Introduction
Glioblastoma (GBM) is a most aggressive glioma, and
it is the most common brain tumor in adult and second most common
in children. Survival rate remains poor although it has improved
over the last five years (1,
2), is usually associated with a
2-year survival rate of only 10–25% (3). One major strategy to anticancer
growth is to induce tumor apoptosis. Multiple apoptotic signals
involved in the intrinsic pathway may result in alterations of
permeability of the mitochondrial outer membrane (MOM) and cause
the release of apoptogenic factors, including cytochrome c
(4). By following, cytochrome
c can promote formation of apoptosomes. Once the initiator
caspase-9 is activated by apoptosome, effector caspase
(caspase-3/7) will be activated to launch apoptosis. The signaling
posterior to death receptor and ligand binding is further
transducing and activates initiator caspase-8 to trans-activate
effector caspases (5). The
intrinsic pathway is controlled by interactions of Bcl2 family
members (6). According to the
protein structure, the Bcl2 family members have at least one of the
Bcl2 homology (BH) domains (7).
Moreover, proteins in Bcl2 family can be divided into three
functional groups based on their domain composition. One is the
anti-apoptotic group, such as Bcl2, Bcl-xL, Bcl-W, Mcl-1 and A1,
containing BH1-4 domains. These proteins can sequester
pro-apoptotic protein or BH3-only activator to facilitate their
anti-apoptosis function. The pro-apoptotic group, Bax and Bak,
containing BH1-3 and transmembrane (TM) domain. These groups of
proteins are essential effectors responsible for controlling
permeability of MOM. The third group is the BH3-only group that
proteins harbor only BH3 domain. The BH3-only proteins can be
subdivided into activators (e.g., Bid and Bim) and sensitizers
(e.g., Bad, Bik, Bmf, Hrk, Noxa and PUMA) (8).
Upon apoptosis, the BH3s activate Bax and Bak to
mediate cytochrome c efflux, leading to caspase activation
(9–14). Conversely, the anti-apoptotic Bcl2,
Bcl-xL, and Mcl-1 sequester Bax/Bak or activator BH3s into inert
complexes, thus preventing Bax/Bak activation (9,11,14,15).
The remaining BH3s do not activate Bax/Bak directly but instead
prevent the anti-apoptotic Bcl2 members from sequestering the
Bax/Bak or the activator BH3s (11,14–18).
A stepwise activation model of Bax and Bak is driven by activator
BH3s. The α1 helix of Bax stabilizes and thereby controls the
engagement of the α9 helix in the dimerization pocket. Activator
BH3s bind transiently to the α1 helix of Bax to unleash
auto-inhibition, which allows for the structural reorganization by
exposing both N and C terminus. The C-terminal transmembrane domain
hence becomes available for insertion into the MOM. The activator
BH3s remain associated with the N-terminally exposed Bax to drive
the homo-oligomerization of mitochondrial localized Bax. Bak, an
integral mitochondrial membrane protein, constitutively exposes its
α1 helix and requires activator BH3s to trigger its
homo-oligomerization. The homo-oligomerization of Bax or Bak
appears to involve the interaction between the BH3 domain of one
molecule and the canonical dimerization pocket of the other monomer
(19). Mutations in BH3 domain
abolish their homo-oligomerization, and the same strategy is
utilized for the heterodimerization between BH3s and anti-apoptotic
Bcl2 members (20,21).
The Bcl2-like 12 (Bcl2L12) gene was discovered and
cloned by Scorilas et al (22) in 2001, as a newly identified member
of the Bcl2 family, containing a highly conserved BH2 domain, a
BH3-like domain and a proline-rich region. Currently, two
functional splicing variants of the Bcl2L12 gene are known: one
consisting of seven coding exons and producing a 334-amino acid
protein with a molecular mass of 36.8 kDa and another one resulting
from alternative splicing and giving rise to a protein of 176 amino
acids, a splice variant called Bcl2L12A which lacks exon 3 (143 bp)
(22). Expression of the
full-length mRNA transcript has been observed in many tissues,
including breast, thymus, prostate, fetal liver, colon, placenta,
pancreas, small intestine, spinal cord, kidney and bone marrow,
whereas the Bcl2L12A is mainly expressed in fetal liver, spinal
cord and skeletal muscle (22).
Previous studies showed that Bcl2L12 localized in both nucleus and
the cytosol (23,24). The biological role of Bcl2L12 is
yet completely understood. Bcl2L12 exhibits pro-apoptotic activity
in breast tumor and gastric cancer (25–27).
A 3-fold increase of Bcl2L12 levels was demonstrated in
non-cancerous compared to cancerous stomach tissues (27). In breast cancer, both proteins
highly expressed in normal breast tissue, and it was documented
that Bcl2L12 as a favorable prognosis marker and the knock down of
its expression leads to a cisplatin-resistance of a breast cancer
line MDA-MB231 (25). In
nasopharyngeal cancer, Bcl2L12 expression status was also found to
be positively associated with the presence of distant metastases.
Moreover, Bcl2L12 expression is an unfavorable and independent
prognostic indicator of short-term relapse in nasopharyngeal
carcinoma, indicating that Bcl2L12 mRNA expression may constitute a
novel biomarker for the prediction of short-term relapse in
nasopharyngeal carcinoma (?). In contrast, both two Bcl2L12 are
ubiquitously overexpressed in primary human GBMs and may be
associated with the resistance to chemotherapeutic agent-induced
apoptosis that is an important hallmark of this disease (24). Furthermore, it has been reported
that Bcl2L12 is an inhibitor of post-mitochondrial effector caspase
activation, as it binds to and inhibits caspase-7 and upregulates
the caspase-3-specific inhibitor αB-crystallin (CRYAB). Bcl2L12
attenuates endogenous p53-directed transcriptomic changes after
genotoxic stress and inhibits p53-dependent DNA damage-induced
apoptosis (24).
Gliomas have been shown to exhibit disrupted
intrinsic and extrinsic apoptosis signaling pathways, and the
resultant inefficient caspase activation is thought to contribute
to gliomagenesis and chemo-/radiotherapy resistance (28). Using TMZ, it was reported that
ROS-dependent autophagy induction may abrogate its effectiveness in
triggering apoptosis in glioma (29). On the other hand, GBM overexpressed
with Bcl2L12 also creates an unfavorable environment to be
intervened by DNA damaging agents. In this study, we explored
whether Bcl2L12 orchestrates its anti-apoptosis function through
its interacting with Bcl2 family members by the BH3-like domain.
Besides, ABT-737, a BH3 mimetic agent, was tested for its possible
combination with TMZ to provide a better outcome of glioma
treatment.
