Introduction
Renal cell carcinoma (RCC) is the most common
neoplasm of the adult kidney. In this disease, cancer cells form in
the tubules of the kidney and approximately 80% of RCC patients are
diagnosed with the clear cell RCC subtype (1). Up to 30% of RCC patients present at
advanced stages, and approximately 40% of patients who undergo
curative surgical resection experience recurrence during subsequent
follow-up (2,3). The five-year survival rate of
advanced RCC is 5–10% (4). RCC is
resistant to radiotherapy and chemotherapy (5,6).
Targeted therapies such as sunitinib, sorafenib, everolimus and
temsirolimus have been developed and have been used widely in
first- and second-line treatments, extending the period of
progression-free-survival (7,8).
However, these treatments are insufficient for patients who have
developed relapse or metastasis. Therefore, increased understanding
of the molecular mechanisms of RCC progression and metastasis is
needed using the latest approaches to genomic analysis.
RNA can be divided into two categories: protein
coding RNA and non-coding RNA (ncRNA). It is important to examine
the functions of ncRNAs and their association with human disease,
including cancer. microRNAs (miRNAs) are endogenous small ncRNA
molecules (19–22 bases in length) that regulate protein coding gene
expression by repressing translation or cleaving RNA transcripts in
a sequence-specific manner (9). A
growing body of evidence suggests that miRNAs are aberrantly
expressed in many human cancers, and that they play significant
roles in their initiation, development, and metastasis (10). Some highly expressed miRNAs could
function as oncogenes by repressing tumor suppressors, whereas low
level miRNAs could function as tumor suppressors by negatively
regulating oncogenes (11).
We previously identified many tumor suppressive
miRNAs based on our miRNA expression signatures of various types of
cancer, such as RCC, bladder cancer, prostate cancer, maxillary
sinus squamous cell carcinoma and hypopharyngeal squamous cell
carcinoma (12–17). In oncogenic pathways, normal
regulatory mechanisms are disrupted by aberrant expression of tumor
suppressive or oncogenic miRNAs. Therefore, identification of
miRNA-regulated pathways is important for further development in
human cancer research. Thus, we have been investigating how tumor
suppressive miRNA regulates novel cancer pathways. For example, the
miR-1/miR-133a cluster regulates several oncogenic genes,
including transgelin-2 (TAGLN2), prothymosin α (PTMA)
and purine nucleoside phosphorylase (PNP) (14,15,18).
More recently, we constructed a miRNA expression
signature of RCC clinical specimens and successfully identified
tumor suppressive miR-1285 targeting transglutaminase 2
(TGM2) (12). Among the
signatures, several miRNAs were significantly downregulated in RCC
specimens as promising candidate of tumor suppressors. In this
study, we focused on miR-138. This miRNA was downregulated
in our previous signature, and downregulation of miR-138 has
been observed in several malignancies, including anaplastic thyroid
carcinoma (19) and lung cancer
(20).
The aim of the study was to investigate the
functional significance of miR-138 and identify its target
genes in RCC cells. To identify miR-138-regulated cancer
pathways, we undertook both a genome-wide gene expression analysis
(miR-138 transfectants and RCC clinical specimens) and an
in silico study. The results showed that vimentin
(VIM) was a promising candidate target gene of
miR-138. It is well known that VIM is one of the most widely
expressed mammalian intermediate filament proteins. Studies have
shown that VIM functions in cell adhesion, migration, survival, and
cell signaling processes via dynamic assembly/disassembly in cancer
cells (21). The existence of a
tumor suppressive miR-138-mediated cancer pathway provides
new insights into the potential mechanisms of RCC oncogenesis and
metastasis.
Materials and methods
Clinical specimens
A total of 33 pairs of clear cell renal cell
carcinoma (ccRCC) and adjacent non-cancerous specimens were
collected from patients who had undergone radical nephrectomies at
Kagoshima University Hospital. The samples were processed and
stored in RNAlater (Qiagen, Valencia, CA, USA) at −20°C until RNA
extraction. The patient information is summarized in Table I. These samples were staged
according to the American Joint Committee on Cancer-Union
Internationale Contre le Cancer (UICC) tumour-node-metastasis
classification and histologically graded (22). Our study was approved by the
Bioethics Committee of Kagoshima University; written prior informed
consent and approval were given by the patients.
 | Table IPatient characteristics of RT-PCR
experiments. |
Table I
Patient characteristics of RT-PCR
experiments.
| No. of patients
(%) |
|---|
| Total number | 33 | |
| Age (average) | 36–83 | (65.6) |
| Gender | | |
| Male | 22 | (66.7) |
| Female | 11 | (33.3) |
| Pathological tumor
stage | | |
| pT1a | 12 | (36.4) |
| pT1b | 14 | (42.4) |
| pT2 | 2 | (6.1) |
| pT3a | 3 | (9.1) |
| pT3b | 2 | (6.1) |
| pT4 | 0 | (0.0) |
| Grade | | |
| G1 | 5 | (15.2) |
| G2 | 26 | (78.8) |
| G3 | 0 | (0.0) |
| Unknown | 2 | (6.1) |
| Infiltration | | |
| α | 12 | (36.4) |
| β | 21 | (63.6) |
| γ | 0 | (0.0) |
| Venous
invasion | | |
| v (−) | 24 | (72.7) |
| v (+) | 9 | (27.3) |
Cell culture and RNA extraction
We used two human RCC cell lines, A498 and 786-O,
obtained from the American Type Culture Collection (Manassas, VA,
USA). The cell lines were incubated in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS) and maintained in a
humidified incubator (5% CO2) at 37°C. Total-RNA was
extracted, as previously described (12).
Quantitative real-time RT-PCR
TaqMan probes and primers for VIM (P/N:
Hs00185584_m1: Applied Biosystems) were assay-on-demand gene
expression products. All reactions were performed in duplicate, and
a negative control lacking cDNA was included. We followed the
manufacturer’s protocol for PCR conditions. Stem-loop RT-PCR
(TaqMan MicroRNA Assays; P/N: 002284 for miR-138; Applied
Biosystems) was used to quantitate miRNAs according to the earlier
published conditions (23). To
normalize the data for quantification of VIM mRNA and the
miRNAs, we used human GUSB (P/N: Hs99999908_m1; Applied
Biosystems) and RNU6B (P/N: 001973; Applied Biosystems),
respectively, and we used the ΔΔCt method to calculate the
fold-change. As a control RNA, we used Premium total-RNA from
normal human kidney (AM 7976; Applied Biosystems).
