Introduction
Non-melanoma skin cancer is the most common type of
cancer in Caucasian populations (1,2). In
the United States alone, two million people are diagnosed with
non-melanoma skin cancer every year, with squamous cell carcinoma
(SCC) and basal cell carcinoma (BCC) accounting for the majority of
cases (3). Surgical removal is the
standard therapy for the treatment of SCC and BCC, but it may cause
morbidity in high risk individuals and have negative cosmetic
outcomes. Thus, the development of alternative modalities for the
treatment of non-melanoma skin cancer remains highly desirable.
Non-steroidal anti-inflammatory drugs (NSAIDs) have
demonstrated significant efficacy in the chemoprevention of colon
(4) and skin cancer (5,6).
However, their use is limited by gastrointestinal toxicity that
does not justify prolonged use in healthy individuals (7). Prompted by these concerns, our group
has synthesized novel phospho-derivatives of conventional NSAIDs
(8–18). Phospho-NSAIDs show superior
anticancer efficacy and reduced gastrointestinal toxicity compared
to conventional NSAIDs in preclinical models. P-S (OXT-328), a
phospho-derivative of sulindac, is a potent inhibitor of colon
cancer. The present study examined the anticancer activity of P-S
towards skin cancer.
Topical treatment regimens are considered to be
effective alternatives for non-melanoma skin cancer. Skin delivery
is non-invasive; achieves high local levels of drug; minimizes
systemic exposure; and is more acceptable to patients. In our
previous investigations, the effective delivery of P-S to tumors
was limited by its inactivation by carboxylesterases (9,12).
We reasoned that the local delivery of P-S by topical application
will bypass the liver and the intestinal tract, major sites of
carboxylesterase expression, and hence, will be more effective than
delivery via the oral route.
In this report, we demonstrate that P-S is a potent
inhibitor of non-melanoma skin cancer cells, and topical delivery
of P-S resulted in strong inhibition of skin cancer xenografts in
mice. These findings suggest that topical P-S is a promising
strategy for the treatment of non-melanoma skin cancer.
Materials and methods
Reagents
P-S (OXT-328) was a gift from Medicon
Pharmaceuticals Inc. (Setauket, NY, USA). Cell culture reagents
were purchased from Cellgro (Herndon, VA, USA). Other reagents,
unless otherwise stated, were obtained from Sigma-Aldrich (St.
Louis, MO, USA).
Cell culture
The human epidermoid carcinoma cell line (A431) was
obtained from American Type Culture Collection (ATCC), and
maintained in DMEM media containing 10% fetal bovine serum and
penicillin/streptomycin. All experiments were performed with cells
between passages 1 to 10.
Topical hydrogel preparation
A mixture of Pluronic P123 and P-S, dissolved in
tetrahydrofuran (1:10, w/w), was dialyzed for 24 h at room
temperature through a membrane (molecular weight cutoff of 3,500
Da) in phosphate-buffered saline, which was replaced three times.
The dialysis bag was then wiped with absorbent paper and placed
under solid PEG (MW 900,000) to absorb and concentrate the solution
inside the bag until gel formation.
The final drug loading onto the gel was 1.4% (w/w),
while the polymer constituted 27–30% w/v of the gel. Control gel
was prepared by a cold method where 28% w/v of pluronic P123 was
dispersed slowly in PBS at 2–5°C.
In vitro cytokinetic analyses
Cell viability was measured with the MTT assay
(Roche Diagnostics) and cell proliferation with the
5-bromo-2′-deoxyuridine (BrdU; BD Immunocytometry Systems) assay,
according to the manufacturer’s instructions. Apoptosis and
necrosis were assessed by staining cells with Annexin V and
propidium iodide (PI) and analyzing them by flow cytometry
(19).
