Introduction
The latest definition of sepsis comes from a
consensus reached by the international medical community in 2016,
specifically the 'Sepsis-3' definition, which characterizes sepsis
as 'a life-threatening organ dysfunction caused by a dysregulated
host response to infection' (1).
Sepsis is a leading cause of mortality in the intensive care unit
(ICU) and poses a threat to human health (2-4).
Over decades of development in critical care medicine, effective
early identification and therapeutic interventions, including early
infection control, maintenance of hemodynamic stability and
modulation of the host response, have successfully reduced
mortality rates during the acute phase of sepsis, leading to an
increase in the number of sepsis survivors (5-7).
However, among survivors, the risk of developing persistent
acquired weakness syndromes is markedly increased (8). Therefore, the long-term prognosis
of sepsis remains unsatisfactory. The muscle weakness induced by
sepsis is a common complication that often leads to a state of
chronic critical illness (9).
This condition is characterized by a decline in muscle strength,
clinically referred to as ICU-acquired weakness (ICU-AW), which
includes sepsis-induced myopathy (SIM) (8-10). The atrophy of limb skeletal
muscles can lead to decreased limb strength, and if respiratory
muscles are affected, it may result in respiratory weakness,
increasing the risk of severe complications such as pulmonary
infections and respiratory failure in critically ill patients
(10). A meta-analysis on
sarcopenia and the risk of mortality in patients with sepsis
indicated that, compared with patients without sarcopenia, those
with sarcopenia have higher early mortality risks (during
hospitalization or within 1 month of illness), as well as increased
mortality risks at 3-6 months after sepsis (11). This highlights the profound
impact of ICU-AW on the long-term recovery of patients with sepsis,
as well as its contribution to prolonged hospital stays.
SIM refers to clinically diagnosed weakness in
critically ill patients that cannot be attributed to non-critical
illness causes, with an incidence rate as high as 40% among
critically ill patients in the ICU (8,12). SIM is characterized by its
systemic and symmetrical nature, presenting with weakness in the
limbs (more proximal than distal) and respiratory muscles, while
facial and ocular muscles are generally unaffected (13-15). William Osler first described
sepsis-associated muscle atrophy in 1892 as 'the rapid wasting of
flesh'. However, due to the limitations of medical knowledge at the
time, the primary focus was on patient survival rather than
long-term complications (16).
With the rapid advancements in critical care medicine at present,
there is a clearer understanding of the risk factors,
pathophysiology, diagnosis and treatment strategies for SIM.
Nevertheless, effective therapeutic options for SIM remain
unavailable, with prevention remaining paramount (8-10). The importance of early avoidance
or treatment of sepsis, anti-inflammatory strategies, nutritional
support and subsequent rehabilitation is emphasized in previous
studies. Nutritional support and rehabilitation training are often
considered to be supportive interventions rather than therapeutic
measures. The main purpose of nutritional support is to alleviate
malnutrition and promote muscle protein synthesis, while the
purpose of rehabilitation training is to maintain muscle function
and prevent further atrophy. However, developing precise and
effective treatments remains a challenge (13,17).
Glucagon-like peptide-1 (GLP-1) is a peptide hormone
comprising 30 or 31 amino acids, and is secreted by L cells in the
distal gut, α-cells in the pancreas, and the hypothalamus and
nucleus of the solitary tract in the central nervous system
(18). Endogenous GLP-1 exists
in two forms: A proglucagon corresponding to the amidation of
C-terminal Arg, GLP-1(7-36) amide, consisting of 30 amino
acids, which is the main form of GLP-1 in the human body and has a
high biological activity. The other is longer and unaminated,
GLP-1(7-37), which consists of 31 amino acids
(18). GLP-1 effectively reduces
glycosylated hemoglobin A1c and fasting plasma glucose levels,
while also promoting weight loss and decreasing systolic blood
pressure, with a low incidence of hypoglycemia (18). Therefore, GLP-1 receptor (GLP-1R)
agonists (GLP-1RAs) are recommended as early and ongoing treatment
options for patients with type 2 diabetes mellitus (T2DM) in the
clinical practice guidelines provided by the American Association
of Clinical Endocrinologists and the American Diabetes
Association/European Association for the Study of Diabetes
(19,20). GLP-1RAs reduce weight through
various mechanisms, including appetite suppression, reduced hunger
and increased satiety, resulting in decreased energy intake
(21). Therefore, GLP-1RAs are
often utilized as weight-loss medications (21). Additionally, GLP-1 exerts various
biological effects, including reducing neuroinflammation, promoting
nerve growth, improving cardiac function, delaying gastric
emptying, modulating lipid metabolism and reducing fat deposition
(22). As a result, GLP-1 has
promising applications in the treatment of various diseases, such
as sepsis and muscular atrophy (19-22). Research indicates that GLP-1R
represents a novel therapeutic target for sepsis, capable of
modulating the immune response and the release of inflammatory
factors in critically ill patients, particularly those with sepsis,
thereby providing organ protection (23). One study has also indicated that
GLP-1RAs can be utilized in the treatment of skeletal muscle
atrophy in diabetic patients with chronic liver disease (CLD)
(24). These findings suggest
that GLP-1RAs may have substantial potential in the treatment of
SIM, prompting a review of the physiological functions of GLP-1 and
its therapeutic potential in sepsis and SIM.
Physiological functions of GLP-1
GLP-1 is a 30- or 31-amino acid peptide hormone,
derived from the proglucagon gene, and expressed by specific
intestinal endocrine L cells, pancreatic α-cells and a subset of
neurons within the nucleus of the solitary tract (NTS) (18). This peptide exists in two active
forms, GLP-1(7-36) amide and GLP-1(7-37), both of which are functionally
equivalent, although the former is more abundant (25-28). Due to enzymatic degradation and
renal clearance (29),
endogenous GLP-1 has a short half-life of 1-2 min (30,31). The primary mechanisms involved in
GLP-1 degradation are enzymatic cleavage and hepatic metabolism
(30,31). Dipeptidyl peptidase-4 (DPP-4) is
the main enzyme responsible for GLP-1 breakdown, and cleaves GLP-1
to produce inactive forms, including GLP-1(9-36) amide or GLP-1(9-37), reducing its biological activity
(22,32,33). Most circulating GLP-1 is degraded
upon entry into the DPP-4-enriched mucosal vasculature, leaving
only 10-15% of GLP-1 to reach systemic circulation, where it
undergoes hepatic metabolism (30,33,34). Renal clearance primarily
eliminates inactive fragments or intact GLP-1 via glomerular
filtration. Thus, patients with renal impairment may experience
delayed elimination of GLP-1 metabolites; however, this does not
substantially impact its biological effects (35).
GLP-1 is predominantly secreted by enteroendocrine L
cells located in the distal ileum and colon, with food intake
serving as a principal physiological stimulant. Plasma GLP-1 levels
rise within minutes postprandially, even before nutrients reach L
cells in the small intestine, indicating that GLP-1 secretion is
primarily regulated by neural and endocrine stimuli (18). Plasma levels of GLP-1 are low in
the fasting state, in the range of 5-10 pmol/l, and increase
rapidly after eating, reaching 15-50 pmol/l (29). GLP-1 exerts its effects through
GLP-1R, a member of the class B G protein-coupled receptor family,
mainly by activating the cAMP-protein kinase A (PKA) pathway to
promote insulin release and improve insulin resistance (30,36). GLP-1R is widely expressed across
various tissues, including the pancreas, lungs, kidneys, central
nervous system, heart, vascular smooth muscle, gastrointestinal
tract and vagus nerve (22,25,33,37,38). One study has also identified the
liver, skeletal muscle and adipose tissue as additional GLP-1
targets (39), further
highlighting its diverse and significant biological functions.
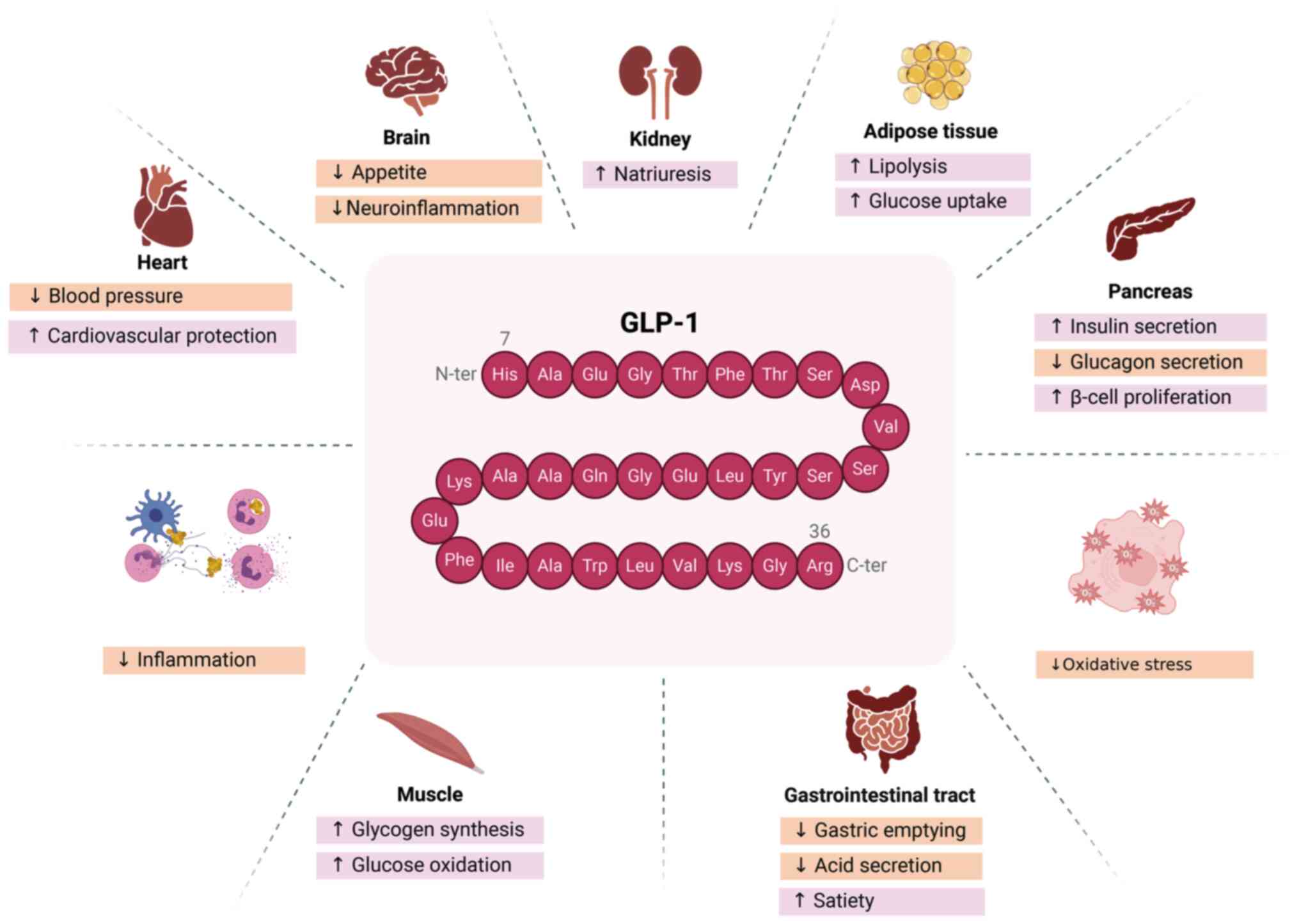
GLP-1 possesses multiple biological functions and
substantial pharmacological potential, including but not limited to
lowering blood sugar and weight control (40-49) (Fig. 1). As an incretin hormone, it
stimulates insulin synthesis and secretion in the pancreas via PKA
and exchange protein directly activated by cAMP signaling pathways,
thereby enhancing glucose metabolism (40-42). In certain cases, it can also
stimulate β-cell proliferation and inhibit β-cell apoptosis
(43). GLP-1 reduces blood
glucose by inhibiting glucagon secretion, an effect that is as
significant as its insulinotropic action in lowering blood glucose
levels, as demonstrated in a study on patients with T2DM (44). Additionally, GLP-1 suppresses
food intake, contributing to weight reduction (21). Long-acting GLP-1RAs have been
shown to influence body weight by modulating food intake and energy
expenditure through various hypothalamic pathways (45). By acting on GLP-1R, GLP-1 also
delays gastric emptying and enhances satiety by inhibiting
postprandial gastrointestinal motility and gastric acid secretion,
making GLP-1 analogues promising anti-obesity agents (33,46). Furthermore, GLP-1 has been
reported to reduce water intake independently of its effect on food
intake (47). Pharmacological
evidence suggests that GLP-1 serves a key role in stress response
regulation, with central GLP-1R signaling being critical in the
acute response to stress and aversive stimuli (48,49). Additionally, GLP-1 enhances
cognitive function, improves memory and exerts neuroprotective
effects (50,51). In addition, GLP-1 promotes
lipolysis and glucose uptake, reduces lipid deposition, and
exhibits various functions, including anti-inflammatory effects,
blood pressure regulation, cardiovascular protection and
antioxidative stress properties (22,33).
GLP-1 analogues have been widely used to treat T2DM
and obesity (52,53). Liraglutide and exendin-4 (Ex-4)
are commonly prescribed GLP-1 analogues, which reduce
cardiovascular-related mortality, non-fatal myocardial infarction
and non-fatal stroke in hospitalized patients with T2DM (52). Liraglutide also reduces the
incidence of diabetic nephropathy in T2DM (53). An animal study demonstrated that
Exendin-4 treatment reduced infarct volume and improved functional
deficits while inhibiting oxidative stress, the inflammatory
response and cell death after reperfusion (54). Furthermore, GLP-1 analogues have
been demonstrated to alter metabolic, bioenergetic and contractile
characteristics within risk and non-risk myocardial regions
following ischemic injury, exerting cardioprotective effects
(55). Research has further
demonstrated the protective role of GLP-1 in organs such as the
gastrointestinal tract (56),
lungs (57) and liver (58). A study has also reported that
GLP-1RAs can treat diabetes-induced skeletal muscle atrophy by
promoting glycogen synthesis and glucose oxidation (24). Additionally, GLP-1RA has emerged
as a novel therapeutic target for sepsis, protecting organs by
modulating immune responses and inflammatory cytokine release in
patients with sepsis (23,59-64).