Mature miRNA and siRNA transfection
As described elsewhere (23), the RCC cell lines were transfected
with Lipofectamine™ RNAiMAX transfection reagent (Invitrogen,
Carlsbad, CA, USA) and Opti-MEM™ (Invitrogen) with 10 nM mature
miRNA molecules. Pre-miR™ (Applied Biosystems) and negative-control
miRNA (Applied Biosystems) were used in the gain-of-function
experiments, whereas VIM siRNA (Cat nos. SASI_ Hs01_00044033
and SASI_HS01_00044036, Sigma-Aldrich, St. Louis, MO, USA) and
negative control siRNA (D-001810-10; Thermo Fisher Scientific,
Waltham, MA, USA) were used in the loss-of-function experiments.
Cells were seeded in 10-cm dishes for protein extraction
(8×105 cells per dish), 6-well plates for wound healing
assays (20×104 cells per well), in 24-well plates for
the mRNA extraction and Matrigel invasion assays (5×104
cells per well) and in 96-well plates for the XTT assays (3,000
cells per well).
Cell morphology
Cells were transfected with miR-138 and
si-VIM for 72 h and were then examined by an inverted
microscope (CK2-BIP2, Olympus).
Cell proliferation, migration and
invasion assays
Cell proliferation was determined using an XTT assay
(Roche Applied Science, Tokyo, Japan) that was performed according
to the manufacturer’s instructions. Cell migration activity was
evaluated with a wound healing assay. Cells were plated in 6-well
dishes and the cell monolayer was scraped using a P-20 micropipette
tip. The initial gap length (0 h) and the residual gap length 24 h
after wounding were calculated from photomicrographs. A cell
invasion assay was carried out using modified Boyden Chambers
consisting of Transwell-precoated Matrigel membrane filter inserts
with 8-mm pores in 24-well tissue cultures plates (BD Bioscience,
Bedford, MA, USA). Minimum essential medium containing 10% FBS in
the lower chamber served as the chemoattractant as described
previously (24). All experiments
were performed in triplicate.
Screening of miR-138-regulated genes by
microarray
Oligomicroarray Human 60K (Agilent) was used for
expression signature in miR-138-transfected A498 cells in
comparison with the miR-negative control transfectant, as
previously described (23).
Briefly, hybridization and washing steps were performed in
accordance with the manufacturer’s instructions. The arrays were
scanned using a Packard GSI Lumonics ScanArray 4000 (PerkinElmer,
Boston, MA, USA). The data obtained were analyzed with DNASIS array
software (Hitachi Software Engineering, Tokyo, Japan) that
converted the signal intensity. Data from each microarray study
were normalized by global normalization.
Expression signature of RCC clinical
specimens by microarray
Oligo-microarray Human 60K (Agilent) was used for
expression signature in 5 pairs of RCC clinical specimens compared
with adjacent non-cancerous tissues. Their age ranged from 42 to 77
years; 3 were G1 and 2 were G2 in their tumor grading; and all were
pT1N0M0 tumors.
Western blot analysis
After three days of transfection, protein lysates
(40 μg) were separated by NuPAGE on 4–12% bis-tris gels
(Invitrogen) and transferred onto polyvinylidene fluoride
membranes. Immunoblotting was done with diluted (1:500) polyclonal
VIM antibody (HPA001762; Sigma-Aldrich) and GAPDH antibody (MAB374;
Chemicon, Temecula, CA, USA). The membrane was washed and then
incubated with goat anti-rabbit IgG (H+L)-HRP conjugate (Bio-Rad,
Hercules, CA, USA). Specific complexes were visualized with an
echochemiluminescence (ECL) detection system (GE Healthcare, Little
Chalfont, UK), and the expression levels of these genes were
evaluated by ImageJ software (ver. 1.43; http://rsbweb.nih.gov/ij/index.html).
Immunohistochemistry
A tissue microarray of 67 RCC samples and 10 normal
kidney samples was obtained from US Biomax Inc. (KD806; Rockville,
MD, USA). Detailed information on all tumor specimens can be found
at http://www.biomax.us/index.php. Patient
characteristics are summarized in Table III. Immunostaining was done on
the tissue microarray following the manufacturer’s protocol by
UltraVision Detection System (Thermo Scientific). The primary
rabbit polyclonal antibodies against VIM (Sigma-Aldrich) were
diluted 1:500. The slides were treated with biotinylated goat
anti-rabbit. Diaminobenzidine hydrogen peroxidase was the
chromogen, and the counterstaining was done with 0.5% hematoxylin.
Immunostaining was evaluated according to a scoring method
described previously (14). Each
case was scored on the basis of the intensity and area of staining.
The intensity of staining was graded on the following scale: 0, no
staining; 1+, mild staining; 2+, 30–60% stained positive; 3+,
>60% stained positive. A combined staining score (intensity +
extent) of <2 was low expression, a score between 3 and 4 was
moderate expression, and a score between 5 and 6 was high
expression.
| Table IIIPatient characteristics of
immunohistochemistry. |
Table III
Patient characteristics of
immunohistochemistry.
| No. of patients
(%) |
|---|
| Total number | 67 | |
| Age (average) | 30–80 | (54.4) |
| Gender | | |
| Male | 45 | (67.2) |
| Female | 22 | (32.8) |
| Pathological tumor
stage | | |
| pT1 | 15 | (22.4) |
| pT2 | 28 | (41.8) |
| pT3 | 22 | (32.8) |
| pT4 | 2 | (3.0) |
| Grade | | |
| G1 | 52 | (77.6) |
| G2 | 14 | (20.9) |
| G3 | 1 | (1.5) |
| Normal tissue | 10 | |
Statistical analysis
The relationships between two variables and
numerical values were analyzed using the Mann-Whitney U test, and
the relationship between three variables and the numerical values
was analyzed using the Bonferroni-adjusted Mann-Whitney U test.
Expert Stat View analysis software (ver. 4; SAS institute Inc.,
Cary, NC, USA) was used in both analyses. In the comparison of
three variables, a non-adjusted statistical level of significance
of P<0.05 corresponded to the Bonferroni-adjusted level of
P<0.0167.
Results
Effect of miR-138 transfection on cell
proliferation, migration, and invasion activity of RCC cell
lines
In this study, we firstly observed that restoration
of miR-138 in RCC cell lines (A498 and 786-O) changed the
bleb-like cell morphology characteristic of the
epithelial-mesenchymal transition (EMT) (Fig. 1A). A morphological change of cancer
cells by miRNA transfection is an important discovery and it
suggested that miR-138 functions as a tumor suppressor in
RCC cells. To explore that possibility, the following experiments
were conducted.
We evaluated the expression levels of miR-138
in two RCC cell lines, A498 and 786-O. RNA was extracted and miRNA
expression levels of miR-138 were determined by real-time
RT-PCR. The expression levels of miR-138 were significantly
lower in both RCC cell lines compared with normal kidney RNA
(relative to normal kidney RNA, 0.090±0.008 and 0.102±0.009,
respectively) (Fig. 1B).