A431 xenografts
Female NOD/SCID mice (6–7 weeks-old) were purchased
from Harlan Sprague-Dawley (Indianapolis, IN, USA). At 7–8 weeks of
age, the mice were inoculated intradermally on both flanks with
A431 cells (2x106 each) suspended in 100 μl complete
DMEM medium. When the average tumor size reached 120±40
mm3, the animals were divided into four groups (n=6–7),
and were given the following treatments, respectively: i) none; ii)
topical plain hydrogel (3 times per day); iii) P-S (150 mg/kg/d,
p.o.); and iv) topical P-S hydrogel (50 mg/kg/d, 3 times per day).
The tumors were measured twice a week with a digital microcaliper,
and tumor volumes were calculated using the following formula:
tumor volume = [length × width × (length + width/2) × 0.56]. After
treatment for 2 weeks, the animals were sacrificed and their tumors
were removed. The levels of P-S and its metabolites in the tumors
were determined by HPLC. This animal study was approved by the
Institutional Animal Care and Use Committee of Stony Brook
University.
HPLC analysis
The HPLC system consisted of a Waters Alliance 2695
Separations Module equipped with a Waters 2998 photo-diode array
detector (328 nm) (Waters, Milford, MA, USA) and a Thermo BDS
Hypersil C18 column (150×4.6 mm, particle size 3 μm) (Thermo Fisher
Scientific, Waltham, MA, USA). The mobile phase consisted of a
gradient between buffer A [H2O, acetonitrile, formic
acid, 95:4.9:0.1 (v/v/v)] and 100% acetonitrile.
Immunohistochemistry
Cell death and proliferation of paraffin-embedded
A431 xenograft tissue sections were determined using the terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and
Ki-67 immunohistochemical staining, respectively, as previously
described (19).
Statistical analyses
Data are expressed as the mean ± SEM. Statistical
analyses were performed by ANOVA. P-values <0.05 were considered
statistically significant.
Results
Topical P-S inhibits the growth of A431
xenografts
To assess the in vivo efficacy of P-S against
skin cancer, we treated SCID mice bearing A431 xenografts (n=6–7)
topically with vehicle hydrogel, or P-S hydrogel, or orally with
P-S, whereas the last group was left untreated. At equivalent
dosage, topical P-S more effectively inhibited the growth of A431
tumors (p=0.017) compared to oral P-S (Fig. 1). Topical P-S caused regression of
tumors during the first week. At the end of the experiment, topical
P-S reduced tumor growth by 70.5% (p<0.001) relative to the
control. Oral P-S (150 mg/kg/day), on the other hand, did not cause
any tumor regression and modestly inhibited tumor growth by 43.4%
(p<0.01). No difference in the final tumor volume was found
between the vehicle and the untreated group. These findings
indicate that topical P-S causes a profound inhibitory effect on
the growth of human skin cancer xenografts. Body weights showed no
significant difference between our two study groups (data not
shown) and no local or systemic side effects were noted in the
treatment group.
P-S decreases proliferation and induces
apoptosis in vitro and in vivo
To evaluate the effect of P-S on cell growth, we
determined the 24 h-IC50 values of P-S and sulindac in
A431 cells (Table I). P-S
demonstrated a dramatically enhanced cytotoxicity (36-fold)
compared to sulindac. In addition, it was also much more potent
than the sulindac metabolites, sulindac sulfide (8-fold) and
sulindac sulfone (31-fold).
 | Table I.Inhibitory effect of P-S and its
metabolites on A431 cell growth. |
Table I.
Inhibitory effect of P-S and its
metabolites on A431 cell growth.
