Introduction
Mesothelioma is a rare malignant tumor and arises in
the pleura of lung, peritoneum and pericardium (1). It is important to study malignant
mesothelioma because most patients with mesothelioma have worked in
careers such as mining. Many environmental factors such as erionite
and asbestos are related to carcinogenesis of mesothelioma
(2). Especially, asbestos can be
easily inhaled or ingested and it is collected in mesothelial
tissue, and thereby asbestos fibers cause cellular damage that
result in tumor growth (3).
Furthermore, asbestos influences epigenetic status of mesothelioma
(4). Despite treatment with
chemotherapy and radiation therapy, it has a poor prognosis.
Acetylation and deacetylation on histones are
controlled by two enzymes, histone acetyltransferase (HAT) and
histone deacetylase (HDAC), respectively. The aberrant histone
acetylation by the imbalance between HAT and HDAC can lead to
carcinogenesis in many cancer cells including colon and
mesothelioma (5,6). Especially, HDAC1 overexpression
promotes invasion and cell proliferation in cancer cells of the
prostate and ovary (7,8). Emerging evidence has demonstrated
that HDAC inhibitors are a potent therapeutic agent for treatment
of mesothelioma (9,10).
Reactive oxygen species (ROS) have important roles
in gene expression, cell signaling and cell differentiation
(11). However, excessive ROS
production may result in significant damage to cells through
oxidizing DNA, proteins and lipids. For this reason, there are
various antioxidants in the cells. Thioredoxin1 (Trx1), as a small
antioxidant protein increased in mesothelioma (12). It is also demonstrated that
asbestos modulates ROS level and the redox status of Trx and it
finally affects carcinogenesis of mesothelioma (13,14).
Moreover, many studies report that Trx1 is a target molecule for
drug-resistance and therapeutics of cancer (15,16).
Suberoylanilide hydroxamic acid (SAHA), a first HDAC
inhibitor for cutaneous T cell lymphoma treatment has an
anti-cancer effect in diverse cancer cells (17,18).
SAHA induced apoptosis via FLICE-like inhibitory protein
(FLIP)/caspase-8 activation in mesothelioma cells (19). However, little is known about the
molecular mechanism of mesothelioma cell death caused by SAHA in
view of the levels of HDAC1, ROS and Trx1. Therefore, in the
present study we investigated the effects of SAHA on cell death in
various mesothelioma cells with regard to HDAC1, ROS and Trx1
levels.
Materials and methods
Cell culture
Human mesothelial cells (HM69 and HM72) and human
mesothelioma cells (ADA, CON, Hmeso, Mill, Phi, REN and ROB) were
obtained from the University of Hawaii Cancer Center (Honolulu, HI,
USA). These cells were cultured in Ham's F-12 media containing 10%
fetal bovine serum (FBS; Gibco-BRL, Grand Island, NY, USA) and 1%
penicillin-streptomycin (Gibco-BRL).
Reagents
SAHA purchased from Cayman Chemical Co., (Ann Arbor,
MI, USA) was dissolved in dimethyl sulf-oxide (DMSO; Sigma-Aldrich,
St. Louis, MO, USA). The pan-caspase inhibitor (Z-VAD-FMK),
caspase-3 inhibitor (Z-DEVD-FMK), caspase-8 inhibitor (Z-IETD-FMK)
and caspase-9 inhibitor (Z-LEHD-FMK) were obtained from R&D
Systems (Minneapolis, MN, USA) and were dissolved in DMSO. NAC and
Vit.C obtained from Sigma-Aldrich Chemical were dissolved in 20 mM
HEPES (pH 7.0) buffer and water, respectively. Based on previous
studies (20,21), cells were pretreated with 15 μM
caspase inhibitors, 2 mM NAC or 0.4 mM Vit.C for 1 h before SAHA
treatment.