The XTT assay revealed that cell proliferation was
significantly inhibited in miR-138 transfectants in
comparison with the transfectant reagent only (mock) and the
miR-control transfectants. The percentages of cell proliferation
for A498 were 94.6±0.9, 100.0±0.8 and 100.0±1.0, respectively, each
P=0.0008. For 786-O, the percentages were 83.5±1.1, 100.0±0.4 and
100.3±0.6, respectively, P<0.0001 (Fig. 1C).
The wound healing assay demonstrated that
significant inhibition of cell migration occurred in the
miR-138 transfectants in comparison with mock and the
miR-control transfectants. The percentages of wound closure for
A498 were 6.5±2.3, 100.0±2.7 and 104.8±4.9, respectively, each
P<0.0001. For 786-O, the percentages were 30.7±3.8, 100.0±4.4
and 95.9±5.4, respectively, each P<0.0001 (Fig. 1D).
The Matrigel invasion assay demonstrated that the
number of invading cells significantly decreased in the
miR-138-transfectants in comparison with mock and the
miR-control transfectants. The percentages of cell invasion for
A498 were 0.9±0.4, 100.0±8.6 and 83.1±7.4, respectively, each
P<0.0001. For 786-O, the values were 10.9±1.1, 100.0±4.4 and
75.3±6.2, respectively, each P<0.0001 (Fig. 1E).
miR-138 regulation of molecular targets
assessed by genome-wide gene expression analysis
To confirm that miR-138 regulated molecular
targets in RCC cells, we performed genome-wide gene expression
analysis using miR-138 transfectants compared with
miRNA-control transfectants in A498 cells. A total of 99 genes were
downregulated in miR-138 transfectants. Among them, 24 genes
had putative target site(s) in their 3′ untranslated region (3′UTR)
according to the TargetScan miRNA program (Table II).
| Table IIDownregulated genes in
microRNA-138 transfectants. |
Table II
Downregulated genes in
microRNA-138 transfectants.
| Entrez gene ID | Symbol | Average | Target site |
|---|
| 3569 | IL6 | −5.35 interleukin 6
(interferon, β 2) | (−) |
| 4856 | NOV | −5.22
nephroblastoma overexpressed gene | (−) |
| 84448 | ABLIM2 | −4.97 actin binding
LIM protein family, member 2 | (−) |
| 3773 | KCNJ16 | −4.86 potassium
inwardly-rectifying channel, subfamily J, member 16 | (−) |
| 6352 | CCL5 | −4.44 chemokine
(C-C motif) ligand 5 | (−) |
| 4316 | MMP7 | −4.1 matrix
metallopeptidase 7 (matrilysin, uterine) | (−) |
| 3038 | HAS3 | −4.03 hyaluronan
synthase 3 | (+) |
| 91543 | RSAD2 | −3.99 radical
S-adenosyl methionine domain containing 2 | (−) |
| 5806 | PTX3 | −3.85
pentraxin-related gene, rapidly induced by IL-1 β | (−) |
| 64220 | STRA6 | −3.85 stimulated by
retinoic acid gene 6 homolog (mouse) | (+) |
| 84419 | C15orf48 | −3.8 chromosome 15
open reading frame 48 | (−) |
| 144406 | WDR66 | −3.67 WD repeat
domain 66 | (−) |
| 4493 | MT1E | −3.66
metallothionein 1E | (−) |
| 718 | C3 | −3.65 complement
component 3 | (−) |
| 10964 | IFI44L | −3.64
interferon-induced protein 44-like | (−) |
| 3990 | LIPC | −3.64 lipase,
hepatic | (−) |
| 9121 | SLC16A5 | −3.61 solute
carrier family 16, member 5 (monocarboxylic acid transporter
6) | (−) |
| 4490 | MT1B | −3.6
metallothionein 1B | (−) |
| 8091 | HMGA2 | −3.56 high mobility
group AT-hook 2 | (−) |
| 1803 | DPP4 | −3.49
dipeptidyl-peptidase 4 | (−) |
| 6288 | SAA1 | −3.48 serum amyloid
A1 | (−) |
| 4502 | MT2A | −3.44
metallothionein 2A | (−) |
| 8638 | OASL | −3.43
2′-5′-oligoadenylate synthetase-like | (−) |
| 9582 | APOBEC3B | −3.39
apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like
3B | (−) |
| 4500 | MT1L | −3.32
metallothionein 1L (gene/pseudogene) | (−) |
| 3437 | IFIT3 | −3.3
interferon-induced protein with tetratricopeptide repeats 3 | (−) |
| 9076 | CLDN1 | −3.05 claudin
1 | (−) |
| 8743 | TNFSF10 | −3.03 tumor
necrosis factor (ligand) superfamily, member 10 | (−) |
| 3433 | IFIT2 | −2.94
interferon-induced protein with tetratricopeptide repeats 2 | (−) |
| 2172 | FABP6 | −2.91 fatty acid
binding protein 6, ileal | (−) |
| 23586 | DDX58 | −2.89 DEAD
(Asp-Glu-Ala-Asp) box polypeptide 58 | (−) |
| 4982 | TNFRSF11B | −2.89 tumor
necrosis factor receptor superfamily, member 11b | (−) |
| 259307 | IL4I1 | −2.88 interleukin 4
induced 1 | (−) |
| 6590 | SLPI | −2.88 secretory
leukocyte peptidase inhibitor | (−) |
| 5174 | PDZK1 | −2.88 PDZ domain
containing 1 | (−) |
| 51015 | ISOC1 | −2.86
isochorismatase domain containing 1 | (+) |
| 3434 | IFIT1 | −2.84
interferon-induced protein with tetratricopeptide repeats 1 | (−) |
| 22822 | PHLDA1 | −2.79 pleckstrin
homology-like domain, family A, member 1 | (−) |
| 2537 | IFI6 | −2.76 interferon,
α-inducible protein 6 | (−) |
| 392636 | TMEM195 | −2.76 transmembrane
protein 195 | (−) |
| 81610 | FAM83D | −2.73 family with
sequence similarity 83, member D | (+) |
| 26154 | ABCA12 | −2.66 ATP-binding
cassette, sub-family A (ABC1), member 12 | (−) |
| 4940 | OAS3 | −2.65
2′-5′-oligoadenylate synthetase 3, 100 kDa | (+) |
| 5359 | PLSCR1 | −2.65 phospholipid
scramblase 1 | (−) |
| 6236 | RRAD | −2.61 Ras-related
associated with diabetes | (−) |