| Compound | 24-h IC50,
μM mean ± SD | Enhancement
(fold) |
|---|
| P-S | 60.4±0.2 | - |
| Sulindac | 2,210±90 | 36 |
| Sulindac sulfide | 461±62 | 8 |
| Sulindac sulfone | 1,860±60 | 31 |
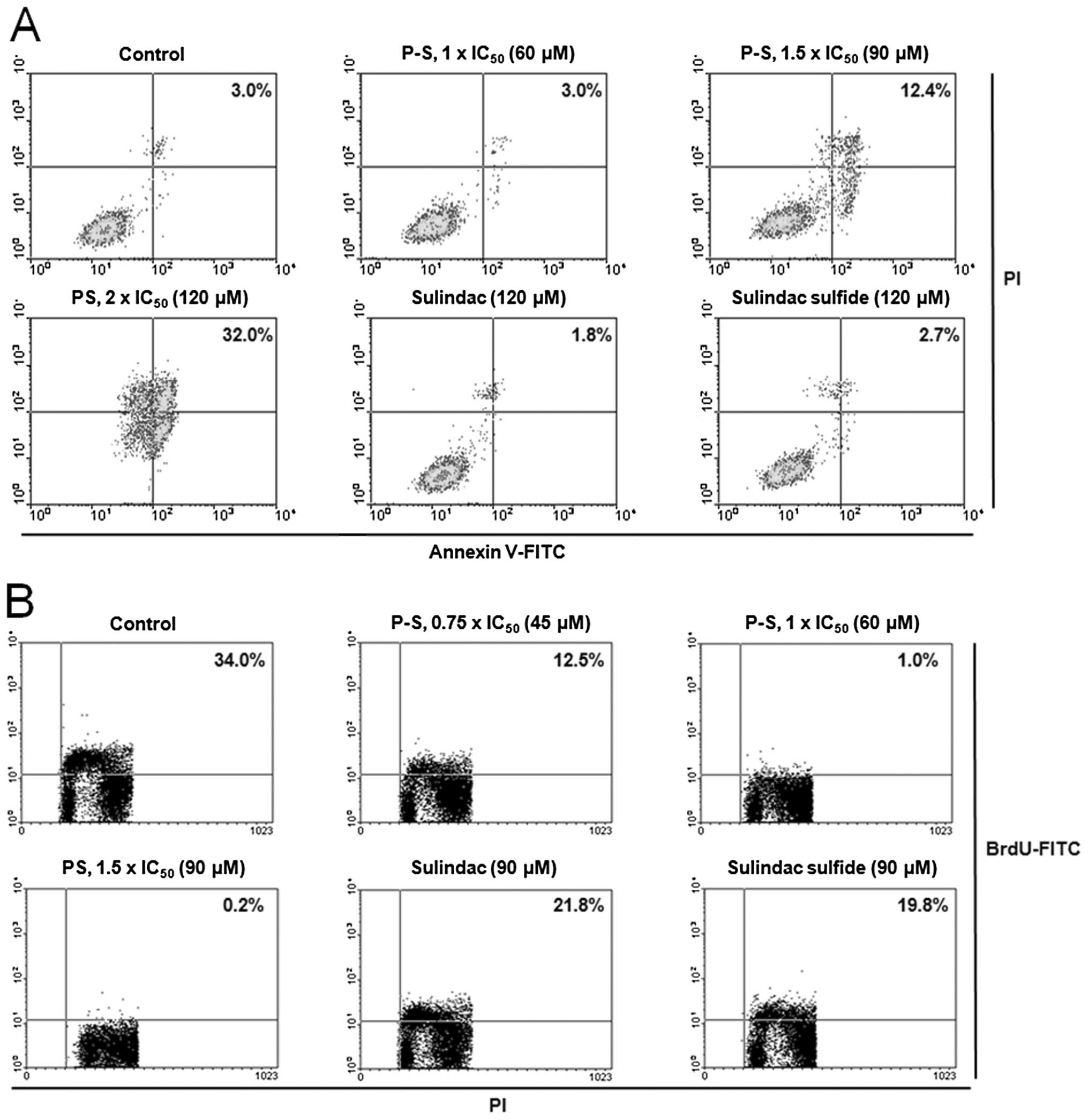
The potent growth inhibitory effect of P-S results
from its cytokinetic effect (Fig.