Growth inhibition assay
The effect of SAHA on growth inhibition in human
mesothelioma cells was determined by measuring the absorbance of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich) dye absorbance as previously described (22). Briefly, 5×103 cells were
incubated with the indicated concentrations of SAHA with or without
each caspase inhibitor, HDAC siRNA, NAC, Vit.C or Trx1 siRNA for 24
h.
Western blot analysis
The protein expression levels were evaluated by
western blot analysis. Briefly, 1×106 cells were
incubated with 5 μM SAHA for 24 h. Total protein (30 μg) was
resolved by 4–20% SDS-PAGE gels, and then transferred to
Immobilon-P PVDF membranes (Merck Millipore, Darmstadt, Germany) by
electroblotting. Then membranes were probed with anti-HDAC1,
anti-acetylated H4, anti-PARP, anti-cleaved PARP, anti-cleaved
caspase-3 (Cell Signaling Technology, Danvers, MA, USA), anti-Trx1,
anti-GAPDH and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz,
CA, USA). Membranes were incubated with fluorescence-conjugated
secondary antibodies.
Measurement of HDAC activity
The HDAC activity was measured by using a HDAC assay
kit according to the manufacturer's instructions (Merck Millipore).
Briefly, 1×106 cells were incubated with 5 μM SAHA for
24 h. Total protein (30 μg) was used to measure the HDAC
activity.
Annexin V-FITC/ PI staining for apoptosis
detection
Apoptosis was detected by staining cells with
AnnexinV-fluoresceinisothiocyanate(FITC;Invitrogen-Life
Technologies, Eugene, OR, USA; Ex/Em=488 nm/519 nm) and propidium
iodide (PI; Sigma-Aldrich; Ex/Em=488 nm/617 nm) as previously
described (23). Briefly,
1×106 cells were incubated with the indicated
concentrations of SAHA with or without each caspase inhibitor, NAC,
Vit.C, HDAC1 and Trx1 siRNAs for 24 h. Annexin V/PI staining was
analyzed with the Accuri C6 flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA).
Measurement of lactate dehydrogenase
(LDH) activity
Necrosis in cells was evaluated by an LDH kit
according to the manufacturer's instructions (Sigma-Aldrich).
Briefly, 1×106 cells were incubated with 5 μM SAHA with
or without each caspase inhibitor, NAC or Vit.C for 24 h. LDH
release was expressed as the percent of extracellular LDH activity
compared with the control cells.
Measurement of MMP (ΔΨm)
MMP (ΔΨm) levels were measured using JC-1 dyes (Enzo
Life Sciences, Plymouth Metting, PA, USA; Ex/Em=515 nm/529 nm).
Briefly, 5×104 cells were incubated with 5 μM SAHA for
24 h. Cells were washed twice with PBS and incubated with 10 μg/ml
JC-1 at 37°C for 30 min. Then cells were washed three times with
PBS and analyzed with the Accuri C6 flow cytometer (BD
Biosciences). Green fluorescence indicates a monomeric state at the
low ΔΨm and red fluorescence presents an aggregate state at the
high ΔΨm.
Sub-G1 analysis
Sub-G1 analysis was determined by PI (Sigma-Aldrich)
staining as previously described (23). Briefly, 1×106 cells were
incubated with 5 μM SAHA with or without each caspase inhibitor,
HDAC1 and Trx1 siRNAs for 24 h. Sub-G1 DNA content cells were
measured and analyzed with Accuri C6 flow cytometer (BD
Biosciences).
Transfection of cells with HDAC1 and Trx1
siRNAs
Gene silencing of HDAC1 and Trx1 was performed by
using small interference RNA (siRNA) delivery system. A
non-specific control siRNA duplex [5′-CCUACGCCACCAAUUUCGU
(dTdT)-3′], HDAC1 siRNA duplex [5′-GAGUCAAAACAGA GGAUGA(dTdT)-3′]
and Trx1 siRNA duplex [5′-GCAUGCC AACAUUCCAGUU(dTdT)-3′] were
purchased from the Bioneer Corp. (Daejeon, South Korea). In brief,
2.5×105 cells were incubated in RPMI-1640 supplemented
with 10% FBS. The next day, cells (~30–40% confluence) in each well
were transfected with the control, HDAC1 siRNA or Trx1 siRNA [100
pmol in Opti-MEM (Gibco-BRL)] using Lipofectamine 2000 according to
the manufacturer's instructions (Invitrogen-Life Technologies). One
day later, cells were treated with or without 5 μM SAHA for
additional 24 h. The transfected cells were collected and used for
western blot analysis, cell growth, sub-G1, Annexin V-FITC and
O2•− level measurements.