| 4496 | MT1H | −2.56
metallothionein 1H | (−) |
| 4814 | NINJ1 | −2.54 ninjurin
1 | (+) |
| 11309 | SLCO2B1 | −2.5 solute carrier
organic anion transporter family, member 2B1 | (+) |
| 158158 | RASEF | −2.47 RAS and
EF-hand domain containing | (−) |
| 259 | AMBP | −2.47
α-1-microglobulin/bikunin precursor | (−) |
| 2669 | GEM | −2.47 GTP binding
protein overexpressed in skeletal muscle | (−) |
| 3656 | IRAK2 | −2.42 interleukin-1
receptor-associated kinase 2 | (−) |
| 3880 | KRT19 | −2.41 keratin
19 | (−) |
| 1978 | EIF4EBP1 | −2.41 eukaryotic
translation initiation factor 4E binding protein 1 | (+) |
| 7431 | VIM | −2.39 vimentin | (+) |
| 57568 | SIPA1L2 | −2.38
signal-induced proliferation-associated 1 like 2 | (−) |
| 7913 | DEK | −2.35 DEK
oncogene | (+) |
| 123 | PLIN2 | −2.34 perilipin
2 | (−) |
| 4501 | MT1X | −2.33
metallothionein 1X | (−) |
| 654346 | LGALS9C | −2.31 lectin,
galactoside-binding, soluble, 9C | (+) |
| 4489 | MT1A | −2.3
metallothionein 1A | (−) |
| 3669 | ISG20 | −2.28 interferon
stimulated exonuclease gene 20 kDa | (−) |
| 2920 | CXCL2 | −2.28 chemokine
(C-X-C motif) ligand 2 | (−) |
| 2274 | FHL2 | −2.27 four and a
half LIM domains 2 | (−) |
| 157506 | RDH10 | −2.27 retinol
dehydrogenase 10 (all-trans) | (−) |
| 25937 | WWTR1 | −2.26 WW domain
containing transcription regulator 1 | (−) |
| 3690 | ITGB3 | −2.26 integrin, β 3
(platelet glycoprotein IIIa, antigen CD61) | (+) |
| 196513 | DCP1B | −2.24 DCP1
decapping enzyme homolog B (S. cerevisiae) | (−) |
| 9518 | GDF15 | −2.24 growth
differentiation factor 15 | (−) |
| 1364 | CLDN4 | −2.23 claudin
4 | (−) |
| 23643 | LY96 | −2.2 lymphocyte
antigen 96 | (−) |
| 10561 | IFI44 | −2.2
interferon-induced protein 44 | (−) |
| 84141 | FAM176A | −2.19 family with
sequence similarity 176, member A | (−) |
| 6281 | S100A10 | −2.17 S100 calcium
binding protein A10 | (−) |
| 7088 | TLE1 | −2.17
transducin-like enhancer of split 1 (E(sp1) homolog,
Drosophila) | (−) |
| 81553 | FAM49A | −2.17 family with
sequence similarity 49, member A | (−) |
| 4599 | MX1 | −2.17 myxovirus
(influenza virus) resistance 1, interferon-inducible protein p78
(mouse) | (−) |
| 6850 | SYK | −2.17 spleen
tyrosine kinase | (−) |
| 7364 | UGT2B7 | −2.17 UDP
glucuronosyltransferase 2 family, polypeptide B7 | (−) |
| 5366 | PMAIP1 | −2.17
phorbol-12-myristate-13-acetate-induced protein 1 | (−) |
| 50515 | CHST11 | −2.16 carbohydrate
(chondroitin 4) sulfotransferase 11 | (+) |
| 2982 | GUCY1A3 | −2.16 guanylate
cyclase 1, soluble, α 3 | (+) |
| 6273 | S100A2 | −2.15 S100 calcium
binding protein A2 | (+) |
| 54478 | FAM64A | −2.15 family with
sequence similarity 64, member A | (−) |
| 3428 | IFI16 | −2.14 interferon,
γ-inducible protein 16 | (−) |
| 9615 | GDA | −2.14 guanine
deaminase | (−) |
| 7849 | PAX8 | −2.13 paired box
8 | (−) |
| 10550 | ARL6IP5 | −2.11
ADP-ribosylation-like factor 6 interacting protein 5 | (+) |
| 23286 | WWC1 | −2.1 WW and C2
domain containing 1 | (+) |
| 9636 | ISG15 | −2.08 ISG15
ubiquitin-like modifier | (+) |
| 896 | CCND3 | −2.07 cyclin
D3 | (+) |
| 5329 | PLAUR | −2.07 plasminogen
activator, urokinase receptor | (−) |
| 4853 | NOTCH2 | −2.06 Notch homolog
2 (Drosophila) | (+) |
| 55652 | SLC48A1 | −2.05 solute
carrier family 48 (heme transporter), member 1 | (+) |
| 23476 | BRD4 | −2.04 bromodomain
containing 4 | (+) |
| 2012 | EMP1 | −2.03 epithelial
membrane protein 1 | (+) |
| 3429 | IFI27 | −2.02 interferon,
α-inducible protein 27 | (−) |
| 79710 | MORC4 | −2.01 MORC family
CW-type zinc finger 4 | (−) |
| 80820 | EEPD1 | −2.01
endonuclease/exonuclease/phosphatase family domain containing
1 | (+) |
Furthermore, we performed gene expression analysis
using RCC clinical specimens (5 pairs of RCC and adjacent
non-cancerous tissues). Several protein-coding genes were
differentially expressed in this signature (data not shown). We
selected 99 genes that were downregulated in miR-138
transfectants and demonstrated their expression levels in a heatmap
diagram (Fig. 2A). Entries from
the microarray data were approved by the Gene Expression Omnibus
(GEO), and were assigned GEO accession numbers GSE 36951 (RCC
clinical specimens) and GSE 37119 (miR-138
transfectants).
The two expression signatures in this study
(miR-138 transfectants and RCC clinical specimens) revealed
that VIM was a promising putative target gene in
miR-138 in RCC. Thus, we focused on the VIM gene and
investigated the functional significance of VIM in RCC
cells.
VIM as a direct target of repression by
miR-138 in RCC cells
The mRNA and protein expression levels of VIM were
markedly downregulated in miR-138 transfectants (A498 and
768-O) in comparison with the mock and miRNA-control transfectants
(Fig. 2B and C). The predicted
target site of miR-138 in VIM in the 3′UTR is shown
in Fig. 2D.
Silencing of VIM in RCC cell lines and
the effect on cell proliferation, migration and invasion
First, we assessed the expression level of
VIM in cells to be used for functional analysis of VIM.