Materials and methods
Cloning
For vector construction of Bcl2L12, Bcl-xL, Bcl2,
Bax into pACT2/pAS2-1 for yeast two-hybrid assay or pEGFP-C1 for
overexpression, PCR was used to generate DNA fragments containing
desired genes using the primer containing XhoI and
BamHI restriction enzyme recognition site. For gene-specific
PCR, 100 ng genomic DNA was used as template, 2.5 μl, 10X reaction
buffer (Takara Bio Inc., Shiga, Japan), 4 μl, 2.5 mM dNTPs (Takara
Bio Inc.), 0.2 μl DNA polymerase (5 U/μl) (Takara Bio Inc.), 1 μl
of 10 μM primers were added into a 25-μl reaction mix. The PCR
conditions were denaturation at 95°C, 30 sec; followed by 30 sec
annealing, extension at 72°C for 35 cycles on GeneAmp 9700 PCR
System (Applied Biosystems, Foster City, CA, USA). The PCR products
were separated on 2.0% agarose gel and visualized by staining with
0.5 μg/ml ethidium bromide. The PCR products with correct sizes
were further purified using PCR clean-up kit (GeneMark Technology
Co., Ltd., Tainan, Taiwan) and subjected to restriction enzyme
digestion. Plasmid DNA was also double-strand sequenced to confirm
its correctness (Mission Biotech, Taipei, Taiwan).
Yeast two-hybrid assay
Yeast (YRG2) was inoculated into 5 ml YPD medium and
grown at 28°C, 240 rpm for 20 h. YPD medium (45 ml) was added into
original yeast culture to refresh its growth and further incubated
for 4 h. To detect whether gene A interacts with gene B, the pACT2
vector/gene A (1.2 μg) was pre-mixed with pAS2-1 vector/gene B (1.2
μg) then further added with 10 μl boiled salmon sperm and 800 μl of
aforementioned yeast solution. Thereafter, the reaction was vortex
completely and incubated at 28°C, 240 rpm for 30 min, then added
with 80 μl DMSO (Sigma-Aldrich Corp., St. Louis, MO, USA), the
reaction was heat-shocked at 42°C for 15 min then placed on ice for
2–5 min. The reaction was centrifuged at 3,000 rpm, for 3 min and
the supernatant was discarded. Sterilized water (200 μl) was used
to re-suspend the transformed yeast pellet, then 100 μl of the
reaction mixture was spread on nutritional deficiency plates G2 and
G3, respectively. The G2 or G3 plates were incubated at 28°C for
colony growing. Sterilized filter papers were used to cover the
G2/G3 plates. The filter papers attached the yeast colonies and
were further dipped into liquid nitrogen for 40 sec to break cell
walls of the attached yeast. A 1,600 μl of X-gal solution was added
onto another filter and incubated at 28°C for detecting whether
bait protein would interact with prey protein.
Site-directed mutagenesis
The specific primers containing desired mutations
[h1, h2, h3, charge and h4, partly as designated as previous report
(30)] and the complementary
sequences surrounding to mutation sites were designed and
synthesized. After PCR amplification, 1.5 μl DpnI (10 U/μl)
was added for digestion at 37°C for 10 min. DpnI treated DNA
(2 μl) was transferred into 45 μl of prepared competent cells.
After incubation on ice for 30 min, heat shock was carried out at
42°C water bath for 30 sec. Then the reaction mixture was left on
ice for 2 min. Then 500 μl LB medium was added and incubated at
37°C, 200 rpm for 1 h. After incubation, bacterial cultures were
spread on selective plates containing antibiotics and incubated at
37°C for 16–18 h. Several colonies were picked up for plasmid
preparation, restriction enzyme digestion and sequencing to confirm
the desired mutations.
Protein structure prediction
I-TASSER server is a web-based program for protein
structural and functional predictions. It allows users to
automatically generate predictions of 3D structure and biological
function of protein molecules from their amino acid sequences. When
an amino acid sequence is submitted, the server tries to retrieve
template proteins of similar folds (or super-secondary structures)
from the PDB library by LOMETS, a locally installed meta-threading
approach (31). Therefore, we
combined the amino acid sequences with Bcl2L12, Bcl2, Bcl-xL and
Bax, to obtain protein secondary and 3D structure for
cross-sectional comparisons.
Cell culture, transfection and
treatments
Human glioblastoma U87MG (HTB-14) and T98G
(CRL-1690) cell lines were purchased from American Type Culture
Collection (ATCC). Cells were cultured using α-MEM and MEM
supplemented with 10% FBS and penicillin with streptomycin (100
IU/l) at 37°C, 5% CO2 in air atmosphere. For transient
transfections, cells were seeded into 100-mm diameter culture
dishes (Becton-Dickinson, San Jose, CA, USA) at a density of
1×107 cells per dish. DNA was transfected into cells
using Lipofectamine 2000 transfection reagent (Invitrogen,
Carlsbad, CA, USA). For apoptosis induction, STS (Sigma-Aldrich
Corp.) and TMZ were used. After transfection for 12–16 h, 0.5 μM
STS and 400 μM TMZ were added into culture dish and incubated for
another 24 h (STS) or 48 h (TMZ) to trigger apoptosis (STS was the
positive control of apoptosis induction).
Quantitative PCR
cDNA was synthesized from total RNA for each of the
studied groups using an Impron II reverse transcriptase kit
(Promega Corp., Madison, WI, USA). All primer pairs with respect to
Bcl2L12, LC3B, Beclin-1 and GAPDH were designed using a web-based
program provided by GeneScript.com. All were qualified and
demonstrated: i) high amplification efficiency (>96%) across a
wide range of cDNA dilutions; ii) specific single products in
dissociation curve analysis; and iii) melting temperatures similar
to those predicted by oligonucleotide software. Quantitative PCR
was performed with Power SYBR Green dye and an ABI StepOne Plus
Real-Time PCR system (Applied Biosystems). The difference in the
cycle threshold (dCT) for target gene (LC3B, Beclin-1 and Bcl2L12)
mRNA expression was calculated by subtracting the geometric mean of
the cycle threshold for the reference gene (GAPDH) from the cycle
threshold of the target gene mRNA. Because this dCT represents the
log2-transformed expression ratio of the target
transcript to the geometric mean of the reference gene, the
relative expression level of the target gene mRNA was determined as
2-dCT. The relative target gene expression of the studied groups is
shown as a fold increasing pattern with standard deviations.
Western blotting
Lysis buffer was used to prepare the total lysates
from cultured cells. For detecting cytochrome c efflux, the
cytosolic fraction was prepared by Mitochondria/Cytosol
Fractionation kit (Merck Millipore, Billerica, MA, USA) according
to the manufacturer’s protocol. Protein concentration was
determined using Protein Assay reagent (Bio-Rad, Hercules, CA,
USA). Protein lysates were separated on 12% SDS-PAGE and then
transferred onto methanol-treated PVDF membrane. The transferred
membranes were blocked by 5% (w/v) non-fat dry milk in TBST with
gentle shaking for 1 h. Then the membrane was incubated with
primary antibodies: anti-LC3B, anti-Atg5, anti-Beclin-1, anti-full
length PARP, anti-PARP (cleaved form), anti-caspase-9/-3 (cleaved
form), anti-cytochrome c, anti-Bax and anti-Bcl-xL (Cell
Signaling Technology, Beverly, MA, USA), anti-GFP, and anti-β-actin
(Santa Cruz Biotechnology, Santa Cruz, CA, USA). The primary
antibodies were used in 1% non-fat dry milk or 5% BSA dilute with
TBST. The PVDF membranes were washed 5 times in TBST, 5 min each.