Detection of intracellular ROS
levels
Intracellular ROS were detected by a fluorescent
probe dye, 2′,7′-dichlorodihydro-fluorescein diacetate
(H2DCFDA, Ex/Em=495 nm/529 nm; Invitrogen-Molecular
Probes) as previously described (23). Dihydroethidium (DHE, Ex/Em=518
nm/605 nm; Invitrogen-Molecular Probes) is a fluorogenic probe that
is highly selective for O2•− among ROS.
Briefly, 1×106 cells were incubated with 5 μM SAHA with
or without Trx1 siRNA for 24 h. DCF and DHE fluorescence were
detected by using the Accuri C6 flow cytometer (BD
Biosciences).
Real-time PCR analysis
Total RNA was extracted by using E.Z.N.A. Total RNA
kit (Omega Bio-Tek Inc., Norcross, GA, USA) according to the
manufacturer's instruction. cDNA was obtained from 0.8 μg of total
RNA by using High-Capacity cDNA reverse transcription kit (Life
Technologies). Real-time PCR was performed using a SYBR-Green
SuperMix (Quanta Bioscience, Gaithersburg, MD, USA) in real-time
PCR cycler, LightCyclerR 480 instrument (Roche Diagnostics,
Mannheim, Germany), setting the cycles as follows: 10 min/95°C PCR
initial activation step; 40 cycles of denaturation for 15 sec/95°C
and annealing/extenstion step for 25 sec/58°C. Trx1 (P115235) and
hypoxanthine-guanine phosphoribosyltransferase (HPRT, P160523) were
obtained from the Bioneer Corp. The changes in mRNA level were
determined by the formula 2−ΔΔCT. The relative amount of
mRNA in the sample was normalized to HPRT mRNA.
Statistical analysis
The results represent the mean of at least three
independent experiments (mean ± SD). The data were analyzed using
an Instat software (GraphPad Prism5; GraphPad Software, Inc., San
Diego, CA, USA). The Student's t-test or one-way analysis of
variance (ANOVA) with post hoc analysis using Tukey's multiple
comparison test was used for parametric data. Statistical
significance was defined as P<0.05.
Results
Effects of SAHA on cell growth and HDAC
activities in human mesothelioma cells
SAHA inhibited the growth of Phi and ROB
mesothelioma cells at 24 h (Fig.
1A). Treatment with 5 μM SAHA reduced the growth of Phi and ROB
cells ~30–40% compared to control cells (Fig. 1A). However, 5 μM SAHA did not
significantly affect the growth of ADA and Mill cells at 24 h
(Fig. 1A). The basal levels of
HDAC1 were increased in Phi, REN and ROB cells, whose cells were
sensitive to SAHA (Fig. 1B). ADA
and Mill cells resistant to SAHA showed lower levels of HDAC1
(Fig. 1B). The HDAC1 levels were
different between the two normal mesothelial cells (Fig. 1B).
After the exposure of mesothelioma cells to SAHA for
24 h, SAHA strongly decreased the activity of HDAC in Phi cells but
this agent did not change the activity of HDAC in ADA cells
(Fig. 1C). Furthermore, the levels
of acetylated-H4 were increased in SAHA-treated Hmeso, Phi, REN and
ROB cells (Fig. 1D). However, SAHA
did not alter the levels of the acetylated-H4 in ADA, CON and Mill
cells (Fig. D).
