Introduction
Breast cancer remains the most frequent tumor and
the leading cause of cancer-related deaths among the female
population worldwide (1). Breast
cancers are classified on the basis of three protein expression
markers: estrogen receptor (ER), progesterone receptor (PgR), and
human epidermal growth factor receptor 2 (HER2). In the standard
chemotherapies, therefore, agents targeting anti-hormone, anti-HER2
or other expression of various receptors have been selected
(2,3). Hormonal therapies can be effective in
treating tumors that express the hormone receptors ER or PgR
(2), and HER2-directed therapies
are useful for tumors that express HER2 (4). However, the treatment options for
triple-negative breast cancers (TNBC), which do not express ER, PgR
or HER2 and are detected in 15% of breast cancers (5), are restricted to chemotherapy, even
for radiation and surgery (6).
Actually, only a limited number of effective treatments are
available to patients with advanced-stage TNBC (7), and prognosis in patients with TNBC is
much worse than that in patients with non-TNBC. For example, the
3-year disease-free rate after certain therapy in patients with
TNBC was 63%, significantly lower than the 76% in those with
non-TNBC (8). Recent studies have
identified molecular subtypes in TNBC from the understanding of
biologic heterogeneity that are leading to additional potential
therapeutic targets for evaluation (9). However, in this era of personalized
medicine (10), there have been no
trials of targeted agents demonstrating significant benefit for
patients with any subtype of TNBC (11). Local treatment has been the focus
recently as a next step in obtaining further favorable treatments
for TNBC. Indeed, ablation therapy with radiofrequency energy
(12) or cryotherapy (13,14)
is an emerging minimally invasive therapy for small and localized
breast cancers. However, the rate of overall local response is
still low, at 48% even when combined with liposomal doxorubicin for
unresectable chest wall recurrences of breast cancer (15). Further, problematic complications
of skin fibrosis after radiofrequency ablation continue to occur
(6), indicating the difficulty in
selecting a satisfactory device.
Menadione (vitamin K3: VK3) is chemically
synthesized to have anticancer effects against various cancers
(16,17), but its mechanism remains unclear.
In vivo studies revealed that menadione acts as a strong
chemo-sensitizer when administered in combination with various
conventional chemotherapeutic agents in different cancer models
(18), and it is active against
doxorubicin-resistant leukemia cells in rats (19). On squamous cells, VK3 was found to
have limited cytotoxic effects on malignant cells, but not normal
tissue, from the observation in which the half maximal inhibitory
concentration (IC50) for cancer and normal cells was
8.45 and 98.5 μM, respectively (18). In several trials of modulated
chemotherapy, VK3 has so far been combined with the alkylating
agent mitomycin C (20–22). According to the clinical results of
23 patients with advanced lung cancer, serious complications over
grade 3, such as hematologic toxicity or pneumonia, were detected
in 31%, despite an objective response rate of just 9% (21). In addition, in 43 cases of
gastrointestinal cancer, 27% of patients had side effects over
grade 3, such as hemolytic anemia or uremic syndrome, without any
objective responses (22). Because
of its strong toxicity, VK3 has been shown to cause a variety of
side effects when administered systemically. However, local
treatment is still expected to lead to satisfactory outcomes for
body surface-type cancers such as breast cancer, even for TNBC. To
evaluate the significance of this novel treatment concept, the
biological responses induced by VK3 should be considered with the
understanding of the features of breast cancer. Therefore, in the
present study, the development of ablation therapy combined with
VK3 will be estimated from not only observation of the mechanism
but also by specific factors to predict the anti-tumor effect of
this therapy.
Materials and methods
Cell lines and culture conditions
Cells from the MCF-7, SK-BR3, BT474 and MDA-MB-231
human breast carcinoma cell lines were obtained from the American
Type Culture Collection (ATCC, Manassas, VA, USA). Cell were
cultured in DMEM medium (Wako, Osaka, Japan) supplemented with 10%
heat-inactivated fetal bovine serum (FBS), 4 mM non-essencial amino
acid (NEAA), 4 mM L-glutamine and 1%
penicillin-streptomycin-amphotericin solution (all from
Sigma-Aldrich, St. Louis, MO, USA) in a humidified atmosphere of 5%
CO2/95% air at 37°C. Cells were passaged once a
week.
Cell proliferation assay
Cell growth was assessed by a standard 3-(4,
5-dimethyl-thiazol-2-yl)-2,5-dephenyltetrazolium bromide (MTT)
assay (23,24), which detects the dehydrogenase
activity in viable cells. A total of 1×104 MDA-MB-231
cells were seeded into each of the 96-well culture plates overnight
and kept in a humidified atmosphere of 5% CO2 and 95%
air at 37°C. After 24-h incubation, diluted menadione (5–100 μM)
was added to each well of the culture medium. After 24, 48 and 72
h, the culture medium was removed, and 100 μl of a 0.5-mg/ml
solution of MTT (Sigma-Aldrich) was added to each well. The plates
were then incuvated for 4 h at 37°C. The culture medium was
replaced with 100 μl of dymethyl sulfoxide (Wako) per well, and the
absorbance at the 540-nm wavelength was measured using a 2104
EnVision Multilabel Reader (Perkin-Elmer, Waltham, MA, USA).