Then secondary antibodies conjugated with horseradish peroxidase
were added in 1% non-fat dry milk and incubated at room temperature
for another 1 h, followed by 5 washes with TBST. The protein
signaling was developed using ECL reagent (GE Healthcare,
Piscataway, NJ, USA). The chemiluminescence signal was recorded on
X-ray film (Fuji, Tokyo, Japan) and imaging using developer and
fixer agents (Kodak, Rochester, NY, USA).
Statistical analysis
Data are expressed as mean ± standard deviation.
Statistical significance between the groups was examined by either
Student’s t-test or Wilcoxon’s Rank-Sum test. P-values <0.05
were considered as a statistically significant difference.
Results
Bcl2L12 can interact with Bcl-xL and
Bcl2; in addition, Bcl2L12192–220 may be important to
its interaction with Bcl-xL
According to results from yeast two-hybrid system
shown in Table I, we revealed that
Bcl2L1270–334 can interact with Bcl-xL and Bcl2. To
narrow down the region responsible for these interactions on
Bcl2L12, different truncated Bcl2L12 fragments were generated to
determine whether they can interact with either Bcl-xL or Bcl2 or
both. As shown in Table I,
Bcl2L12192–240 interacts with Bcl-xL, but not
Bcl2L1270–191. Bcl2L1290–220 also interacts
with Bcl-xL; therefore, it is assumed that
Bcl2L12220–240 may be not critical for its interaction
with Bcl-xL. Shorter Bcl2L12 truncated fragment (e.g.,
Bcl2L12192–240) showed weaker interaction compared to
that of the long Bcl2L12 truncated fragments (e.g.,
Bcl2L1270–334). Altogether, it was revealed that
Bcl2L12192–220 as a minimal region may play an important
role involved in the interaction between Bcl2L12 and Bcl-xL.
 | Table IThe results of yeast two hybrid
system to detect the minimal region responsible for interaction
with BclxL, Bcl2 and Bax. |
Table I
The results of yeast two hybrid
system to detect the minimal region responsible for interaction
with BclxL, Bcl2 and Bax.
| Protein interaction
with indicated construct in BD vector pAS2-1 |
|---|
|
|
|---|
| Bcl2L12 fragments
in AD vector pACT2 |
BclxL1–209 |
Bcl21–211 |
Bax1–171 | pAS2-1 |
|---|
|
Bcl2L1270–334 | B+ | B+ | B+ | - |
|
Bcl2L1270–266 | B+ | B+ | B+ | - |
|
Bcl2L1270–240 | B+ | B+ | B+ | - |
|
Bcl2L1290–229 | B+ | B+ | B+ | - |
|
Bcl2L1290–220 | B+ | B+ | B+ | - |
|
Bcl2L1270–117 | - | - | - | - |
|
Bcl2L1270–152 | - | - | - | - |
|
Bcl2L1270–191 | - | - | - | - |
|
Bcl2L12192–240 | B+ | B+ | B+ | - |
| pACT-2 | - | - | - | - |
Bcl2L12211–226 may contain a
BH3-like domain, which is response for the interactions with Bcl-xL
and Bcl2
Using a web-based program identifying
protein-protein interaction sites (ISIS) (32), we preferentially predicted the
possible crucial residues within the Bcl2L12192–220 that
are responsible for protein-protein interactions. In addition, we
predicted the protein 3D structure of Bcl2L12 (Fig. 1A). The residue of 1–173 contains
seven α-helixes, whereas residue of 174–334 contains nine
α-helixes. The predicted BH3-like domain corresponded to residue of
211–226. Moreover, we homologically overlapped the whole protein
structure of the studied Bcl2 family proteins (Fig. 1B), it was observed that Bcl2L12 was
only restrictedly overlapped with Bcl2, Bcl-xL and Bax in its
C-terminal region. Hence, we further magnified the overlapped
region of BH3 domain of these four proteins (Fig. 1C). As a result, BH3-like domain of
Bcl2L12 can homologically be overlapped with Bcl-xL and Bax, but
not Bcl2. The closed view of Bcl2L12 BH3-like domain revealed that
it comprised of an approximately seven-turn α-helix structure
(Fig. 1D). Based on multiple
sequence alignment and the protein structural simulations, we
conclude the region corresponding to α9 helix of Bcl2L12 may be
important and highly similar to the hydrophobic region (α2 helix,
BH3 domain) of the other Bcl2 family proteins (Table II). Further, we narrowed the
region down to Bcl2L12211–226 as BH3-like domain
(20) of Bcl2L12 and LXXXAE as
core motif other than canonical LXXXXD (33). We suspected this region may be
important and/or mediate the interactions with respect to Bcl-xL
and Bcl2 as shown in Fig. 2.
 | Figure 1The structural simulation of Bcl2L12
and comparison of BH3 Domain of studied Bcl2 family proteins. (A)
The predicted protein 3D structure of Bcl2L12. The N-terminal
protein of Bcl2L12 (homology modeling, 1–173 amino residues)
(orange), harboring a unique folding pattern with extra
coiled-coiled domain compared to others Bcl2 family proteins; in
addition, this region was suspected to be involved in Bcl2L12
biofunction. The BH3 domain of Bcl2L12 (BH3 domain, 211–226th amino
residues) (red). The region spanning from residues 174 to 334
(green). N, N-terminus, C, C-terminus. (B) The whole protein
structure of studied Bcl2 family proteins were homologically
overlapped, it was observed that Bcl2L12 only restrictedly
overlapped with Bcl2, Bcl-xL and Bax in its C-terminal region. (C)
The structure simulation of four Bcl2 family proteins using the
amino acid residues corresponding to α2 or α9 helix. The core
residues of BH3 domain of BAX [PDB ID, 1F16, 54–71 amino residues
(37)] (blue), the BH3 domain of
Bcl-xL [PDB ID, 1R2D, 90–98 amino residues (44)] (colored in magentas), the BH3
domain of Bcl2 [PDB ID, 2XA0, 93–108 amino residues (36)] (yellow) and the BH3 domain of
Bcl2L12 [homology modeling, 211–226 amino residues as BH3-like
motif as previously described (6)]
(red). (D) The closed view of Bcl2L12 BH3-like domain revealed that
it is comprised of approximately seven turns α-helix structure
(red). |
 | Figure 2Sequence of Bcl2L12 BH3-like domain
was aligned with BH3 domain of Bcl2 family proteins and the
interacting patterns between Bcl2L12 wt and Bcl2L12 mutants were
compared using yeast two-hybrid system. (A) Bcl2L12 harbors BH2 and
BH3-like domain, and the domain composition is different from other
members in Bcl2 family. However, the amino acid sequence of
BH3-like domain shares a certain level of similarity to BH3 domain
of different Bcl2 family proteins. The core motif of LXXXAE is
similar to LXXXXD. The chosen residues for performing site-directed
mutagenesis were indicated as I213 (h1), L217 (h2), L220 (h3), E222
(charge) and E224 (h4). Of note, pro-apoptotic Bcl2 members usually
sequestered by anti-apoptotic members through the interactions of
their BH3 domain with hydrophobic groove of anti-apoptotic Bcl2
family proteins. (B) Bcl2L1270–266 was used to perform
site-directed mutagenesis at the above sites, which correspond to
h1, h2, h3, charge and h4 in the BH3-like domain to determine
whether, and which site, is crucial for Bcl2L12 interacting with
Bcl-xL and Bcl2 using the yeast two hybrid system. B+,
positive interaction; −, no interaction. |
| Table IIComparing the domain structure of
Bcl2 family proteins in this study. |
Table II
Comparing the domain structure of
Bcl2 family proteins in this study.
