear. The role of pendrin in
health and disease. Dossena S and Paulmichl M: Springer
International Publishing; Switzerland: pp. 7–22. 2017, View Article : Google Scholar
|
|
18
|
Wangemann P, Nakaya K, Wu T, Maganti RJ,
Itza EM, Sanneman JD, Harbidge DG, Billings S and Marcus DC: Loss
of cochlear HCO3-secretion causes deafness via endolymphatic
acidification and inhibition of Ca2+ reabsorption in a Pendred
syndrome mouse model. Am J Physiol Renal Physiol. 292:F1345–F1353.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nishio A, Ito T, Cheng H, Fitzgerald TS,
Wangemann P and Griffith AJ: Slc26a4 expression prevents
fluctuation of hearing in a mouse model of large vestibular
aqueduct syndrome. Neuroscience. 329:74–82. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wen Z, Zhu H, Li Z, Zhang S, Zhang A,
Zhang T, Fu X, Sun D, Zhang J and Gao J: A knock-in mouse model of
Pendred syndrome with Slc26a4 L236P mutation. Biochem Biophys Res
Commun. 515:359–365. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Taylor JP, Metcalfe RA, Watson PF, Weetman
AP and Trembath RC: Mutations of the PDS gene, encoding pendrin,
are associated with protein mislocalization and loss of iodide
efflux: Implications for thyroid dysfunction in Pendred syndrome. J
Clin Endocrinol Metab. 87:1778–1784. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rotman-Pikielny P, Hirschberg K, Maruvada
P, Suzuki K, Royaux IE, Green ED, Kohn LD, Lippincott-Schwartz J
and Yen PM: Retention of pendrin in the endoplasmic reticulum is a
major mechanism for Pendred syndrome. Hum Mol Genet. 11:2625–2633.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dai P, Stewart AK, Chebib F, Hsu A,
Rozenfeld J, Huang D, Kang D, Lip V, Fang H, Shao H, et al:
Distinct and novel SLC26A4/Pendrin mutations in Chinese and U.S.
patients with nonsyndromic hearing loss. Physiol Genomics.
38:281–290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choi BY, Stewart AK, Madeo AC, Pryor SP,
Lenhard S, Kittles R, Eisenman D, Kim HJ, Niparko J, Thomsen J, et
al: Hypo-functional SLC26A4 variants associated with nonsyndromic
hearing loss and enlargement of the vestibular aqueduct:
Genotype-phenotype correlation or coincidental polymorphisms? Hum
Mutat. 30:599–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wasano K, Takahashi S, Rosenberg SK,
Kojima T, Mutai H, Matsunaga T, Ogawa K and Homma K: Systematic
quantification of the anion transport function of pendrin (SLC26A4)
and its disease-associated variants. Hum Mutat. 41:316–331. 2020.
View Article : Google Scholar
|
|
26
|
de Moraes VCS, Bernardinelli E, Zocal N,
Fernandez JA, Nofziger C, Castilho AM, Sartorato EL, Paulmichl M
and Dossena S: Reduction of cellular expression levels is a common
feature of functionally affected pendrin (SLC26A4) protein
variants. Mol Med. 22:41–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roesch S, Bernardinelli E, Nofziger C,
Tóth M, Patsch W, Rasp G, Paulmichl M and Dossena S: Functional
testing of SLC26A4 Variants-clinical and molecular analysis of a
cohort with enlarged vestibular aqueduct from Austria. Int J Mol
Sci. 19:2092018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shepshelovich J, Goldstein-Magal L,
Globerson A, Yen PM, Rotman-Pikielny P and Hirschberg K: Protein
synthesis inhibitors and the chemical chaperone TMAO reverse
endoplasmic reticulum perturbation induced by overexpression of the
iodide transporter pendrin. J Cell Sci. 118:1577–1586. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee K, Hong TJ and Hahn JS: Roles of
17-AAG-induced molecular chaperones and Rma1 E3 ubiquitin ligase in
folding and degradation of Pendrin. FEBS Lett. 586:2535–2541. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jung J, Kim J, Roh SH, Jun I, Sampson RD,
Gee HY, Choi JY and Lee MG: The HSP70 co-chaperone DNAJC14 targets
misfolded pendrin for unconventional protein secretion. Nat Commun.
7:113862016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nanami M, Pham TD, Kim YH, Yang B, Sutliff
RL, Staub O, Klein JD, Lopez-Cayuqueo KI, Chambrey R, Park AY, et
al: The role of intercalated cell Nedd4-2 in BP regulation, Ion
transport, and transporter expression. J Am Soc Nephrol.