Detection of reactive oxygen species
(ROS)
We used the Total ROS detection kit® to
detect intracellular ROS. A total of 6×105 MDA-MB-231
cells were seeded into each of the 6-well culture plates overnight
and kept in a humidified atmosphere of 5% CO2 and 95%
air at 37°C. After 24-h incubation, the cells were simultaneously
treated with an experimental test agent (or control) and loaded
with the same volume of the ROS detection solution. The ROS
detection mix was carefully removed from the glass slides by gently
tapping against layers of paper towel, or from the tissue culture
plates The cells were washed cells twice with 1× wash buffer,
adding a few drops of 1× wash buffer, and immediately overlayed
with a cover slip and observed under a fluorescence/confocal
microscope using standard excitation/emission filter sets.
Oxidative stress detection requires a filter set compatible with
fluorescein (Ex/Em: 490/525 nm).
Western blot analysis and antibodies
Treatment of the specimen was as described
previously (25–27). The cell lysate was boiled in sample
buffer solution (Wako). Total cell protein extracts (20 μg/lane)
were separated by sodium dodecylsulface-polyacrylamide gel
electrophoresis using SuperSep™ (Wako) and were electrophoretically
transfected onto polyvinyl difluoride menmbranes. The membranes
were blocked with PVDF blocking reagent (Toyobo, Osaka, Japan) for
1 h. The membrane was then incubated with primary antibodies
against β-actin, phosphorylated type of extracellular
signal-regulated kinase (p-ERK), c-jun N-terminal kinase (p-JNK),
p-AKT, mammalian target of rapamycin (p-mTOR), hepatocyte growth
factor receptor (p-cMet), epithelial growth factor receptor
(p-EGFR) and caspase-3 (1:5,000; Cell Signaling Technology,
Danvers, MA, USA) and EGFR inhibitor (1:5,000; Calbiochem 324674,
Merck KGaA, Germany) overnight at 4°C. The primary antibodies were
diluted with Can Get Signal Solution 1 (Toyobo). The membrane were
then washed with Dako washing buffer (Dako, Glostrup, Denmark) and
incubated with the appropriate secondary antibodies (1:25,000;
Millipore, Darmstadt, Germany), which were diluted with Can Get
Signal Solution 2. The immunoreactive proteins were visualized by
chemiluminescence using a LAS-4000 (Fuji film, Tokyo, Japan) and
analyzed using ImageJ software (NIH, Bethesda, MD, USA).
Animals and in vivo experiments
Female 5-week-old BALB/c nu/nu mice were purchased
from SRL (Hamamatsu, Japan) and housed in the animal facilities of
the Division of Animal Experiment, Life Science Research Center,
Gifu University (Gifu, Japan) with free access to water and food. A
subcutaneous tumor model of breast cancer was created by injection
of 2.0×107 MDA-MB-231 cells into the back of nude mice
as described previously (28). At
21 days after injection, menadione was injected into subcutaneous
tumors and the tumor size was measured each day. After 3–4 weeks,
the subcutaneous xenografts were injected with PBS, ethanol or
menadione, removed and evaluated by western blot analysis in an
immunohistochemical study. Animal experiments in this study were
performed in compliance with the guidelines of the Institute for
Laboratory Animal Research, Gifu University Graduate School of
Medicine, and the UCCCR Guidelines for the Welfare of Animal in
Experimental Neoplasia.
Statistical analysis
The data were examined using the Student t-test,
Chi-square test, and ANOVA or Kruskal-Wallis test (with appropriate
post hoc analysis for multiple comparisons) to determine
statistical significances. The p-values <0.05 were regarded as
statistically significant.