VIM mRNA expression levels in A498 and 786-O were significantly
higher than those in normal human kidney RNA (relative to normal
kidney RNA, 4.879±0.131 and 9.298±0.255, respectively, each
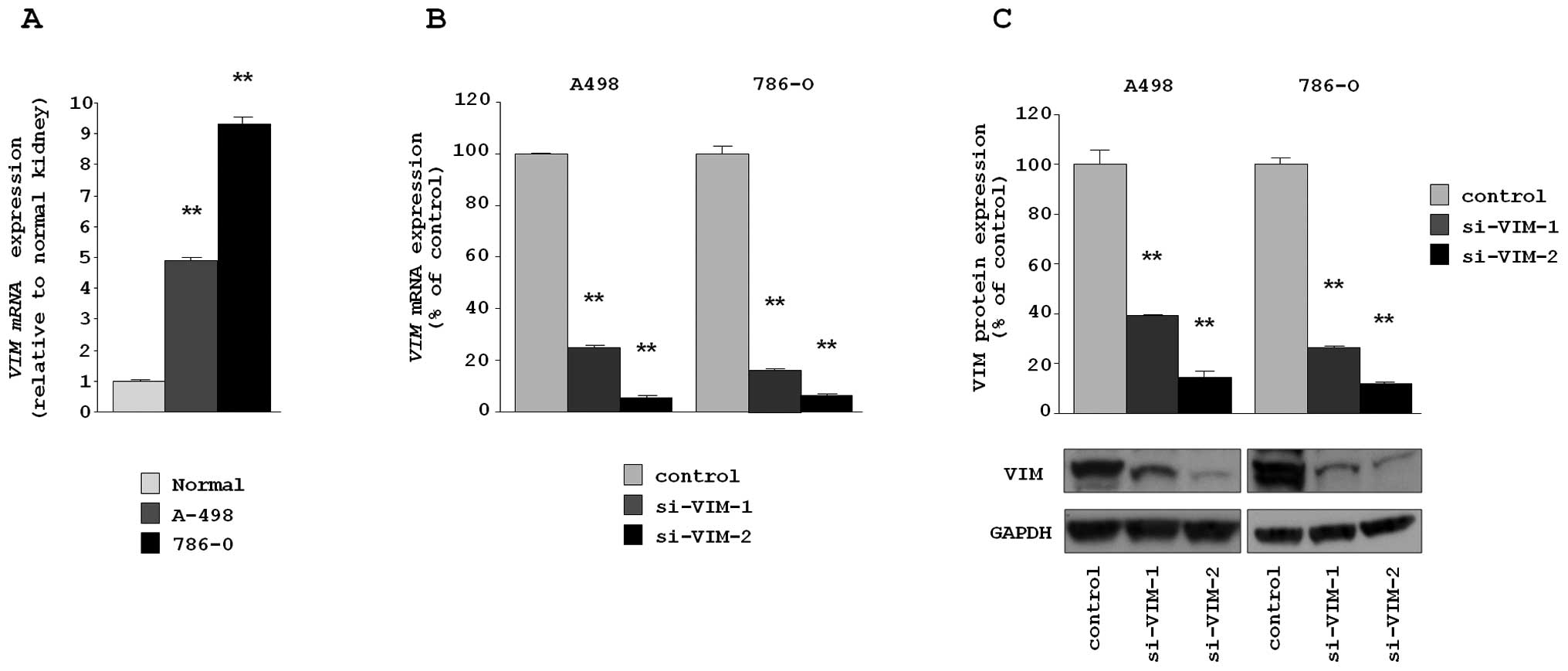
P<0.0001) (Fig. 3A).
To examine the functional role of VIM, we performed
loss-of-function studies using two different siRNAs,
si-VIM-1 and si-VIM-2 transfected into A498 and 768-O
cell lines. The mRNA and protein expression levels of VIM were
markedly downregulated in both si-VIM-1 and si-VIM-2
transfectants (A498 and 768-O) in comparison with the siRNA-control
transfectants (Fig. 3B and C).
This result shows that two siRNA were useful for loss-of-function
assays in this study.
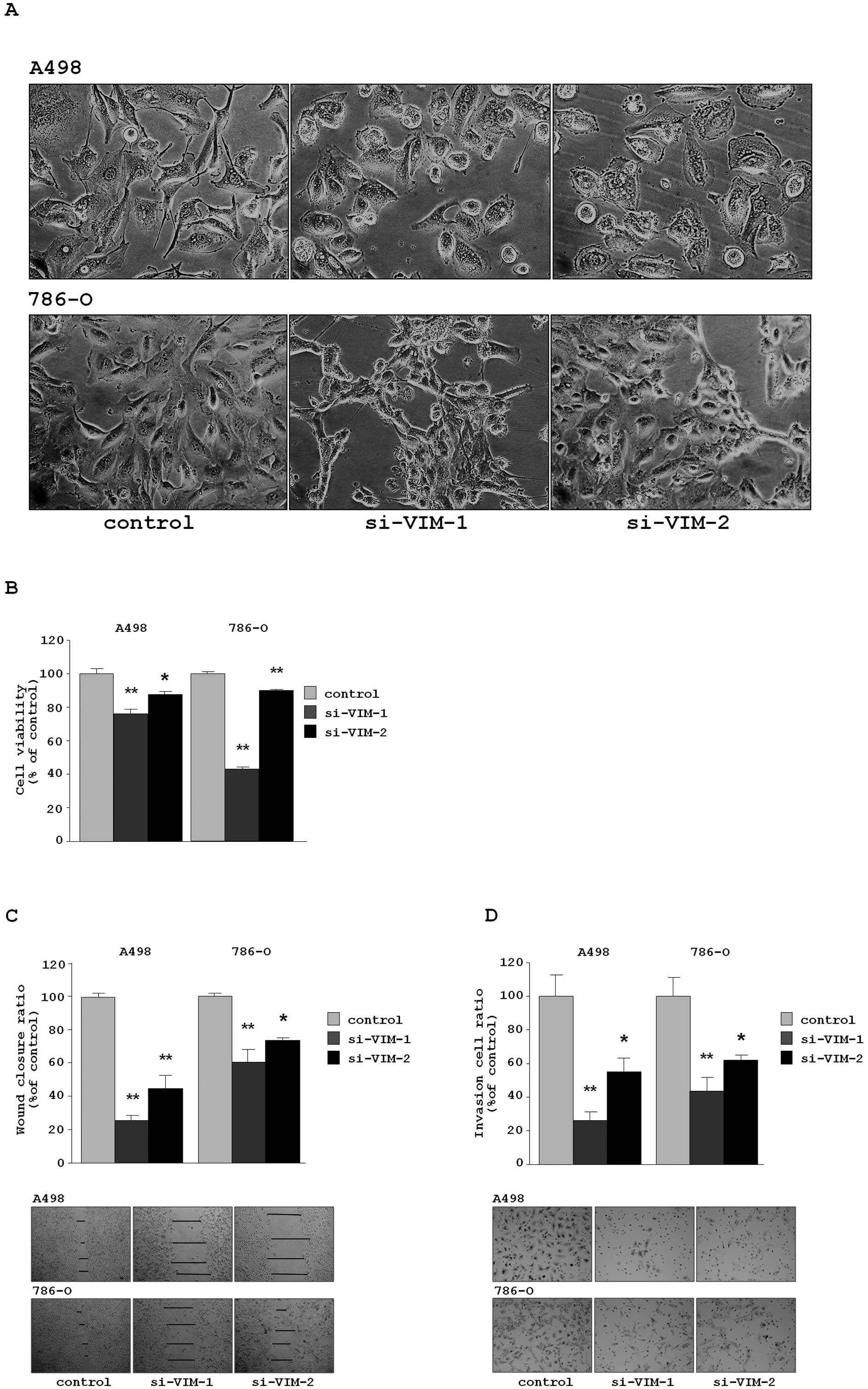
Transfection of si-VIM-1 and si-VIM-2
in the RCC cell lines (A498 and 768-O) caused EMT-like changes in
cell morphology, as that observed when cells were transfected with
miR-138 (Fig. 4A).
