Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of
the deadliest of all solid malignant tumors and the fourth leading
cause of cancer death in the Western world, with an overall 5-year
survival rate of only 6% (1). One
reason for this poor prognosis is the propensity of PDAC cells to
invade adjacent tissues and to metastasize, even during the early
stage (2). PDAC often harbors
multiple molecular alterations in cancer cells, including
activating KRAS mutations and loss-of-function mutations in
the P16/CDKN2A, TP53, and SMAD4/DPC4 genes (2). Mutations in these four genes are
recognized as ‘driver mutations’ in PDAC because they drive
neoplastic transformation and tumor progression (3). A high percentage of PDACs also
overexpress a number of growth factors and their receptors,
including epidermal growth factor (EGF), EGF receptor (EGFR), human
epidermal growth factor (HER)-2/c-erbB2, transforming growth factor
(TGF)-α, CRIPTO, TGF-β1, vascular endothelial growth factor (VEGF),
basic fibroblast growth factor (bFGF/FGF-2), acidic FGF
(aFGF/FGF-1), FGF-5, FGF-7 [also known as keratinocyte growth
factor (KGF)], and KGF receptor (KGFR)/FGFR2IIIb (4–11).
The multiple stepwise alterations in oncogenes and tumor suppressor
genes, in conjunction with the overexpression of mitogenic growth
factors and their receptors, may contribute to the formation of
precancerous lesions in the pancreas (PanIN) and the biological
aggressiveness of PDAC (2,12,13).
KGF is a member of the FGF group of heparin-binding
polypeptides, which was initially identified in human embryonic
lung fibroblasts (14,15). KGF is synthesized by mesenchymal
cells and T lymphocytes, and acts predominantly on epithelial cells
in a paracrine manner (16). KGF
is expressed in a variety of tissues, including the lung, prostate,
mammary gland, digestive tract, bladder, and skin, and has been
implicated in organ development and homeostasis (16). KGF also stimulates the growth of
the gastrointestinal tract mucosa, and KGF-expressing transgenes
exhibit pancreatic ductal hyperplasia (17,18).
KGF mRNA levels have been substantially higher in pancreatic and
colorectal cancer specimens than in the corresponding normal
tissues (11,19).
KGF binds to a specific cell-surface receptor, KGFR,
also known as the FGFR2 IIIb isoform (20,21).
KGFR and FGFR2IIIc are splicing variants of the FGFR2 gene, and
they differ from each other in the carboxy-terminal half of the
third immunoglobulin-like region of the extracellular domain
(22). KGFR is localized in
epithelial cells, while FGFR2 IIIc is mainly localized in
mesenchymal cells. KGFR and FGFR2 IIIc exhibit different
ligand-binding specificities. FGF-1, -3, -7, -10, and -22
reportedly bind to FGFR2 IIIb with high affinity, while FGF-1, -2,
-4, -6, -9, -17, and -18 bind to FGFR2 IIIc with high affinity
(20,21). KGFR mRNA is expressed in many
organs, including breast, colon, stomach and esophagus, pancreas,
prostate, oral mucosa, and uterus (23,24).
Loss of FGFR2 IIIb expression has been associated with the
activation of FGFR2 IIIc expression, and/or a shift to more
virulent behavior (25).
Degradation of basement membranes and extracellular
matrices is an essential process in the invasion and metastasis of
malignant tumors. Matrix metalloproteinases (MMPs), are potent
proteolytic enzymes that play key roles in this process (26). One of the first steps of cancer
invasion is basement membrane degradation. MMP-2 (gelatinase A) and
MMP-9 (gelatinase B) play important roles in destroying the
basement membrane of tumor vessels, and their expression correlates
with metastasis (27). MMP-9
cleaves type IV collagen and gelatin, which are the principal
structural components of the vascular basement membrane. Expression
of MMP-9 has been reported in breast, colon, and lung cancers, as
well as skin tumors, and the expression of both MMP-2 and MMP-9 has
been correlated with local invasion of the tumor, lymph node
metastasis, and survival rates (28). In PDAC, MMP-9 expression is
correlated with lymph node involvement and distant metastasis
(29). Furthermore, MMP-9
expression is reportedly correlated with (or tends to correlate
with) shorter overall survival in PDAC patients (30,31).
We previously reported that the co-expression of KGF
and KGFR in PDAC is associated with venous invasion and poor
prognosis (32). Although a
previous report has shown that VEGF-A expression is closely
involved in the KGF/KGFR pathway in PDAC, there have been no
reports about the relation between KGF/KGFR and MMPs (32). In this study, we investigated the
expression and roles of KGF/KGFR and MMP-9 in human PDAC cell lines
and tissues. We now report that the expression of KGF/KGFR and
MMP-9 is correlated with venous invasion, and that MMP-9 expression
is regulated by KGF/KGFR. The KGF/KGFR pathway might be a potent
therapeutic target for PDAC metastasis.
Materials and methods
Materials
The following were purchased: Isogen from Nippon
Gene (Tokyo, Japan); a Takara RNA PCR kit (AMV) Ver. 3.0 and
pBAsi-hU6 Neo DNA vector from Takara Biotech. (Tokyo, Japan);
RNeasy mini kit from Qiagen GmbH (Hilden, Germany); Transcriptor
First Strand cDNA Synthesis kit and LightCycler FastStart DNA
Master SYBR Green I, FuGENE HD transfection reagent from Roche
Diagnostics GmbH (Mannheim, Germany); goat polyclonal anti-KGF
antibodies and recombinant human KGF (rhKGF) from R&D Systems
Inc. (Westerville, OH); a Histofine Simple Stain Max PO (G), (R),
or (M) kit from Nichirei Biosciences, Inc. (Tokyo, Japan); mouse
monoclonal anti-MMP-9 antibodies from Daiichi Fine Chemical Co.,
Ltd. (Toyama, Japan); Human Tissue Microarray 1 and Human Digestive
Tissue Sets from Novagen (Darmstadt, Germany); Silane-coated slides
and malinol mounting medium from Muto Pure Chemicals Co., Ltd.
(Tokyo, Japan); Transwell permeable supports from Life Sciences
(Acton, MA); Matrigel from BD Biosciences (Franklin Lakes, NJ). All
other chemicals and reagents were purchased from Sigma Chemical
Corp. (St. Louis, MO).
Patients and tissues
Tissues from 63 patients with invasive PDAC were
obtained for this study. These patients received treatment at
Nippon Medical School Hospital (Tokyo, Japan) from 1995 to 2004.
None of the patients received preoperative chemotherapy or
radiotherapy. The patients consisted of 40 males and 23 females,
whose median age was 63, range, 35–84 years. The clinicopathologic
stage was determined according to the TNM classification system of
the International Union Against Cancer (UICC), and additionally
characterized using the Japan Pancreas Society classification
(Table I). Thirty-two patients did
not receive postoperative chemotherapy, and 31 patients received
adjuvant chemotherapy after surgery. Thirteen patients received
Uracil/Tegafur (UFT) and 18 patients received gemcitabine (GEM).