Results
In vitro experiments for the VK3-induced
signal pathway
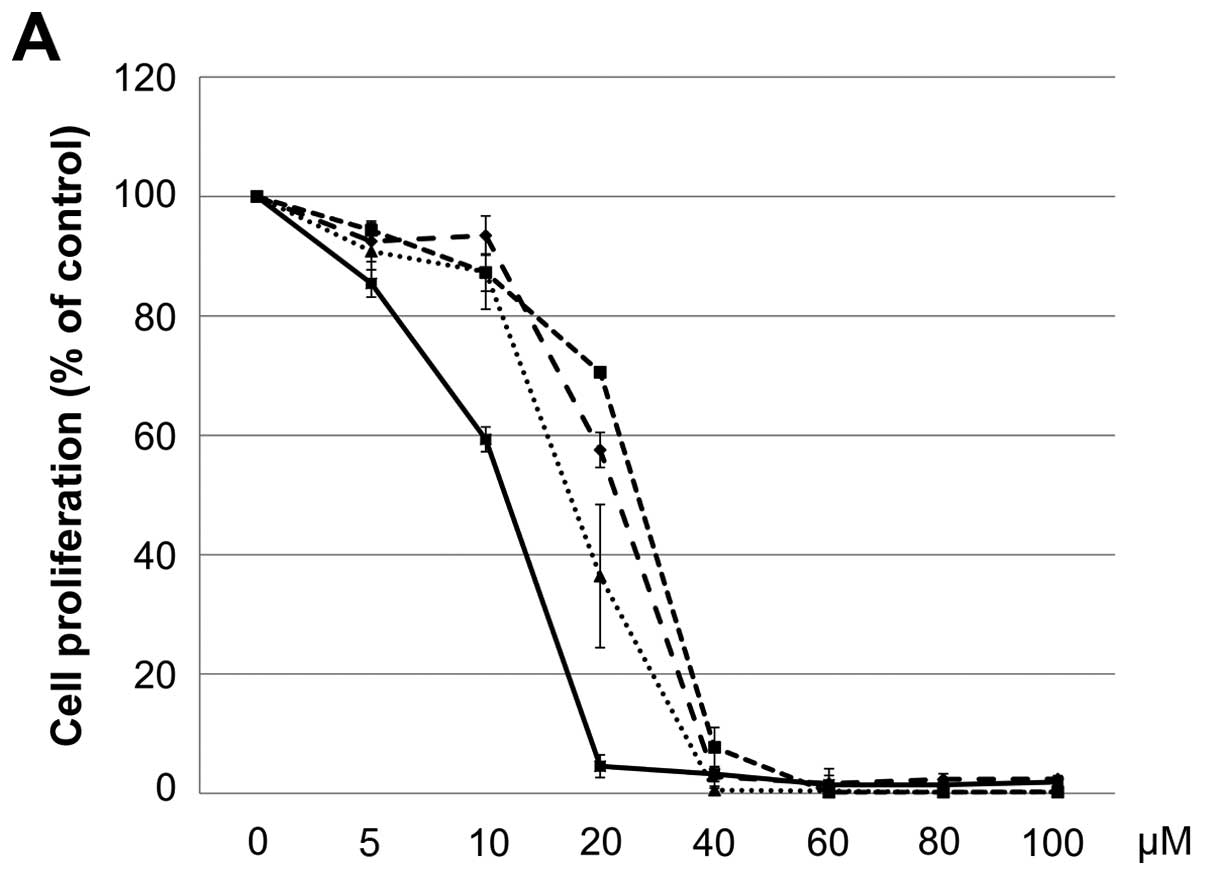
The IC50 (μM) of VK3 was calculated as
25.1±0.2, 22.0±0.7, 11.3±0.3 and 17.0±2.7 on MCF7, BT474, SK-BR3
and MDA-MB-231, respectively (Fig.
1A). Of the treated cell lines, MDA-MB-231 was typed as TNBC
based on PCR experiments (Fig. 1B)
and protein expression by western blotting (data not shown). Then,
the VK3-mediated signaling pathway on the receptors and cellular
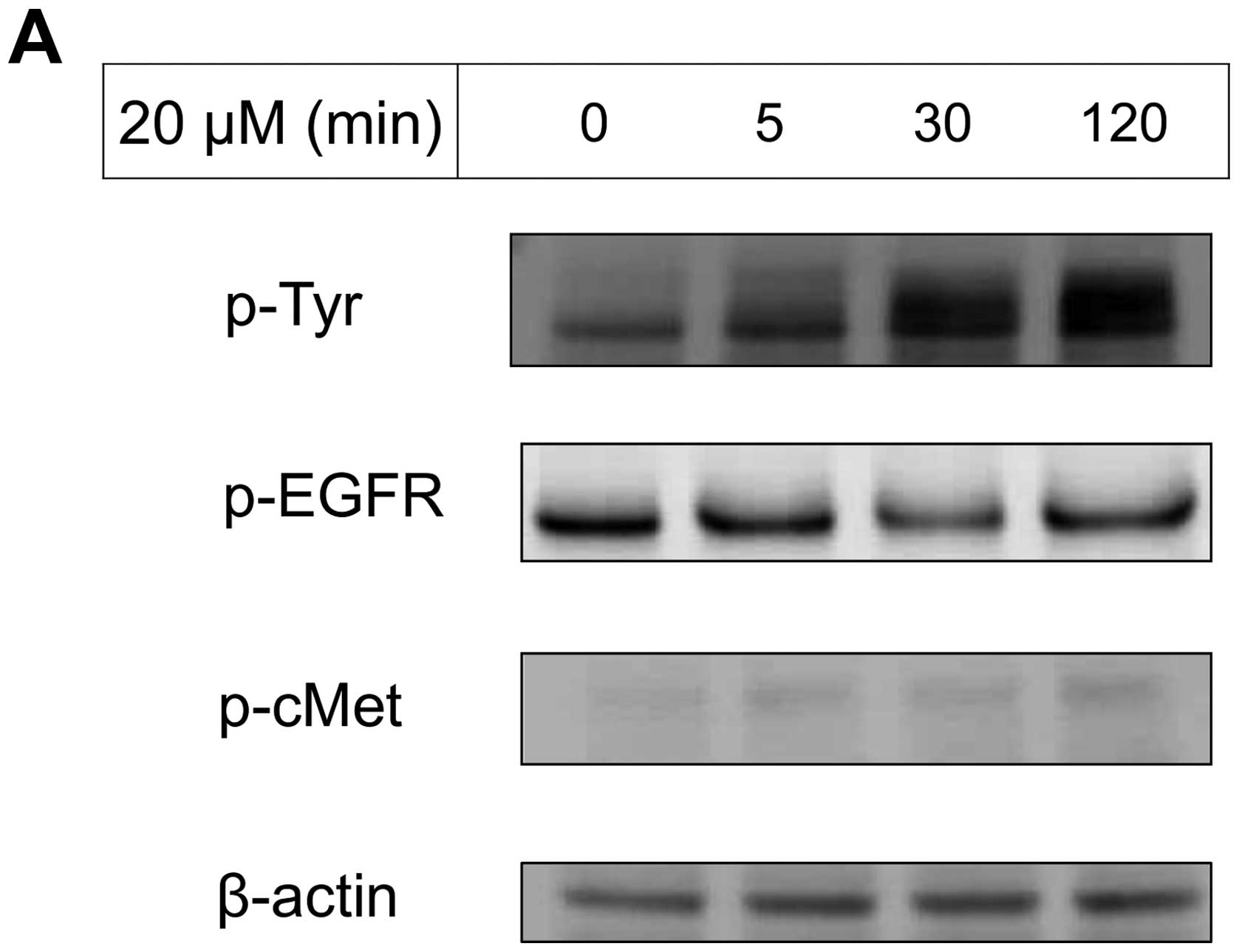
factors were examined for MDA-MB-231 (Fig. 2A). VK3 induced phosphorylation of
whole tyrosine in a time-dependent manner, 1.9±0.1-fold at 5 min,
3.5±0.2-fold at 30 min and 4.2±0.3-fold at 120 min, compared to the
control. Epidermal growth factor receptor (EGFR) was also activated
1.1-fold at 5 min, but c-Met was not phosphorylated. Of the cell
signaling factors, ERK was activated in a time-dependent manner,
3.4±0.1-fold at 5 min to 4.5±0.1-fold at 120 min (Fig. 2B). The phosphorylation of both JNK
and mTOR peaked at 1.3-fold at 5 min, and Akt remained at a plateau
of 1.5-fold from 5 to 120 min. Regarding cell cycle-related
factors, accumulation of cyclin D1 was detected at 1.2-fold the
control from 12 h by VK3, whereas cyclin B1 was reduced by
0.65-fold (Fig. 2C). The
expression of inactivated caspase-3 was decreased 0.7-fold, and
that of the activated type of cleaved PARP was increased by