Pathophysiology of sepsis-associated muscle
atrophy
Sarcopenia is a syndrome marked by a progressive
reduction in skeletal muscle mass, quality and strength, often
exacerbated by aging and reduced activity (65). Various systemic diseases,
including cancer, renal failure, chronic obstructive pulmonary
disease, sepsis and trauma, also accelerate sarcopenia progression
(66). In sepsis,
pathophysiological changes include sympathetic nervous system
activation, and excessive release of catabolic hormones
(catecholamines and glucocorticoids) and pro-inflammatory
cytokines. This catabolic state leads to anabolic resistance,
tipping the balance between protein synthesis and degradation
towards net catabolism in critically ill patients (67,68). As muscle mass declines, muscle
strength deteriorates, ultimately resulting in ICU-AW (67). The pathogenesis of SIM is complex
and remains incompletely understood. This condition primarily
arises from an imbalance between muscle protein synthesis and
degradation, alongside reduced force-generating capacity, often
secondary to neuropathy, myofibrillar disruption, sarcoplasmic
reticulum (SR) impairment, altered excitability and mitochondrial
dysfunction (68).
Increased proteolysis in skeletal
muscle
Protein degradation in skeletal muscle cells
primarily involves four proteolytic systems: The
ubiquitin-proteasome system (UPS), calpains, caspases and the
autophagy-lysosome pathway (13,68,69). The UPS serves a critical role in
degrading most damaged proteins, which, after being ubiquitinated,
are broken down in the 26S proteasome. Key E3 ubiquitin ligases in
this system, muscle RING finger 1 (MuRF1) and muscle atrophy F-box
(MAFbx/atrogin-1), are essential in skeletal muscle degradation
(68,69). MuRF1 hydrolyzes myosin in thick
filaments, while MAFbx/atrogin-1 likely suppresses protein
synthesis by downregulating eukaryotic translation initiation
factor 3 subunit F (eIF3f; an essential protein synthesis
initiator) and MyoD (a crucial muscle differentiation transcription
factor) (70-72). The NF-κB pathway and its
inhibitors interact to regulate UPS activation (68,69). During sepsis, elevated
inflammatory cytokine levels enhance ubiquitination and the
degradation of NF-κB inhibitors (IkB), which in turn activates the
UPS pathway (73). Excessive
expression of TNF and NF-κB increases the levels of muscle-specific
E3 ligases (MuRF1 and atrogin-1), ultimately leading to proteolysis
in respiratory and limb muscles, manifesting as difficulties in
ventilator weaning and muscle weakness in patients (73,74).
Calpain activation is associated with SIM (75,76). A study has indicated that calpain
activation in septic muscle leads to increased protein degradation,
while concurrently downregulating Akt activity, thereby reducing
protein synthesis (68). The
PI3K/Akt signaling pathway, which maintains protein balance, is
inhibited under catabolic conditions such as sepsis, inflammation
and immobilization, promoting UPS activation and protein
degradation (68). Reduced Akt
activity alleviates inhibition of FoxO, a transcription factor that
upregulates the expression of the muscular atrophy factor
(atrogin-1/MAFbx), and the upregulation of muscle atrophy factor
accelerates muscle atrophy (68). Low PI3K activity induces BAX
expression, which subsequently releases cytochrome c from
mitochondria into the cytosol, thereby activating caspase-3
(68). Caspases, while widely
recognized for their role in apoptosis, are now being implicated in
sepsis-induced myopathies (68,77). Studies in infected animal models
have demonstrated that caspases, by cleaving cytoskeletal proteins
in the diaphragm, led to muscle weakness (68,77). Similarly, the autophagy-lysosome
pathway, crucial for skeletal muscle degradation, becomes
hyperactivated under catabolic, oxidative stress and
pro-inflammatory conditions, which exacerbates muscle atrophy
through excessive protein breakdown (78). Autophagy is regulated by multiple
factors, including hypoxia, infection and stress (79,80). Hussain et al (79) demonstrated that prolonged
mechanical ventilation led to disuse atrophy of the diaphragm,
which further triggered the oxidative stress response and activated
the FoxO1 transcription factor, thereby inducing skeletal muscle
protein degradation through the autophagy lysosomal pathway
(80).
Decreased protein synthesis
Protein synthesis is primarily regulated through two
signaling pathways, the insulin/insulin-like growth factor 1
(IGF-1) and mTOR signaling pathways, both of which are essential
for anabolic processes. The insulin/IGF-1-Akt pathway enhances
protein synthesis by activating the mTOR pathway, while inhibiting
FoxO (73,81). Specifically, the
IGF-1-PI3K-Akt-mTOR cascade promotes protein synthesis and
suppresses protein degradation by phosphorylating downstream FoxO,
thereby inhibiting E3 ligases (MuRF1 and atrogin-1) (73,81). In addition to activating the
IGF-1/Akt pathway for protein synthesis, IGF-1 also stimulates the
MAPK cascade, promoting myoblast proliferation (68,69). However, during sepsis and muscle
atrophy, IGF-1 levels in the muscle decline, which reduces protein
synthesis (81-83). FoxO promotes the expression of
atrophy-related genes (atrogenes), and under critical conditions
such as sepsis, decreased IGF-1-PI3K-Akt-mTOR signaling increases
FoxO-mediated expression of atrogenes, reducing protein synthesis
(81-83). Nystrom et al (84) have confirmed this, showing that
IGF-1 administration in septic mice enhanced muscle protein
synthesis and alleviated sepsis-induced muscle atrophy.
The mTOR pathway, another critical regulator of
protein synthesis, is activated by growth factors, nutrients,
insulin and other signals, facilitating anabolic processes and
regulating biosynthesis, including protein synthesis (69,85). Upon activation, mTOR
phosphorylates downstream targets, including eukaryotic translation
initiation factor 4E-binding protein 1 (4E-BP1) and S6K1, to
promote protein translation (69,85). A study has shown that sepsis
inhibited mTOR activity, and suppressed the phosphorylation of
4E-BP1 and S6K1 (86).
Non-phosphorylated 4E-BP1 binds tightly to eukaryotic translation
initiation factor 4E (eIF4E), a subunit of the eukaryotic
initiation factor 4F complex responsible for mRNA cap binding,
thereby inhibiting eIF4E-mediated initiation of protein synthesis
(85). Similarly,
dephosphorylation of S6K1, which activates the S6 protein of the
40S ribosomal subunit, inhibits translation (85). Sepsis-induced mTOR suppression
may, in part, be attributed to excessive pro-inflammatory cytokines
(87).
Mitochondrial dysfunction
Mitochondrial dysfunction is a key factor in the
development of critical illness myopathy, especially in sepsis.
Mitochondria are central to energy conversion, biosynthesis and
signaling, with ATP production being essential for muscle
contraction. Mitochondria also regulate calcium homeostasis,
reactive oxygen species (ROS) production, intracellular
communication and apoptosis. Structural damage and dysfunction in
mitochondria lead to ATP deficiency, ROS overproduction and
cytochrome c release, potentially resulting in organ failure
(88,89). Research indicates that the
accumulation of damaged mitochondria contributes to motor neuron
and muscle fiber degeneration (88,89). During sepsis, excessive ROS
production exacerbates mitochondrial dysfunction, creating a
feedback loop that worsens oxidative damage (13,90). Studies have indicated reduced
expression and activity of respiratory chain complexes I, III and
IV in muscle tissue of critically ill patients, partially
explaining the muscle weakness observed in critically ill patients
(13,90).
Calcium homeostasis dysregulation
Calcium level fluctuations affect protein
degradation, since calpain activity and UPS activation are
calcium-dependent (69).
Additionally, calcium ions are crucial for muscle contraction,
influencing myosin ATPase activity, glycolysis and oxidative
metabolism (69). The SR serves
a role in maintaining calcium homeostasis (91,92). Friedrich et al (91) demonstrated that serum from
critically ill patients affected SR Ca2+ release in
mammalian muscle fibers, altering membrane excitability and
excitation-contraction coupling. Similarly, Zink et al
(92) reported decreased SR
Ca2+ release in skeletal muscle fibers during sepsis,
suggesting that disrupted Ca2+ homeostasis may
contribute to muscle dysfunction in sepsis.
Neuropathy and membrane excitability
impairment
Sepsis induces microvascular changes in the
endoneurium, increasing vascular permeability and allowing toxic
factors to infiltrate nerve endings (13,93). Increased permeability leads to
endoneurial edema, which can impair energy transmission along
axons, ultimately resulting in axonal death (13,93). Under physiological conditions,
cellular permeability to Na+ and K+ ions
forms the basis for electrophysiological stability. In sepsis,
alterations in Na+ pump function can lead to
dysregulated membrane excitability. Notably, inflammatory cytokines
exert neurotoxic effects, causing persistent membrane
depolarization, a phenomenon referred to as 'denervation' (13,69,93,94). Na+ channel
inactivation in ICU-AW may lead to rapid, reversible hypo- or
non-excitability, impairing both neuronal and muscular function
(13,69,93,94).
GLP-1RAs and septic multi-organ
dysfunction
Research on the extrapancreatic effects of GLP-1RAs
is a rapidly developing field. As GLP-1Rs are distributed in the
pancreas, kidneys, lungs, heart, intestines and other organs, an
increasing number of studies have focused on the protective effects
of GLP-1RAs on organ function (22,23,63). Sepsis is a complex syndrome
characterized by a systemic inflammatory response leading to severe
multi-organ dysfunction (95).
Numerous studies (23,59-64) have demonstrated that GLP-1RAs
exert immunomodulatory and protective effects in sepsis-related
organ dysfunction, including in the lungs, brain, heart, kidneys,
liver, gastrointestinal tract and vasculature (Fig. 2).
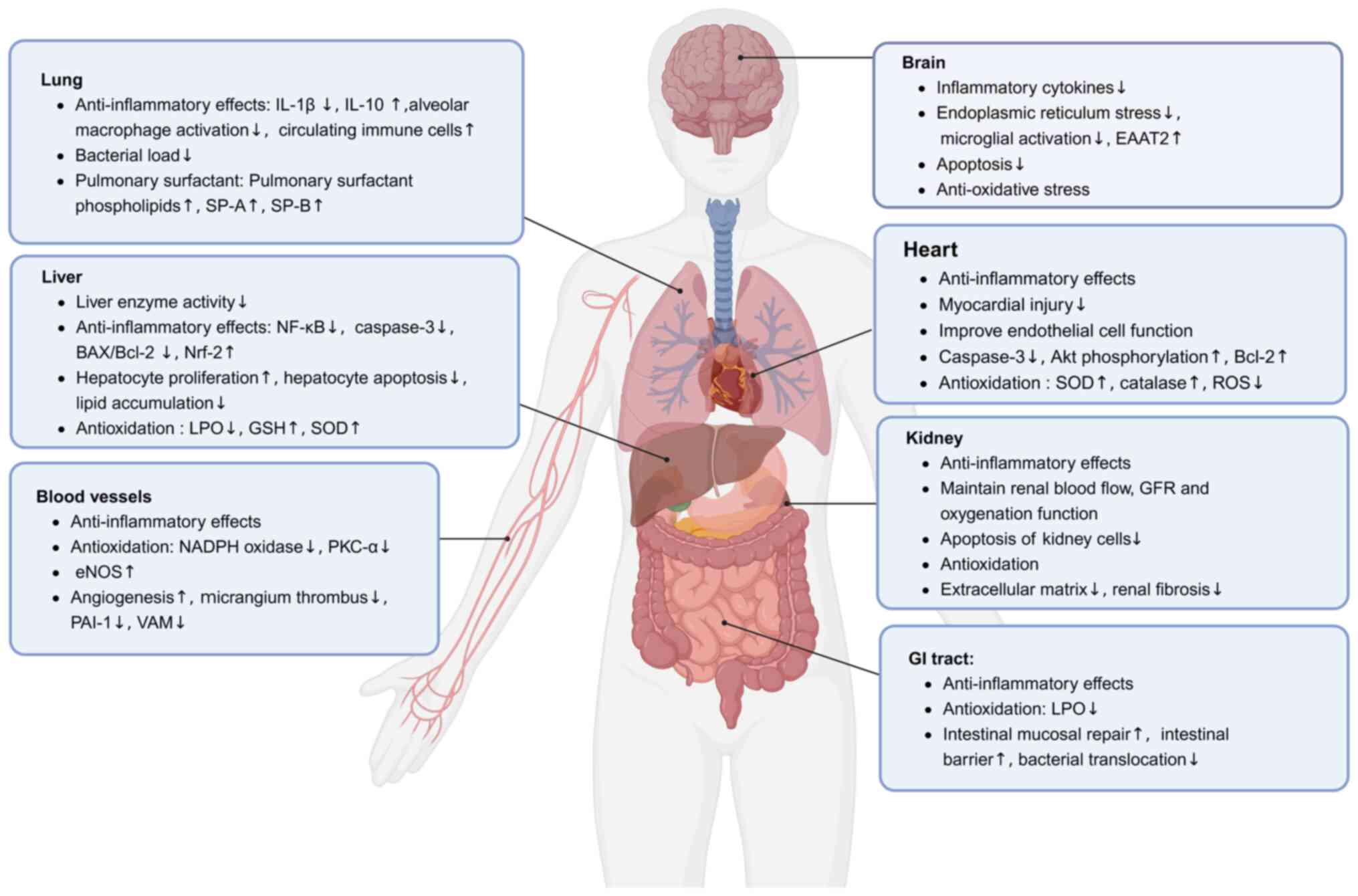 | Figure 2Protective mechanisms of GLP-1RAs in
septic multi-organ dysfunction. Due to the widespread distribution
of GLP-1 receptors in organs such as the pancreas, kidneys, lungs,
heart and intestines, a growing number of studies have focused on
the protective effects of GLP-1RAs on organ function during sepsis
(23,59-64). In preclinical settings, GLP-1RAs
possess potential immunomodulatory and protective effects in
sepsis-related organ dysfunction, including in the lungs, brain,
heart, kidneys, liver, GI tract and vasculature. EAAT2, excitatory
amino acid transporter-2; eNOS, endothelial nitric oxide synthase;
GFR, glomerular filtration rate; GI, gastrointestinal; GLP-1,
glucagon-like peptide-1; GLP-1RAs, GLP-1 receptor agonists; GSH,
glutathione; LPO, lipid peroxidation; Nrf2, nuclear factor
erythroid 2-related factor 2; PAI-1, plasminogen activator
inhibitor type 1; PKC-α, protein kinase C-α; ROS, reactive oxygen
species; SOD, superoxide dismutase; SP-A, surfactant protein A;
SP-B, surfactant protein B; VAM, vascular adhesion molecules. |
GLP-1RAs and septic lung injury
Sepsis often affects the respiratory system, leading
to acute respiratory distress syndrome (ARDS), the most common
cause of which is sepsis (96).