The median follow-up period was 14.7 months. This study was
conducted in accordance with the principles embodied in the 2008
Declaration of Helsinki, and informed consent for the usage of
pancreatic tissues was obtained from each patient. Normal
pancreatic tissues were obtained from human digestive tissue sets
and Human Tissue Microarray 1.
 | Table ICorrelation of clinicopathological
features and KGF, KGFR, MMP-9 or co-expression of KGF and KGFR in
pancreatic cancers. |
Table I
Correlation of clinicopathological
features and KGF, KGFR, MMP-9 or co-expression of KGF and KGFR in
pancreatic cancers.
| | KGF | KGFR | MMP-9 | KGF and KGFR |
|---|
| |
|
|
|
|
|---|
| Variables | No. | No. (%) | P-value | No. (%) | P-value | No. (%) | P-value | No. (%) | P-value |
|---|
| Gender | | | | | | | | | |
| Male | 40 | 16 (40) | NS | 18 (45) | NS | 21 (53) | NS | 10 (25) | NS |
| Female | 23 | 11 (48) | | 5 (22) | | 14 (61) | | 4 (17) | |
| Age | | | | | | | | | |
| <65 | 30 | 13 (43) | NS | 9 (30) | NS | 16 (53) | NS | 4 (13) | NS |
| ≥65 | 33 | 14 (42) | | 14 (42) | | 19 (58) | | 10 (30) | |
| UICC
classification | | | | | | | | | |
| T-primary
tumor | | | | | | | | | |
| T1 | 4 | 2 (50) | NS | 2 (50) | NS | 2 (50) | NS | 1 (25) | NS |
| T2 | 4 | 1 (25) | | 1 (25) | | 2 (50) | | 1 (25) | |
| T3 | 18 | 11 (61) | | 7 (39) | | 11 (61) | | 5 (28) | |
| T4 | 37 | 13 (35) | | 13 (35) | | 20 (54) | | 7 (19) | |
| N-regional lymph
nodes | | | | | | | | | |
| N0 | 23 | 12 (52) | NS | 11 (48) | NS | 15 (65) | NS | 6 (26) | NS |
| N1 | 40 | 15 (38) | | 12 (30) | | 20 (50) | | 8 (20) | |
| M-distant
metastasis | | | | | | | | | |
| M0 | 61 | 26 (43) | NS | 22 (36) | NS | 34 (56) | NS | 13 (21) | NS |
| M1 | 2 | 1 (50) | | 1 (50) | | 1 (50) | | 1 (50) | |
| G-histological
grading | | | | | | | | | |
| G1 | 34 | 11 (32) | NS | 14 (41) | NS | 20 (59) | NS | 6 (18) | NS |
| G2 | 26 | 13 (50) | | 8 (31) | | 12 (46) | | 7 (27) | |
| G3 | 3 | 3 (100) | | 1 (33) | | 3 (100) | | 1 (33) | |
| G4 | 0 | 0 | | 0 | | 0 | | 0 | |
| Stage | | | | | | | | | |
| I or II | 11 | 6 (55) | NS | 5 (45) | NS | 5 (45) | NS | 4 (36) | NS |
| III or IV | 52 | 21 (40) | | 18 (35) | | 30 (58) | | 10 (19) | |
| Other tumor
characteristics | | | | | | | | | |
| Lymphatic
invasion | | | | | | | | | |
| Negative | 8 | 4 (50) | NS | 3 (38) | NS | 5 (63) | NS | 2 (25) | NS |
| Positive | 55 | 23 (42) | | 20 (36) | | 30 (55) | | 12 (22) | |
| Venous
invasion | | | | | | | | | |
| Negative | 40 | 12 (30) | 0.0065 | 11 (28) | 0.05 | 19 (48) | 0.0082 | 5 (13) | 0.014 |
| Positive | 23 | 15 (65) | | 12 (52) | | 16 (70) | | 9 (39) | |
| Nerve invasion
(intrapancreatic) | | | | | | | | | |
| Negative | 14 | 8 (57) | NS | 6 (43) | NS | 9 (64) | NS | 3 (21) | NS |
| Positive | 49 | 19 (39) | | 17 (35) | | 26 (53) | | 11 (22) | |
Human PDAC cell lines
PANC-1, MIA PaCa-2, KLM-1, PK-1, PK-8, PK-9 and
PK-59 PDAC cell lines were obtained from the Cell Resource Center
for Biomedical Research, Institute of Development, Aging and
Cancer, Tohoku University (Sendai, Japan), and Capan-1 was
purchased from American Type Culture Collection (ATCC). The cells
were grown in RPMI-1640 medium containing 10% fetal bovine serum
(FBS), 200 U/ml penicillin, and 200 μg/ml kanamycin at 37°C under a
humidified 5% CO2 atmosphere. Capan-1 was grown in the
same medium containing 15% FBS.
Quantitative real-time PCR analysis
Total RNA extraction from tumor cells was performed
using the RNeasy Mini kit. cDNA synthesis was performed using the
Transcriptor First Strand cDNA Synthesis kit following the
manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was
performed using a LightCycler-FastStart DNA Master SYBR Green I
system. The primers used for MMP-9 corresponded to nucleotides
192–214 (5′-CAG-AGA-TGC-GTG-GAG-AGT-CGA-AA-3′) and nucleotides
426–445 (5′-GGC-AAA-GGC-GTC-GTC-AAT-CA-3′) of the human MMP-9 cDNA
(254 bp, NM_004994). The primers used for 18S rRNA (RS-18)
corresponded to nucleotides 184–207
(5′-AAA-GCA-GAC-ATT-GAC-CTC-ACC-AAG-3′) and nucleotides 319–341
(5′-AGG-ACC-TGG-CTG-TAT-TTT-CCA-TC-3′) of the human RS-18 cDNA (158
bp, NM_022551). PCR reaction mixture contaning 2 μl of template
cDNA, 3 mM MgCl2, 0.5 μM primers, and
LightCycler-FastStart DNA Master SYBR Green I mix was applied to
the capillary tube (Roche). qRT-PCR was conducted in a LightCycler
(Roche) and the PCR products were analyzed using LightCycler Data
Analysis software version 3.5 (Roche). The optimized program
involved denaturation at 95°C for 10 min, followed by 45 cycles of
amplification: 95°C for 10 sec, 60°C for 10 sec, and 72°C for 10
sec for MMP-9; and 95°C for 10 sec, 65°C for 10 sec, and 72°C for 7
sec for RS-18. To confirm amplification specificity, PCR products
were subjected to a melting curve analysis. Results were expressed
as MMP-9/RS-18, as an internal standard concentration ratio. Each
experiment was performed twice, and gene expression measurements
were performed in triplicate.