1.2-fold at 6 h (Fig. 2D). As
shown in Fig. 3, the activation of
ERK or JNK was not affected by the EGFR inhibitor even at a
concentration adequate to block EGFR phosphorylation. Next, the
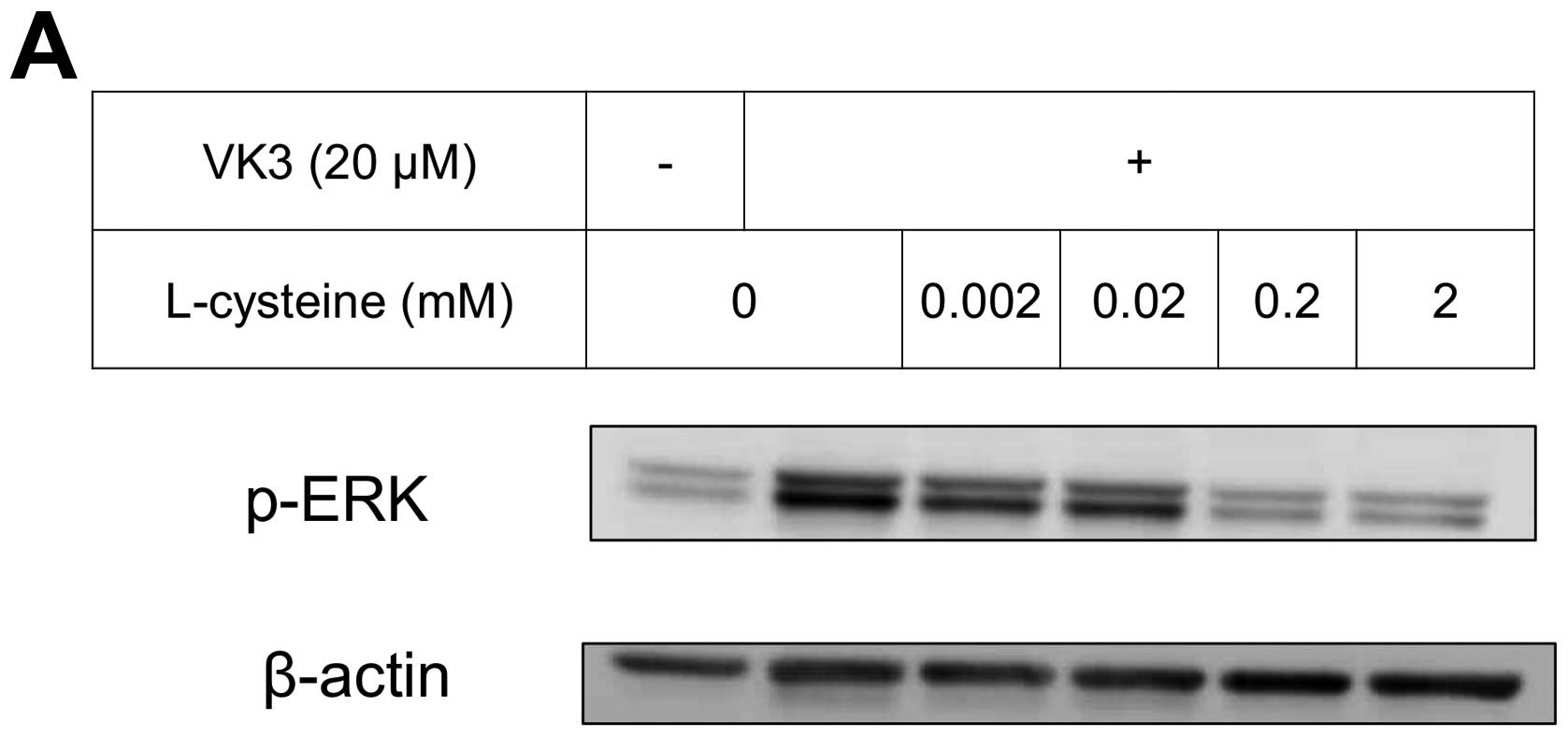
effects of antioxidants on VK3-induced cell signaling and growth
were examined (Fig. 4). L-cysteine
was found to inhibit VK3-induced ERK phosphorylation in a
dose-dependent manner; especially, concentrations of 0.2 mM or more
blocked ERK phosphorylation completely, and its growth inhibitory
action was also inhibited. In contrast, another antioxidant agent,
catalase, showed no effect on either ERK phosphorylation or growth
inhibition, and VK3 generated reactive oxygen species (ROS), as
assessed by fluorescence microscopy compared with the negative
control (Fig. 5).
In vivo experiments in xenograft tumors
in mice
On built-up tumors under the skin of mice created by
injection of 1.0×107 MDA-MB-231 cells, local treatment
with VK3 was effective time- and dose-dependently (Fig. 6A). VK3 at 20 and 50 mM decreased
the tumor size to 170.8±96.1 and 161.3±25.0 on day 1 and 93.3±45.3
and 81.7±34.0 on day 3, respectively. The experiments for total
tumor volume also showed a dose-dependent effect of VK3 (Fig. 6B). At 30 min after injection of
VK3, the expression of phosphorylated ERK was clearly increased
10.9-fold the control in tumor tissue, whereas ethanol itself had
no effect.
Discussion
Drug therapies for breast cancer have been selected
on the basis of the expression of hormonal receptors or HER2, but
therapy for TNBC remains difficult because of its high potential
for malignancy and its drug-resistant ability. In the present
study, VK3 caused cell growth inhibition regardless of the
molecular subtypes from expression of the receptors. VK3 could
disrupt cellular function via two distinct chemical pathways, by
generating ROS and by modifying cellular nucleophiles such as
cysteine residues on proteins. According to these mechanisms, redox
cycling, and arylating potential (27), the mechanisms of the cellular
growth inhibitory effects of VK3 will be argued.
Mitochondria undergo an initial priming phase
associated with hyperpolarization, which leads to an effector phase
during apoptosis. Clinical anticancer drugs can induce cytotoxicity
and the abrogation of proliferative signals though ROS production
in cancer cells (29). ROS are
well known to mediate mitochondrial dysfunction and apoptosis with
the activation of caspase-3 (30),
despite the loss of mitochondrial membrane potential (31). Indeed, activation of caspase-3 was
detected in VK3-treated glioblastoma cells with ROS production
(32). Mitochondrial damage
through ROS production not only induces apoptosis, but it also
reduces cell survival factors (33). Of them, survivin is highly
expressed in a variety human cancer cells but not in normal cells.