Effects of SAHA on cell death and
mitochondrial membrane potential (MMP; ΔΨm) in ADA and Phi
cells
Treatment with 5 μM SAHA increased the numbers of
Annexin V-FITC cells in Phi cells (Fig. 2A). In addition, SAHA induced the
cleavages of PARP and caspase-3 in Phi cells (Fig. 2B). Moreover, this agent
significantly increased LDH release in Phi cells (Fig. 2C). These results implied that
SAHA-induced Phi cell death occurred via apoptosis as well as
necrosis. However, SAHA did not influence the percent of Annexin
V-FITC cells, apoptosis-related protein levels and LDH release in
ADA cells (Fig. 2A–C). The loss of
MMP (ΔΨm) can lead to cell death. As shown in Fig. 2D, red fluorescence of JC-1
indicating the high ΔΨm was decreased in 5 μM SAHA-treated Phi
cells whereas green fluorescence of JC-1 indicating the low ΔΨm was
increased in these cells (Fig.
2D). SAHA affected neither red fluorescence nor green
fluorescence of JC-1 in ADA cells (Fig. 2D). Carbonyl cyanide m-chlorophenyl
hydrazine (CCCP) was used as a positive control to induce the loss
of ΔΨm.
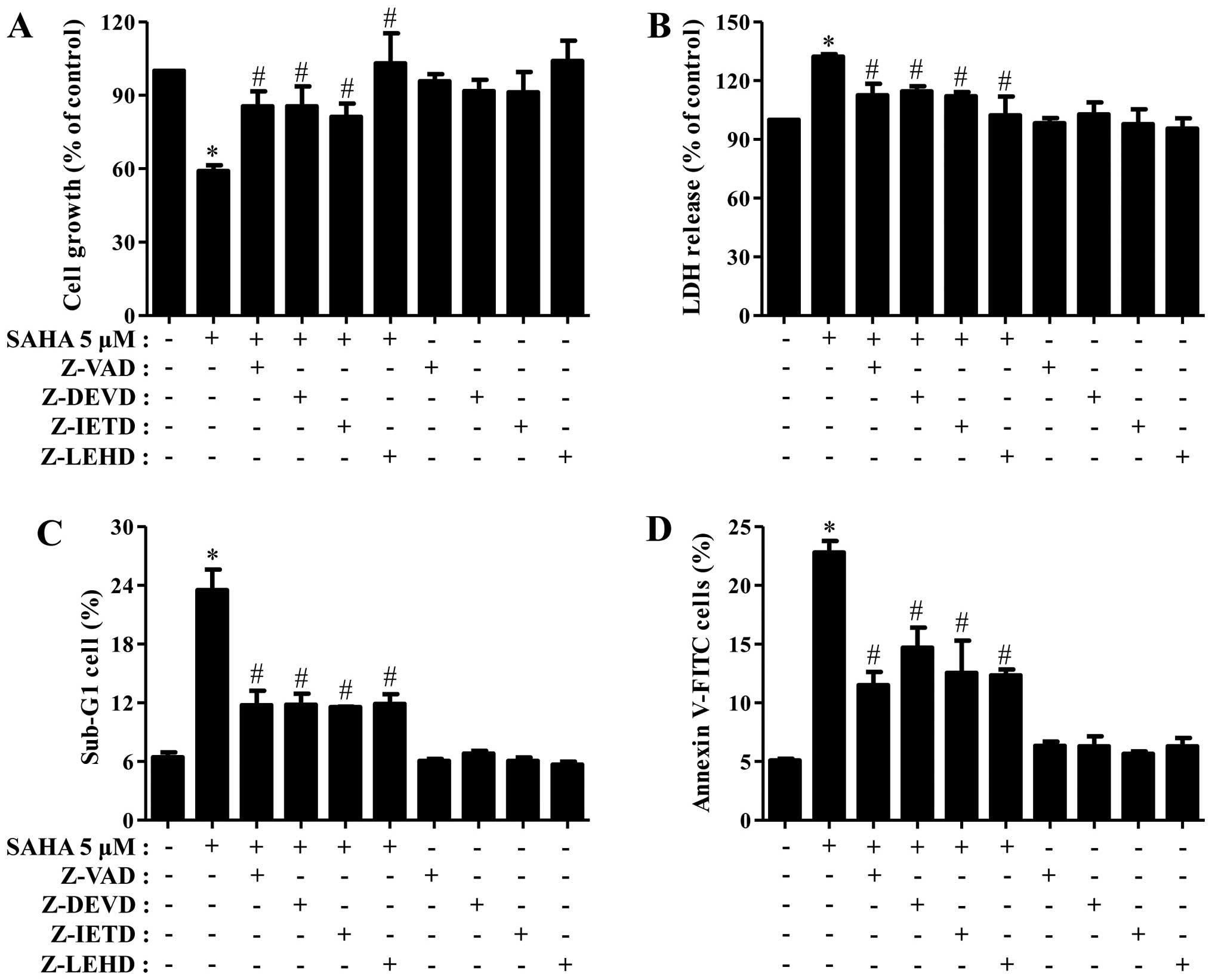
Next, we determined which caspase is involved in Phi
cell death and growth inhibition induced by SAHA. As shown Fig. 3A and B, all the tested caspase