Immunohistochemistry
Paraffin-embedded tissue sections (3.5 μm) were
subjected to immunostaining using the Histofine Simple Stain Max PO
(G), (M), or (R) kit. After deparaffinization, endogenous
peroxidase activity was blocked by incubation with 0.3% hydrogen
peroxide in methanol for 30 min, and the sections were incubated
with the appropriate antibody overnight at 4°C (1:50 dilution for
the anti-KGF antibody, 1:1000 dilution for the anti-KGFR antibody,
and 1:50 dilution for the anti-MMP-9 antibody) using PBS containing
1% BSA. The anti-KGFR antibody used in this study was an
affinity-purified rabbit polyclonal antibody raised against a
peptide corresponding to an amino acid sequence from the human KGFR
protein (32). Bound antibodies
were detected with Simple Stain Max PO (G), (M), or (R) reagents
using diaminobenzidine tetrahydrochloride (DAB) as the substrate,
and the sections were counterstained with Mayer’s hematoxylin.
Negative control studies were performed by omitting the primary
antibodies. The immunohistochemical results for KGF, KGFR and MMP-9
were evaluated as follows: when staining was noted in the cytoplasm
and/or membrane of more than 30% of the tumor cells, regardless of
the intensity of staining, the cells were designated positive
(32). Two investigators (K.C. and
T.I.) separately evaluated all specimens in a blinded manner.
Effects of rhKGF on MMP-9 mRNA
expression
MIA PaCa-2 cells (1×105/well) plated in
6-well plates were grown in 2 ml of RPMI-1640 medium with 10% FBS
for 24 h, and then cultured in serum-free medium for 48 h.
Subsequently, the cells were cultured in serum-free RPMI-1640
medium in the absence or presence of 10 ng/ml rhKGF for 3 h.
Transient transfection of short hairpin
RNA (shRNA) for KGF
The shRNA for KGF (KGF shRNA) and negative control
shRNA were obtained as previously described (32). Twenty-four hours before
transfection, KLM-1 cells (2×105/well) were seeded in
6-well plates and grown in 2 ml of RPMI-1640 medium with 10% FBS.
Transfection of KGF shRNA and control shRNA were performed using
FuGENE HD transfection reagent, according to the manufacturer’s
instructions. The medium from KLM-1 cells was replaced with a
serum-free medium 48 h after transfection, and cells were incubated
for an additional 48 h. The mRNA levels for KGF and MMP-9 in KLM-1
cells were measured by qRT-PCR. All shRNA experiments were
conducted twice, with triplicate determinations for each
experiment.
Cell migration and invasion assay
The cell migration assay was performed in 24-well
plates using Transwell permeable supports; pore size was 8.0 μm and
pore density was 1×105 pores per cm2. Cells
(1×105) were resuspended in serum-free medium. The cell
suspension (500 μl) was placed in the upper compartment, and the
lower compartment was immediately filled with 750 μl of RPMI-1640
medium containing 5% FBS and 0 or 20 ng/ml rhKGF. Migrating cells
were fixed in 50% methanol and stained with 0.05% crystal violet.
The membranes were mounted on glass slides and manually counted in
nine different fields under a light microscope at 400×
magnification. Each experiment was performed in triplicate.
Invasion assays were conducted using Matrigel-coated
Transwell permeable supports. Subsequently, 1×105 cells
were resuspended in serum-free medium. The cell suspension (500 μl)
was placed in the upper compartment, and the lower compartment was
immediately filled with 750 μl of RPMI-1640 medium containing 5%
FBS and 0 or 20 ng/ml rhKGF. After 24 h of incubation, the
non-invading cells were removed from the upper surface of the
separating membrane by gentle scrubbing with a cotton swab. The
incubation, staining, counting, and photography procedures were
performed as for the migration assay. Each experiment was performed
in triplicate.
Statistical analysis
Whenever indicated, the χ2 test and
Fisher’s exact test were used to analyze the correlation between
KGFR, KGF, or MMP-9 expression and clinicopathologic features.
Cumulative survival rate was calculated using the Kaplan-Meier
method, and the significance of differences in survival rate was
analyzed using the log-rank test; p<0.05 was considered
significant in all analyses. Computations were performed using the
StatView J software package, version 5.0 (SAS Institute, Inc.,
Cary, NC, USA).
Results
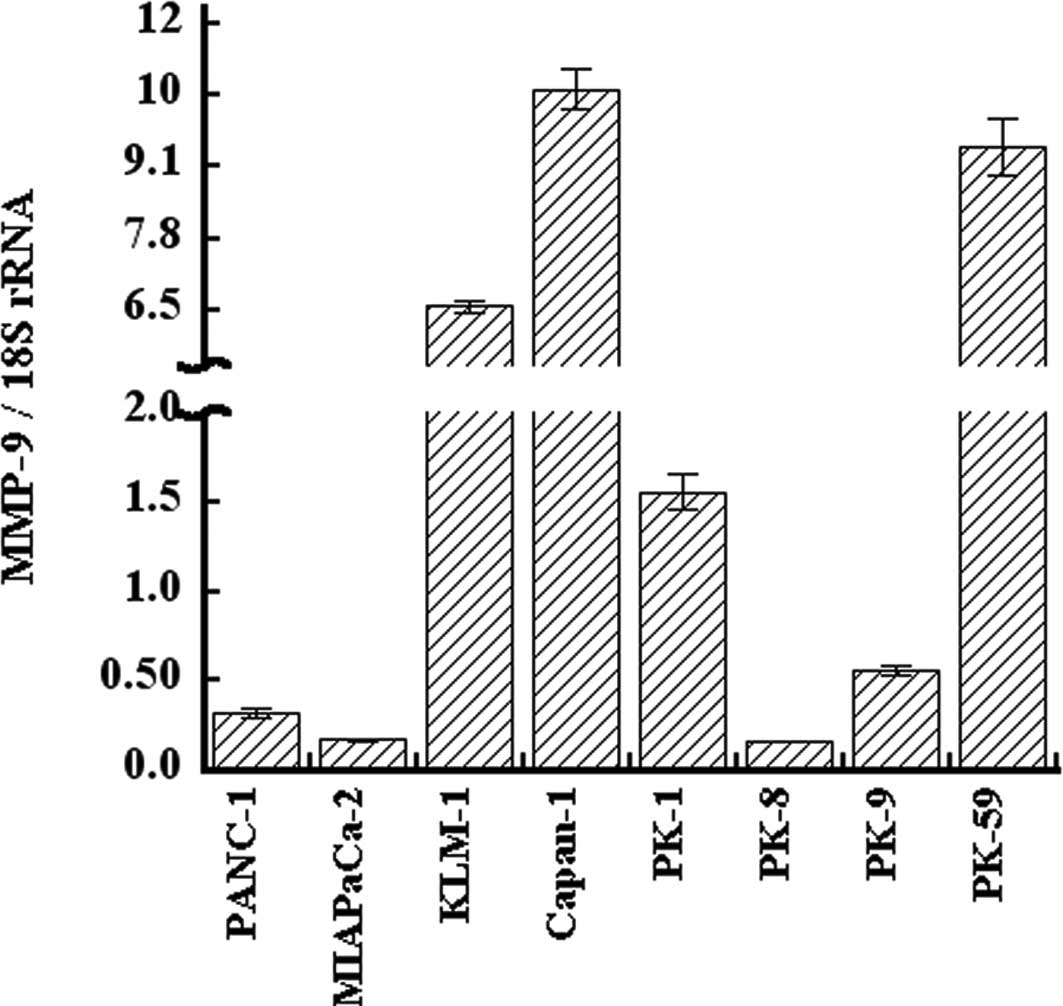
qRT-PCR analysis of MMP-9 in PDAC cell
lines
Expression of MMP-9 was examined in PANC-1, MIA
PaCa-2, KLM-1, Capan-1, PK-1, PK-8, PK-9, and PK-59 cell lines.