29:1706–1719. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Galietta LJ, Haggie PM and Verkman AS:
Green fluorescent protein-based halide indicators with improved
chloride and iodide affinities. FEBS Lett. 499:220–224. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
34
|
DiCiommo DP, Duckett A, Burcescu I,
Bremner R and Gallie BL: Retinoblastoma protein purification and
transduction of retina and retinoblastoma cells using improved
alphavirus vectors. Invest Ophthalmol Vis Sci. 45:3320–3329. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Procino G, Milano S, Tamma G, Dossena S,
Barbieri C, Nicoletti MC, Ranieri M, Di Mise A, Nofziger C, Svelto
M, et al: Co-regulated pendrin and aquaporin 5 expression and
trafficking in Type-B intercalated cells under potassium depletion.
Cell Physiol Biochem. 32:184–199. 2013. View Article : Google Scholar
|
|
36
|
Pera A, Dossena S, Rodighiero S, Gandía M,
Bottà G, Meyer G, Moreno F, Nofziger C, Hernández-Chico C and
Paulmichl M: Functional assessment of allelic variants in the
SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA.
Proc Natl Acad Sci USA. 105:18608–18613. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fugazzola L, Cirello V, Dossena S,
Rodighiero S, Muzza M, Castorina P, Lalatta F, Ambrosetti U,
Beck-Peccoz P, Bottà G and Paulmichl M: High phenotypic
intrafamilial variability in patients with Pendred syndrome and a
novel duplication in the SLC26A4 gene: Clinical characterization
and functional studies of the mutated SLC26A4 protein. Eur J
Endocrinol. 157:331–338. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dror AA, Politi Y, Shahin H, Lenz DR,
Dossena S, Nofziger C, Fuchs H, Hrabé de Angelis M, Paulmichl M,
Weiner S and Avraham KB: Calcium oxalate stone formation in the
inner ear as a result of an Slc26a4 mutation. J Biol Chem.
285:21724–21735. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dossena S, Bizhanova A, Nofziger C,
Bernardinelli E, Ramsauer J, Kopp P and Paulmichl M: Identification
of allelic variants of pendrin (SLC26A4) with loss and gain of
function. Cell Physiol Biochem. 28:467–476. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dossena S, Nofziger C, Brownstein Z,
Kanaan M, Avraham KB and Paulmichl M: Functional characterization
of pendrin mutations found in the Israeli and Palestinian
populations. Cell Physiol Biochem. 28:477–484. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bernardinelli E, Costa R, Nofziger C,
Paulmichl M and Dossena S: Effect of known inhibitors of ion
transport on pendrin (SLC26A4) activity in a human kidney cell
line. Cell Physiol Biochem. 38:1984–1998. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dossena S, Rodighiero S, Vezzoli V,
Bazzini C, Sironi C, Meyer G, Fürst J, Ritter M, Garavaglia ML,
Fugazzola L, et al: Fast fluorometric method for measuring pendrin
(SLC26A4) Cl-/I-transport activity. Cell Physiol Biochem. 18:67–74.
2006. View Article : Google Scholar
|
|
43
|
Dossena S, Rodighiero S, Vezzoli V,
Nofziger C, Salvioni E, Boccazzi M, Grabmayer E, Bottà G, Meyer G,
Fugazzola L, et al: Functional characterization of wild-type and
mutated pendrin (SLC26A4), the anion transporter involved in
Pendred syndrome. J Mol Endocrinol. 43:93–103. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tsukada K, Nishio SY, Hattori M and Usami
S: Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their
origin and a literature review. Ann Otol Rhinol Laryngol. 124(Suppl
1): 61S–76S. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr
M, Hijlkema KJ, Coppes RP, Engedal N, Mari M and Reggiori F:
Chloroquine inhibits autophagic flux by decreasing
autophagosome-lysosome fusion. Autophagy. 14:1435–1455. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Van Goor F, Hadida S, Grootenhuis PD,
Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J,
Olson ER, et al: Correction of the F508del-CFTR protein processing
defect in vitro by the investigational drug VX-809. Proc Natl Acad
Sci USA. 108:18843–18848. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Keating D, Marigowda G, Burr L, Daines C,
Mall MA, McKone EF, Ramsey BW, Rowe SM, Sass LA, Tullis E, et al:
VX-445-Tezacaftor-ivacaftor in patients with cystic fibrosis and
one or two phe508del alleles. N Engl J Med. 379:1612–1620. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Aslam AA, Sinha IP and Southern KW:
Ataluren and similar compounds (specific therapies for premature
termination codon class I mutations) for cystic fibrosis. Cochrane
Database Syst Rev. 3:CD0120402023.PubMed/NCBI
|
|
49
|
Hosoya M, Saeki T, Saegusa C, Matsunaga T,
Okano H, Fujioka M and Ogawa K: Estimating the concentration of
therapeutic range using disease-specific iPS cells: Low-dose
rapamycin therapy for Pendred syndrome. Regen Ther. 10:54–63. 2019.