Because its high level correlates with more aggressive behavior,
decreased response to chemotherapeutic agents and shortened
survival time (34), the blockage
of survivin provides an important strategy for cancer therapy. Poly
(ADP-ribose) polymerase (PARP) is also one of the DNA repair
proteins that has been shown to be cleaved when cells undergo
apoptosis. ROS production was demonstrated to lead to both survivin
inhibition and PARP protein cleavage (33). The mitochondria-related action is a
main path for cell death (35),
indicating that ROS generation plays one of the critical roles in
the VK3-induced cellular effect (36). The present study showed that VK3
activated the apoptotic pathway through caspase-3 or PARP cleavage
and arrested the cell cycle (Fig.
2). VK3 was also reported to lead not only to the generation of
ROS but also to growth inhibition with the phosphorylation of ERK
(25,26). The agent, and ROS, that provokes
the sustained activation of ERK can induce the progressive
accumulation of death-promoting factors. Sustained cytoplasmic ERK
activity might promote senescence or autophagy, whereas sustained
nuclear sequestration of ERK activity might trigger apoptosis
(37). From the evidence that
prolonged ERK activation promotes death of human cancer cell lines
(38), the relationship of ERK to
cell growth inhibition was indicated to support the present
results.
Protein tyrosine phosphorylation is regulated by a
balance between the action of protein tyrosine phosphatase (PTPase)
and protein tyrosine kinase (PTK) activities. Ligand-induced
phosphorylation usually leads to PTK activity, whereas inhibition
of PTPase also induces phosphorylation of cellular signal protein
due to binding at the active site of PTPase (39). According to a previous report
(25), VK3 activates the signaling
pathway by inhibition of PTPase in hepatoma cells without causing
any ligand stimulation, and MAPKs are commonly activated by the
upstream action including the receptor. However, the present study
demonstrated that ERK and JNK were phosphorylated even under the
blocked condition of the receptor by its inhibitor. Taken together,
the phosphorylation of MAPKs might be mediated by a direct effect
on their PTPase activity. VK3 was also detected to lead to MAPKs
phosphorylation by a dual system with or without the receptor
signaling pattern. From the experiments with antioxidant agents,
VK3-mediated ERK phosphorylation was completely inhibited by the
thiol antioxidant L-cysteine but not by the non-thiol agent
catalase (Fig. 4), indicating that
the mechanism of sulfhydryl arylation (40) is critical. The growth inhibitory
action of VK3 was also antagonized completely by the thiol
antioxidant. Of the cellular signaling factors, ERK is usually
recognized to relate to cell proliferation (39), whereas ERK phosphorylation was
shown to promote apoptosis or cell cycle arrest and lead to growth
inhibition (26,41). In contrast, ERK phosphorylation
also focuses on ROS-independent DNA damage that leads to the
inhibition of breast cancer cell growth (29,37).
Actually, in melanoma cells, inhibiting the appearance of a
ubiquitin, which is required for protein ubiquitination related to
a key biological event, appeared to arrest tumorigenesis through
ERK phosphorylation (42). A more
recent study (43) showed some
keratins, which are a major component of the cellular cytoskeletal
structure, to have a dependent role with ERK, and blockage of their
activation was found to delay the onset of VK3-induced growth
inhibition. Furthermore, because of their effect on promoting cell
growth, cancer cells are well known to include greatly more ERK
protein than normal cells (44).
Thus, VK3 might be more effective against vigorous cell growth,
indicating that VK3 might have an expected therapeutic potential
even for TNBC. To reduce the harmful toxicity of VK3 by considering
the drug delivery system, a VK3 analog compound has been developed
for breast cancer therapy on mice in an in vivo experiment
(33). In parallel with these
novel trials, with the aim to improve the problems of VK3 toxicity
systemically, the present novel concept of VK3 as a local treatment
against TNBC may be accepted clinically as a favorable treatment
modality in the near future.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Williams C and Lin CY: Oestrogen receptors
in breast cancer: Basic mechanisms and clinical implications. E
Cancer Medical Sci. 7:3702013.
|
|
3
|
Oostra DR and Macrae ER: Role of
trastuzumab emtansine in the treatment of HER2-positive breast
cancer. Breast Cancer (Dove Med Press). 6:103–113. 2014.
|
|
4
|
Zelnak AB and Wisinski KB: Management of
patients with HER2-positive metastatic breast cancer: Is there an
optimal sequence of HER2-directed approaches? Cancer. 121:17–24.
2015. View Article : Google Scholar
|
|
5
|
Abramson VG, Lehmann BD, Ballinger TJ and
Pietenpol JA: Subtyping of triple-negative breast cancer:
Implications for therapy. Cancer. 121:8–16. 2015. View Article : Google Scholar
|
|
6
|
O’Toole SA, Beith JM, Millar EK, West R,
McLean A, Cazet A, Swarbrick A and Oakes SR: Therapeutic targets in
triple negative breast cancer. J Clin Pathol. 66:530–542. 2013.