MMP-9 mRNA was expressed in all eight PDAC cell lines at varying
levels. The expression level of MMP-9 mRNA was high in Capan-1,
PK-59, and KLM-1 cells, and low in PK-8, MIA PaCa-2, and PANC-1
cells (Fig. 1). The expression
level of MMP-9 mRNA in Capan-1 was 66-fold higher than that of
PK-8. KGF expression was also high in KLM-1 and Capan-1 cells, and
low in MIA PaCa-2 and PANC-1 cells, as previously reported
(32). Furthermore, KGFR
expression was high in KLM-1 cells and low in MIA PaCa-2 and PANC-1
cells.
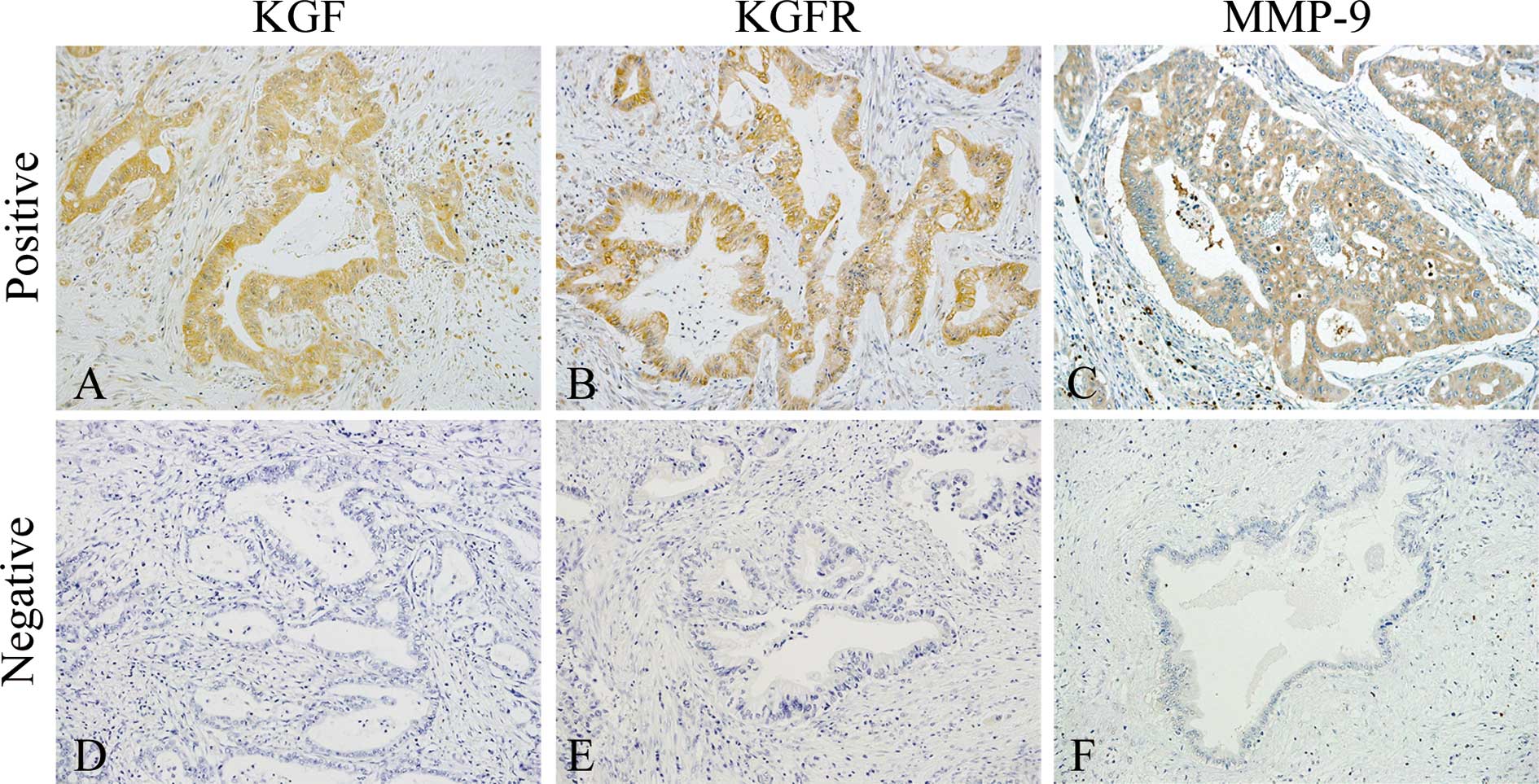
Immunohistochemical analysis of KGF,
KGFR, and MMP-9 in PDAC tissues
Immunohistochemical analysis of the PDAC samples was
conducted next to determine whether there was a correlation between
cellular KGF, KGFR, and MMP-9 expression and clinicopathological
features. KGF was localized in the cytoplasm of cancer cells in 27
of 63 (42.9%) patients (Fig. 2A),
while KGFR immunoreactivity was detected in the cytoplasm and/or
cell membrane of cancer cells in 23 of 63 (36.5%) patients
(Fig. 2B). There was a
statistically significant correlation between the presence of
venous invasion and either KGF (p=0.0065) or KGFR (p=0.050)
immunoreactivity (Table I).
Moreover, concomitant expression of KGF and KGFR was observed in 14
of 63 (22.2%) samples, and this concomitant expression also
correlated with venous invasion (p=0.014, Table I). These results were consistent
with our previous report (in a smaller number of cases) that
KGF/KGFR expression in human PDAC tissues correlated with venous
invasion (32).