2). P-S concentration-dependently decreased A431 cell
proliferation, reaching 97% and a near complete (>99%)
inhibition at 1×IC50 and 1.5×IC50,
respectively. Equimolar concentration (1.5×IC50 PS) of
sulindac or sulindac sulfide only weakly inhibited cell growth
(<40%). Cell cycle analysis revealed that PS treatment induced
cell cycle arrest at G2/M phase (Table II).
| Table II.The effect of P-S on the cell cycle
distribution of A431 cells. |
Table II.
The effect of P-S on the cell cycle
distribution of A431 cells.
|
G0/G1 | S %total, mean ±
SD | G2/M |
|---|
| Control | 43±3 | 26±4 | 30±2 |
| P-S 60 μM | 46±3 | 19±2 | 34±5 |
| P-S 120 μM | 28±2 | 29±6 | 42±7 |
| Sulindac 120 μM | 47±2 | 26±3 | 27±4 |
| Sulindac sulfide 120
μM | 49±5 | 24±3 | 26±2 |
Moreover, treatment of A431 cells with P-S at
1.5×IC50 and 2×IC50 for 24 h induced
significant apoptosis to 4.1- and 10.7-fold higher than that of the
control, respectively. Equimolar concentration (2×IC50
PS) of sulindac or sulindac sulfide had no measurable impact on
cell proliferation or apoptosis.
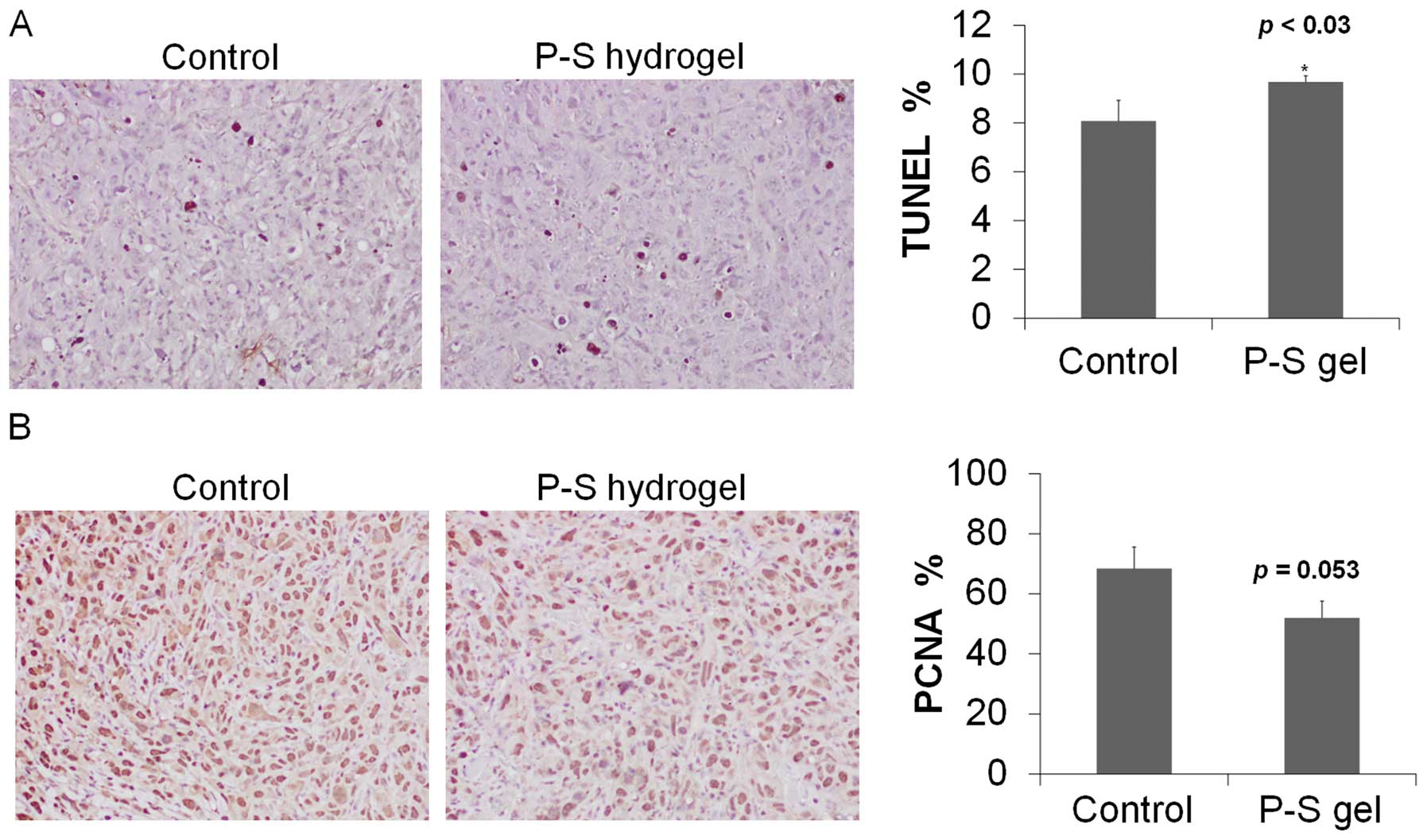
We next determined cell proliferation and apoptosis
in tumor tissue sections. Compared to control, topical P-S
decreased cell proliferation (determined by PNCA staining) by 25%
and induced apoptosis (TUNEL) by 30% (p<0.05) (Fig. 3). These findings indicate that P-S