| Anti-apoptotic | Pro-apoptotic | |
|---|
|
|
|
|---|
| Bcl-xL | Bcl2 | Bax | Bcl2L12 |
|---|
|
|
|
|---|
| BH-domain | Location of α-helix
(a.a.) | | BH-domain | Location of α-helix
(a.a.) | BH-domain | | Location of α-helix
(a.a.) |
|---|
| | | | | | | α1 | 12–15 |
| | | | | | | α2 | 20–31 |
| | | | | | | α3 | 37–41 |
| | | | | | | α4 | 84–91 |
| | | | | | | α5 | 95–110 |
| | | | | | | α6 | 127–146 |
| | | | | | | α7 | 160–163 |
| α1 | BH4 | 4–19 | 11–26 | | 22–37 | BH4-like | α8 | 175–190 |
| | | | | | domain? | | |
| α2 | BH3 | 84–99 | 91–106 | BH3 | 57–72 |
BH3-like | α9 | 211–226 |
| α3 | | 104–112 | 111–119 | | 77–85 | | α10 | 22–235 |
| α4 | BH1 | 120–132 | 127–139 | BH1 | 89–101 | BH1-like | α11 | 243–255 |
| | | | | | domain? | | |
| α5 | | 137–156 | 144–163 | | 107–126 | | α12 | 260–279 |
| α6 | | 162–176 | 169–183 | | 132–146 | | α13 | 285–299 |
| α7 | BH2 | 180–183 | 187–190 | BH2 | 150–153 | BH2 | α14 | 303–306 |
| α8 | | 188–195 | 195–202 | | 158–165 | | α15 | 311–322 |
| TM | | 213–231 | 219–237 | | 169–188 | | α16 | 326–328 |
For delineating whether Bcl2L12 BH3-like domain
serves its function similarly to Bax BH3 domain, we performed
site-directed mutagenesis on several residues within the helix
interacting surface (h1, h2, h3 and h4) (34) and we firstly predicted interacting
residue (20) corresponding to Bax
BH3 domain since mutations on these residues in Bax have been
reported to reduce Bax-Bcl-xL interaction. We compared the
interacting patterns of Bcl2L12 wild-type (wt) and mutants with
respect to Bcl-xL and Bcl2; in addition, the interacting pattern of
Bax BH3 mutant was investigated and compared. As expected, the
interacting patterns between Bax BH3 domain and Bcl2L12 BH3-like
domain did show high similarity (Table III), but Bcl2L12 may have
different binding manner between Bcl-xL and Bcl2, indicating that
BH3 domain may not be equally responsible for Bcl2L12-BclxL and
Bax-Bcl-xL interaction (Table
III, BclxL1–209 L90A in lines 1 and 3 compare to
Bcl21–211 L97A in lines 1 and 3). Moreover, it was
revealed that h2 residue in BH3-like domain is important for
Bc2L1270–266 interacting with Bcl-xL1–209 and
possibly with Bcl21–211. Besides, the h2 residue is also
crucial for Bax1–171 interacting with
Bcl21–211. It is of note that when expressed these
proteins, in yeast colonies took 3–5 days to grow to an appropriate
size for filter assay. The colonies also showed a high variety in
growth speed and we presumed this phenomenon may due to, in part,
either to weaker interactions of tested proteins or a negative side
effect of yeast growth. The 3D structure simulation showed that
structure computerized using sequences from Bcl2L12 BH3-like to BH2
domain was similar to that of Bcl-xL, Bcl2 and Bax (35–37),
indicating that BH3-like domain may have the same function as BH3
domain of Bcl2 family proteins, especially Bax.
| Table IIIYeast two hybrid system detection of
the critical residue affecting Bcl2L12 and/or Bax interaction with
BclxL and Bcl2. |
Table III
Yeast two hybrid system detection of
the critical residue affecting Bcl2L12 and/or Bax interaction with
BclxL and Bcl2.