We next sought to determine whether KGF/KGFR
correlated with MMP-9 expression in human PDAC tissues, because
MMP-9 is known to participate in the degradation of type IV
collagen, which is a major component of vascular basement
membranes. In the normal pancreas, MMP-9 was occasionally detected
in the cytoplasm of pancreatic ductal cells, islet cells, and
acinar cells, as previously reported (30) (data not shown). In the PDAC
samples, MMP-9 immunoreactivity was observed in the cytoplasm of
cancer cells in 35 of 63 (55.6%) patients (Fig. 2C). The presence of MMP-9 in cancer
cells correlated with venous invasion (p=0.0082, Table I). Moreover, there was a
statistically significant correlation between the presence of liver
metastasis and either KGF (p=0.00030) or MMP-9 (p=0.022)
immunoreactivity (Table II). KGFR
expression in PDAC cells did not correlate with liver
metastasis.
| Table IICorrelation of liver metastasis with
KGF and MMP-9 expression. |
Table II
Correlation of liver metastasis with
KGF and MMP-9 expression.
| Liver
metastasis | P-value |
|---|
| Negative
(n=32) | Positive
(n=28) |
|---|
| KGF expression |
| Negative
(n=36) | 26 | 10 | 0.0003 |
| Positive
(n=24) | 6 | 18 | |
| MMP-9
expression |
| Negative
(n=33) | 22 | 11 | 0.022 |
| Positive
(n=27) | 10 | 17 | |
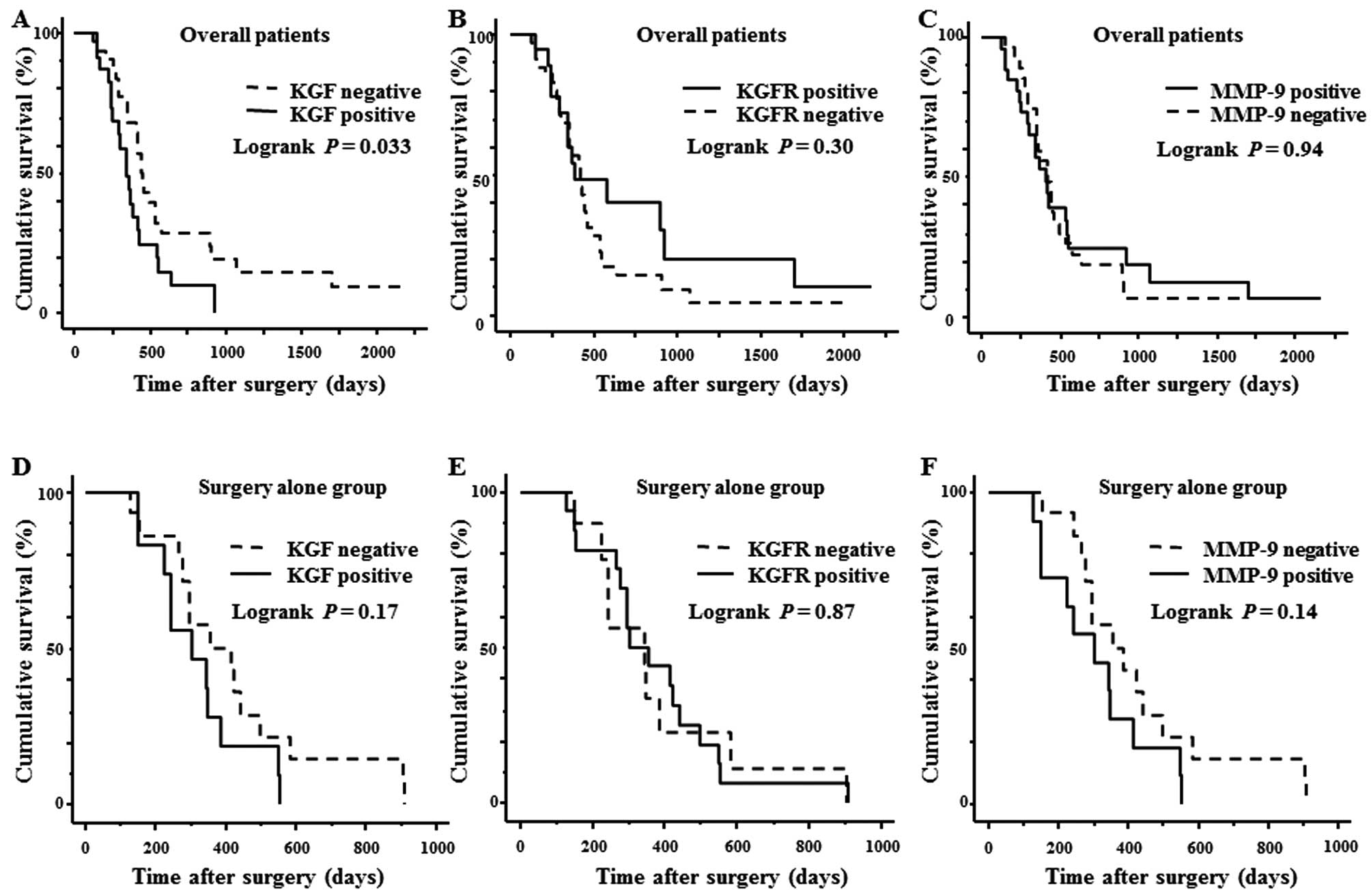
Cumulative Kaplan-Meier survival curve
and multivariate analysis
The overall 2-year survival rate of all 63 PDAC
cases was 15.9%. The survival duration of the KGF-positive group
was significantly shorter than that of the KGF-negative group
(p=0.033; Fig. 3A). The survival
rates of the KGFR-positive group and KGFR-negative group were not
significantly different (p=0.30; Fig.
3B). Likewise, the survival rates of PDAC patients whose cancer
cells were positive for MMP-9 and negative for MMP-9 did not differ
significantly (p=0.94; Fig. 3C).
Next, the survival rates of patients who had surgery but did not
receive any adjuvant chemotherapy were analyzed in a similar
manner. In these patients, there was no statistically significant
difference between the KGF-, KGFR-, or MMP-9-positive groups and
the corresponding negative groups (Fig. 3D-F).
Effect of KGF on MMP-9 expression in PDAC
cells
Immuno-staining and survival results pointed to a
close relationship between the KGF/KGFR pathway and MMP-9.
Therefore, two different types of experiments were performed to
assess the relationship between the KGF/KGFR pathway and MMP-9. In
the first set of experiments, MIA PaCa-2 cells, which expressed
KGFR and negligible levels of KGF, were incubated for 3 h in the
absence or presence of rhKGF, and MMP-9 mRNA levels were determined
by qRT-PCR. rhKGF caused a significant increase in MMP-9 mRNA
levels (p<0.05; Fig. 4A),
indicating that exogenous KGF effectively induces MMP-9 expression
in PDAC cells.
Next, to establish a better link between KGF and
MMP-9 expression, we conducted experiments using KGF shRNA. KGF
shRNA and control shRNA were transfected into KLM-1 cells, which
expressed the highest KGF levels among all the tested PDAC cell
lines (32). qRT-PCR revealed that
KGF shRNA significantly reduced KGF and MMP-9 mRNA levels in KLM-1
cells compared with control shRNA (p<0.001 and p<0.05,
respectively; Fig. 4B). These
results demonstrate that a KGF/KGFR signaling pathway is implicated
in MMP-9 regulation.