The XTT assay revealed that cell proliferation was
inhibited in both si-VIM-transfectants in comparison with
the si-control transfectants. The percentages of cell proliferation
for A498 were 76.1±2.6, 87.5±1.9 and 100.0±3.1, respectively,
P<0.0001 and P =0.0038. For 786-O, the values were 43.1±1.0,
89.9±0.7 and 100.0±1.1, respectively, P<0.0001 (Fig. 4B).
The wound healing assay demonstrated that
significant inhibition of cell migration occurred in the
si-VIM-transfectants in comparison with the si-control
transfectants. The percentages of wound closure for A498 were
26.0±10.6, 44.7±8.4 and 100.0±2.6, respectively, each P<0.0001.
For 786-O, the values were 60.8±7.3, 73.0±1.7 and 100.0±7.1,
respectively, P<0.0001 and P=0.0002 (Fig. 4C).
The Matrigel invasion assay demonstrated that the
number of invading cells significantly decreased in the
si-VIM-transfectants in comparison with the si-control
transfectants. For A498, the percentages of cells invading were
26.5±4.7, 54.8±8.3 and 100.0±12.4, respectively, P<0.0001 and
P=0.0019. For 786-O, the values were 43.6±8.3, 61.6±3.3 and
100.0±11.1, respectively, P<0.0001 and P=0.0034 (Fig. 4D).
Expression levels of miR-138 and VIM mRNA
in RCC clinical specimens
Quantitative stem-loop RT-PCR demonstrated that the
expression levels of miR-138 were significantly reduced in
33 RCC samples (Table I) in
comparison with adjacent non-cancerous specimens (clinical RCC
specimens, 0.346±0.201 versus adjacent normal tissues, 2.983±0.715,
P<0.0001) (Fig. 5A).
On the other hand, the mRNA expression level of
VIM was significantly higher in RCC than adjacent
non-cancerous specimens (clinical RCC specimens, 6.017±0.622,
adjacent normal tissues; 1.316±0.224, P<0.0001) (Fig. 5B).
The mRNA expression of ≥T2 specimens (n=7) was
significantly higher than that of T1 (n=26) (T1, 5.230±0.607; ≥T2,
8.773±1.349, P=0.0277) (Fig.
5C).
Immunohistochemistry of VIM in tissue
microarray
VIM was detected by immunohistochemical staining.
Fig. 6A–D shows representative
results of immunohistochemical staining of VIM. VIM was strongly
expressed in tumor lesions (Fig.
6A–C), whereas no expression was observed in normal tissue
(Fig. 6D). The expression score of
VIM was significantly higher in 67 RCC specimens in comparison with
ten normal kidney specimens (Fig.
6E). The VIM expression of ≥T2 specimens (n=52) was higher than
that of T1 (n=15). There was a trend but no significant difference
in the expression score of VIM between T1 and ≥T2 specimens
(P=0.053, Fig. 6F). While there
was a trend between VIM expression and grade, the difference was
not significant. Patient characteristics are summarized in Table III.
Discussion
The incidence of RCC has increased over the last few
decades, and it currently represents approximately 2% of all
cancer-related deaths (25).
Although two-thirds of RCC patients have clinically localized
disease and will undergo curative surgery, up to 40% of patients
develop distant metastasis and their outcomes are poor (2–4).
Many studies have indicated that cell adhesion and extra-cellular
matrix proteins contribute to the cells’ acquired abilities for
invasion, migration and metastasis (26).
EMT is an embryologically conserved genetic program
that is an essential step enabling cancer cell invasion and
metastasis. For example, epithelial cells lose intercellular tight
junctions and polarity (27).
Recent data indicate that the microRNA-200 family
(miR-200a, -200b, -200c, -141 and -429) is
downregulated in aggressive human cancers. Moreover, it plays
critical roles in the inhibition of key regulators of EMT and
β-catenin/Wnt signaling (28,29).
Interestingly, our miRNA expression signature of RCC showed that
all miR-200 family members were reduced in clinical
specimens and that restoration of the miR-200 family
inhibited cancer cell migration and invasion (data not shown).
Our previous study showed that miR-138 was
reduced in RCC miRNA expression signature (12), we validated the down-regulation of
miR-138 in RCC clinical specimens in this study. Aberrant
expression of miR-138 has been observed in several types of
cancer such as head and neck squamous cell carcinoma (HNSCC)
(30), anaplastic thyroid
carcinoma (19) and lung cancer
(20). Two miR-138
precursor genes, miR-138-1 and miR-138-2, have
identical sequences in the mature miRNA and map to human
chromosomes 3p21.33 and 16q13, respectively. Although it is
believed that genomic deletion or epigenetic silencing of miRNA in
cancer cells, the molecular mechanism of downregulated miRNAs in
RCC is not clear. In the human chromosomal region 3p, LOH is
frequently observed in many cancers including RCC (31,32).
This problem can be solved by genome-based high throughput analysis
in each clinical case.
Importantly, we found significant morphologic change
in RCC cell lines (A498 and 786-O) by miR-138 transfection.
Furthermore, restoration of miR-138 significantly inhibited
cancer cell migration and invasion in RCC cells. These data
suggested that miR-138 functions as a tumor suppressor that
inhibits RCC invasion and metastasis. miRNAs are unique in their
ability to regulate many protein-coding genes. A single miRNA is
capable of targeting a number of genes to regulate biological
processes globally. Bioinformatic predictions suggest that miRNAs
regulate more than 30% of protein coding genes (9). The elucidation of new molecular
pathways regulated by tumor suppressive miR-138 is important
for our understanding of human RCC invasion and metastasis. Based
on this view, we performed molecular target searches for
miR-138 in cancer cells by combining two genome-wide gene
expression studies (miR-138 transfectants and RCC mRNA
clinical signature) and in silico analysis.