profoundly suppresses proliferation and induces apoptosis in A431
cells, which rationalizes its potent growth inhibitory effect.
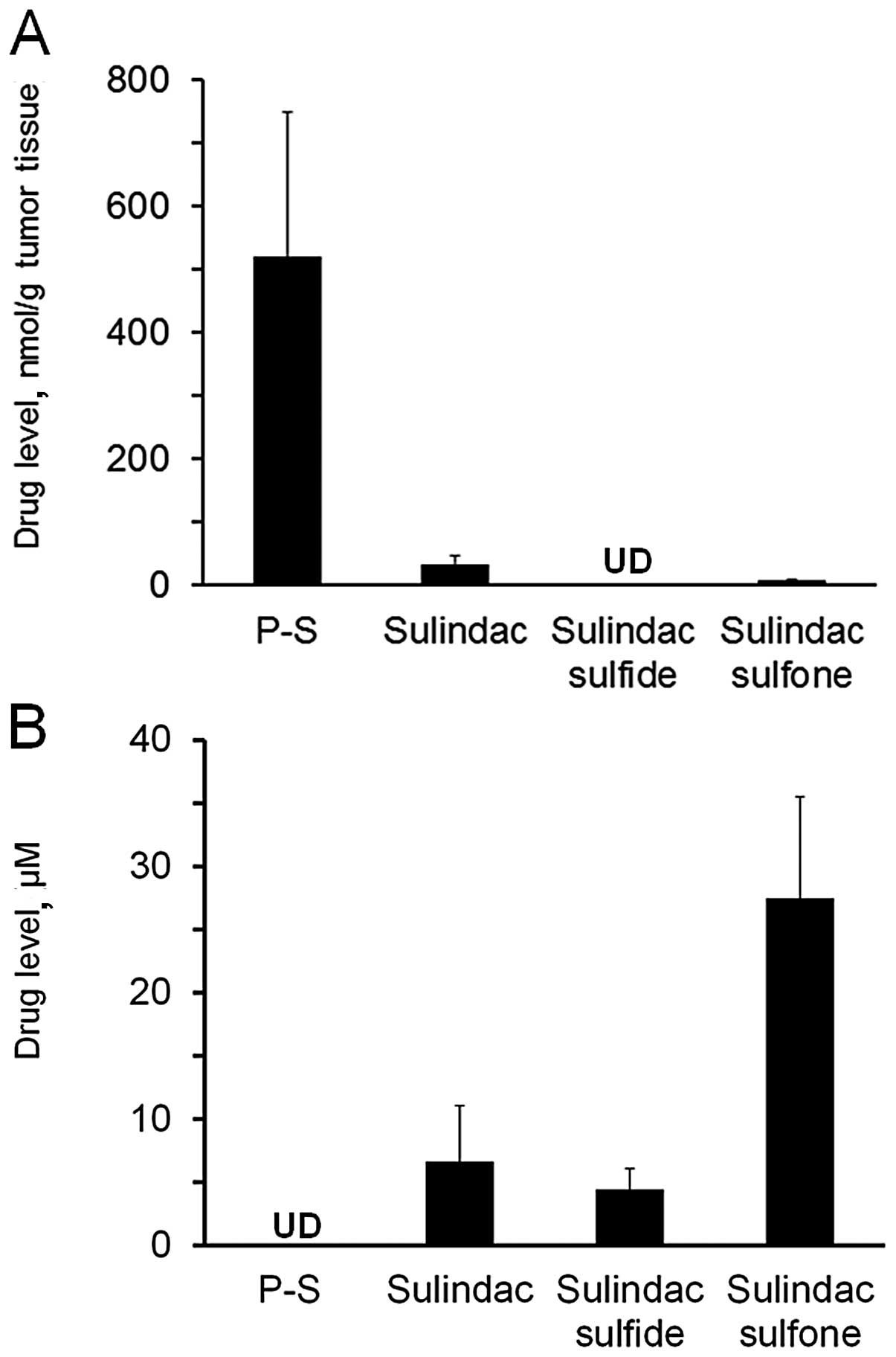
P-S hydrogel delivers substantial amount
of intact drug in vivo
To evaluate the delivery of P-S via the topical
route, we measured blood and tumor drug levels one hour after drug
administration (Fig. 4). Topical
P-S generated significant levels (>500 nmol/g tumor tissue) of
intact P-S in the tumors, accounting for 92.5% of the total
metabolites in the A431 xenografts. Sulindac, sulindac sulfide and
sulindac sulfone were also detected in tumors, but at much lower
levels.
No intact P-S could be detected in the blood of the
topical P-S treated animals. Sulindac is the major metabolite found
in the blood, albeit at lower levels (7-fold lower) compared to
those after oral administration (12). These data indicate that topical
delivery of P-S results in high levels of intact P-S in skin
tumors, which may contribute to its higher antitumor efficacy
compared to oral delivery.
Discussion
Our study demonstrates that P-S is a strong
inhibitor of nonmelanoma skin cancer in pre-clinical models.
Topical P-S strongly suppresses the growth of A431 skin cancer
xenografts in mice, an effect mediated by i) the potent cytokinetic
effect of P-S on A431 skin cancer cells, and ii) direct delivery to
the skin tumors of intact P-S, the biologically most active