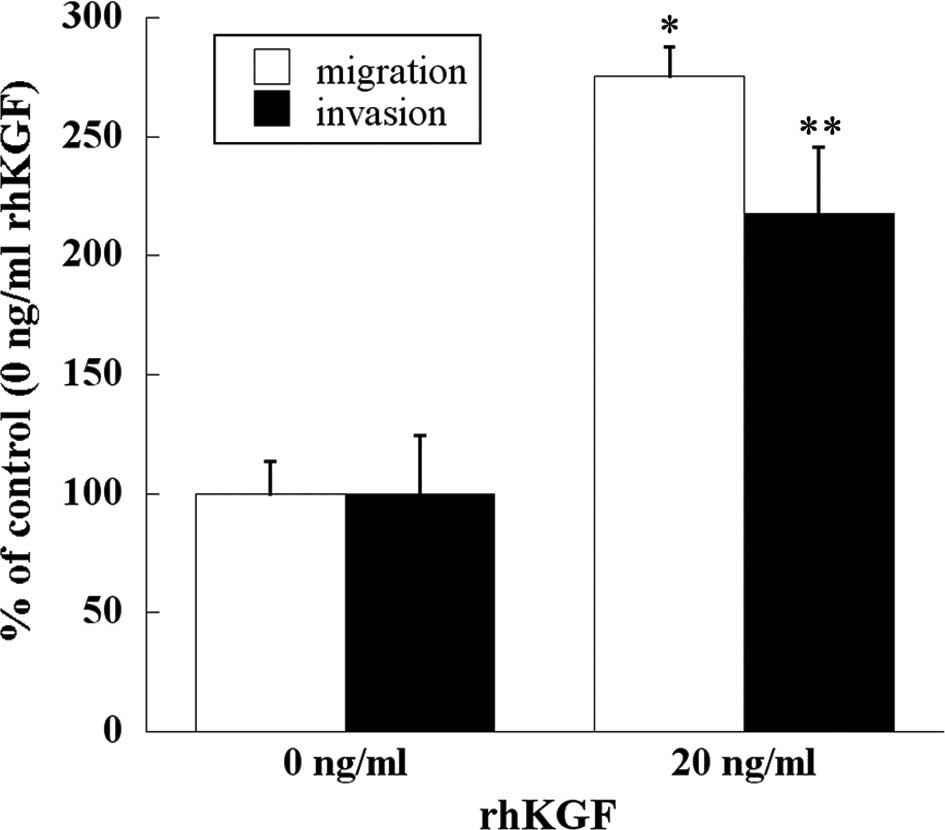
Effect of KGF on migration and invasion
of human PDAC cells
Migration and invasion assays were performed to
examine whether KGF stimulates the migration and invasion of human
PDAC cells. Addition of rhKGF significantly enhanced migration and
invasion through the extracellular matrices of MIA PaCa-2 cells
(p<0.001 and p<0.05, respectively; Fig. 5).
Discussion
The strong correlations between MMPs at the protein
and mRNA levels and the malignancies of various human cancers have
been reported in a number of recent papers (33–37).
The presence or elevated expression of MMPs (including MMP-1, -2,
-3, -7, -9, -13, and -14) in both primary tumors and/or metastases
are positively associated with tumor progression, poor tumor grade,
invasive stage of cancer, poor prognosis, metastasis to secondary
organs, and shorter survival time (26).
MMP-2 and MMP-9 degrade type IV collagen, and have
important roles in the vascular invasion of cancer cells (27). MMP-2 and MMP-9 expression have each
been reported in PDAC tissues (38). MMP-9 mRNA was expressed in cancer
epithelial cells and stromal cells, while MMP-2 mRNA was
predominantly expressed in tumor stromal cells (39). We also examined
immunohistochemically the expression of MMP-2 in PDAC tissues.
Consistent with previous reports, MMP-2 was mainly localized in
stromal tissues adjacent to PDAC cells, but was not localized or
weakly localized in PDAC cells. Therefore, we focused on MMP-9 to
clarify the relationship between the KGF/KGFR pathway and MMPs in
PDAC cells. In the present study, we observed MMP-9 expression at
varying levels in PDAC cell lines. KLM-1 cells were established
from PK-1 cells as a highly metastatic variant, and the KLM-1 cells
metastasize more often than PK-1 cells to the liver of nude mice
(40). Notably, MMP-9 expression
was higher in KLM-1 cells than in parental PK-1 cells in this
study. Furthermore, Capan-1 cells exhibited extremely high MMP-9
levels, and the cells originate from liver metastases in PDAC
patients.
In the present study, KGF, KGFR, co-expression of
KGF/KGFR, and MMP-9 expression in PDAC tissues are correlated with
venous invasion; however, they do not correlate with all other
clinicopathological factors. Positive correlation between venous
invasion and KGF, KGFR, and co-expression of KGF and KGFR was
consistent with our previous study, which contained a smaller
number of patients (32). The
evidence that expression of KGF and MMP-9 was correlated with liver
metastasis may indicate that the KGF/KGFR pathway induces MMP-9
expression, and MMP-9 in turn destroys the basement membrane of
tumor vessels to lead to liver metastasis.
To confirm this hypothesis, we examined the effect
of KGF on MMP-9 expression using two sets of experiments. First, we
exogenously added rhKGF and examined the MMP-9 expression levels in
MIA PaCa-2 cells, which express negligible levels of endogenous
KGF, but express KGFR. Next, we transfected shRNA-targeting KGF
transcripts into KLM-1 cells, which express high levels of KGF and
KGFR. Recombinant KGF induced MMP-9 mRNA expression; however, KGF
shRNA reduced MMP-9 expression in PDAC cells. These findings
suggest that KGF regulates the MMP-9 expression level in PDAC
cells. MMPs are considered to be regulated by a variety of
cytokines, growth factors, steroid hormones, and phorbol esters
(41). However, the
transcriptional activation mechanisms underlying this regulation
have not been fully understood. Interleukin (IL)-1α, IL-1β, IL-8,
TGF β-1, and tumor necrosis factor reportedly induce MMP expression
(28). Growth factors implicated
in stimulating MMP regulation include basic fibroblast growth
factor (bFGF), EGF, and VEGF (42–44).
KGF induced MMP-9 expression through NF-kappaB in
immortalized/non-tumorigenic human pancreatic ductal epithelial
cells (40). To our knowledge,
this is a first report that KGF induces MMP-9 expression in PDAC
cells. That KGF also stimulates VEGF-A expression in PDAC may
suggest that KGF is a strong inducer of tumor vascular angiogenesis
and simultaneously stimulates migration and the destruction of
incomplete tumor vessels, through the induction of MMP-9 (32,45).