In this study, we focused on VIM as a
putative candidate of miR-138 in RCC cells. We chose
VIM for the following reasons. First, downregulation of
VIM was recognized in the expression signature of
miR-138 transfectants. Second, overexpression of VIM
was observed in RCC clinical specimens. Third, VIM has a
putative miR-138 target site in its 3′ untranslated region.
Our data demonstrated that restoration of miR-138
significantly inhibited both mRNA and protein expression levels of
VIM in RCC cells, suggesting VIM was regulated by
tumor suppressive miR-138. It is well known that VIM is an
essential constituent of cytoskeletal proteins of mesenchymal cells
and VIM is a marker of EMT (21).
During EMT, cytoskeletal proteins are changed from keratin-rich
networks to VIM-rich networks connected to focal adhesions.
Morphological changes of RCC cells and accelerated cell migration
and invasion is caused by the reduction of miR-138 and the
upregulation of VIM pathways. Interestingly, it has been
shown that miR-138 regulated cell migration and invasion by
targeting RhoC, ROCK, ZEB2, EZH2 and
VIM in HNSCC cells (33,34).
Importantly, restoration of miR-138 in an HNSCC cell line
changed the EMT-like cell morphology and suppressed cell migration
and invasion (34). This result is
in accord with the data obtained in our study of RCC. Furthermore,
overexpression of miR-138 reduced cell viability and colony
formation in HCC cell lines targeting CCND3(35). The report also showed that protein
expression of CCND3 was negatively correlated with miR-138
expression in HCC tissues. Our data of miR-138 transfectants
in RCC cell lines demonstrated that CCND3 is a putative
target of miR-138 in RCC, suggesting that miR-138
regulation of the CCND3 pathway is important for RCC
oncogenesis.
The results of this study and previous data indicate
that VIM is a functional target of tumor suppressive
miR-138, and this pathway contributes to cancer cell
migration, invasion, and metastasis. In this study, we also
demonstrated overexpression of VIM in clinical specimens of RCC.
Previous studies indicated that VIM was a sensitive and specific
marker for conventional RCCs (36,37).
The combination of VIM and CD9 staining was found to distinguish
clear cell RCC and chromophobe RCC (37). Our tissue microarray data showed a
positive correlation between VIM expression and tumor grade in RCC
specimens. In this analysis, we were not able to obtain a
correlation of VIM expression and metastasis in RCC
patients. Silencing of VIM in RCC cell lines changed cell
morphology and significantly inhibited cell migration and invasion
in this study. Thus, we propose that overexpression of VIM
participates in metastasis of RCC. Studies of a large number of
samples with balanced pathological backgrounds are needed to
elucidate the precise correlation between VIM and/or
miR-138 expression and clinicopathological parameters.
In summary, the reduction of miR-138 and the
increased expression of VIM are frequent events in RCC
clinical specimens. Restoration of miR-138 in RCC cells
changed the EMT-like morphology and suppressed cell migration and
invasion. The tumor suppressive miR-138-mediated cancer
pathway provides new insights into the potential mechanisms of RCC
oncogenesis and metastasis.
Acknowledgements
This research was supported by the
Ministry of Education, Science, Sports and Culture Grant-in-Aid for
Scientific Research (C), 20591861 and 21592187.
References
|
1
|
Karumanchi SA, Merchan J and Sukhatme VP:
Renal cancer: molecular mechanisms and newer therapeutic options.
Curr Opin Nephrol Hypertens. 11:37–42. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Janzen NK, Kim HL, Figlin RA and
Belldegrun AS: Surveillance after radical or partial nephrectomy
for localized renal cell carcinoma and management of recurrent
disease. Urol Clin North Am. 30:843–852. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Koul H, Huh JS, Rove KO, Crompton L, Koul
S, Meacham RB and Kim FJ: Molecular aspects of renal cell
carcinoma: a review. Am J Cancer Res. 1:240–254. 2011.PubMed/NCBI
|
|
4
|
Hadoux J, Vignot S and De La Motte Rouge
T: Renal cell carcinoma: focus on safety and efficacy of
temsirolimus. Clin Med Insights Oncol. 4:143–154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang Y, Dai Y, Yang J, Chen T, Yin Y,
Tang M, Hu C and Zhang L: Microarray analisis of microRNA
expression in renal cell carcinoma. Eur J Surg Oncol. 35:1119–1123.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Motzer RJ, Bander NH and Nanus DM:
Renal-cell carcinoma. N Engl J Med. 335:865–875. 1996. View Article : Google Scholar
|
|
7
|
Motzer RJ, Aqarwal N, Beard C, Bhayani S,
Bolger GB, Carducci MA, Chang SS, Choueiri TK, Hancock SL, Hudes
GR, Jonasch E, Josephson D, Kuzel TM, Lavine EG, Lin DW, Margolin
KA, Michaelson MD, Olencki T, Pili R, Ratliff TW, Redman BG,
Robertson CN, Ryan CJ, Sheinfeld J, Spiess PE, Wang J and Wilder
RB: Kidney cancer. J Natl Compr Canc Netw. 9:960–977.
2011.PubMed/NCBI
|
|
8
|
Agarwala SS and Case S: Everolimus
(RAD001) in the treatment of advanced renal cell carcinoma: a
review. Oncologist. 15:236–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fillipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nelson KM and Weiss GJ: MicroRNAs and
cancer: past, present, and potential future. Mol Cancer Ther.
7:3655–3660. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Esquela-Kerscher A and Slack FJ:
OncomiRs-microRNAs with a role in cancer. Nat Rev Cancer.