The pathogenesis of ARDS primarily involves excessive inflammation
and damage to the alveolar-capillary barrier, resulting in the
accumulation of inflammatory cells, increased levels of
pro-inflammatory factors and injury to alveolar epithelial cells.
The activation and recruitment of inflammatory cells, including
macrophages and neutrophils in the pulmonary circulation, serve
crucial roles in the development and progression of septic lung
injury (97,98). Neutrophils activate Toll-like
receptors and CD14+ T cells, promoting the release of
inflammatory mediators [IL-1β, TNF-α, nitric oxide (NO) and ROS]
(97,98). Key pathophysiological changes in
acute lung injury caused by sepsis include infiltration of
inflammatory cells, release of inflammatory mediators, thrombus
formation in pulmonary capillaries, interstitial pulmonary edema
and pulmonary fibrosis, with clinical manifestations primarily
being progressive dyspnea and persistent hypoxemia secondary to
sepsis (99).
Baer et al (97) demonstrated that liraglutide, a
GLP-1RA, could mitigate sepsis-induced acute lung injury by
reducing various inflammatory markers in the lungs, including the
number of immune cells and the concentration of pro-inflammatory
factors. The anti-inflammatory effects of liraglutide may be
mediated through the inhibition of the NLR family pyrin domain
containing 3/IL-1β inflammasome and the stimulation of IL-10
(97). Animal experiments
indicated that liraglutide could decrease the transcription of
pro-inflammatory factors such as IL-1β at the 6-h time point in
septic mice, while increasing IL-10 mRNA expression (97). The study also found that
liraglutide not only promoted the resolution of sepsis-induced
inflammation but also did not impair the ability of the host to
clear bacteria (97). GLP-1RAs
may reduce excessive inflammation and damage to the
alveolar-capillary barrier, possibly by inhibiting its inflammatory
response and subsequent immune cell recruitment, or through direct
effects on alveolar macrophages and circulating immune cells,
altering their migration, proliferation and phenotype/activation
(97). The beneficial effects of
GLP-1RAs on septic lung injury were similarly confirmed by Guo
et al (60), who found
that liraglutide treatment in mice with sepsis induced by bacterial
pneumonia could reduce the bacterial load, improve animal survival
rates, decrease lung inflammatory cells and cytokines, and enhance
the production of pulmonary surfactant phospholipids and surfactant
proteins A and B, thereby alleviating acute lung injury and
mortality due to pneumonia-induced sepsis. Additionally,
liraglutide can modulate pulmonary surfactant through autophagy
inhibition to mitigate sepsis-induced acute lung injury (57).
GLP-1RAs and sepsis-associated
encephalopathy (SAE)
The central nervous system is one of the organs most
susceptible to sepsis, with 50-70% of septic patients experiencing
SAE. The pathogenesis of SAE primarily involves neuroinflammation,
blood-brain barrier (BBB) damage, cerebral microcirculatory
dysfunction and mitochondrial dysfunction (61,100,101). Clinical manifestations of SAE
range from behavioral disturbances to altered consciousness,
including delirium and coma, complicating its management due to a
lack of specific diagnostic criteria (61,100,101).
Endogenous GLP-1 can also be produced by neurons in
the NTS of the brain stem, helping to regulate insulin resistance,
food intake, neuroprotection, and learning and memory dysfunction
(33). Neuroinflammation and
excessive activation of microglia are neuropathological features of
SAE (100,102). The breakdown of the BBB during
sepsis facilitates the entry of inflammatory mediators and other
substances from the periphery into the brain, leading to microglial
activation and subsequent neuronal dysfunction (100,102). Research by Yi et al
(61) in a mouse model of sepsis
indicated that the GLP-1/GLP-1R pathway could regulate the
progression of SAE. Activation of GLP-1/GLP-1RA may inhibit
endoplasmic reticulum stress, microglial activation, production of
inflammatory cytokines and hippocampal cell apoptosis, thereby
improving survival rates and cognitive deficits in cecal ligation
and puncture mice. The authors also demonstrated that liraglutide
protected microglia through modulation of the cAMP/PKA/cAMP
response element binding protein (CREB) signaling pathway (61). Fang et al (103) demonstrated that recombinant
human GLP-1 improved neurological deficits and reduced infarct
volume in rats with middle cerebral artery occlusion by inhibiting
oxidative stress and apoptosis, and promoting excitatory amino acid
transporter-2 expression.
GLP-1RAs and sepsis-associated myocardial
injury
Sepsis-induced myocardial injury (SIMI) is one of
the most common complications of sepsis and a leading cause of
death. The pathogenic factors of SIMI include the release of
myocardial depressant factors, upregulation of NO, impairment of
myocardial calcium homeostasis and mitochondrial dysfunction
(104,105). SIMI is primarily characterized
by myocardial ischemia and hypoxia, impaired contractile function,
and reduced left ventricular ejection fraction, leading to
inadequate tissue and organ perfusion (104,105). Research suggests that the
occurrence of SIMI may be related to the activation of Bax and
caspase-3, and the inhibition of Bcl-2, which induces apoptosis of
myocardial cells during sepsis. The apoptosis involved in SIMI
encompasses the activation or inhibition of multiple pathways,
including TNF-α and MAPK-related, PI3K/AKT/mTOR, and NF-κB
signaling pathways, with the release of various inflammatory
factors that directly lead to myocardial damage, resulting in
increased secretion of inducible NO synthase and excessive
production of NO (98,104). Excessive NO inhibits type I
calcium channels and mitochondrial function in myocardial cells,
resulting in impaired contractile function and decreased cardiac
output (98,104).
The cardioprotective effects of GLP-1RAs primarily
include anti-inflammatory actions, reduction of myocardial ischemic
injury, alteration of lipid synthesis and secretion, and
improvement of endothelial dysfunction (106). A study has revealed that
liraglutide increased cAMP formation in a GLP-1R-dependent manner
in vitro and reduced the activation of caspase-3 in
myocardial cells (107).
GLP-1RAs engage pro-survival pathways in the mouse heart, improving
outcomes following myocardial infarction and enhancing survival
(107). Exenatide, a GLP-1
analog, has been shown by Timmers et al (108) to reduce myocardial infarction
size, and prevent deterioration of both systolic and diastolic
heart function. Following exenatide treatment, the levels of
phosphorylated Akt and Bcl-2 were increased, while active caspase-3
expression was decreased. There was also a reduction in nuclear
oxidative stress, and an increase in the activity of superoxide
dismutase (SOD) and catalase, which could reduce infarct size in
patients with acute myocardial infarction (108). Another study has also confirmed
the beneficial effects of GLP-1 analogs in myocardial infarction
(109). Compared with the
placebo group, treatment with the GLP-1 analog liraglutide reduced
the rates of non-fatal myocardial infarction, non-fatal stroke and
hospitalization for heart failure (52). Additionally, Chang et al
(110) demonstrated that
exenatide exerted cardioprotective effects against oxidative
stress-induced injury both in vitro and in vivo,
which was potentially attributed to the clearance of oxidative
stress products (such as ROS), increased concentrations of
antioxidant defense enzymes and suppression of myocardial cell
apoptosis. The authors also suggested that the anti-apoptotic
effects of exenatide were at least partially related to the
activation of the PI3K/Akt signaling pathway (110).
GLP-1RAs and sepsis-induced kidney
injury
Acute kidney injury (AKI) occurs in 40-50% of
patients with sepsis, increasing the in-hospital mortality risk by
6-8 times (111). The
pathophysiology of sepsis-induced AKI is characterized by
microvascular dysfunction, inflammation and cellular responses to
inflammatory injury (111). The
excessive release of inflammatory mediators during sepsis severely
disrupts the renal microenvironment (112,113). The pathophysiology of AKI in
sepsis is complex and multifactorial, encompassing intrarenal
hemodynamic changes, endothelial dysfunction, infiltration of
inflammatory cells within the renal parenchyma, and obstruction of
renal tubules by necrotic cells and debris (112,113). Furthermore, inflammatory
mediators can induce the release of tissue factors and activate the
extrinsic coagulation pathway, leading to the formation of
microthrombi in the renal microcirculation (98). Ischemia and hypoxia contribute to
an increase in ROS within renal tissue, resulting in increased
mitochondrial permeability, a decrease in the mitochondrial
membrane potential, mitochondrial dysfunction and exacerbation of
renal injury (98).
Studies have demonstrated that GLP-1RAs exert renal
protective effects through various mechanisms (114-117). For instance, Xiang et al
(114) demonstrated that early
GLP-1 treatment after severe trauma preserved renal function in
obese Zucker rats. The authors found that GLP-1 treatment reduced
IL-6 levels post-trauma, while maintaining normal renal blood flow,
glomerular filtration rate and oxygenation (114). Elkhoely (115) indicated that liraglutide could
exert renal protection effects by alleviating oxidative stress,
inflammation and apoptosis. This effect was mediated by the
upregulation of PKA/CREB signaling and the downregulation of the
Notch/Hes-1 pathway, leading to enhanced peroxisome
proliferator-activated receptor γ coactivator 1α expression, thus
improving gentamicin-induced renal injury in rats (115). Xu et al (116) found that liraglutide could
mitigate cisplatin-induced nephrotoxicity by inhibiting
nuclear-cytoplasmic translocation and release of high mobility
group box 1 (HMGB1), reducing inflammatory cytokine levels and
HMGB1 receptor expression. Additionally, Li et al (117) demonstrated that GLP-1R
activation inhibited the TGF-β1/Smad3 and ERK1/2 signaling
pathways, preventing epithelial-to-mesenchymal transition, thus
reducing extracellular matrix secretion and deposition, and
alleviating renal fibrosis. These studies collectively suggest that
GLP-1 serves a protective role in kidney injury.
GLP-1RAs and sepsis-related liver
dysfunction
During sepsis, systemic inflammatory responses,
dysregulated immune responses, hepatic microcirculatory dysfunction
and cellular hypoxia can lead to various forms of acute liver
dysfunction, including hypoxic hepatitis, sepsis-associated
cholestasis or acute liver failure (118). The liver acts both as a
mediator and regulator of the immune response, characterized by an
increase in acute phase proteins and inflammatory cytokine
production. Simultaneously, the liver itself becomes a target organ
for injury in sepsis. In sepsis, Kupffer cells, the resident
macrophages in the liver sinusoids, become activated and continue
to synthesize pro-inflammatory factors (such as TNF-α, IL-6, IL-18
and IL-1β), secrete chemokines and leukotrienes, and initiate ROS
and NO production. The locally released pro-inflammatory factors
induce inflammatory responses in the liver sinusoids, activating
pro-apoptotic signaling cascades (118-120). Additionally, neutrophils,
natural killer T cells and sinusoidal endothelial cells contribute
to the development of sepsis-related liver dysfunction, ultimately
leading to the destruction of hepatocytes and the liver biliary
system (118-120).
Overall, the inflammatory cascade is the primary
cause of liver injury induced by sepsis (118-120). Studies have shown that
liraglutide exerts protective effects during acute liver injury
(121,122). Specifically, liraglutide
reduced lipid peroxidation (LPO) levels, increased reduced
glutathione levels and enhanced SOD activity in a mouse model of
acute liver injury, thereby reducing oxidative stress (121). Additionally, liraglutide
inhibited carbon tetrachloride-induced hepatic lipid accumulation,
promoted hepatocyte proliferation and suppressed apoptosis in the
liver, enhancing mitochondrial respiratory function and ultimately
providing liver protection effects (121,122). Abdelaziz et al (123) demonstrated that liraglutide
reduced hepatic enzyme activity, oxidative stress, NF-κB expression
and related inflammatory markers, while increasing the expression
of the antioxidant transcription factor nuclear factor erythroid
2-related factor 2 (Nrf2) and downstream phosphorylated CREB,
thereby enhancing Nrf2 activity. Furthermore, liraglutide
pretreatment resulted in decreased expression/activity of caspase-3
and a lower BAX/Bcl-2 ratio (123). Similarly, Zhu et al
(124) suggested that
liraglutide exerted liver protective effects by modulating the
expression of Nrf2 and antioxidant enzymes in hepatocytes.
Additionally, Atef et al (62) proposed that the protective
effects of liraglutide against hepatotoxicity occur through the
improvement of oxidative stress, inflammation and necrotic
apoptosis.
GLP-1RAs and sepsis-related
gastrointestinal barrier dysfunction
Sepsis alters the intestinal microenvironment,
promoting epithelial cell barrier disruption, inflammation and
bacterial translocation, leading to enteric infections. The
intestine is regarded as a key player in sepsis; damage to the
intestinal barrier can result in the entry of numerous bacteria and
toxins into the systemic circulation, thereby perpetuating the
sepsis-induced inflammatory response (125-127). During sepsis, excessive
inflammatory responses and oxidative stress cause microvascular
thrombosis, neutrophil-endothelial adhesion and tissue perfusion
deficits, resulting in varying degrees of gastrointestinal injury
(125-127). GLP-1 can suppress inflammation
and promote mucosal integrity. It has been identified as an early
biomarker of intestinal mucosal injury. In lipopolysaccharide
(LPS)-induced intestinal injury mouse models, the intestine rapidly
responds to ischemic damage, characterized by increased GLP-1
secretion following intestinal barrier injury (56). This highlights the importance of
GLP-1 in the response to injury and inflammation (56). Furthermore, exogenous GLP-1 can
protect the intestine from oxidative damage by inhibiting
neutrophil infiltration, and can reduce inflammatory mediators that
induce LPO production, regulate intestinal homeostasis through
local effects and restore intestinal integrity, thereby reducing
bacterial translocation caused by intestinal barrier dysfunction
(128).
GLP-1RAs and sepsis-related coagulation
disorders
Coagulation dysfunction is prevalent during sepsis,
with the pathogenesis primarily involving the release of numerous
inflammatory mediators, endothelial cell injury, activation of the
extrinsic coagulation pathway, and suppression of anticoagulant and
fibrinolytic systems (129).