|
BclxL1–209 |
BclxL1–209 L90A |
Bcl21–211 |
Bcl21–211 L97A |
|---|
|
Bcl2L1270–266 | B+ | B+ | B+ | - |
|
Bcl2L1270–266 L213A | - | - | - | - |
|
Bax1–171 | B+ | B+ | B+ | - |
| Bax1–171
L63A | - | - | - | - |
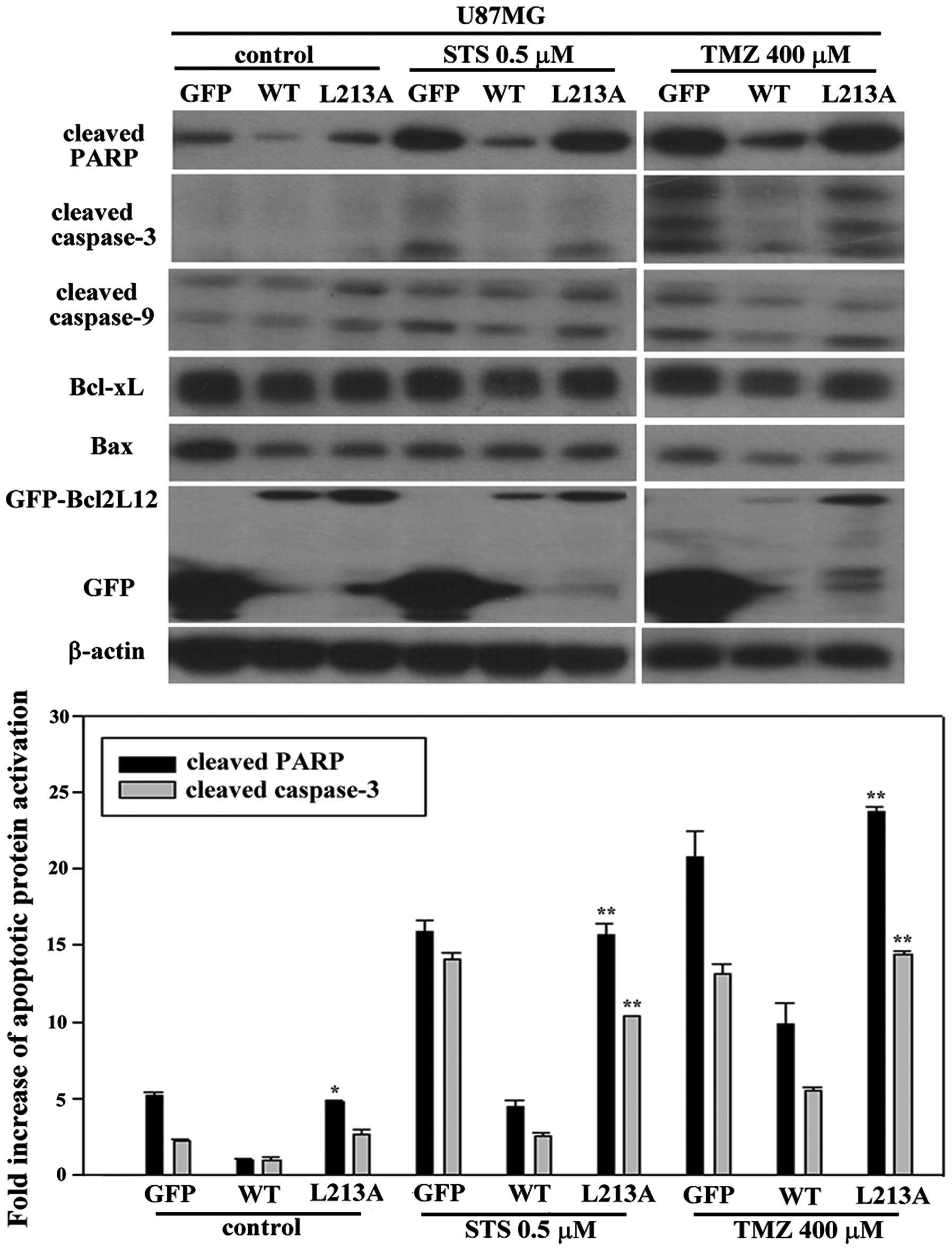
Bcl2L12 L213A and L217A mutant can
abolish its anti-apoptosis role and cause re-activations of
apoptotic markers in GBM cell line
U87MG cell line with expression of GFP alone,
GFP-Bcl2L12 wt and GFP-Bcl2L12 L213A (h1 mutant), were treated with
either STS (0.5 μM) or TMZ (400 μM) for 24 and 48 h, respectively,
to induce apoptosis. Regardless of the treatment drug, Bcl2L12 wt
showed a lower activation of cleaved-PARP than other groups. When
overexpressed Bcl2L12 L213A, apoptotic markers were significantly
reactivated during treatment with either STS or TMZ (Fig. 3). This result is consistent with
the reported anti-apoptotic role of Bcl2L12 in GBM. Additionally,
mutation on one of the residues (L213) of core motif did abolish
anti-apoptotic role of Bcl2L12, which caused a significant
elevation of apoptotic markers including cleaved-PARP and capase-3
in U87MG (Fig. 3). No inter-group
difference was found between Bcl2L12 wt and L213 mutant group in
expression levels with respect to Bcl-xL, Bax and ratio of
Bax/Bcl-xL. To confirm this finding, we further overexpressed
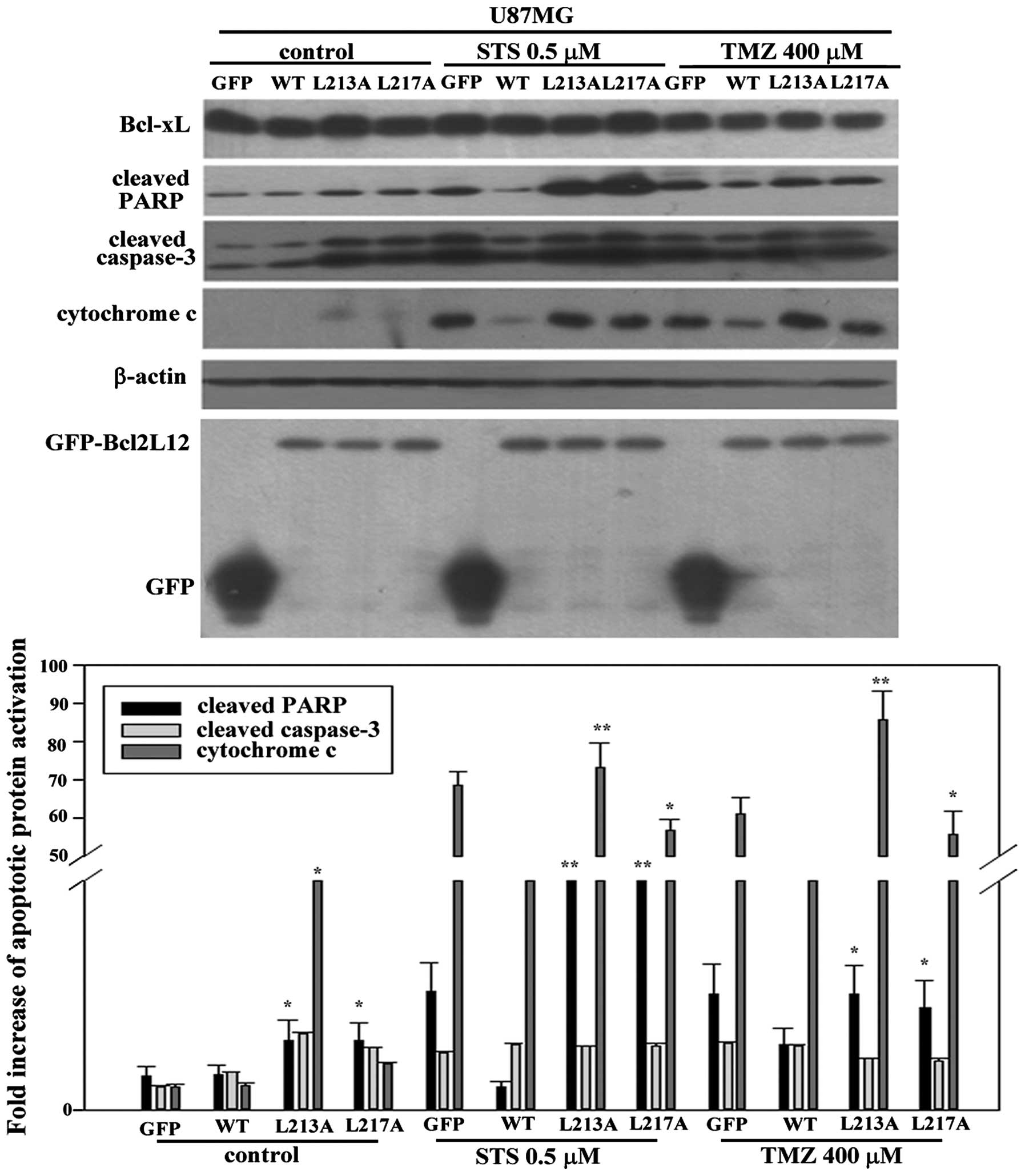
another BH3-like mutant domain, h2 mutant (L217A) in U87MG cells of
appropriate size. As shown in Fig.