6:259–269. 2006. View Article : Google Scholar
|
|
12
|
Hidaka H, Seki N, Yoshino H, Yamasaki T,
Yamada Y, Nohata N, Fuse M, Nakagawa M and Enokida H: Tumor
suppressive microRNA-1285 regulates novel molecular targets:
Aberrant expression and functional significance in renal cell
carcinoma. Oncotarget. 3:44–57. 2012.PubMed/NCBI
|
|
13
|
Chiyomaru T, Enokida H, Tatarano S,
Kawahara K, Uchida Y, Nishiyama K, Fujimura L, Kikkawa N, Seki N
and Nakagawa M: miR-145 and miR-133a function as tumour suppressors
and directly regulate FSCN1 expression in bladder cancer. Br J
Cancer. 102:883–891. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoshino H, Chiyomaru T, Enokida H,
Kawakami K, Tatarano S, Nishiyama K, Nohata N, Seki N and Nakagawa
M: The tumoursuppressive function of miR-1 and miR-133a targeting
TAGLN2 in bladder cancer. Br J Cancer. 104:808–818. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kojima S, Chiyomaru T, Kawakami K, Yoshino
H, Enokida H, Nohata N, Fuse M, Ichikawa T, Naya Y, Nakagawa M and
Seki N: Tumour suppressors miR-1 and miR-133a target the oncogenic
function of purine nucleoside phosphorylase (PNP) in prostate
cancer. Br J Cancer. 106:405–413. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nohata N, Hanazawa T, Kikkawa N, Sakurai
D, Fujimura L, Chiyomaru T, Kawakami K, Yoshino H, Enokida H,
Nakagawa M, Katayama A, Harabuchi Y, Okamoto T and Seki N: Tumor
suppressive microRNA-874 regulates novel cancer networks in
maxillary sinus squamous cell carcinoma. Br J Cancer. 105:833–841.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kikkawa N, Hanazawa T, Fujimura L, Nohata
N, Suzuki H, Chazono H, Sakurai D, Horiguchi S, Okamoto Y and Seki
N: miR-489 is a tumour-suppressive miRNA target PTPN11 in
hypopharyngeal squamous cell carcinoma (HSCC). Br J Cancer.
103:877–884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yamasaki T, Yoshino H, Enokida H, Hidaka
H, Chiyomaru T, Nohata N, Kinoshita T, Fuse M, Seki N and Nakagawa
M: Novel molecular targets regulated by tumor suppressors
microRNA-1 and microRNA-133a in bladder cancer. Int J Oncol.
40:1821–1830. 2012.PubMed/NCBI
|
|
19
|
Mitomo S, Maesawa C, Ogasawara S, Iwaya T,
Shibazaki M, Yashima-Abo A, Kotani K, Oikawa H, Sakurai E, Izutsu
N, Kato K, Komatsu H, Ikeda K, Wakabayashi G and Masuda T:
Downregulation of miR-138 is associated with overexpression of
human telomerase reverse transcriptase protein in human anaplastic
thyroid carcinoma cell lines. Cancer Sci. 99:280–286. 2008.
View Article : Google Scholar
|
|
20
|
Seike M, Goto A, Okano T, Bowman ED,
Schetter AJ, Horikawa I, Mathe EA, Jen J, Yang P, Sugimura H, Gemma
A, Kudoh S, Croce CM and Harris CC: MiR-21 is an EGFR-regulated
anti-apoptotic factor in lung cancer in never-smokers. Proc Natl
Acad Sci USA. 106:12085–12090. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sobin LH and Wittekind C: TNM
classification of malignant tumours In: International Union Against
Cancer (UICC). 6th edition. John Wiley & Sons; Hoboken, NJ: pp.
255–257. 2009
|
|
23
|
Nohata N, Hanazawa T, Kikkawa N, Mutallip
M, Fujimaru L, Yoshino H, Kawakami K, Chiyomaru T, Enokida H,
Nakagawa M, Okamoto T and Seki N: Caveolin-1 mediates tumor cell
migration and invasion and its regulation by miR-133a in head and
neck squamous cell carcinoma. Int J Oncol. 38:209–217.
2011.PubMed/NCBI
|
|
24
|
Nohata N, Sone Y, Hanazawa T, Fuse M,
Kikkawa N, Yoshino H, Chiyomaru T, Kawakami K, Enokida H, Nakagawa
M, Shozu M, Okamoto T and Seki N: miR-1 as a tumor suppressive
microRNA targeting TAGLN2 in head and neck squamous cell carcinoma.
Oncotarget. 2:29–44. 2011.PubMed/NCBI
|
|
25
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
26
|
Friedl P and Alexander S: Cancer invasion
and the microenvironment: plasticity and reciprocity. Cell.
147:992–1009. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Soini Y: Tight junctions in lung cancer
and lung metastasis: a review. Int J Clin Exp Pathol. 5:126–136.
2012.PubMed/NCBI
|
|
28
|
Brabletz S and Brabletz T: The ZEB/miR-200
feedback loop - a motor of cellular plasticity in development and
cancer? EMBO Rep. 11:670–677. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mongroo PS and Rustgi AK: The role of the
miR-200 family in epithelial-mesenchymal transition. Cancer Biol
Ther. 10:219–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li X, Jiang L, Wang A, Yu J, Shi F and
Zhou X: MicroRNA-138 suppresses invasion and promotes apoptosis in
head and neck squamous cell lines. Cancer Lett. 286:217–222. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hogg RP, Honorio S, Martinez A,
Agathanggelou A, Dallol A, Fullwood P, Weichselbaum R, Kuo MJ,
Maher ER and Latif F: Frequent 3p allele loss and epigenetic
inactivation of the RASSF1A tumour suppressor gene from region
3p21.3 in head and neck squamous cell carcinoma. Eur J Cancer.
38:1585–1592. 2002. View Article : Google Scholar
|
|
32
|
Sukosd F, Kuroda N, Beothe T, Kaur AP and
Kovacs G: Deletion of chromosome 3p14.2-p25 involving the VHL and
FHIT genes in conbentional renal cell carcinoma. Cancer Res.
63:455–457. 2003.PubMed/NCBI
|
|
33
|
Jiang L, Liu X, Kolokythas A, Yu J, Wang
A, Heidbreder CE, Shi F and Zhou X: Downregulation of the Rho
GTPase signaling pathway is involved in the microRNA-138-mediated
inhibition of cell migration and invasion in tongue squamous cell
carcinoma. Int J Cancer. 127:505–512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu X, Wang C, Chen Z, Jin Y, Wang Y,
Kolokythas A, Dai Y and Zhou X: MicroRNA-138 suppresses
epithelial-mesenchymal transition in squamous cell carcinoma cell
lines. Biochem J. 440:23–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang W, Zhao LJ, Tan YX, Ren H and Qi ZT:
Mir-138 induces cell cycle arrest by targeting cyclin D3 in
hepatocellular carcinoma. Carcinogenesis. 33:1113–1120. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Young AN, Amin MB, Moreno CS, Lim SD,
Cohen C, Petros JA, Marshall FF and Neish AS: Expression profiling
of renal epithelial neoplasms: a method for tumor classification
and discovery of diagnostic molecular markers. Am J Pathol.
158:1639–1651. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Williams AA, Higgins JP, Zhao H, Ljunberg
B and Brooks JD: CD9 and vimentin distinguish clear cell from
chromophobe renal cell carcinoma. BMC Clin Pathol. 9:92009.
View Article : Google Scholar : PubMed/NCBI
|