inhibitors partially recovered the growth inhibition and LDH
release of SAHA-treated Phi cells. In addition, all the inhibitors
strongly reduced the percents of sub-G1 cells and Annexin V-FITC
cells in these cells (Fig. 3C and
D).
Effects of HDAC1 siRNA on cell growth and
death in SAHA-treated Phi cells
Because the basal levels of HDAC1 were different
between SAHA-sensitive and SAHA-resistant mesothelioma cells
(Figs. 1 and 2), the status of HDAC1 might influence
mesothelioma cell death caused by SAHA. To investigate whether
HDAC1 protein affects SAHA-induced cell death in mesothelioma, the
mRNA level of HDAC1 was knocked downed by the administration of
siRNA. The knockdown of HDAC1 successfully occurred in Phi cells
via its siRNA (Fig. 4A). HDAC1
siRNA significantly promoted cell growth inhibition in SAHA-treated
Phi cells (Fig. 4B). In addition,
HDAC1 siRNA increased the numbers of sub-G1 cells and Annexin
V-positive cells in these cells (Fig.
4C and D). HDAC1 siRNA alone induced cell death in
SAHA-untreated Phi control cells (Fig.
4D).
Effects of SAHA on intracellular ROS and
Trx1 levels in SAHA-treated Phi cells
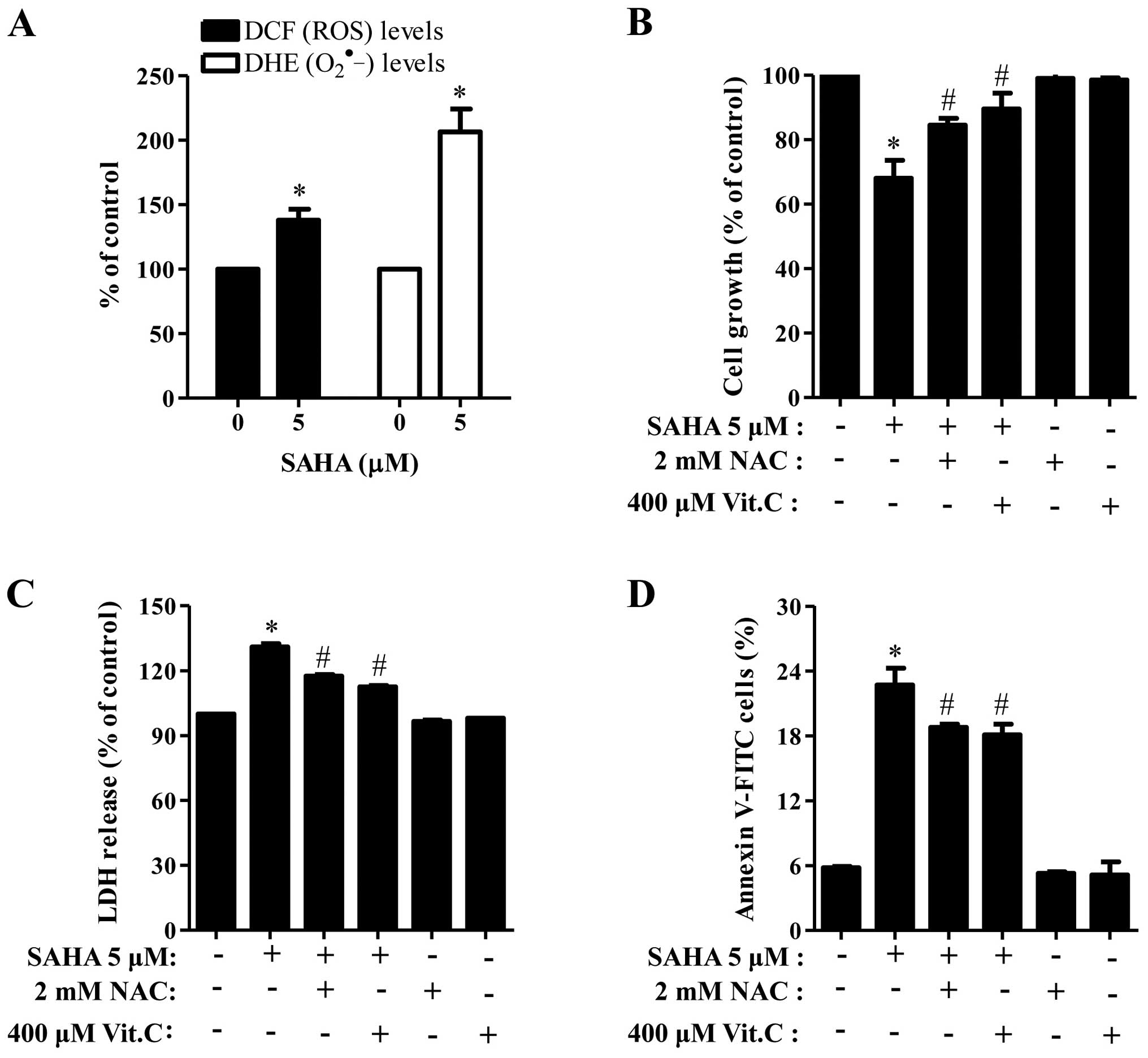
When we measured the intracellular ROS levels by