Endothelium, the single-cell layer lining blood vessels, serves a
crucial role in various vascular processes, including vascular
homeostasis, smooth muscle cell proliferation and thrombus
formation/lysis balance (130).
GLP-1RAs exhibit protective effects on vascular endothelium
(131). In vitro, a
study has shown that liraglutide reduced the expression of
plasminogen activator inhibitor type 1 and vascular adhesion
molecules in human vascular endothelial cells, while increasing
endothelial NO synthase (eNOS) levels in endothelial cells and
reducing the expression of intercellular adhesion molecule-1,
thereby improving endothelial function (131). Liraglutide exerts antioxidant
and anti-inflammatory effects on endothelial cells by inhibiting
protein kinase C-α, NADPH oxidase and NF-κB signaling, while
upregulating protective antioxidant enzymes (132). GLP-1RAs have been shown to
enhance endothelial function by modulating the expression of genes
involved in endothelial apoptosis, angiogenesis, inflammation and
thrombosis, specifically by upregulating eNOS and promoting the
expression of angiogenesis-related genes, as observed for Ex-4
(133). Steven et al
(134) found that GLP-1RAs
activated GLP-1Rs in platelets via a cAMP/PKA-dependent mechanism,
mitigating endotoxemia-induced microvascular thrombosis and
mortality, thereby preventing systemic inflammation, vascular
dysfunction and end-organ damage.
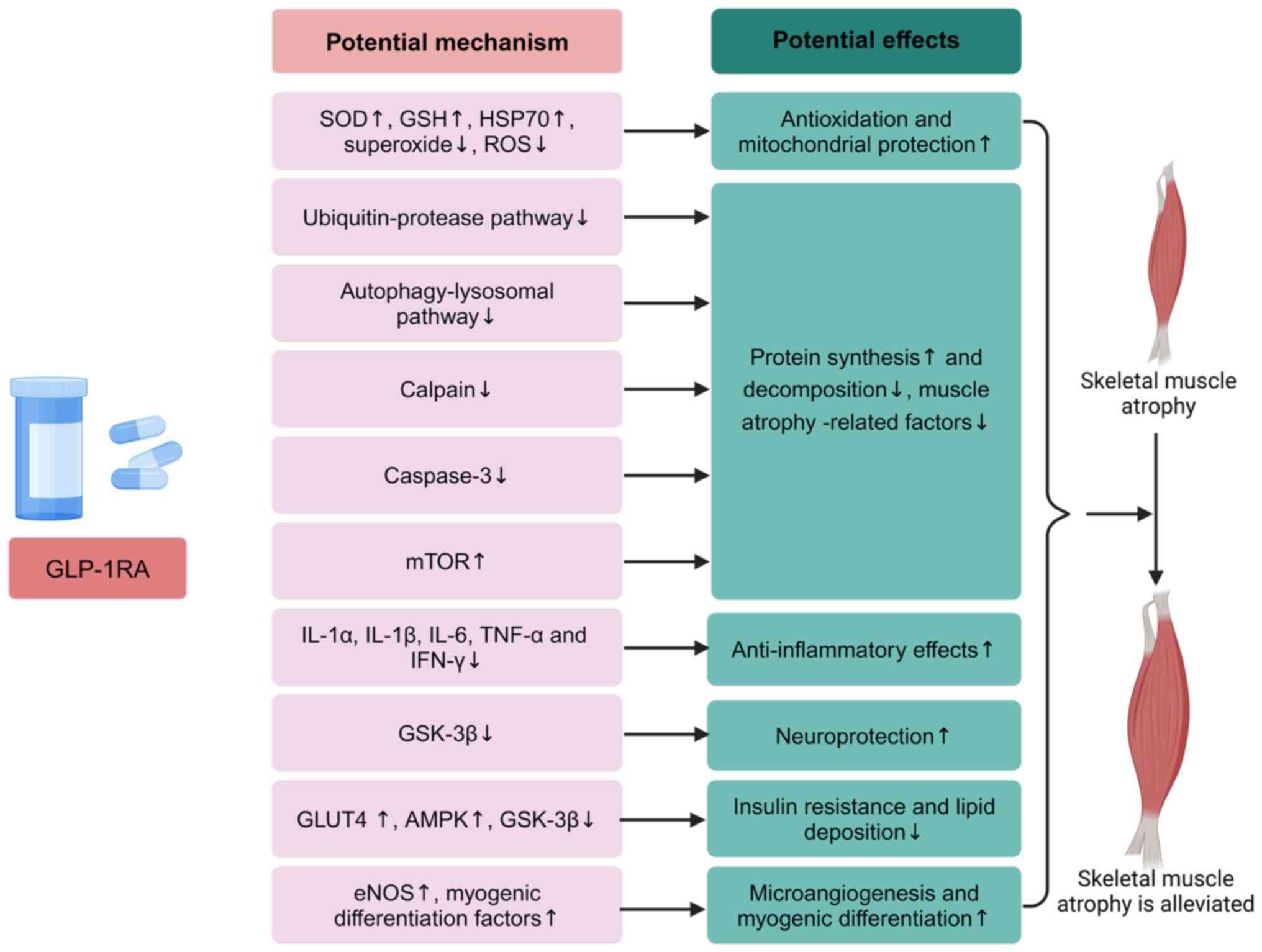
Potential mechanisms of GLP-1RAs in treating
sarcopenia induced by sepsis
As aforementioned, during critical illnesses such
as sepsis, the body is in a state of oxidative stress, where
factors such as excessive release of inflammatory cytokines,
mitochondrial dysfunction, dysregulation of calcium homeostasis,
neuropathy and abnormal cell membrane depolarization collectively
contribute to muscle protein imbalance. Therefore, the present
review, starting from the etiology of SIM, explored the potential
therapeutic mechanism of GLP-1RAs in SIM. These mechanisms include
inhibition of the expression of factors related to muscular
atrophy, anti-inflammatory and antioxidant effects,
neuroprotection, improvement of metabolism, and promotion of
angiogenesis and myogenic differentiation (Fig. 3).
Inhibition of muscle atrophy-related
factor expression
The E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1
serve key roles in skeletal muscle degradation (70). The former induces muscle atrophy
by hydrolyzing myosin within myofibrils, while the latter may
inhibit protein synthesis by downregulating the expression of eIF3f
and MyoD, leading to muscle atrophy (71,72). Gurjar et al (135) revealed that long-acting GLP-1
analogs regulated the expression of myostatin through
GLP-1R-mediated PKA and Akt signaling pathways, increasing the
expression of the myogenic regulatory factors, MyoD and MyoG, while
inhibiting the expression of muscle atrophy-related factors MAFbx
and MuRF1, thereby promoting muscle tissue growth and repair, and
alleviating muscle atrophy. The activated Akt signaling pathway
activates downstream mTOR via the Akt-mTOR axis to promote protein
synthesis, while also inhibiting NF-κB and the binding of myostatin
promoter, reducing myostatin expression to mitigate muscle atrophy
(136-138). Myostatin, a member of the TGF-β
family, is expressed and secreted in skeletal muscle cells,
functioning as a negative regulator of muscle growth. Its gene
expression can be inhibited to induce an increase in muscle mass
(139). Myostatin primarily
mediates the upregulation of MAFbx gene expression through the
transcription factors Smad2/3, thereby promoting protein
degradation (140,141). Inhibition of the
myostatin-Smad2/3 signaling pathway not only alleviates inhibition
of the IGF-1-PI3K-Akt-mTOR signaling pathway but also downregulates
MAFbx expression (140,141). Hong et al (136) found that Ex-4 improved muscle
atrophy by inhibiting the expression of myostatin and muscle
atrophy factors, while enhancing muscle growth factors through the
GLP-1R-mediated signaling pathway. GLP-1RAs can downregulate the
expression of the myostatin gene in C2C12 myotubes, reversing the
upregulation of atrophy factors and downregulation of myogenic
factors (MyoD/G) (136).
Silveira et al (142)
reported that activated cAMP/PKA signaling inhibited the UPS in
skeletal muscle, while activated Akt signaling induced protein
synthesis in skeletal muscle. On one hand, GLP-1RAs increase the
expression levels of MyoD and MyoG, which are major transcription
factors involved in satellite cell muscle tissue repair (143); on the other hand, PKA-mediated
CREB is activated by muscle injury and promotes muscle regeneration
(144). Fan et al
(145) found that liraglutide
treatment reduced body weight in diabetic mice and improved the
condition of skeletal muscle, accompanied by decreased expression
levels of MuRF1 and MAFbx in skeletal muscle.
Anti-inflammatory effects
During sepsis, excessive activation of inflammatory
factors leads to protein imbalance, ultimately resulting in
increased protein degradation and reduced synthesis, which causes
SIM. Therefore, anti-inflammatory interventions can effectively
alleviate the onset of SIM (65-68). The NF-κB and Nrf2 pathways are
two important pathways that mediate cellular homeostasis by
controlling neuroinflammation, while the MAPK pathway is a
classical pathway that can indirectly or directly initiate the
production of inflammatory mediators and activation of NF-κB
(146). Ma et al
(147) revealed that GLP-1RAs
reduced the serum levels of TNF-α, IL-1β and IL-6 in the sciatic
nerve of rats, and inhibited the mRNA expression levels of
pro-inflammatory cytokines. The anti-inflammatory effect of
GLP-1RAs may be mediated through the inhibition of p38 MAPK/NF-κB
pathway activation, as activated p38 MAPK/NF-κB promotes the gene
expression of TNF-α, IL-6 and IL-1β (147). The study suggests that the p38
MAPK/NF-κB pathway could provide targeted therapeutic approaches
for preventing inflammatory changes in diabetic peripheral
neuropathy (147). GLP-1RAs
inhibit TNF-α-induced NF-κB activation and reduce the expression of
adhesion molecules, thereby inhibiting the adhesion of monocytes
induced by TNF-α and LPS, exerting anti-inflammatory effects, while
reducing vascular endothelial damage (148). GLP-1Rs are expressed on
eosinophils and neutrophils, and GLP-1RAs can attenuate eosinophil
activation and LPS-induced expression of pro-inflammatory
cytokines, including IL-1α, IL-1β, IL-6, TNF-α and IFN-γ (23,149,150). Steven et al (134) demonstrated that GLP-1RAs
activated GLP-1Rs on platelets via the AMP/PKA pathway, inhibiting
sepsis-induced microvascular thrombosis, serving an important role
in preventing systemic inflammation and vascular dysfunction, and
protecting against organ injury.
Mitochondrial protection and antioxidant
effects
During sepsis, the body is in a state of stress,
with elevated blood glucose levels and increased ROS levels
(88,89,151). Notably, the treatment of
animals with mitochondrial antioxidants was able to effectively
protect the diaphragm from muscle fiber atrophy and contractile
dysfunction induced by mechanical ventilation, further confirming
the critical role of ROS in mediating muscle atrophy (151). The production of ROS
exacerbates muscle atrophy, as ROS can activate the AMP-activated
protein kinase (AMPK)-FoxO3 pathway, leading to the activation of
the UPS and lysosomal-autophagy system (152). GLP-1RA treatment increases
serum antioxidant enzymes (such as SOD and glutathione), and
reduces circulating oxidized low-density lipoprotein particles and
oxidative stress markers (153,154). In cardiomyocytes, GLP-1RAs
improve mitochondrial fragmentation, while restoring mitochondrial
morphology and function in diabetes (153,154). Additionally, it has been
reported that GLP-1RAs reduced cardiac oxidative stress in
hypertensive mice by lowering ROS levels in whole blood, superoxide
and hydrogen peroxide levels in the heart, and
3-nitrotyrosine-positive proteins in the heart (155). Timper et al (156) demonstrated that GLP-1R
signaling was essential for maintaining mitochondrial integrity and
function in astrocytes; a lack of GLP-1R signaling in hypothalamic
astrocytes mildly impaired mitochondrial function and induced
cellular stress responses.
The glucocorticoid pathway is another signaling
cascade involved in proteolysis, activating two major proteolytic
cascades: Caspase-3 and the UPS (157,158). Furthermore, glucocorticoids
enhance myostatin expression, thereby negatively impacting skeletal
muscle (69,158). Research indicates that Ex-4 can
upregulate the expression of the glucocorticoid receptor (GR)
inhibitory complex, promoting its binding with the GR, which
reduces the binding of glucocorticoids to GR and their transport
into the nucleus, thereby diminishing the negative effects of
glucocorticoids on skeletal muscle (136). On the other hand, heat shock
protein 70 (HSP70) is part of the GR inhibitory complex, which can
alleviate oxidative stress and act as an anti-inflammatory agent in
pro-inflammatory environments by inhibiting the activation of
NF-κB, thus counteracting muscle atrophy (159-161). Senf et al (162) found that the upregulation of
HSP70 could inhibit the expression of muscle atrophy-related
factors MAFbx and MuRF1, while also suppressing the transcriptional
activity of atrophy-related factors controlled by FoxO and NF-κB
genes.
Neuroprotection
One of the causes of muscle atrophy is the
dysfunction of the nervous system, leading to denervation atrophy
of muscles (13,93). Sadek et al (163) demonstrated that GLP-1RAs could
activate the bifurcated PI3K/Akt/GSK-3β pathway, which affects
cellular function on multiple levels. On the one hand, it improved
pathological states associated with neurodegeneration, oxidative
stress and neuroinflammation by promoting the phosphorylation of
GSK-3β, resulting in its inactivation. On the other hand, GLP-1RA
could also increase the level of Nrf2, activate antioxidant and
anti-inflammatory mechanisms in the body, and serve an important
neuroprotective role (163). In
the process of denervation, muscle proteins associated with
proteolysis, including atrogin-1/MAFbx and MuRF1, are increased,
while expression of proteins associated with protein synthesis,
including AKT/mTOR, decreases (70,137,164). Therefore, GLP-1RAs may promote
protein synthesis and alleviate denervation atrophy by activating
the IGF-1-PI3K-Akt-mTOR signaling pathway (70,137,164). Additionally, Mohiuddin et
al (165) indicated that
GLP-1RAs exerted neuroprotective effects through direct action on
root ganglion neurons.