4, without drug treatment, both mutant groups already showed a
significantly higher expression of cleaved-PARP compared to Bcl2L12
wt group. However, only h1 mutant showed an evidently higher
expression of cytochrome c efflux in untreated control.
Using either STS or TMZ caused severe activation of apoptotic
markers including cleaved-PARP and cytochrome c, but not
caspase-3 in GFP alone group. In contrast, overexpressed Bcl2L12 wt
prohibited activation of these apoptotic markers. Further, as
expectated, overexpression of either h1 or h2 mutant exhibited a
reactivation of cleaved-PARP and cytochrome c efflux
compared to Bcl2L12 wt group (Fig.
4, lower panel) while Beclin-1 expression in each group seems
unaltered.
Combination of TMZ and ABT-737 exerts a
superior apoptosis triggering effect in the glioma cell line
For the purpose of evaluating whether Bcl2L12 is
promising for target therapy, we introduced ABT-737, a BH3 mimetic
agent, to counteract the anti-apoptotic role of Bcl2L12 in glioma
since: i) glioma was usually detected with overexpression of
Bcl2L12; ii) we speculated Bcl2L12 harbors a functional BH3-like
domain, and may play a role similar to that reported for
hydrophobic groove of anti-apoptotic members in Bcl2 family. The
results in Fig. 5, indicate no
inter-group difference. Consistently, using either TMZ or ABT-737,
overexpressed h2 mutant caused an elevated expression of
cleaved-PARP compared to Bcl2L12 wt group. However, using ABT-737
alone, it successfully triggered cleaved caspase-3 expression
although it had not reached significant level. The combination with
TMZ and ABT-737, it activated approximately 2- and 3-fold
enhancement of cleaved-PARP and cleaved caspase-3 expression,
respectively. When compared to single drug treatment, it triggered
significantly higher activation of apoptotic markers compared to
other Bcl2L12 wt overexpressing groups. These data revealed that
mutation on L217 may partially destroy the linked functionalities
of BH3-like domain of Bcl2L12 through a dominant-negative machinery
resulting in marginal benefit on re-activation of apoptotic markers
when treated with TMZ. Implicating ABT-737 may supply another tool
to minimize the functional consequences of BH3-like domain of
Bcl2L12 as well as the hydrophobic groove of others anti-apoptotic
Bcl2 members. The combination of TMZ and ABT-737 provide a good
microenvironment of ABT-737 and TMZ to take action resulting in the
best outcome of triggering apoptosis in the U87MG cell line.
Discussion
In this study, we demonstrated, for the first time,
that Bcl2L12 plays an anti-apoptotic role in a glioma cell line
partly due to harboring a BH3-like domain and through its
interaction with Bcl-xL and Bcl2. This finding is independent of
the previously reported mechanisms of p53-dependent or caspase-3/7
inactivation (24,38–40).
Mutagenesis on either h1 or h2 residue of BH3-like domain within
the hydrophobic groove of Bcl2L12 caused re-activation of apoptotic
markers in U87MG cell line with or without treatment with STS or
TMZ. These data may provide a new strategy to intervene in those
GBM with highly expressing Bcl2L12 through attenuating its BH3-like
domain, such as implying the BH3 mimetic agent ABT-737, ABT-263 and
analogs corresponding to the Bcl2L12211–226. The
possible interaction between Bcl2L12, Bcl-xL and Bcl2 may be
important to the anti-apoptotic the role of Bcl2L12; however, this
event is unable to deny that p53 may associated with role playing
of Bcl2L12 since the reactivation of apoptotic markers of h1 mutant
is more evident in U87MG cells (with wild-type p53) than in T98G
cells (with p53 codon 237 homozygous mutant) (data not shown). Our
finding of h1, h2 and h4 as important residues in the core motif
(position −4, 0, and +7) of BH3-like domain of Bcl2L12 may reflect
that they serve to interact with Bcl-xL and Bcl2. Interestingly,
mutations on corresponding residues of Bax also disrupted the
interaction with Bcl2 pro-survival members leading to failure in
inducing apoptosis (30).
According to results from the yeast two-hybrid
system, full-length Bcl2L12 was unable to interact with Bcl-xL,
Bcl2 and Bax until depleting residues 1–70 of its N-terminal. The
failure of interaction may be attributed to the Bcl2L12 stereo
structural barrier, which similarly to the situation of Bcl2L12
interacts with GSK3β (23). This
region of Bcl2L121–70 perhaps has some regulatory role
on controlling the Bcl2L12 activity and/or interaction. In this
study, we did not examine all the interactions between Bcl2L12,
Bcl-xL, Bcl2 and Bax due to problems attributed to secondary
generation yeast two-hybrid assay. Some of the proteins, such as
Bcl-xL and Bcl2, can only be cloned into BD vector and further
tested with other proteins. When cloned into the AD vector, the
ability of some proteins was reported to be decreased
significantly. The problems of directional specific interaction of
some proteins should be noted when performing yeast two-hybrid
system to avoid both false-negative and -positive results (41). The incomplete evaluation of the
interaction between Bcl2L12, Bcl-xL, Bcl2 and Bax remains as an
experimental limitation in this study.
Protein levels with respect to Bcl-xL and Bax did
not show a consistent result with apoptotic markers in our western
blotting, and these results may be attributed to the mechanism of
STS. STS is the broad-spectrum kinase inhibitor that is frequently
used as apoptosis inducer in the cell-based assay. However, it may
inhibit the phosphorylation on Bcl2 family members to affect the
level of these proteins. Moreover, STS induce more abundantly
caspase-dependent apoptotic events in the primary cortical neurons
(42). This can be part of the
reason why the effect attributed to protein-protein interaction of
Bcl2 family proteins is lacking. Therefore, other apoptosis inducer
may be used to study variations on the Bcl2 family protein level
consequent on overexpressed Bcl2L12 wt and BH3-like domain
mutants.