DCFDA and DHE dyes, 5 μM SAHA significantly increased ROS levels
including O2•− in Phi cells (Fig. 5A). Moreover, well-known
antioxidants, NAC and Vit.C effectively blocked cell growth
inhibition and LDH release in SAHA-treated Phi cells (Fig. 5B and C) and both of them
significantly reduced the number of Annexin V-positive cells in the
cells (Fig. 5D).
It is reported that SAHA decreased Trx1 in cancer
cells (24). Cellular antioxidants
can change intracellular ROS levels and affect cell death such as
apoptosis. As shown in Fig. 6A and
B, 5 μM SAHA decreased the mRNA and protein levels of Trx1 in
Phi and ROB cells. While SAHA decreased the protein level of Trx1
in REN cells, this agent did not alter the mRNA level of Trx1 in
these cells (Fig. 6A and B). Next,
it was determined whether Trx1 knockdown affects cell growth, cell
death and ROS levels in SAHA-treated Phi cells. Administration of
Trx1 siRNA markedly decreased the level of Trx1 (Fig. 6C) and this siRNA enhanced cell
growth inhibition in SAHA-untreated and -treated Phi cells
(Fig. 6D). Trx1 siRNA also
increased the percents of sub-G1 cells and apoptotic cells in these
cells (Fig. 6E and F).
Furthermore, Trx1 siRNA increased the O2•−
levels in SAHA-untreated and -treated Phi cells (Fig. 6G).