Improvement of insulin resistance and
reduction of lipid accumulation in skeletal muscle
Systemic insulin resistance often accompanies
trauma or severe illness (166). As aforementioned, GLP-1RAs can
improve insulin resistance and promote insulin secretion.
Semaglutide, a type of GLP-1RA, was found by Sadek et al
(163) to inhibit GSK-3β
activity through the PI3K/Akt/GSK-3β pathway, promoting glycogen
synthesis in muscle. Glucose transporter 4 (GLUT4) is an
insulin-dependent glycoprotein that is expressed in muscle, fat and
bone, and its expression and displacement affect glucose uptake in
muscle and adipose tissue (167). Research shows that exenatide
and liraglutide can activate the AMPK pathway, promoting GLUT4
translocation in an insulin-independent manner, thus increasing
glucose uptake in skeletal muscle (168). A study has indicated that high
glucose levels activated JNK and p38 MAPK after 12 weeks without
causing cell damage. However, oxidative stress activated ERK and
p38 MAPK after 8 weeks, leading to cell damage (169). p38 MAPK can activate chromatin
remodeling proteins and myogenic transcription factors, serving an
important role in muscle differentiation (170). Additionally, in rat
cardiomyocytes, the p38 MAPK/myocyte enhancer factor 2 axis is a
strong inducer of GLUT4 expression (171). However, liraglutide inhibits
the p38-MAPK/NF-κB pathway (147). Therefore, we hypothesized that
p38 MAPK may enhance skeletal muscle mass by regulating myocyte
differentiation and promoting glucose uptake, improving the energy
metabolism of skeletal muscle. On the other hand, GLP-1RAs can
inhibit the excessively activated p38 MAPK/NF-κB pathway, exerting
antioxidant effects (147).
Excessive lipid accumulation in skeletal muscle
cells, and the free fatty acids produced from their breakdown,
inhibit glucose utilization in skeletal muscle (172). AMPK also serves a role in
regulating lipid and protein metabolism by inhibiting the
generation of transcription factors that bind to sterol regulatory
elements, suppressing the synthesis of lipid biosynthetic enzymes
in tissues, inhibiting fat synthesis, increasing fatty acid
oxidation and reducing lipid accumulation (173). Cao et al (174) demonstrated that Ex-4 reduced
lipid accumulation and insulin resistance in the skeletal muscle of
obese mice induced by a high-fat diet by activating AMPK and
upregulating insulin signaling pathways.
Recruitment of skeletal muscle
microvasculature and induction of myogenic differentiation
GLP-1RAs can increase microvascular perfusion in
skeletal muscle through various pathways (175-178). For example, as aforementioned,
GLP-1RAs are present in vascular endothelial cells. GLP-1 analogs
can increase microvascular perfusion in skeletal muscle through the
PKA-NO pathway, and they can also enhance microvascular perfusion
independently of NO (175).
Furthermore, GLP-1RAs improve insulin resistance. Insulin, by
binding to its receptor, activates the PI3K-Akt-eNOS pathway to
produce NO, mediating vasodilation (176). Improved microcirculation in
skeletal muscle aids in the delivery and exchange of oxygen,
nutrients and insulin, thus nourishing muscle tissue (176). Subaran et al (177) indicated that acute GLP-1
infusion not only dilated blood vessels but also recruited skeletal
and cardiac microvascular systems. Chai et al (178) demonstrated that GLP-1 infusion
increased PKA activity, promoting the phosphorylation of eNOS,
subsequently increasing plasma NO levels, enhancing muscle
oxygenation and glucose uptake, and recruiting the microvascular
system via NO-dependent mechanisms, which increased glucose
utilization in muscles. Gurjar et al (135) also reported that liraglutide
could induce C2C12 myoblast differentiation through cAMP-dependent
signaling events, promoting myogenesis and alleviating muscle
atrophy. In vitro experiments demonstrated that GLP-1RAs
promoted myogenic differentiation in a dose-dependent manner, while
concurrently reducing the transcriptional expression of MAFbx and
MuRF1 in myoblasts in a dose-dependent manner (145). A further in vitro study
on myogenic differentiation confirmed that liraglutide could
promote myogenic differentiation, alleviate myotube atrophy, and
inhibit the upregulation of atrophy-induced MAFbx and MuRF1 mRNA
expression, ultimately contributing to the preservation of muscle
mass (145).
Clinical safety and limitations
GLP-1RAs are commonly used medications for diabetes
treatment and have gained attention due to their weight loss
effects (19-21). Studies have shown that the
regulatory effects of GLP-1RAs on metabolism and inflammatory
responses may offer potential benefits for sepsis and associated
muscle atrophy (23,59-64). However, there are still certain
limitations when using GLP-1RAs to treat SIM. Relevant reviews have
reported serious adverse events associated with GLP-1RAs, among
which gastrointestinal diseases (commonly nausea, diarrhea,
vomiting and constipation) are the most frequently reported;
however, most gastrointestinal events are of mild to moderate
severity, transient and do not require permanent discontinuation of
treatment (179,180). Other reported adverse reactions
include allergic reactions, cardiovascular, psychiatric and
thyroid-related events, as well as biliary diseases and
pancreatitis (179,180). Additionally, small sample
sizes, short durations and selective participant recruitment limit
the identification of rare or long-term adverse events. Tobaiqy
(179) noted that clinicians
must weigh the risks and benefits, monitor patients for adverse
events, and conduct ongoing long-term safety assessments,
especially regarding the cumulative effects and susceptibilities in
specific populations. Although numerous studies (135,145) suggest that GLP-1RAs may have
protective effects against sepsis and sepsis-induced muscle
atrophy, most studies (135,145) primarily focus on animal models,
with limited clinical research available. Currently, to the best of
our knowledge, there are no clinical reports of GLP-1RA treatment
for SIM, and the long-term efficacy and safety in patients with SIM
have not been sufficiently validated. For instance, some studies
have found that GLP-1RAs can cause or exacerbate muscle atrophy,
leading to a decrease in muscle mass, this contradicts the
hypothesis that GLP-1RAs have therapeutic potential in SIM
(181,182). Therefore, further verification
of the long-term efficacy and safety in clinical settings is
necessary. Additionally, sepsis is a systemic inflammatory response
syndrome induced by infection, often accompanied by multiple organ
system failures (1). The
mechanisms of muscle atrophy in sepsis are complex and not yet
fully elucidated (69). Due to
the multifactorial and complex nature of sepsis-induced muscle
atrophy, treatment typically requires multidisciplinary approaches.
Patients with sepsis often have multiple comorbidities, such as
diabetes and hepatic or renal insufficiency. These factors can
affect the pharmacokinetics and pharmacodynamics of GLP-1Ras
(1). Thus, in some cases,
GLP-1RAs may not be suitable for certain patients. Currently, there
are no effective treatments for SIM, with a focus on prevention,
commonly involving blood glucose control, early anti-inflammation
treatment, early enteral nutrition, and early exercise and
rehabilitation (13). Therefore,
GLP-1RA treatment alone may not address all relevant mechanisms and
is more likely to serve as an adjunct therapy. Recent reviews
indicate that mesenchymal stem cells and melatonin hold certain
potential in the treatment of sepsis-related muscle atrophy
(10,98). Consequently, if GLP-1RAs are used
in the treatment of sepsis-related muscle atrophy, they may need to
be combined with other medications.
Outlook and conclusion
Sepsis is a systemic inflammatory response syndrome
caused by infection, characterized by organ dysfunction and
metabolic imbalance, and often accompanied by muscle atrophy.
Muscle atrophy is common in patients with sepsis, leading to
prolonged recovery times, reduced quality of life and even an
increased risk of mortality. Currently, there are no specific
therapies available for SIM, a situation that may be attributed to
several factors. On the one hand, the pathophysiological mechanisms
underlying SIM are complex, involving processes such as
inflammation, oxidative stress and metabolic dysregulation, which
pose challenges to the development of targeted treatments. On the
other hand, SIM typically occurs in critically ill patients with
sepsis or other systemic diseases, potentially limiting the
feasibility of conducting large-scale clinical trials. Nutritional
support and rehabilitation training are generally regarded as
supportive rather than curative interventions. The primary goal of
nutritional support is to alleviate malnutrition and promote muscle
protein synthesis, while rehabilitation training aims to preserve
muscle function and prevent further atrophy. However, these
approaches have shown limited efficacy in reversing SIM, as they do
not directly address the underlying mechanisms driving muscle
atrophy. This underscores the urgent need for the development of
novel therapeutic strategies.
GLP-1RAs have emerged as a potential therapeutic
approach. Although clinical research on GLP-1RAs in SIM is still
limited, some preliminary evidence (135,145) supports the improvement of
muscle mass and function by GLP-1 analogs. Additionally, GLP-1RAs
exhibit organ-protective effects on the heart, brain, kidneys,
lungs, liver, intestines and vasculature in the context of septic
multi-organ dysfunction. However, there is currently a lack of
focused reviews on the relationship between GLP-1RAs and
sepsis-related muscle atrophy. The present review may help fill the
existing gaps in the literature and provide directions for future
research.
Numerous studies (135-178) have indicated that GLP-1RAs can
reduce oxidative stress, inflammatory responses, apoptosis and
mitochondrial dysfunction, improve insulin resistance, and induce
myogenic differentiation, thereby showing potential in the
treatment of SIM. Consequently, the present review discusses the
possibilities of GLP-1RA therapy for SIM in detail and elaborates
on its potential therapeutic mechanisms. Although GLP-1RAs exhibit
certain potential in some metabolic and inflammatory diseases,
their use as a treatment option for sepsis-induced muscle atrophy
still faces numerous limitations, particularly concerning
insufficient clinical evidence, complex therapeutic mechanisms,
drug side effects and individual variability. Therefore, the
application of GLP-1RAs in sepsis-related muscle atrophy requires
more clinical research and practice to validate their efficacy and
safety, and GLP-1RAs may need to be combined with other therapeutic
approaches to achieve maximum effectiveness.
Availability of data and materials
Not applicable.
Authors' contributions
XZ, YL, DW, TL, ZX, ZL, XB and YW contributed to
the design of the study and writing of the manuscript. XZ, YL, DW,
TL and ZX performed the literature research. XZ and YW wrote the
main manuscript text and prepared figures. ZL, XB and YW revised
the article critically for important intellectual content and
provided final approval of the version to be submitted. All authors
reviewed the manuscript. Data authentication is not applicable. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from Hubei Provincial
Natural Science Foundation of China (grant no. 2023AFB825), and
Tongji Hospital, Tongji Medical College, Huazhong University of
Science and Technology (grant no. 2023A15).
References
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mushtaq A and Kazi F: Updates in sepsis
management. Lancet Infect Dis. 22:242022. View Article : Google Scholar
|
|
3
|
Vincent JL, Jones G, David S, Olariu E and
Cadwell KK: Frequency and mortality of septic shock in Europe and
North America: A systematic review and meta-analysis. Crit Care.
23:1962019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abraham E: New definitions for sepsis and
septic shock: Continuing evolution but with much still to be done.
JAMA. 315:757–759. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bracht H, Hafner S and Weiß M: Sepsis
update: Definition and epidemiology. Anasthesiol Intensivmed
Notfallmed Schmerzther. 54:10–20. 2019.In German. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vincent JL: Current sepsis therapeutics.
EBioMedicine. 86:1043182022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leviner S: Post-sepsis syndrome. Crit Care
Nurs Q. 44:182–186. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Callahan LA and Supinski GS:
Sepsis-induced myopathy. Crit Care Med. 37(10 Suppl): S354–S367.
2009. View Article : Google Scholar
|
|
9
|
Gardner AK, Ghita GL, Wang Z,
Ozrazgat-Baslanti T, Raymond SL, Mankowski RT, Brumback BA, Efron
PA, Bihorac A, Moore FA, et al: The development of chronic critical
illness determines physical function, quality of life, and
long-term survival among early survivors of sepsis in surgical
ICUs. Crit Care Med. 47:566–573. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Y, Wang D, Li T, Xu L, Li Z, Bai X,
Tang M and Wang Y: Melatonin: A potential adjuvant therapy for
septic myopathy. Biomed Pharmacother. 158:1142092023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu W, Hu C and Zhao S: Sarcopenia and
mortality risk of patients with sepsis: A meta-analysis. Int J Clin
Pract. 2022:49744102022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mankowski RT, Laitano O, Clanton TL and
Brakenridge SC: Pathophysiology and treatment strategies of acute
myopathy and muscle wasting after sepsis. J Clin Med. 10:18742021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen J and Huang M: Intensive care
unit-acquired weakness: Recent insights. J Intensive Med. 4:73–80.
2023. View Article : Google Scholar
|
|
14
|
Piva S, Fagoni N and Latronico N:
Intensive care unit-acquired weakness: Unanswered questions and
targets for future research. F1000Res. 8:F1000Faculty Rev-508.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Latronico N, Herridge M, Hopkins RO, Angus
D, Hart N, Hermans G, Iwashyna T, Arabi Y, Citerio G, Ely EW, et
al: The ICM research agenda on intensive care unit-acquired
weakness. Intensive Care Med. 43:1270–1281. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schefold JC, Bierbrauer J and
Weber-Carstens S: Intensive care unit-acquired weakness (ICUAW) and
muscle wasting in critically ill patients with severe sepsis and
septic shock. J Cachexia Sarcopenia Muscle. 1:147–157. 2010.
View Article : Google Scholar
|
|
17
|
Farhan H, Moreno-Duarte I, Latronico N,
Zafonte R and Eikermann M: Acquired muscle weakness in the surgical
intensive care unit: Nosology, epidemiology, diagnosis, and
prevention. Anesthesiology. 124:207–234. 2016. View Article : Google Scholar
|
|
18
|
Graaf Cd, Donnelly D, Wootten D, Lau J,
Sexton PM, Miller LJ, Ahn JM, Liao J, Fletcher MM, Yang D, et al:
Glucagon-like peptide-1 and its class B G protein-coupled
receptors: A long march to therapeutic successes. Pharmacol Rev.