View Article : Google Scholar
|
|
50
|
Dossena S, Vezzoli V, Cerutti N, Bazzini
C, Tosco M, Sironi C, Rodighiero S, Meyer G, Fascio U, Fürst J, et
al: Functional characterization of wild-type and a mutated form of
SLC26A4 identified in a patient with Pendred syndrome. Cell Physiol
Biochem. 17:245–256. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rapid UBIquitination detection. http://old.protein.bio.unipd.it/rubi/.
2013 accessed 5 July 2024
|
|
52
|
Wang L, Hoang A, Gil-Iturbe E, Laganowsky
A, Quick M and Zhou M: Mechanism of anion exchange and
small-molecule inhibition of pendrin. Nat Commun. 15:3462024.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ward CL, Omura S and Kopito RR:
Degradation of CFTR by the ubiquitin-proteasome pathway. Cell.
83:121–127. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen YA, Peng YJ, Hu MC, Huang JJ, Chien
YC, Wu JT, Chen TY and Tang CY: The Cullin 4A/B-DDB1-Cereblon E3
ubiquitin ligase complex mediates the degradation of CLC-1 chloride
channels. Sci Rep. 5:106672015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hsu PH, Ma YT, Fang YC, Huang JJ, Gan YL,
Chang PT, Jow GM, Tang CY and Jeng CJ: Cullin 7 mediates
proteasomal and lysosomal degradations of rat Eag1 potassium
channels. Sci Rep. 7:408252017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Iwai C, Li P, Kurata Y, Morikawa K,
Maharani N, Higaki K, Sasano T, Notsu T, Ishido Y, Miake J, et al:
Hsp90 prevents interaction between CHIP and HERG proteins to
facilitate maturation of wild-type and mutant HERG proteins.
Cardiovasc Res. 100:520–528. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gozzetti A, Papini G, Candi V, Brambilla
CZ, Sirianni S and Bocchia M: Second generation proteasome
inhibitors in multiple myeloma. Anticancer Agents Med Chem.
17:920–926. 2017. View Article : Google Scholar
|
|
58
|
Zeniya M, Mori T, Yui N, Nomura N, Mandai
S, Isobe K, Chiga M, Sohara E, Rai T and Uchida S: The proteasome
inhibitor bortezomib attenuates renal fibrosis in mice via the
suppression of TGF-β1. Sci Rep. 7:130862017. View Article : Google Scholar
|
|
59
|
Ikeda T, Fujii H, Nose M, Kamogawa Y,
Shirai T, Shirota Y, Ishii T and Harigae H: Bortezomib treatment
induces a higher mortality rate in lupus model mice with a higher
disease activity. Arthritis Res Ther. 19:1872017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pohl C and Dikic I: Cellular quality
control by the ubiquitin-proteasome system and autophagy. Science.
366:818–822. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sheridan C: Drug makers target ubiquitin
proteasome pathway anew. Nat Biotechnol. 33:1115–1117. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sarikas A, Hartmann T and Pan ZQ: The
cullin protein family. Genome Biol. 12:2202011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bulatov E and Ciulli A: Targeting
Cullin-RING E3 ubiquitin ligases for drug discovery: Structure,
assembly and small-molecule modulation. Biochem J. 467:365–386.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Guedat P and Colland F: Patented small
molecule inhibitors in the ubiquitin proteasome system. BMC
Biochem. 8(Suppl 1): S142007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Landre V, Rotblat B, Melino S, Bernassola
F and Melino G: Screening for E3-ubiquitin ligase inhibitors:
Challenges and opportunities. Oncotarget. 5:7988–8013. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Younger JM, Chen L, Ren HY, Rosser MF,
Turnbull EL, Fan CY, Patterson C and Cyr DM: Sequential
quality-control checkpoints triage misfolded cystic fibrosis
transmembrane conductance regulator. Cell. 126:571–582. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu J, Ye J, Zou X, Xu Z, Feng Y, Zou X,
Chen Z, Li Y and Cang Y: CRL4A(CRBN) E3 ubiquitin ligase restricts
BK channel activity and prevents epileptogenesis. Nat Commun.
5:39242014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Mijnders M, Kleizen B and Braakman I:
Correcting CFTR folding defects by small-molecule correctors to
cure cystic fibrosis. Curr Opin Pharmacol. 34:83–90. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tomati V, Sondo E, Armirotti A, Caci E,
Pesce E, Marini M, Gianotti A, Jeon YJ, Cilli M, Pistorio A, et al:
Genetic inhibition of the ubiquitin ligase Rnf5 attenuates
phenotypes associated to F508del cystic fibrosis mutation. Sci Rep.
5:121382015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sondo E, Falchi F, Caci E, Ferrera L,
Giacomini E, Pesce E, Tomati V, Mandrup Bertozzi S, Goldoni L,
Armirotti A, et al: Pharmacological inhibition of the ubiquitin
ligase RNF5 rescues F508del-CFTR in cystic fibrosis airway
epithelia. Cell Chem Biol. 25:891–905.e8. 2018. View Article : Google Scholar : PubMed/NCBI
|