We assumed that, in GBM, the Bcl2L12 is
overexpressed and plays an anti-apoptotic role due to, in part,
through interacting with Bcl-xL and Bcl2 via a novel BH3-like
domain (in this study) to reinforce the integrity of the
mitochondria outer membrane. A previous study mentioned that TMZ
treatment in GBM may cause autophagy and high ROS production due to
ERK1 signaling-mediated autophagy induction (29). We have repeated this phenomenon,
and assumed that TMZ-induced both apoptosis and autophagy in a
time-dependent manner, but with a cell type specificity as shown in
Fig. 6. In U87MG cell line, the
elevated expression of Beclin-1 and LC3B was detected at 12 and 24
h, respectively. The expression level of Bcl-xL also gradually
accumulated from 12 to 72 h. In contrast, the expression level of
cleaved-PARP and cleaved caspase-9 was enhanced until 36 h. The
TMZ-induced autophagy to apoptosis shift can be observed in U87MG
cell line, but not in T98G when treated with TMZ. To confirm this
finding, we perform time-course treatment of TMZ using U87MG cell
line, and the TMZ-induced autophagy to apoptosis shift was robustly
demonstrated (Fig. 7, upper
panel), in which ATG5, Beclin-1 and LC3B all induced expression at
early period from 12 to 36 h, but cleaved-PARP was gradually
activated beyond 24 h and highest expressed at 72 h. In addition,
we checked mRNA expression pattern with respect to LC3B, Beclin-1
and Bcl2L12 during the trajectory of TMZ treatment showing a
consistent rhythm (Fig. 7, lower
panel), which reached the highest expression at 36 h. These data
indicated that Bcl2L12 may be involve in TMZ-induced autophagy. In
contrast to previous finding, overexpressed Bcl2L12 wt may cause
elevation of Beclin-1 and LC3B, but not cleaved caspase-9 (Fig. 8). Moreover, this phenomenon of
autophagic marker induction can be reversed by Bcl2L12 h1 and h2
mutant (Fig. 8), which indicated
that BH3-like domain, in addition to apoptosis regulation, was also
involved in TMZ-induced autophagy.
In this study, we tested the beneficial outcome for
the combination of TMZ and ABT-737. As expected, it provided a
superior apoptosis triggering effect than used alone. Moreover, it
successfully counteracted that Bcl2L12 wt overexpressing background
and produced the best result. Our above data demonstrated that h2
mutant may partially destroy the linked functionalities of BH3-like
domain of Bcl2L12 through a dominant-negative machinery, which
probably raised a functional competition with the endogenous
Bcl2L12. When treated with ABT-737, the functionality of BH3-like
domain of Bcl2L12, hydrophobic groove of other Bcl2 family protein
or BH3 targets may be blocked, and thus an advanced apoptosis
inducing effect was observed. The combination of TMZ and ABT-737
provides a good microenvironment to take actions resulting in the
best outcome of triggering apoptosis in the U87MG cell line. This
phenomenon can be rationalized as follows: i) in U87MG cell line
with high Bcl2L12 expression, the TMZ may not work well due to: a)
Bcl2L12 helps glioma cells to abrogate the p53-dependent DNA
damaging apoptosis event, b) Bcl2L12 is involved in TMZ-induced
autophagy and lead to an acquired resistance of TMZ, and c) Bcl2L12
mediates shut-down of caspase-3/-7 activation and upregulated the
expression of CRYAB, and thus the caspase-dependent apoptosis might
not be easy to launch. However, we found that TMZ can decrease the
expression Mcl-1 (data not shown), which alternatively created a
good environment for ABT-737 to take action without interfering.
The extent of the high Mcl-1 expression would restrict the
therapeutic effect of ABT-737 as described (43). Likewise, siRNA-based knock down
Mcl-1 expression or implicated pharmaceutical-based (using platinum
agent) drug all improved ABT-737 resistance; ii) ABT-737 as BH3
mimetic agent, may be suitable to counteract an anti-apoptotic role
of Bcl2L12 in glioma that retains an acquired resistance to TMZ.
Abolishing the self-defence of glioma due to Bcl2L12
overexpression, it would be beneficial for TMZ to damage the tumor
genome; iii) ABT-737 is broadly antagonists to BH3 targets through
blocking the interaction between anti-apoptotic Bcl2 family
proteins and BH3 provides advantages in triggering more dominant
apoptosis. Altogether, the special characteristics of GBM
overexpressing Bcl2L12 retain an acquired resistance of TMZ
providing ideal conditions for combination use of TMZ and ABT-737.
Nonetheless, further insight into the Bcl2L12 multifaceted role in
GBM is still needed since it is involved in regulation of apoptosis
and autophagy. Eventually, a practical and effective regimen can be
developed based on better knowledge of Bcl2L12.
In conclusion, we demonstrated that Bcl2L12 has a
BH3-like domain, and it functions similarly to BH3 domain of Bcl2
family proteins in binding. Interrupted Bcl2L12 interaction with
Bcl2 family members can modulate its anti-apoptotic role based on
re-activation of apoptotic markers in GBM cell line. The
combination TMZ and ABT-737 exerts a better apoptosis induction
than each used alone. We conclude that ABT-737 has potential in
combination with TMZ to sensitize the drug responsiveness of
glioma.
Acknowledgements
We thank Dr Lynda Chin for providing us with the
full length of Bcl2L12 plasmid. This study was sponsored by
National Science Council-101-2320-B-037-036-MY3, National Science
Council-102-2633-B-037-001, National Health Research
Intitute-EX100-9809SI (Taiwan), National Sun Yet-San
University-Kaohsiung Medical University Joint Research
Project-102-P037 and -103-P025 to Yi-Ren Hong, National Science
Council-101-2314-B-037-017 (Taiwan) to Shen-Long Howng and
103-CMRPG8C0891 to An-Kuo Chou.
Abbreviations:
|
GBM
|
glioblastoma
|
|
MOM
|
mitochondrial outer membrane
|
|
BH
|
Bcl2 homology
|
|
TM
|
transmembrane
|
|
STS
|
staurosporine
|
|
TMZ
|
temolozomide
|
|
h
|
hydrophobic
|
|
Bcl2L12
|
Bcl2-like-protein 12
|
|
PxxP
|
proline-rich region
|
|
CRYAB
|
αB-crystallin
|
References
|
1
|
Van Meir EG, Hadjipanayis CG, Norden AD,
Shu H-K, Wen PY and Olson JJ: Exciting new advances in
neuro-oncology: the avenue to a cure for malignant glioma. CA
Cancer J Clin. 60:166–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Surawicz TS, McCarthy BJ, Kupelian V,
Jukich PJ, Bruner JM and Davis FG: Descriptive epidemiology of
primary brain and CNS tumors: results from the Central Brain Tumor
Registry of the United States, 1990–1994. Neuro Oncol. 1:14–25.