68:954–1013. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Garber AJ, Abrahamson MJ, Barzilay JI,
Blonde L, Bloomgarden ZT, Bush MA, Dagogo-Jack S, Davidson MB,
Einhorn D, Garvey WT, et al: AACE comprehensive diabetes management
algorithm 2013. Endocr Pract. 19:327–336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Inzucchi SE, Bergenstal RM, Buse JB,
Diamant M, Ferrannini E, Nauck M, Peters AL, Tsapas A, Wender R,
Matthews DR, et al: Management of hyperglycemia in type 2 diabetes:
A patient-centered approach: position statement of the American
diabetes association (ADA) and the European association for the
study of diabetes (EASD). Diabetes Care. 35:1364–1379. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gutzwiller JP, Drewe J, Göke B, Schmidt H,
Rohrer B, Lareida J and Beglinger C: Glucagon-like peptide-1
promotes satiety and reduces food intake in patients with diabetes
mellitus type 2. Am J Physiol. 276:R1541–R1544. 1999.PubMed/NCBI
|
|
22
|
Zhao X, Wang M, Wen Z, Lu Z, Cui L, Fu C,
Xue H, Liu Y and Zhang Y: GLP-1 receptor agonists: Beyond their
pancreatic effects. Front Endocrinol (Lausanne).
12:7211352021.PubMed/NCBI
|
|
23
|
Yang F, Zeng F, Luo X, Lei Y, Li J, Lu S,
Huang X, Lan Y and Liu R: GLP-1 receptor: A new target for sepsis.
Front Pharmacol. 12:7069082021.PubMed/NCBI
|
|
24
|
Iwai S, Kaji K, Nishimura N, Kubo T,
Tomooka F, Shibamoto A, Suzuki J, Tsuji Y, Fujinaga Y, Kitagawa K,
et al: Glucagon-like peptide-1 receptor agonist, semaglutide
attenuates chronic liver disease-induced skeletal muscle atrophy in
diabetic mice. Biochim Biophys Acta Mol Basis Dis.
1869:1667702023.PubMed/NCBI
|
|
25
|
Sandoval DA and D'Alessio DA: Physiology
of proglucagon peptides: Role of glucagon and GLP-1 in health and
disease. Physiol Rev. 95:513–548. 2015.PubMed/NCBI
|
|
26
|
Nian M, Drucker DJ and Irwin D: Divergent
regulation of human and rat proglucagon gene promoters in vivo. Am
J Physiol. 277:G829–G837. 1999.PubMed/NCBI
|
|
27
|
Jin SL, Han VK, Simmons JG, Towle AC,
Lauder JM and Lund PK: Distribution of glucagonlike peptide I
(GLP-I), glucagon, and glicentin in the rat brain: An
immunocytochemical study. J Comp Neurol. 271:519–532.
1988.PubMed/NCBI
|
|
28
|
Han VK, Hynes MA, Jin C, Towle AC, Lauder
JM and Lund PK: Cellular localization of proglucagon/glucagon-like
peptide I messenger RNAs in rat brain. J Neurosci Res. 16:97–107.
1986.PubMed/NCBI
|
|
29
|
Drucker DJ and Nauck MA: The incretin
system: Glucagon-like peptide-1 receptor agonists and dipeptidyl
peptidase-4 inhibitors in type 2 diabetes. Lancet. 368:1696–1705.
2006.PubMed/NCBI
|
|
30
|
Sharma D, Verma S, Vaidya S, Kalia K and
Tiwari V: Recent updates on GLP-1 agonists: Current advancements
& challenges. Biomed Pharmacother. 108:952–962. 2018.PubMed/NCBI
|
|
31
|
Kreymann B, Williams G, Ghatei MA and
Bloom SR: Glucagon-like peptide-1 7-36: A physiological incretin in
man. Lancet. 2:1300–1304. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deacon CF, Johnsen AH and Holst JJ:
Degradation of glucagon-like peptide-1 by human plasma in vitro
yields an N-terminally truncated peptide that is a major endogenous
metabolite in vivo. J Clin Endocrinol Metab. 80:952–957.
1995.PubMed/NCBI
|
|
33
|
Müller TD, Finan B, Bloom SR, D'Alessio D,
Drucker DJ, Flatt PR, Fritsche A, Gribble F, Grill HJ, Habener JF,
et al: Glucagon-like peptide 1 (GLP-1). Mol Metab. 30:72–130. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hansen L, Deacon CF, Orskov C and Holst
JJ: Glucagon-like peptide-1-(7-36)amide is transformed to
glucagon-like peptide-1-(9-36)amide by dipeptidyl peptidase IV in
the capillaries supplying the L cells of the porcine intestine.
Endocrinology. 140:5356–5363. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Meier JJ, Nauck MA, Kranz D, Holst JJ,
Deacon CF, Gaeckler D, Schmidt WE and Gallwitz B: Secretion,
degradation, and elimination of glucagon-like peptide 1 and gastric
inhibitory polypeptide in patients with chronic renal insufficiency
and healthy control subjects. Diabetes. 53:654–662. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Leech CA, Chepurny OG and Holz GG:
Epac2-dependent rap1 activation and the control of islet insulin
secretion by glucagon-like peptide-1. Vitam Horm. 84:279–302. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Göke R, Larsen PJ, Mikkelsen JD and Sheikh
SP: Distribution of GLP-1 binding sites in the rat brain: Evidence
that exendin-4 is a ligand of brain GLP-1 binding sites. Eur J
Neurosci. 7:2294–2300. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Campos RV, Lee YC and Drucker DJ:
Divergent tissue-specific and developmental expression of receptors
for glucagon and glucagon-like peptide-1 in the mouse.
Endocrinology. 134:2156–2164. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cantini G, Mannucci E and Luconi M:
Perspectives in GLP-1 research: New targets, new receptors. Trends
Endocrinol Metab. 27:427–438. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Holz GG: Epac: A new cAMP-binding protein
in support of glucagon-like peptide-1 receptor-mediated signal
transduction in the pancreatic beta-cell. Diabetes. 53:5–13. 2004.
View Article : Google Scholar
|
|
41
|
Drucker DJ, Philippe J, Mojsov S, Chick WL
and Habener JF: Glucagon-like peptide I stimulates insulin gene
expression and increases cyclic AMP levels in a rat islet cell
line. Proc Natl Acad Sci USA. 84:3434–3438. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fehmann HC and Habener JF: Galanin
inhibits proinsulin gene expression stimulated by the
insulinotropic hormone glucagon-like peptide-I(7-37) in mouse
insulinoma beta TC-1 cells. Endocrinology. 130:2890–2896. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Arakawa M, Ebato C, Mita T, Hirose T,
Kawamori R, Fujitani Y and Watada H: Effects of exendin-4 on
glucose tolerance, insulin secretion, and beta-cell proliferation
depend on treatment dose, treatment duration and meal contents.
Biochem Biophys Res Commun. 390:809–814. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hare KJ, Vilsbøll T, Asmar M, Deacon CF,
Knop FK and Holst JJ: The glucagonostatic and insulinotropic
effects of glucagon-like peptide 1 contribute equally to its
glucose-lowering action. Diabetes. 59:1765–1770. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Beiroa D, Imbernon M, Gallego R, Senra A,
Herranz D, Villarroya F, Serrano M, Fernø J, Salvador J, Escalada
J, et al: GLP-1 agonism stimulates brown adipose tissue
thermogenesis and browning through hypothalamic AMPK. Diabetes.
63:3346–3358. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Halim MA, Degerblad M, Sundbom M, Karlbom
U, Holst JJ, Webb DL and Hellström PM: Glucagon-like peptide-1
inhibits prandial gastrointestinal motility through myenteric
neuronal mechanisms in humans. J Clin Endocrinol Metab.
103:575–585. 2018. View Article : Google Scholar
|
|
47
|
McKay NJ, Kanoski SE, Hayes MR and Daniels
D: Glucagon-like peptide-1 receptor agonists suppress water intake
independent of effects on food intake. Am J Physiol Regul Integr
Comp Physiol. 301:R1755–R1764. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Herman JP: Regulation of
hypothalamo-pituitary-adrenocortical responses to stressors by the
nucleus of the solitary tract/dorsal vagal complex. Cell Mol
Neurobiol. 38:25–35. 2018. View Article : Google Scholar :
|
|
49
|
Ghosal S, Packard AEB, Mahbod P, McKlveen
JM, Seeley RJ, Myers B, Ulrich-Lai Y, Smith EP, D'Alessio DA and
Herman JP: Disruption of glucagon-like peptide 1 signaling in sim1
neurons reduces physiological and behavioral reactivity to acute
and chronic stress. J Neurosci. 37:184–193. 2017.PubMed/NCBI
|
|
50
|
Perry T, Lahiri DK, Chen D, Zhou J, Shaw
KT, Egan JM and Greig NH: A novel neurotrophic property of
glucagon-like peptide 1: A promoter of nerve growth factor-mediated
differentiation in PC12 cells. J Pharmacol Exp Ther. 300:958–966.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
During MJ, Cao L, Zuzga DS, Francis JS,
Fitzsimons HL, Jiao X, Bland RJ, Klugmann M, Banks WA, Drucker DJ
and Haile CN: Glucagon-like peptide-1 receptor is involved in
learning and neuroprotection. Nat Med. 9:1173–1179. 2003.PubMed/NCBI
|
|
52
|
Marso SP, Daniels GH, Brown-Frandsen K,
Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR,
Ravn LS, et al: Liraglutide and cardiovascular outcomes in type 2
diabetes. N Engl J Med. 375:311–322. 2016.PubMed/NCBI
|
|
53
|
Mann JFE, Ørsted DD, Brown-Frandsen K,
Marso SP, Poulter NR, Rasmussen S, Tornøe K, Zinman B and Buse JB;
LEADER Steering Committee and Investigators: Liraglutide and renal
outcomes in type 2 diabetes. N Engl J Med. 377:839–848.
2017.PubMed/NCBI
|
|
54
|
Teramoto S, Miyamoto N, Yatomi K, Tanaka
Y, Oishi H, Arai H, Hattori N and Urabe T: Exendin-4, a
glucagon-like peptide-1 receptor agonist, provides neuroprotection
in mice transient focal cerebral ischemia. J Cereb Blood Flow
Metab. 31:1696–1705. 2011.PubMed/NCBI
|
|
55
|
Aravindhan K, Bao W, Harpel MR, Willette
RN, Lepore JJ and Jucker BM: Cardioprotection resulting from
glucagon-like peptide-1 administration involves shifting metabolic
substrate utilization to increase energy efficiency in the rat
heart. PLoS One. 10:e01308942015.PubMed/NCBI
|
|
56
|
Lebrun LJ, Lenaerts K, Kiers D, Pais de
Barros JP, Le Guern N, Plesnik J, Thomas C, Bourgeois T, Dejong
CHC, Kox M, et al: Enteroendocrine L cells sense LPS after gut
barrier injury to enhance GLP-1 secretion. Cell Rep. 21:1160–1168.
2017.PubMed/NCBI
|
|
57
|
Guo J, Zhang X, Pan R, Zheng Y, Chen W and
Wang L: Liraglutide alleviates sepsis-induced acute lung injury by
regulating pulmonary surfactant through inhibiting autophagy.
Immunopharmacol Immunotoxicol. 46:573–582. 2024.PubMed/NCBI
|
|
58
|
Armstrong MJ, Gaunt P, Aithal GP, Barton
D, Hull D, Parker R, Hazlehurst JM, Guo K; LEAN trial team; Abouda
G, et al: Liraglutide safety and efficacy in patients with
non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind,
randomised, placebo-controlled phase 2 study. Lancet. 387:679–690.
2016.
|
|
59
|
Pan HC, Chen JY, Chen HY, Yeh FY, Sun CY,
Huang TT and Wu VC: GLP-1 receptor agonists' impact on cardio-renal
outcomes and mortality in T2D with acute kidney disease. Nat
Commun. 15:59122024.PubMed/NCBI
|
|
60
|
Guo J, Chen X, Wang C, Ruan F, Xiong Y,
Wang L, Abdel-Razek O, Meng Q, Shahbazov R, Cooney RN and Wang G:
Liraglutide alleviates acute lung injury and mortality in
pneumonia-induced sepsis through regulating surfactant protein
expression and secretion. Shock. 61:601–610. 2024.
|
|
61
|
Yi H, Duan Y, Song R, Zhou Y, Cui Y, Liu
C, Mao Z, Hu J and Zhou F: Activation of glucagon-like peptide-1
receptor in microglia exerts protective effects against
sepsis-induced encephalopathy via attenuating endoplasmic reticulum
stress-associated inflammation and apoptosis in a mouse model of
sepsis. Exp Neurol. 363:1143482023. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Atef MM, Abou Hashish NA, Hafez YM, Selim
AF, Ibrahim HA, Eltabaa EF, Rizk FH, Shalaby AM, Ezzat N, Alabiad
MA and Elshamy AM: The potential protective effect of liraglutide
on valproic acid induced liver injury in rats: Targeting HMGB1/RAGE
axis and RIPK3/MLKL mediated necroptosis. Cell Biochem Funct.
41:1209–1219. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Seufert J and Gallwitz B: The
extra-pancreatic effects of GLP-1 receptor agonists: A focus on the
cardiovascular, gastrointestinal and central nervous systems.
Diabetes Obes Metab. 16:673–688. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Glorie LLF, Verhulst A, Matheeussen V,
Baerts L, Magielse J, Hermans N, D'Haese PC, De Meester I and De
Beuf A: DPP4 inhibition improves functional outcome after renal
ischemia-reperfusion injury. Am J Physiol Renal Physiol.
303:F681–F688. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cruz-Jentoft AJ, Baeyens JP, Bauer JM,
Boirie Y, Cederholm T, Landi F, Martin FC, Michel JP, Rolland Y,
Schneider SM, et al: Sarcopenia: European consensus on definition
and diagnosis: Report of the European working group on sarcopenia
in older people. Age Ageing. 39:412–423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Cohen S, Nathan JA and Goldberg AL: Muscle
wasting in disease: Molecular mechanisms and promising therapies.
Nat Rev Drug Discov. 14:58–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Preiser JC, Ichai C, Orban JC and
Groeneveld AB: Metabolic response to the stress of critical
illness. Br J Anaesth. 113:945–954. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bloch S, Polkey MI, Griffiths M and Kemp
P: Molecular mechanisms of intensive care unit-acquired weakness.