In the present study, administration of recombinant KGF to MIA
PaCa-2 cells resulted in increased cell migration and invasion
through extracellular matrices. These lines of evidence suggest
that the KGF/KGFR pathway is centrally involved in vascular
invasion and metastases in PDAC.
The relationship between MMP-9 and the prognosis of
PDAC patients has been controversial. Some groups have reported no
correlation between MMP-9 expression and PDAC prognosis, while
others have shown a correlation between MMP-9 expression and poor
prognosis (30,31,39,46,47).
Furthermore, the expression and roles of KGF/KGFR have not been
well clarified. In prostate and bladder cancers, the expression of
FGFR2IIIc, another splicing isoform of FGFR2, correlated with more
malignant phenotype, compared with KGFR (48,49).
Class switching from KGFR to FGFR2IIIc occurs during the metastatic
process in prostate cancer. In contrast, KGF/KGFR was closely
correlated with venous invasion and liver metastasis in PDAC. Our
results indicate that KGF-induced VEGF-A and MMP-9 expression
through KGFR might be involved in vascular invasion, liver
metastasis, and poor PDAC prognosis. The expression and alteration
of other FGFs and/or FGFRs might be involved in discrepancies in
the roles of KGF/KGFR in cancer metastasis. Further examination
with larger patient populations and additional assessment of other
forms of FGFs and FGFRs are needed to clarify the roles of the
KGF/KGFR pathway in PDAC metastasis.
In summary, KGF induced MMP-9 expression in
KGFR-positive PDAC cells, and KGF and MMP-9 expression were each
closely correlated with vascular invasion and liver metastasis of
PDAC. The KGF/KGFR pathway may be a novel therapeutic target for
PDAC metastasis.
Acknowledgements
We express our appreciation to Drs. Masahito Hagio
and Tetsushi Yamamoto, Mr. Takenori Fujii, Ms. Taeko Suzuki and Ms.
Kiyoko Kawahara (Departments of Pathology and Integrative
Oncological Pathology, Nippon Medical School) for their excellent
technical assistance. We thank Dr Shin-Ichi Tsuchiya (Division of
Surgical Pathology, Nippon Medical School Hospital) for preparing
tissue blocks. This study was supported by a Grant-in-Aid for Young
Scientists from the Japan Society for the Promotion of Science to
Y. Matsuda (A, no. 22689038), a Grant-in Aid for Challenging
Exploratory Research from the Japan Society for the Promotion of
Science to Y. Matsuda (no. 23650604), and Grants-in Aid for
Scientific Research from the Japan Society for the Promotion of
Science to T. Ishiwata (C, no. 22591531) and Z. Naito (C, no.
23590477).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
2
|
Bardeesy N and DePinho RA: Pancreatic
cancer biology and genetics. Nat Rev Cancer. 2:897–909. 2002.
View Article : Google Scholar
|
|
3
|
Korc M: Driver mutations: a roadmap for
getting close and personal in pancreatic cancer. Cancer Biol Ther.
10:588–591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Itakura J, Ishiwata T, Friess H, et al:
Enhanced expression of vascular endothelial growth factor in human
pancreatic cancer correlates with local disease progression. Clin
Cancer Res. 3:1309–1316. 1997.PubMed/NCBI
|
|
5
|
Yamanaka Y: The immunohistochemical
expressions of epidermal growth factors, epidermal growth factor
receptors and c-erbB-2 oncoprotein in human pancreatic cancer.
Nihon Ika Daigaku Zasshi. 59:51–61. 1992.
|
|
6
|
Kornmann M, Ishiwata T, Beger HG and Korc
M: Fibroblast growth factor-5 stimulates mitogenic signaling and is
overexpressed in human pancreatic cancer: evidence for autocrine
and paracrine actions. Oncogene. 15:1417–1424. 1997. View Article : Google Scholar
|
|
7
|
Friess H, Yamanaka Y, Buchler M, Kobrin
MS, Tahara E and Korc M: Cripto, a member of the epidermal growth
factor family, is over-expressed in human pancreatic cancer and
chronic pancreatitis. Int J Cancer. 56:668–674. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yamanaka Y, Friess H, Buchler M, et al:
Overexpression of acidic and basic fibroblast growth factors in
human pancreatic cancer correlates with advanced tumor stage.
Cancer Res. 53:5289–5296. 1993.PubMed/NCBI
|
|
9
|
Friess H, Yamanaka Y, Buchler M, et al:
Enhanced expression of transforming growth factor beta isoforms in
pancreatic cancer correlates with decreased survival.
Gastroenterology. 105:1846–1856. 1993.PubMed/NCBI
|
|
10
|
Korc M, Chandrasekar B, Yamanaka Y, Friess
H, Buchier M and Beger HG: Overexpression of the epidermal growth
factor receptor in human pancreatic cancer is associated with
concomitant increases in the levels of epidermal growth factor and
transforming growth factor alpha. J Clin Invest. 90:1352–1360.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishiwata T, Friess H, Buchler MW, Lopez ME
and Korc M: Characterization of keratinocyte growth factor and
receptor expression in human pancreatic cancer. Am J Pathol.
153:213–222. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hruban RH, Maitra A and Goggins M: Update
on pancreatic intraepithelial neoplasia. Int J Clin Exp Pathol.
1:306–316. 2008.PubMed/NCBI
|
|
13
|
Garcea G, Neal CP, Pattenden CJ, Steward
WP and Berry DP: Molecular prognostic markers in pancreatic cancer:
a systematic review. Eur J Cancer. 41:2213–2236. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rubin JS, Osada H, Finch PW, Taylor WG,
Rudikoff S and Aaronson SA: Purification and characterization of a
newly identified growth factor specific for epithelial cells. Proc
Natl Acad Sci USA. 86:802–806. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Finch PW, Rubin JS, Miki T, Ron D and
Aaronson SA: Human KGF is FGF-related with properties of a
paracrine effector of epithelial cell growth. Science. 245:752–755.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rubin JS, Bottaro DP, Chedid M, et al:
Keratinocyte growth factor. Cell Biol Int. 19:399–411. 1995.
View Article : Google Scholar
|
|
17
|
Nguyen HQ, Danilenko DM, Bucay N, et al:
Expression of keratinocyte growth factor in embryonic liver of
transgenic mice causes changes in epithelial growth and
differentiation resulting in polycystic kidneys and other organ
malformations. Oncogene. 12:2109–2119. 1996.
|
|
18
|
Playford RJ, Marchbank T, Mandir N, et al:
Effects of keratinocyte growth factor (KGF) on gut growth and
repair. J Pathol. 184:316–322. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Watanabe M, Ishiwata T, Nishigai K,
Moriyama Y and Asano G: Overexpression of keratinocyte growth
factor in cancer cells and enterochromaffin cells in human
colorectal cancer. Pathol Int. 50:363–372. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eswarakumar VP, Lax I and Schlessinger J:
Cellular signaling by fibroblast growth factor receptors. Cytokine
Growth Factor Rev. 16:139–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mohammadi M, Olsen SK and Ibrahimi OA:
Structural basis for fibroblast growth factor receptor activation.