molecule.
P-S is a potent inhibitor of the A431 epidermal skin
cancer cell line in vitro (36-fold more potent than
sulindac). A strong cytokinetic effect underpins the inhibitory
potency of P-S, which is a result of inhibition of cell
proliferation, induction of apoptosis and cell cycle arrest
(G2/M). The induction of apoptosis appears to be the
predominant mechanism, as P-S profoundly triggers apoptosis in A431
cells (4 to 10-fold over control) in vitro; whereas an
equimolar level of sulindac, the parent NSAID, has no significant
effect.
P-S can be incorporated in a pluronic polymer to
form a hydrogel for topical application. In vivo, topical
application of P-S strongly suppresses the growth of A431
xenografts by 70.5%, compared to the control. Oral administration
of P-S, on the other hand, only resulted in a moderate inhibition
of xenograft growth (43.4%). Interestingly, the anti-tumor efficacy
of the topical route is significantly better than that of oral
administration (differs by nearly two fold). Topical P-S also
effectively induces apoptosis in A431 xenografts. These results
indicate that P-S is an efficacious agent against non-melanoma skin
cancer, and topical delivery of P-S appears to confer a significant
therapeutic advantage compared to oral administration. Dilution
effects, intestinal absorption and the metabolism of oral P-S can
account for its lower efficacy.