Eur Respir J. 39:1000–1011. 2012. View Article : Google Scholar
|
|
69
|
Kanova M and Kohout P: Molecular
mechanisms underlying intensive care unit-acquired weakness and
sarcopenia. Int J Mol Sci. 23:83962022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Bodine SC, Latres E, Baumhueter S, Lai VK,
Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K,
et al: Identification of ubiquitin ligases required for skeletal
muscle atrophy. Science. 294:1704–1708. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Csibi A, Cornille K, Leibovitch MP, Poupon
A, Tintignac LA, Sanchez AM and Leibovitch SA: The translation
regulatory subunit eIF3f controls the kinase-dependent mTOR
signaling required for muscle differentiation and hypertrophy in
mouse. PLoS One. 5:e89942010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tintignac LA, Lagirand J, Batonnet S,
Sirri V, Leibovitch MP and Leibovitch SA: Degradation of MyoD
mediated by the SCF (MAFbx) ubiquitin ligase. J Biol Chem.
280:2847–2856. 2005. View Article : Google Scholar
|
|
73
|
Cai D, Frantz JD, Tawa NE Jr, Melendez PA,
Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ and
Shoelson SE: IKKbeta/NF-kappaB activation causes severe muscle
wasting in mice. Cell. 119:285–298. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Klaude M, Fredriksson K, Tjäder I,
Hammarqvist F, Ahlman B, Rooyackers O and Wernerman J: Proteasome
proteolytic activity in skeletal muscle is increased in patients
with sepsis. Clin Sci (Lond). 112:499–506. 2007. View Article : Google Scholar
|
|
75
|
Supinski GS and Callahan LA: Calpain
activation contributes to endotoxin-induced diaphragmatic
dysfunction. Am J Respir Cell Mol Biol. 42:80–87. 2010. View Article : Google Scholar :
|
|
76
|
Smith IJ, Lecker SH and Hasselgren PO:
Calpain activity and muscle wasting in sepsis. Am J Physiol
Endocrinol Metab. 295:E762–E771. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Supinski GS and Callahan LA: Caspase
activation contributes to endotoxin-induced diaphragm weakness. J
Appl Physiol (1985). 100:1770–1777. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hussain SN, Mofarrahi M, Sigala I, Kim HC,
Vassilakopoulos T, Maltais F, Bellenis I, Chaturvedi R, Gottfried
SB, Metrakos P, et al: Mechanical ventilation-induced diaphragm
disuse in humans triggers autophagy. Am J Respir Crit Care Med.
182:1377–1386. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Franco-Romero A and Sandri M: Role of
autophagy in muscle disease. Mol Aspects Med. 82:1010412021.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Latres E, Amini AR, Amini AA, Griffiths J,
Martin FJ, Wei Y, Lin HC, Yancopoulos GD and Glass DJ: Insulin-like
growth factor-1 (IGF-1) inversely regulates atrophy-induced genes
via the phosphatidylinositol 3-kinase/Akt/mammalian target of
rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem. 280:2737–2744.
2005. View Article : Google Scholar
|
|
82
|
Sandri M, Sandri C, Gilbert A, Skurk C,
Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH and Goldberg
AL: Foxo transcription factors induce the atrophy-related ubiquitin
ligase atrogin-1 and cause skeletal muscle atrophy. Cell.
117:399–412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lang CH and Frost RA: Role of growth
hormone, insulin-like growth factor-I, and insulin-like growth
factor binding proteins in the catabolic response to injury and
infection. Curr Opin Clin Nutr Metab Care. 5:271–279. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Nystrom G, Pruznak A, Huber D, Frost RA
and Lang CH: Local insulin-like growth factor I prevents
sepsis-induced muscle atrophy. Metabolism. 58:787–797. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Showkat M, Beigh MA and Andrabi KI: mTOR
signaling in protein translation regulation: Implications in cancer
genesis and therapeutic interventions. Mol Biol Int.
2014:6869842014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lang CH, Frost RA and Vary TC: Regulation
of muscle protein synthesis during sepsis and inflammation. Am J
Physiol Endocrinol Metab. 293:E453–E459. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Frost RA and Lang CH: mTor signaling in
skeletal muscle during sepsis and inflammation: Where does it all
go wrong? Physiology (Bethesda). 26:83–96. 2011.PubMed/NCBI
|
|
88
|
Ferri E, Marzetti E, Calvani R, Picca A,
Cesari M and Arosio B: Role of age-related mitochondrial
dysfunction in sarcopenia. Int J Mol Sci. 21:52362020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wu Y, Yao YM and Lu ZQ: Mitochondrial
quality control mechanisms as potential therapeutic targets in
sepsis-induced multiple organ failure. J Mol Med (Berl).
97:451–462. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Preau S, Vodovar D, Jung B, Lancel S,
Zafrani L, Flatres A, Oualha M, Voiriot G, Jouan Y, Joffre J, et
al: Energetic dysfunction in sepsis: A narrative review. Ann
Intensive Care. 11:1042021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Friedrich O, Hund E, Weber C, Hacke W and
Fink RH: Critical illness myopathy serum fractions affect membrane
excitability and intracellular calcium release in mammalian
skeletal muscle. J Neurol. 251:53–65. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zink W, Kaess M, Hofer S, Plachky J,
Zausig YA, Sinner B, Weigand MA, Fink RH and Graf BM: Alterations
in intracellular Ca2+-homeostasis of skeletal muscle fibers during
sepsis. Crit Care Med. 36:1559–1563. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Bolton CF: Neuromuscular manifestations of
critical illness. Muscle Nerve. 32:140–163. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Friedrich O, Reid MB, Van den Berghe G,
Vanhorebeek I, Hermans G, Rich MM and Larsson L: The sick and the
weak: Neuropathies/myopathies in the critically Ill. Physiol Rev.
95:1025–1109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Evans L, Rhodes A, Alhazzani W, Antonelli
M, Coopersmith CM, French C, Machado FR, Mcintyre L, Ostermann M,
Prescott HC, et al: Surviving sepsis campaign: International
guidelines for management of sepsis and septic shock 2021.
Intensive Care Med. 47:1181–1247. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Bos LDJ and Ware LB: Acute respiratory
distress syndrome: Causes, pathophysiology, and phenotypes. Lancet.
400:1145–1156. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Baer B, Putz ND, Riedmann K, Gonski S, Lin
J, Ware LB, Toki S, Peebles RS Jr, Cahill KN and Bastarache JA:
Liraglutide pretreatment attenuates sepsis-induced acute lung
injury. Am J Physiol Lung Cell Mol Physiol. 325:L368–L384. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wang D, Xu L, Liu Y, Wang C, Qi S, Li Z,
Bai X, Liao Y and Wang Y: Role of mesenchymal stem cells in sepsis
and their therapeutic potential in sepsis-associated myopathy
(Review). Int J Mol Med. 54:922024. View Article : Google Scholar :
|
|
99
|
Martin GS and Bernard GR; International
Sepsis Forum: Airway and lung in sepsis. Intensive Care Med.
27(Suppl 1): S63–S79. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Mazeraud A, Righy C, Bouchereau E,
Benghanem S, Bozza FA and Sharshar T: Septic-associated
encephalopathy: A comprehensive review. Neurotherapeutics.
17:392–403. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Catarina AV, Branchini G, Bettoni L, De
Oliveira JR and Nunes FB: Sepsis-associated encephalopathy: From
pathophysiology to progress in experimental studies. Mol Neurobiol.
58:2770–2779. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li Y, Yin L, Fan Z, Su B, Chen Y, Ma Y,
Zhong Y, Hou W, Fang Z and Zhang X: Microglia: A potential
therapeutic target for sepsis-associated encephalopathy and
sepsis-associated chronic pain. Front Pharmacol. 11:6004212020.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Fang Y, Jiang D, Wang Y, Wang Q, Lv D, Liu
J and Liu C: Neuroprotection of rhGLP-1 in diabetic rats with
cerebral ischemia/reperfusion injury via regulation of oxidative
stress, EAAT2, and apoptosis. Drug Dev Res. 79:249–259. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bi CF, Liu J, Yang LS and Zhang JF:
Research progress on the mechanism of sepsis induced myocardial
injury. J Inflamm Res. 15:4275–4290. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Aneman A and Vieillard-Baron A: Cardiac
dysfunction in sepsis. Intensive Care Med. 42:2073–2076. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Sharma A and Verma S: Mechanisms by which
glucagon-like-peptide-1 receptor agonists and sodium-glucose
cotransporter-2 inhibitors reduce cardiovascular risk in adults
with type 2 diabetes mellitus. Can J Diabetes. 44:93–102. 2020.
View Article : Google Scholar
|
|
107
|
Noyan-Ashraf MH, Momen MA, Ban K, Sadi AM,
Zhou YQ, Riazi AM, Baggio LL, Henkelman RM, Husain M and Drucker
DJ: GLP-1R agonist liraglutide activates cytoprotective pathways
and improves outcomes after experimental myocardial infarction in
mice. Diabetes. 58:975–983. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Timmers L, Henriques JP, de Kleijn DP,
Devries JH, Kemperman H, Steendijk P, Verlaan CW, Kerver M, Piek
JJ, Doevendans PA, et al: Exenatide reduces infarct size and
improves cardiac function in a porcine model of ischemia and
reperfusion injury. J Am Coll Cardiol. 53:501–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Bose AK, Mocanu MM, Carr RD, Brand CL and
Yellon DM: Glucagon-like peptide 1 can directly protect the heart
against ischemia/reperfusion injury. Diabetes. 54:146–151. 2005.
View Article : Google Scholar
|
|
110
|
Chang G, Zhang D, Yu H, Zhang P, Wang Y,
Zheng A and Qin S: Cardioprotective effects of exenatide against
oxidative stress-induced injury. Int J Mol Med. 32:1011–1020. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Gómez H and Kellum JA: Sepsis-induced
acute kidney injury. Curr Opin Crit Care. 22:546–553. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Zarjou A and Agarwal A: Sepsis and acute
kidney injury. J Am Soc Nephrol. 22:999–1006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wan L, Bagshaw SM, Langenberg C, Saotome
T, May C and Bellomo R: Pathophysiology of septic acute kidney
injury: What do we really know? Crit Care Med. 36(4 Suppl):
S198–S203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Xiang L, Thompson MS, Clemmer JS, Mittwede
PN, Khan T and Hester RL: Early treatment with GLP-1 after severe
trauma preserves renal function in obese Zucker rats. Am J Physiol
Regul Integr Comp Physiol. 316:R621–R627. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Elkhoely A: Liraglutide ameliorates
gentamicin-induced acute kidney injury in rats via PGC-1α-mediated
mitochondrial biogenesis: Involvement of PKA/CREB and Notch/Hes-1
signaling pathways. Int Immunopharmacol. 114:1095782023. View Article : Google Scholar
|
|
116
|
Xu C, Lu C, Wang Z, Hu X, Li S, Xie Y, Qiu
Y, Cao R, Li Y and Yang J: Liraglutide abrogates nephrotoxic
effects of chemotherapies. Pharmacol Res. 189:1066802023.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Li YK, Ma DX, Wang ZM, Hu XF, Li SL, Tian
HZ, Wang MJ, Shu YW and Yang J: The glucagon-like peptide-1 (GLP-1)
analog liraglutide attenuates renal fibrosis. Pharmacol Res.
131:102–111. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Kasper P, Tacke F, Steffen HM and Michels
G: Hepatic dysfunction in sepsis. Med Klin Intensivmed Notfmed.
115:609–619. 2020.In German. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Horvatits T, Drolz A, Trauner M and
Fuhrmann V: Liver injury and failure in critical illness.
Hepatology. 70:2204–2215. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Strnad P, Tacke F, Koch A and Trautwein C:
Liver-guardian, modifier and target of sepsis. Nat Rev
Gastroenterol Hepatol. 14:55–66. 2017. View Article : Google Scholar
|
|
121
|
Milani L, Galindo CM, Turin de Oliveira
NM, Corso CR, Adami ER, Stipp MC, Beltrame OC and Acco A: The GLP-1
analog liraglutide attenuates acute liver injury in mice. Ann
Hepatol. 18:918–928. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zhang WY, Hu XF, Wan N, Zhang JF, Yang P,
Wen Q, Chen WJ, Zhu F, Liang ML, Cheng LX and Shu YW: Protective
effect of the glucagon-like peptide-1 analogue liraglutide on
carbon tetrachloride-induced acute liver injury in mice. Biochem
Biophys Res Commun. 514:386–392. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Abdelaziz AI, Mantawy EM, Gad AM, Fawzy HM
and Azab SS: Activation of pCREB/Nrf-2 signaling mediates
re-positioning of liraglutide as hepato-protective for
methotrexate-induced liver injury (MILI). Food Chem Toxicol.
132:1107192019. View Article : Google Scholar
|
|
124
|
Zhu CG, Luo Y, Wang H, Li JY, Yang J, Liu
YX, Qu HQ, Wang BL and Zhu M: Liraglutide ameliorates
lipotoxicity-induced oxidative stress by activating the NRF2
pathway in HepG2 cells. Horm Metab Res. 52:532–539. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Fay KT, Ford ML and Coopersmith CM: The
intestinal microenvironment in sepsis. Biochim Biophys Acta Mol
Basis Dis. 1863:2574–2583. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Mittal R and Coopersmith CM: Redefining
the gut as the motor of critical illness. Trends Mol Med.
20:214–223. 2014. View Article : Google Scholar :
|
|
127
|
Haseeb MA and Salwen MJ: Collateral
damage: Sepsis-induced gut injury. Crit Care Med. 33:2439–2440.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Deniz M, Atasoy BM, Dane F, Can G, Erzik
C, Çetinel Ş and Yeğen BÇ: Radiation-induced oxidative injury of
the ileum and colon is alleviated by glucagon-like peptide-1 and
-2. J Radiat Res Appl Sci. 8:234–242. 2015.
|
|
129
|
Semeraro N, Ammollo CT, Semeraro F and
Colucci M: Coagulopathy of acute sepsis. Semin Thromb Hemost.
41:650–658. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Bonetti PO, Lerman LO and Lerman A:
Endothelial dysfunction: A marker of atherosclerotic risk.
Arterioscler Thromb Vasc Biol. 23:168–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Gaspari T, Liu H, Welungoda I, Hu Y,
Widdop RE, Knudsen LB, Simpson RW and Dear AE: A GLP-1 receptor
agonist liraglutide inhibits endothelial cell dysfunction and
vascular adhesion molecule expression in an ApoE−/− mouse model.