1999.
|
|
3
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
5
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar
|
|
7
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Green DR and Chipuk JE: Apoptosis: stabbed
in the BAX. Nature. 455:1047–1049. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng EH, Wei MC, Weiler S, et al: BCL-2,
BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and
BAK-mediated mitochondrial apoptosis. Mol Cell. 8:705–711. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim H, Rafiuddin-Shah M, Tu H-C, et al:
Hierarchical regulation of mitochondrion-dependent apoptosis by
BCL-2 subfamilies. Nat Cell Biol. 8:1348–1358. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wei MC, Lindsten T, Mootha VK, et al:
tBID, a membrane-targeted death ligand, oligomerizes BAK to release
cytochrome c. Genes Dev. 14:2060–2071. 2000.PubMed/NCBI
|
|
13
|
Wei MC, Zong WX, Cheng EH, et al:
Proapoptotic BAX and BAK: a requisite gateway to mitochondrial
dysfunction and death. Science. 292:727–730. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Willis SN, Chen L, Dewson G, et al:
Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2,
until displaced by BH3-only proteins. Genes Dev. 19:1294–1305.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Walensky LD: Playing fullBAK. Cell Cycle.
12:1333–1334. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Certo M, Del Gaizo Moore V, Nishino M, et
al: Mitochondria primed by death signals determine cellular
addiction to anti-apoptotic BCL-2 family members. Cancer Cell.
9:351–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kuwana T, Bouchier-Hayes L, Chipuk JE, et
al: BH3 domains of BH3-only proteins differentially regulate
Bax-mediated mitochondrial membrane permeabilization both directly
and indirectly. Mol Cell. 17:525–535. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Letai A, Bassik MC, Walensky LD,
Sorcinelli MD, Weiler S and Korsmeyer SJ: Distinct BH3 domains
either sensitize or activate mitochondrial apoptosis, serving as
prototype cancer therapeutics. Cancer Cell. 2:183–192. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim H, Tu H-C, Ren D, et al: Stepwise
activation of BAX and BAK by tBID, BIM, and PUMA initiates
mitochondrial apoptosis. Mol Cell. 36:487–499. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sattler M, Liang H, Nettesheim D, et al:
Structure of Bcl-xL-Bak peptide complex: recognition between
regulators of apoptosis. Science. 275:983–986. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Walensky LD: BCL-2 in the crosshairs:
tipping the balance of life and death. Cell Death Differ.
13:1339–1350. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scorilas A, Kyriakopoulou L, Yousef GM,
Ashworth LK, Kwamie A and Diamandis EP: Molecular cloning, physical
mapping, and expression analysis of a novel gene, BCL2L12, encoding
a proline-rich protein with a highly conserved BH2 domain of the
Bcl-2 family. Genomics. 72:217–221. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chou C-H, Chou A-K, Lin C-C, et al: GSK3β
regulates Bcl2L12 and Bcl2L12A anti-apoptosis signaling in
glioblastoma and is inhibited by LiCl. Cell Cycle. 11:532–542.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stegh AH, Brennan C, Mahoney JA, et al:
Glioma oncoprotein Bcl2L12 inhibits the p53 tumor suppressor. Genes
Dev. 24:2194–2204. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hong Y, Yang J, Wu W, et al: Knockdown of
BCL2L12 leads to cisplatin resistance in MDA-MB-231 breast cancer
cells. Biochim Biophys Acta. 1782:649–657. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thomadaki H, Talieri M and Scorilas A:
Prognostic value of the apoptosis related genes BCL2 and BCL2L12 in
breast cancer. Cancer Lett. 247:48–55. 2007. View Article : Google Scholar
|
|
27
|
Florou D, Papadopoulos IN and Scorilas A:
Molecular analysis and prognostic impact of the novel apoptotic
gene BCL2L12 in gastric cancer. Biochem Biophys Res Commun.
391:214–218. 2010. View Article : Google Scholar
|
|
28
|
Furnari FB, Fenton T, Bachoo RM, et al:
Malignant astrocytic glioma: genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin CJ, Lee CC, Shih YL, et al:
Resveratrol enhances the therapeutic effect of temozolomide against
malignant glioma in vitro and in vivo by inhibiting autophagy. Free
Radic Biol Med. 52:377–391. 2012. View Article : Google Scholar
|
|
30
|
Czabotar PE, Lee EF, Thompson GV, Wardak
AZ, Fairlie WD and Colman PM: Mutation to Bax beyond the BH3 domain
disrupts interactions with pro-survival proteins and promotes
apoptosis. J Biol Chem. 286:7123–7131. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Roy A, Xu D, Poisson J and Zhang Y: A
protocol for computer-based protein structure and function
prediction. JoVE. e32592011.PubMed/NCBI
|
|
32
|
Yachdav G, Kloppmann E, Kajan L, et al:
PredictProtein - an open resource for online prediction of protein
structural and functional features. Nucleic Acids Res.
42:W337–W343. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bhat V, Olenick MB, Schuchardt BJ, Mikles
DC, McDonald CB and Farooq A: Molecular determinants of the binding
specificity of BH3 ligands to BclXL apoptotic repressor.
Biopolymers. 101:573–582. 2014. View Article : Google Scholar
|
|
34
|
Czabotar PE, Westphal D, Dewson G, et al:
Bax crystal structures reveal how BH3 domains activate Bax and
nucleate its oligomerization to induce apoptosis. Cell.
152:519–531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wysoczanski P, Mart RJ, Loveridge EJ, et
al: NMR solution structure of a photo-switchable apoptosis
activating Bak peptide bound to Bcl-xL. J Am Chem Soc.
134:7644–7647. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Petros AM, Medek A, Nettesheim DG, et al:
Solution structure of the antiapoptotic protein bcl-2. Proc Natl
Acad Sci USA. 98:3012–3017. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Suzuki M, Youle RJ and Tjandra N:
Structure of Bax: coregulation of dimer formation and intracellular
localization. Cell. 103:645–654. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stegh AH, Kim H, Bachoo RM, et al: Bcl2L12
inhibits post-mitochondrial apoptosis signaling in glioblastoma.
Genes Dev. 21:98–111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stegh AH, Kesari S, Mahoney JE, et al:
Bcl2L12-mediated inhibition of effector caspase-3 and caspase-7 via
distinct mechanisms in glioblastoma. Proc Natl Acad Sci USA.
105:10703–10708. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stegh AH and DePinho RA: Beyond effector
caspase inhibition: Bcl2L12 neutralizes p53 signaling in
glioblastoma. Cell Cycle. 10:33–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hong YR: False positive: detection and
elimination. Yeast Hybrid Methods. Zhu L and Hannon G: Eaton
publishing; Natick, MA: pp. 101–113. 2000
|
|
42
|
Higgins GC, Devenish RJ, Beart PM and
Nagley P: Autophagic activity in cortical neurons under acute
oxidative stress directly contributes to cell death. Cell Mol Life
Sci. 68:3725–3740. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Simonin K, N’Diaye M, Lheureux S, et al:
Platinum compounds sensitize ovarian carcinoma cells to ABT-737 by
modulation of the Mcl-1/Noxa axis. Apoptosis. 18:492–508. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oltersdorf T, Elmore SW, Shoemaker AR, et
al: An inhibitor of Bcl-2 family proteins induces regression of
solid tumours. Nature. 435:677–681. 2005. View Article : Google Scholar : PubMed/NCBI
|