Cytokine Growth Factor Rev. 16:107–137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Miki T, Bottaro DP, Fleming TP, et al:
Determination of ligand-binding specificity by alternative
splicing: two distinct growth factor receptors encoded by a single
gene. Proc Natl Acad Sci USA. 89:246–250. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kurban G, Ishiwata T, Kudo M, Yokoyama M,
Sugisaki Y and Naito Z: Expression of keratinocyte growth factor
receptor (KGFR/FGFR2 IIIb) in human uterine cervical cancer. Oncol
Rep. 11:987–991. 2004.PubMed/NCBI
|
|
24
|
Yoshino M, Ishiwata T, Watanabe M, et al:
Expression and roles of keratinocyte growth factor and its receptor
in esophageal cancer cells. Int J Oncol. 31:721–728.
2007.PubMed/NCBI
|
|
25
|
Yan G, Fukabori Y, McBride G,
Nikolaropolous S and McKeehan WL: Exon switching and activation of
stromal and embryonic fibroblast growth factor (FGF)-FGF receptor
genes in prostate epithelial cells accompany stromal independence
and malignancy. Mol Cell Biol. 13:4513–4522. 1993.PubMed/NCBI
|
|
26
|
Deryugina EI and Quigley JP: Matrix
metalloproteinases and tumor metastasis. Cancer Metastasis Rev.
25:9–34. 2006. View Article : Google Scholar
|
|
27
|
Jones L, Ghaneh P, Humphreys M and
Neoptolemos JP: The matrix metalloproteinases and their inhibitors
in the treatment of pancreatic cancer. Ann N Y Acad Sci.
880:288–307. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
John A and Tuszynski G: The role of matrix
metalloproteinases in tumor angiogenesis and tumor metastasis.
Pathol Oncol Res. 7:14–23. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bloomston M, Zervos EE and Rosemurgy AS
II: Matrix metalloproteinases and their role in pancreatic cancer:
a review of preclinical studies and clinical trials. Ann Surg
Oncol. 9:668–674. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Harvey SR, Hurd TC, Markus G, et al:
Evaluation of urinary plasminogen activator, its receptor, matrix
metalloproteinase-9, and von Willebrand factor in pancreatic
cancer. Clin Cancer Res. 9:4935–4943. 2003.PubMed/NCBI
|
|
31
|
Kuniyasu H, Ellis LM, Evans DB, et al:
Relative expression of E-cadherin and type IV collagenase genes
predicts disease outcome in patients with resectable pancreatic
carcinoma. Clin Cancer Res. 5:25–33. 1999.PubMed/NCBI
|
|
32
|
Cho K, Ishiwata T, Uchida E, et al:
Enhanced expression of keratinocyte growth factor and its receptor
correlates with venous invasion in pancreatic cancer. Am J Pathol.
170:1964–1974. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fingleton B: Matrix metalloproteinases:
roles in cancer and metastasis. Front Biosci. 11:479–491. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kerkela E and Saarialho-Kere U: Matrix
metalloproteinases in tumor progression: focus on basal and
squamous cell skin cancer. Exp Dermatol. 12:109–125. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mook OR, Frederiks WM and Van Noorden CJ:
The role of gelatinases in colorectal cancer progression and
metastasis. Biochim Biophys Acta. 1705:69–89. 2004.PubMed/NCBI
|
|
36
|
Folgueras AR, Pendas AM, Sanchez LM and
Lopez-Otin C: Matrix metalloproteinases in cancer: from new
functions to improved inhibition strategies. Int J Dev Biol.
48:411–424. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hofmann UB, Houben R, Brocker EB and
Becker JC: Role of matrix metalloproteinases in melanoma cell
invasion. Biochimie. 87:307–314. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Koshiba T, Hosotani R, Wada M, et al:
Detection of matrix metalloproteinase activity in human pancreatic
cancer. Surg Today. 27:302–304. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Maatta M, Soini Y, Liakka A and
Autio-Harmainen H: Differential expression of matrix
metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in
hepatocellular and pancreatic adenocarcinoma: implications for
tumor progression and clinical prognosis. Clin Cancer Res.
6:2726–2734. 2000.
|
|
40
|
Kimura Y, Kobari M, Yusa T, et al:
Establishment of an experimental liver metastasis model by
intraportal injection of a newly derived human pancreatic cancer
cell line (KLM-1). Int J Pancreatol. 20:43–50. 1996.PubMed/NCBI
|
|
41
|
Parsons SL, Watson SA, Brown PD, Collins
HM and Steele RJ: Matrix metalloproteinases. Br J Surg. 84:160–166.
1997. View Article : Google Scholar
|
|
42
|
Kanno N, Nonomura N, Miki T, et al:
Effects of epidermal growth factor on the invasion activity of the
bladder cancer cell line. J Urol. 159:586–590. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Miyake H, Yoshimura K, Hara I, Eto H,
Arakawa S and Kamidono S: Basic fibroblast growth factor regulates
matrix metalloproteinases production and in vitro invasiveness in
human bladder cancer cell lines. J Urol. 157:2351–2355. 1997.
View Article : Google Scholar
|
|
44
|
Lamoreaux WJ, Fitzgerald ME, Reiner A,
Hasty KA and Charles ST: Vascular endothelial growth factor
increases release of gelatinase A and decreases release of tissue
inhibitor of metalloproteinases by microvascular endothelial cells
in vitro. Microvasc Res. 55:29–42. 1998. View Article : Google Scholar
|
|
45
|
Narita K, Fujii T, Ishiwata T, et al:
Keratinocyte growth factor induces vascular endothelial growth
factor-A expression in colorectal cancer cells. Int J Oncol.
34:355–360. 2009.PubMed/NCBI
|
|
46
|
Yamamoto H, Itoh F, Iku S, et al:
Expression of matrix metalloproteinases and tissue inhibitors of
metalloproteinases in human pancreatic adenocarcinomas:
clinicopathologic and prognostic significance of matrilysin
expression. J Clin Oncol. 19:1118–1127. 2001.
|
|
47
|
Koshiba T, Hosotani R, Wada M, et al:
Involvement of matrix metalloproteinase-2 activity in invasion and
metastasis of pancreatic carcinoma. Cancer. 82:642–650. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chaffer CL, Brennan JP, Slavin JL, Blick
T, Thompson EW and Williams ED: Mesenchymal-to-epithelial
transition facilitates bladder cancer metastasis: role of
fibroblast growth factor receptor-2. Cancer Res. 66:11271–11278.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kwabi-Addo B, Ropiquet F, Giri D and
Ittmann M: Alternative splicing of fibroblast growth factor
receptors in human prostate cancer. Prostate. 46:163–172. 2001.
View Article : Google Scholar : PubMed/NCBI
|