Diab Vasc Dis Res. 8:117–124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Shiraki A, Oyama J, Komoda H, Asaka M,
Komatsu A, Sakuma M, Kodama K, Sakamoto Y, Kotooka N, Hirase T and
Node K: The glucagon-like peptide 1 analog liraglutide reduces
TNF-α-induced oxidative stress and inflammation in endothelial
cells. Atherosclerosis. 221:375–382. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Erdogdu Ö, Eriksson L, Nyström T, Sjöholm
Å and Zhang Q: Exendin-4 restores glucolipotoxicity-induced gene
expression in human coronary artery endothelial cells. Biochem
Biophys Res Commun. 419:790–795. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Steven S, Jurk K, Kopp M, Kröller-Schön S,
Mikhed Y, Schwierczek K, Roohani S, Kashani F, Oelze M, Klein T, et
al: Glucagon-like peptide-1 receptor signalling reduces
microvascular thrombosis, nitro-oxidative stress and platelet
activation in endotoxaemic mice. Br J Pharmacol. 174:1620–1632.
2017. View Article : Google Scholar
|
|
135
|
Gurjar AA, Kushwaha S, Chattopadhyay S,
Das N, Pal S, China SP, Kumar H, Trivedi AK, Guha R, Chattopadhyay
N and Sanyal S: Long acting GLP-1 analog liraglutide ameliorates
skeletal muscle atrophy in rodents. Metabolism. 103:1540442020.
View Article : Google Scholar
|
|
136
|
Hong Y, Lee JH, Jeong KW, Choi CS and Jun
HS: Amelioration of muscle wasting by glucagon-like peptide-1
receptor agonist in muscle atrophy. J Cachexia Sarcopenia Muscle.
10:903–918. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Bonaldo P and Sandri M: Cellular and
molecular mechanisms of muscle atrophy. Dis Model Mech. 6:25–39.
2013. View Article : Google Scholar :
|
|
138
|
Choi DH, Yang J and Kim YS: Rapamycin
suppresses postnatal muscle hypertrophy induced by
myostatin-inhibition accompanied by transcriptional suppression of
the Akt/mTOR pathway. Biochem Biophys Rep. 17:182–190.
2019.PubMed/NCBI
|
|
139
|
Lee SJ and McPherron AC: Regulation of
myostatin activity and muscle growth. Proc Natl Acad Sci USA.
98:9306–9311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Sartori R, Milan G, Patron M, Mammucari C,
Blaauw B, Abraham R and Sandri M: Smad2 and 3 transcription factors
control muscle mass in adulthood. Am J Physiol Cell Physiol.
296:C1248–C1257. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Amirouche A, Durieux AC, Banzet S,
Koulmann N, Bonnefoy R, Mouret C, Bigard X, Peinnequin A and
Freyssenet D: Down-regulation of Akt/mammalian target of rapamycin
signaling pathway in response to myostatin overexpression in
skeletal muscle. Endocrinology. 150:286–294. 2009. View Article : Google Scholar
|
|
142
|
Silveira WA, Gonçalves DA, Graça FA,
Andrade-Lopes AL, Bergantin LB, Zanon NM, Godinho RO, Kettelhut IC
and Navegantes LC: Activating cAMP/PKA signaling in skeletal muscle
suppresses the ubiquitin-proteasome-dependent proteolysis:
Implications for sympathetic regulation. J Appl Physiol (1985).
117:11–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Le Grand F and Rudnicki MA: Skeletal
muscle satellite cells and adult myogenesis. Curr Opin Cell Biol.
19:628–633. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Stewart R, Flechner L, Montminy M and
Berdeaux R: CREB is activated by muscle injury and promotes muscle
regeneration. PLoS One. 6:e247142011. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Fan D, Wang Y, Liu B and Yin F:
Hypoglycemic drug liraglutide alleviates low muscle mass by
inhibiting the expression of MuRF1 and MAFbx in diabetic muscle
atrophy. J Chin Med Assoc. 86:166–175. 2023.
|
|
146
|
Sandireddy R, Yerra VG, Areti A,
Komirishetty P and Kumar A: Neuroinflammation and oxidative stress
in diabetic neuropathy: Futuristic strategies based on these
targets. Int J Endocrinol. 2014:6749872014. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Ma J, Shi M, Zhang X, Liu X, Chen J, Zhang
R, Wang X and Zhang H: GLP-1R agonists ameliorate peripheral nerve
dysfunction and inflammation via p38 MAPK/NF-κB signaling pathways
in streptozotocin-induced diabetic rats. Int J Mol Med.
41:2977–2985. 2018.PubMed/NCBI
|
|
148
|
Krasner NM, Ido Y, Ruderman NB and
Cacicedo JM: Glucagon-like peptide-1 (GLP-1) analog liraglutide
inhibits endothelial cell inflammation through a calcium and AMPK
dependent mechanism. PLoS One. 9:e975542014. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Mitchell PD, Salter BM, Oliveria JP,
El-Gammal A, Tworek D, Smith SG, Sehmi R, Gauvreau GM, Butler M and
O'Byrne PM: Glucagon-like peptide-1 receptor expression on human
eosinophils and its regulation of eosinophil activation. Clin Exp
Allergy. 47:331–338. 2017. View Article : Google Scholar
|
|
150
|
Yanay O, Bailey AL, Kernan K, Zimmerman JJ
and Osborne WR: Effects of exendin-4, a glucagon like peptide-1
receptor agonist, on neutrophil count and inflammatory cytokines in
a rat model of endotoxemia. J Inflamm Res. 8:129–135. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Powers SK, Hudson MB, Nelson WB, Talbert
EE, Min K, Szeto HH, Kavazis AN and Smuder AJ:
Mitochondria-targeted antioxidants protect against mechanical
ventilation-induced diaphragm weakness. Crit Care Med.
39:1749–1759. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Romanello V, Guadagnin E, Gomes L, Roder
I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz
M, et al: Mitochondrial fission and remodelling contributes to
muscle atrophy. EMBO J. 29:1774–1785. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Luna-Marco C, Iannantuoni F, Hermo-Argibay
A, Devos D, Salazar JD, Víctor VM and Rovira-Llopis S:
Cardiovascular benefits of SGLT2 inhibitors and GLP-1 receptor
agonists through effects on mitochondrial function and oxidative
stress. Free Radic Biol Med. 213:19–35. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Xie S, Zhang M, Shi W, Xing Y, Huang Y,
Fang WX, Liu SQ, Chen MY, Zhang T, Chen S, et al: Long-term
activation of glucagon-like peptide-1 receptor by dulaglutide
prevents diabetic heart failure and metabolic remodeling in type 2
diabetes. J Am Heart Assoc. 11:e0267282022. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Helmstädter J, Frenis K, Filippou K, Grill
A, Dib M, Kalinovic S, Pawelke F, Kus K, Kröller-Schön S, Oelze M,
et al: Endothelial GLP-1 (glucagon-like peptide-1) receptor
mediates cardiovascular protection by liraglutide in mice with
experimental arterial hypertension. Arterioscler Thromb Vasc Biol.
40:145–158. 2020. View Article : Google Scholar :
|
|
156
|
Timper K, Del Río-Martín A, Cremer AL,
Bremser S, Alber J, Giavalisco P, Varela L, Heilinger C, Nolte H,
Trifunovic A, et al: GLP-1 receptor signaling in astrocytes
regulates fatty acid oxidation, mitochondrial integrity, and
function. Cell Metab. 31:1189–1205.e13. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Menconi M, Fareed M, O'Neal P, Poylin V,
Wei W and Hasselgren PO: Role of glucocorticoids in the molecular
regulation of muscle wasting. Crit Care Med. 35(9 Suppl):
S602–S608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Combaret L, Taillandier D, Dardevet D,
Béchet D, Rallière C, Claustre A, Grizard J and Attaix D:
Glucocorticoids regulate mRNA levels for subunits of the 19 S
regulatory complex of the 26 S proteasome in fast-twitch skeletal
muscles. Biochem J. 378:239–246. 2004. View Article : Google Scholar
|
|
159
|
Turturici G, Sconzo G and Geraci F: Hsp70
and its molecular role in nervous system diseases. Biochem Res Int.
2011:6181272011. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Petry ÉR, Dresch DF, Carvalho C, Medeiros
PC, Rosa TG, de Oliveira CM, Martins LAM, Schemitt E, Bona S, Guma
FCR, et al: Oral glutamine supplementation attenuates inflammation
and oxidative stress-mediated skeletal muscle protein content
degradation in immobilized rats: Role of 70 kDa heat shock protein.
Free Radic Biol Med. 145:87–102. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Hutchison KA, Dittmar KD, Stancato LF and
Pratt WB: Ability of various members of the hsp70 family of
chaperones to promote assembly of the glucocorticoid receptor into
a functional heterocomplex with hsp90. J Steroid Biochem Mol Biol.
58:251–258. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Senf SM, Dodd SL, McClung JM and Judge AR:
Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional
activities and prevents skeletal muscle atrophy. FASEB J.
22:3836–3845. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Sadek MA, Kandil EA, El Sayed NS, Sayed HM
and Rabie MA: Semaglutide, a novel glucagon-like peptide-1 agonist,
amends experimental autoimmune encephalomyelitis-induced multiple
sclerosis in mice: Involvement of the PI3K/Akt/GSK-3β pathway. Int
Immunopharmacol. 115:1096472023. View Article : Google Scholar
|
|
164
|
Ribeiro CB, Christofoletti DC, Pezolato
VA, de Cássia Marqueti Durigan R, Prestes J, Tibana RA, Pereira EC,
de Sousa Neto IV, Durigan JL and da Silva CA: Leucine minimizes
denervation-induced skeletal muscle atrophy of rats through
akt/mtor signaling pathways. Front Physiol. 6:732015. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Mohiuddin MS, Himeno T, Inoue R,
Miura-Yura E, Yamada Y, Nakai-Shimoda H, Asano S, Kato M, Motegi M,
Kondo M, et al: Glucagon-like peptide-1 receptor agonist protects
dorsal root ganglion neurons against oxidative insult. J Diabetes
Res. 2019:94260142019. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Bell KE, von Allmen MT, Devries MC and
Phillips SM: Muscle disuse as a pivotal problem in
sarcopenia-related muscle loss and dysfunction. J Frailty Aging.
5:33–41. 2016.PubMed/NCBI
|
|
167
|
Govers R: Molecular mechanisms of GLUT4
regulation in adipocytes. Diabetes Metab. 40:400–410. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Andreozzi F, Raciti GA, Nigro C, Mannino
GC, Procopio T, Davalli AM, Beguinot F, Sesti G, Miele C and Folli
F: The GLP-1 receptor agonists exenatide and liraglutide activate
glucose transport by an AMPK-dependent mechanism. J Transl Med.
14:2292016. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Purves T, Middlemas A, Agthong S, Jude EB,
Boulton AJ, Fernyhough P and Tomlinson DR: A role for
mitogen-activated protein kinases in the etiology of diabetic
neuropathy. FASEB J. 15:2508–2514. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Lluís F, Perdiguero E, Nebreda AR and
Muñoz-Cánoves P: Regulation of skeletal muscle gene expression by
p38 MAP kinases. Trends Cell Biol. 16:36–44. 2006. View Article : Google Scholar
|
|
171
|
Montessuit C, Rosenblatt-Velin N,
Papageorgiou I, Campos L, Pellieux C, Palma T and Lerch R:
Regulation of glucose transporter expression in cardiac myocytes:
p38 MAPK is a strong inducer of GLUT4. Cardiovasc Res. 64:94–104.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Guebre-Egziabher F, Alix PM, Koppe L,
Pelletier CC, Kalbacher E, Fouque D and Soulage CO: Ectopic lipid
accumulation: A potential cause for metabolic disturbances and a
contributor to the alteration of kidney function. Biochimie.
95:1971–1979. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Ruderman N and Prentki M: AMP kinase and
malonyl-CoA: Targets for therapy of the metabolic syndrome. Nat Rev
Drug Discov. 3:340–351. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Cao HY, Xu F, Chen ZL, Lin BS, Zheng XB,
Yuan SH, Liang H and Weng JP: Effect of exendin-4 on lipid
deposition in skeletal muscle of diet-induced obese mice and its
underlying mechanism. Zhonghua Yi Xue Za Zhi. 97:131–136. 2017.In
Chinese. PubMed/NCBI
|
|
175
|
Smits MM, Muskiet MH, Tonneijck L, Kramer
MH, Diamant M, van Raalte DH and Serné EH: GLP-1 receptor agonist
exenatide increases capillary perfusion independent of nitric oxide
in healthy overweight men. Arterioscler Thromb Vasc Biol.
35:1538–1543. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Montagnani M, Chen H, Barr VA and Quon MJ:
Insulin-stimulated activation of eNOS is independent of
Ca2+ but requires phosphorylation by Akt at Ser(1179). J
Biol Chem. 276:30392–30398. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Subaran SC, Sauder MA, Chai W, Jahn LA,
Fowler DE, Aylor KW, Basu A and Liu Z: GLP-1 at physiological
concentrations recruits skeletal and cardiac muscle
microvasculature in healthy humans. Clin Sci (Lond). 127:163–170.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Chai W, Dong Z, Wang N, Wang W, Tao L, Cao
W and Liu Z: Glucagon-like peptide 1 recruits microvasculature and
increases glucose use in muscle via a nitric oxide-dependent
mechanism. Diabetes. 61:888–896. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Tobaiqy M: A review of serious adverse
events linked with GLP-1 agonists in type 2 diabetes mellitus and
obesity treatment. Pharmacol Rep. 76:981–990. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Wilding JPH, Batterham RL, Calanna S,
Davies M, Van Gaal LF, Lingvay I, McGowan BM, Rosenstock J, Tran
MTD, Wadden TA, et al: Once-weekly semaglutide in adults with
overweight or obesity. N Engl J Med. 384:989–1002. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Huang HH, Wang YJ, Jiang HY, Yu HW, Chen
YQ, Chiou A and Kuo JC: Sarcopenia-related changes in serum GLP-1
level affect myogenic differentiation. J Cachexia Sarcopenia
Muscle. 15:1708–1721. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Sargeant JA, Henson J, King JA, Yates T,
Khunti K and Davies MJ: A review of the effects of glucagon-like
peptide-1 receptor agonists and sodium-glucose cotransporter 2
inhibitors on lean body mass in humans. Endocrinol Metab (Seoul).
34:247–262. 2019. View Article : Google Scholar : PubMed/NCBI
|