Discussion
Mesothelioma is a rare form of cancer derived from
the pleura, peritoneum or pericardium. Epigenetic changes affect
drug resistance and pathogenesis of mesothelioma. In the present
study, we determined an anticancer effect of SAHA on various
mesothelioma cells in view of HDAC1 and Trx1 levels. SAHA inhibited
the growth of Phi and ROB mesothelioma cells. This result supports
that HDAC inhibitor shows anti-tumor effect on mesothelioma
(9,25). However, this drug did not affect
the growth of ADA and Mill mesothelioma cells. Interestingly,
SAHA-sensitive Phi and ROB cells strongly expressed the basal level
of HDAC1 protein whereas SAHA-resistant ADA and Mill cells showed
lower basal levels of HDAC1. Moreover, SAHA notably inhibited HDAC
activity and induced acetylation of H4 in SAHA-sensitive Phi cells.
This result implies that SAHA as an HDAC inhibitor especially works
well in mesothelioma cells having overexpressed HDAC1 protein. In
addition, the different levels of HDAC1 among mesothelioma cells
seemed to differently influence the sensitivity of mesothelioma
cells to HDAC inhibitor. Many studies have reported that inhibition
of HDAC1 enhances cell death in various cancer cells including
ovarian and liver (26,27). Likewise, HDAC1 knockdown enhanced
Phi cell death caused by SAHA. Taken together, HDAC1 might be a
target for the mesothelioma therapy.
SAHA induced apoptosis in Phi cells which was
accompanied by the cleavages of PARP and caspase-3 and the loss of
MMP (ΔΨm). In addition, all the tested caspase inhibitors strongly
prevented cell death in these cells. Therefore, it seems that
apoptosis occurs via intrinsic and extrinsic pathways and that cell
death is the main mechanism for the inhibition of cell growth by
SAHA. We also observed that SAHA induced LDH release in
SAHA-treated Phi cells. However, NecroX-2 and necrostatin1,
necrosis inhibitors did not significantly attenuate cell death in
SAHA-treated Phi cells (data not shown). Therefore, necrosis seems
to be in part related to SAHA-induced Phi cell death.
Oxidative stress is an important cause in
mesothelioma cell death and antioxidant contributes the drug
resistance of mesothelioma cells. Likewise, SAHA increased the
intracellular ROS levels including O2•− in
Phi cells. Both NAC and Vit.C prevented the growth inhibition and
cell death in SAHA-treated Phi cells. SAHA did not change ROS level
in ADA cells which was resistant to this drug (data not shown).
Therefore, these results suggest that oxidative stress induced by
SAHA leads to apoptotic cell death in Phi mesothelioma cells.
Trx is an important antioxidant protein in cells and
it protects the cell from oxidative stress damage by facilitating
the reduction of other oxidative proteins via cysteine
thiol-disulfide exchange (28).
HDAC inhibitor changes the redox state of Trx (29). In the present study, SAHA
downregulated the mRNA and protein levels of Trx1 in Phi and ROB
cells. In addition, Trx1 siRNA sensitized Phi cells to SAHA and it
alone induced apoptosis in SAHA-untreated Phi cells. These results
imply that Trx1 has a critical role in cell death in mesothelioma
cells. In regard to ROS levels, Trx1 siRNA intensified the
O2•− level in SAHA-treated and untreated Phi
cells. This result suggests that Trx1 also act as a strong
antioxidant in mesothelioma cells.
In conclusion, SAHA inhibited the growth of Phi and
ROB cells among the tested human mesothelioma cells. These cells
relatively have higher levels of HDACs. SAHA-induced Phi cell death
was related to oxidative stress and Trx1 levels.
Acknowledgements
We thank Professor Peter R. Hoffmann and Dr Pietro
Bertino for providing mesothelioma cells. The present study was
supported by the National Research Foundation of Korea (NRF) grant
funded by the Korea government (MSIP) (no. 2008-0062279) and
supported by the Basic Science Research Program through the NRF
funded by the Ministry of Education (2013006279).