An important contributing factor to the potent
activity of topical P-S is the improved delivery of intact drug to
tumors in vivo. P-S is considerably (8- to 36-fold) more
cytotoxic towards A431 cells than its metabolites, sulindac,
sulindac sulfide and sulindac sulfone (Table I), or the diethylphosphate linker
(15). However, our previous
investigations have shown that P-S, when given orally, was rapidly
metabolized, which primarily gave rise to the above three
metabolites in plasma (9,12) with minimal distribution of P-S to
tumors.
Carboxylesterase 1, a broadly-specific
carboxylesterase highly expressed in the intestine, liver and
plasma, is primarily responsible for the hydrolytic inactivation of
P-S (9,12). The presence of carboxylesterases in
these organs compromises drug efficacy by converting P-S into its
significantly less active metabolites. Rodent skin, on the other
hand, has esterase activity over 10-fold lower than that of the
liver and plasma (20). Consistent
with the low carboxylesterase activity in the skin, we demonstrated
that the topical application of P-S resulted in very high local
levels of intact P-S (>90%) in A431 xenografts. Correspondingly,
topical P-S exerts a considerably more potent growth inhibitory
effect compared to P-S given orally.
Topical drug delivery is a valuable strategy of
limiting systemic exposure, thereby lowering the risk of
undesirable side effects. Indeed, topical therapy has been proposed
to reduce the potential gastrointestinal and cardiovascular side
effects of conventional NSAIDs in the treatment of actinic
keratosis (21), pain (22) and arthritis (23). The topical administration of P-S to
mice bearing human skin cancer xenografts resulted in 4- to 5-fold
lower levels of sulindac and its metabolites (<35 μM) compared
to those after oral administration (>200 μM) (12).
In conclusion, topical application of
P-S-incorporating pluronic hydrogel is effective in inhibiting the
growth of nonmelanoma skin cancer, and has superior efficacy
compared to oral administration. Our results indicate that direct
skin delivery of P-S is a promising modality for the treatment of
skin cancer which merits further investigation.
Acknowledgements
This study was supported by grants
from the National Institutes of Health, National Cancer Institute
(R01CA09242308, R01CA139454 and R01CA154172) and the Department of
Defense (W81XWH1010873).
References
|
1
|
Trakatelli M, Ulrich C, del Marmol V,
Euvrard S, Stockfleth E and Abeni D: Epidemiology of nonmelanoma
skin cancer (NMSC) in Europe: accurate and comparable data are
needed for effective public health monitoring and interventions. Br
J Dermatol. 156(Suppl 3): 1–7. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim RH and Armstrong AW: Nonmelanoma skin
cancer. Dermatol Clin. 30:125–139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rogers HW, Weinstock MA, Harris AR, et al:
Incidence estimate of nonmelanoma skin cancer in the United States,
2006. Arch Dermatol. 146:283–287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baron JA, Cole BF, Sandler RS, et al: A
randomized trial of aspirin to prevent colorectal adenomas. N Engl
J Med. 348:891–899. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gravitz L: Chemoprevention: First line of
defence. Nature. 471:S5–S7. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Elmets CA, Viner JL, Pentland AP, et al:
Chemoprevention of nonmelanoma skin cancer with celecoxib: a
randomized, double-blind, placebo-controlled trial. J Natl Cancer
Inst. 102:1835–1844. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cuzick J, Otto F, Baron JA, et al: Aspirin
and non-steroidal anti-inflammatory drugs for cancer prevention: an
international consensus statement. Lancet Oncol. 10:501–507. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nie T, Wong CC, Alston N, Aro P,
Constantinides PP and Rigas B: Phospho-ibuprofen (MDC-917)
incorporated in nanocarriers: anticancer activity in vitro and in
vivo. Br J Pharmacol. Dec 5–2011, (Epub ahead of print).