View Article : Google Scholar
|
|
7
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: Clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liedtke C, Mazouni C, Hess KR, André F,
Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B,
Green M, et al: Response to neoadjuvant therapy and long-term
survival in patients with triple-negative breast cancer. J Clin
Oncol. 26:1275–1281. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsimberidou AM, Iskander NG, Hong DS,
Wheler JJ, Falchook GS, Fu S, Piha-Paul S, Naing A, Janku F, Luthra
R, et al: Personalized medicine in a phase I clinical trials
program: The MD Anderson Cancer Center initiative. Clin Cancer Res.
18:6373–6383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gelmon K, Dent R, Mackey JR, Laing K,
McLeod D and Verma S: Targeting triple-negative breast cancer:
Optimising therapeutic outcomes. Ann Oncol. 23:2223–2234. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nguyen T, Hattery E and Khatri VP:
Radiofrequency ablation and breast cancer: A review. Gland Surg.
3:128–135. 2014.PubMed/NCBI
|
|
13
|
Hamza A and Elrefaey S: Non-surgical
treatment of early breast cancer: Techniques on the way. Gland
Surg. 3:149–150. 2014.PubMed/NCBI
|
|
14
|
Manenti G, Scarano AL, Pistolese CA,
Perretta T, Bonanno E, Orlandi A and Simonetti G: Subclinical
breast cancer: Minimally invasive approaches Our experience with
percutaneous radiofrequency ablation vs cryotherapy. Breast Care
(Basel). 8:356–360. 2013. View Article : Google Scholar
|
|
15
|
Zagar TM, Vujaskovic Z, Formenti S, Rugo
H, Muggia F, O’Connor B, Myerson R, Stauffer P, Hsu I-C, Diederich
C, et al: Two phase I dose-escalation/pharmacokinetics studies of
low temperature liposomal doxorubicin (LTLD) and mild local
hyperthermia in heavily pretreated patients with local regionally
recurrent breast cancer. Int J Hyperthermia. 30:285–294. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kovacic P and Jacintho JD: Mechanisms of
carcinogenesis: Focus on oxidative stress and electron transfer.
Curr Med Chem. 8:773–796. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dong-Yun S, Yu-Ru D, Shan-Lin L, Ya-Dong Z
and Lian W: Redox stress regulates cell proliferation and apoptosis
of human hepatoma through Akt protein phosphorylation. FEBS Lett.
542:60–64. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suresh S, Raghu D and Karunagaran D:
Menadione (Vitamin K3) induces apoptosis of human oral cancer cells
and reduces their metastatic potential by modulating the expression
of epithelial to mesenchymal transition markers and inhibiting
migration. Asian Pac J Cancer Prev. 14:5461–5465. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baran I, Ionescu D, Filippi A, Mocanu MM,
Iftime A, Babes R, Tofolean IT, Irimia R, Goicea A, Popescu V, et
al: Novel insights into the antiproliferative effects and synergism
of quercetin and menadione in human leukemia Jurkat T cells. Leuk
Res. 38:836–849. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Margolin KA, Akman SA, Leong LA, Morgan
RJ, Somlo G, Raschko JW, Ahn C and Doroshow JH: Phase I study of
mitomycin C and menadione in advanced solid tumors. Cancer
Chemother Pharmacol. 36:293–298. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tetef M, Margolin K, Ahn C, Akman S, Chow
W, Leong L, Morgan RJ Jr, Raschko J, Somlo G and Doroshow JH:
Mitomycin C and menadione for the treatment of lung cancer: A phase
II trial. Invest New Drugs. 13:157–162. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tetef M, Margolin K, Ahn C, Akman S, Chow
W, Coluzzi P, Leong L, Morgan RJ Jr, Raschko J, Shibata S, et al:
Mitomycin C and menadione for the treatment of advanced
gastrointestinal cancers: A phase II trial. J Cancer Res Clin
Oncol. 121:103–106. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Osada S, Tomita H, Tanaka Y, Tokuyama Y,
Tanaka H, Sakashita F and Takahashi T: The utility of vitamin K3
(menadione) against pancreatic cancer. Anticancer Res. 28A:45–50.
2008.
|
|
24
|
Osada S, Sakashita F, Hosono Y, Nonaka K,
Tokuyama Y, Tanaka H, Sasaki Y, Tomita H, Komori S, Matsui S, et
al: Extracellular signal-regulated kinase phosphorylation due to
menadione-induced arylation mediates growth inhibition of pancreas
cancer cells. Cancer Chemother Pharmacol. 62:315–320. 2008.
View Article : Google Scholar
|
|
25
|
Osada S and Carr BI: Mechanism of novel
vitamin K analog induced growth inhibition in human hepatoma cell
line. J Hepatol. 34:676–682. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Osada S, Saji S and Osada K: Critical role
of extracellular signal-regulated kinase phosphorylation on
menadione (vitamin K3) induced growth inhibition. Cancer.
91:1156–1165. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Osada S, Osada K and Carr BI: Tumor cell
growth inhibition and extracellular signal-regulated kinase (ERK)
phosphorylation by novel K vitamins. J Mol Biol. 314:765–772. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Imai H, Saio M, Nonaka K, Suwa T, Umemura
N, Ouyang GF, Nakagawa J, Tomita H, Osada S, Sugiyama Y, et al:
Depletion of CD4+CD25+ regulatory T cells
enhances interleukin-2-induced antitumor immunity in a mouse model
of colon adenocarcinoma. Cancer Sci. 98:416–423. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang S, Konorev EA, Kotamraju S, Joseph J,
Kalivendi S and Kalyanaraman B: Doxorubicin induces apoptosis in
normal and tumor cells via distinctly different mechanisms.
intermediacy of H (2) O (2)- and p53-dependent pathways. J Biol
Chem. 279:25535–25543. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizutani H, Tada-Oikawa S, Hiraku Y,
Oikawa S, Kojima M and Kawanishi S: Mechanism of apoptosis induced
by a new topoisomerase inhibitor through the generation of hydrogen
peroxide. J Biol Chem. 277:30684–30689. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen FH, Zhang LB, Qiang L, Yang Z, Wu T,
Zou MJ, Tao L, You QD, Li ZY, Yang Y, et al: Reactive oxygen
species-mitochondria pathway involved in LYG-202-induced apoptosis
in human hepatocellular carcinoma HepG (2) cells. Cancer Lett.
296:96–105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu J, Chien CC, Yang LY, Huang GC, Cheng
MC, Lin CT, Shen SC and Chen YC: Vitamin K3-2, 3-epoxide induction
of apoptosis with activation of ROS-dependent ERK and JNK protein
phosphorylation in human glioma cells. Chem Biol Interact.
193:3–11. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang CR, Liao WS, Wu YH, Murugan K, Chen C
and Chao JI: CR108, a novel vitamin K3 derivative induces apoptosis
and breast tumor inhibition by reactive oxygen species and
mitochondrial dysfunction. Toxicol Appl Pharmacol. 273:611–622.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Johnson ME and Howerth EW: Survivin: A
bifunctional inhibitor of apoptosis protein. Vet Pathol.
41:599–607. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Akiyoshi T, Matzno S, Sakai M, Okamura N
and Matsuyama K: The potential of vitamin K3 as an anticancer agent
against breast cancer that acts via the mitochondria-related
apoptotic pathway. Cancer Chemother Pharmacol. 65:143–150. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sasaki R, Suzuki Y, Yonezawa Y, Ota Y,
Okamoto Y, Demizu Y, Huang P, Yoshida H, Sugimura K and Mizushina
Y: DNA polymerase gamma inhibition by vitamin K3 induces
mitochondria-mediated cytotoxicity in human cancer cells. Cancer
Sci. 99:1040–1048. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li X, Wang K, Ren Y, Zhang L, Tang XJ,
Zhang HM, Zhao CQ, Liu PJ, Zhang JM and He JJ: MAPK signaling
mediates sino-menine hydrochloride-induced human breast cancer cell
death via both reactive oxygen species-dependent and -independent
pathways: An in vitro and in vivo study. Cell Death Dis.
5:e13562014. View Article : Google Scholar
|
|
38
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death - apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar
|
|
39
|
Osada S and Yoshida K: A novel strategy
for advanced pancreatic cancer - progression of molecular targeting
therapy. Anticancer Agents Med Chem. 9:877–881. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Osada S and Saji S: New approach to cancer
therapy: The application of signal transduction to anti-cancer
drug. Curr Med Chem Anticancer Agents. 3:119–131. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Esakky P, Hansen DA, Drury AM and Moley
KH: Cigarette smoke-induced cell cycle arrest in spermatocytes
[GC-2spd (ts)] is mediated through crosstalk between Ahr-Nrf2
pathway and MAPK signaling. J Mol Cell Biol. 7:73–87. 2015.
View Article : Google Scholar
|
|
42
|
Shah M, Stebbins JL, Dewing A, Qi J,
Pellecchia M and Ronai ZA: Inhibition of Siah2 ubiquitin ligase by
vitamin K3 (menadione) attenuates hypoxia and MAPK signaling and
blocks melanoma tumorigenesis. Pigment Cell Melanoma Res.
22:799–808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Scott GK, Atsriku C, Kaminker P, Held J,
Gibson B, Baldwin MA and Benz CC: Vitamin K3 (menadione)-induced
oncosis associated with keratin 8 phosphorylation and histone H3
arylation. Mol Pharmacol. 68:606–615. 2005.PubMed/NCBI
|
|
44
|
Osada S, Kanematsu M, Imai H, Goshima S
and Sugiyama Y: Evaluation of extracellular signal regulated kinase
expression and its relation to treatment of hepatocellular
carcinoma. J Am Coll Surg. 201:405–411. 2005. View Article : Google Scholar : PubMed/NCBI
|