|
|
9
|
Wong CC, Cheng KW, Xie G, et al:
Carboxylesterases 1 and 2 hydrolyze phospho-NSAIDs: relevance to
their pharmacological activity. J Pharmacol Exp Ther. 340:422–432.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mattheolabakis G, Nie T, Constantinides PP
and Rigas B: Sterically stabilized liposomes incorporating the
novel anti-cancer agent phospho-ibuprofen (MDC-917): preparation,
characterization, and in vitro/in vivo evaluation. Pharm Res. Nov
10–2011, (Epub ahead of print).
|
|
11
|
Huang L, Mackenzie GG, Sun Y, et al:
Chemotherapeutic properties of phospho-nonsteroidal
anti-inflammatory drugs, a new class of anticancer compounds.
Cancer Res. 71:7617–7627. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xie G, Nie T, Mackenzie G, et al: The
metabolism and pharmacokinetics of phospho-sulindac (OXT-328) and
the effect of difluoromethylornithine. Br J Pharmacol.
165:2152–2166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun Y, Huang L, Mackenzie GG and Rigas B:
Oxidative stress mediates through apoptosis the anticancer effect
of phosphononsteroidal anti-inflammatory drugs: implications for
the role of oxidative stress in the action of anticancer agents. J
Pharmacol Exp Ther. 338:775–783. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mackenzie GG, Ouyang N, Xie G, et al:
Phospho-sulindac (OXT-328) combined with difluoromethylornithine
prevents colon cancer in mice. Cancer Prev Res (Phila).
4:1052–1060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xie G, Sun Y, Nie T, et al:
Phospho-ibuprofen (MDC-917) is a novel agent against colon cancer:
efficacy, metabolism, and pharmacokinetics in mouse models. J
Pharmacol Exp Ther. 337:876–886. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang L, Mackenzie G, Ouyang N, et al: The
novel phospho-non-steroidal anti-inflammatory drugs, OXT-328,
MDC-22 and MDC-917, inhibit adjuvant-induced arthritis in rats. Br
J Pharmacol. 162:1521–1533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang L, Zhu C, Sun Y, et al:
Phospho-sulindac (OXT-922) inhibits the growth of human colon
cancer cell lines: a redox/polyamine-dependent effect.
Carcinogenesis. 31:1982–1990. 2010. View Article : Google Scholar
|
|
18
|
Mackenzie GG, Sun Y, Huang L, et al:
Phospho-sulindac (OXT-328), a novel sulindac derivative, is safe
and effective in colon cancer prevention in mice. Gastroenterology.
139:1320–1332. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kozoni V, Tsioulias G, Shiff S and Rigas
B: The effect of lithocholic acid on proliferation and apoptosis
during the early stages of colon carcinogenesis: differential
effect on apoptosis in the presence of a colon carcinogen.
Carcinogenesis. 21:999–1005. 2000. View Article : Google Scholar
|
|
20
|
Ahmed S, Imai T, Yoshigae Y and Otagiri M:
Stereospecific activity and nature of metabolizing esterases for
propranolol prodrug in hairless mouse skin, liver and plasma. Life
Sci. 61:1879–1887. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Akarsu S, Aktan S, Atahan A, Koc P and
Ozkan S: Comparison of topical 3% diclofenac sodium gel and 5%
imiquimod cream for the treatment of actinic keratoses. Clin Exp
Dermatol. 36:479–484. 2011.
|
|
22
|
McCarberg BH and Argoff CE: Topical
diclofenac epolamine patch 1.3% for treatment of acute pain caused
by soft tissue injury. Int J Clin Pract. 64:1546–1553
|
|
23
|
Fuller P and Roth S: Diclofenac sodium
topical solution with dimethyl sulfoxide, a viable alternative to
oral nonsteroidal anti-inflammatories in osteoarthritis: review of
current evidence. J Multidiscip Healthc. 4:223–231. 2011.
View Article : Google Scholar : PubMed/NCBI
|