Abbreviations:
|
SAHA
|
suberoylanilide hydroxamic acid
|
|
HAT
|
histone acetyltransferase
|
|
HDAC
|
histone deacetylase
|
|
ROS
|
reactive oxygen species
|
|
FITC
|
fluorescein isothiocyanate
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
NAC
|
N-acetylcysteine
|
|
Vit.C
|
vitamin C
|
|
LDH
|
lactate dehydrogenase
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
Trx
|
thioredoxin
|
|
siRNA
|
small interfering RNA
|
References
|
1
|
Comertpay S, Pastorino S, Tanji M,
Mezzapelle R, Strianese O, Napolitano A, Baumann F, Weigel T,
Friedberg J, Sugarbaker P, et al: Evaluation of clonal origin of
malignant mesothelioma. J Transl Med. 12:3012014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carbone M and Yang H: Molecular pathways:
Targeting mechanisms of asbestos and erionite carcinogenesis in
mesothelioma. Clin Cancer Res. 18:598–604. 2012. View Article : Google Scholar :
|
|
3
|
Matsuzaki H, Maeda M, Lee S, Nishimura Y,
Kumagai-Takei N, Hayashi H, Yamamoto S, Hatayama T, Kojima Y,
Tabata R, et al: Asbestos-induced cellular and molecular alteration
of immuno-competent cells and their relationship with chronic
inflammation and carcinogenesis. J Biomed Biotechnol.
2012:4926082012. View Article : Google Scholar
|
|
4
|
Christensen BC, Houseman EA, Godleski JJ,
Marsit CJ, Longacker JL, Roelofs CR, Karagas MR, Wrensch MR, Yeh
RF, Nelson HH, et al: Epigenetic profiles distinguish pleural
mesothelioma from normal pleura and predict lung asbestos burden
and clinical outcome. Cancer Res. 69:227–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karczmarski J, Rubel T, Paziewska A,
Mikula M, Bujko M, Kober P, Dadlez M and Ostrowski J: Histone H3
lysine 27 acetylation is altered in colon cancer. Clin Proteomics.
11:242014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kalari S, Moolky N, Pendyala S, Berdyshev
EV, Rolle C, Kanteti R, Kanteti A, Ma W, He D, Husain AN, et al:
Sphingosine kinase 1 is required for mesothelioma cell
proliferation: Role of histone acetylation. PLoS One. 7:e453302012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim NH, Kim SN and Kim YK: Involvement of
HDAC1 in E-cadherin expression in prostate cancer cells; its
implication for cell motility and invasion. Biochem Biophys Res
Commun. 404:915–921. 2011. View Article : Google Scholar
|
|
8
|
Hayashi A, Horiuchi A, Kikuchi N, Hayashi
T, Fuseya C, Suzuki A, Konishi I and Shiozawa T: Type-specific
roles of histone deacetylase (HDAC) overexpression in ovarian
carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates
cell migration with downregulation of E-cadherin. Int J Cancer.
127:1332–1346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Paik PK and Krug LM: Histone deacetylase
inhibitors in malignant pleural mesothelioma: Preclinical rationale
and clinical trials. J Thorac Oncol. 5:275–279. 2010. View Article : Google Scholar
|
|
10
|
Katafygiotis P, Giaginis C, Patsouris E
and Theocharis S: Histone deacetylase inhibitors as potential
therapeutic agents for the treatment of malignant mesothelioma.
Anticancer Agents Med Chem. 13:476–482. 2013.
|
|
11
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tabata C, Terada T, Tabata R, Yamada S,
Eguchi R, Fujimori Y and Nakano T: Serum thioredoxin-1 as a
diagnostic marker for malignant peritoneal mesothelioma. J Clin
Gastroenterol. 47:e7–e11. 2013. View Article : Google Scholar
|
|
13
|
Thompson JK, Westbom CM, MacPherson MB,
Mossman BT, Heintz NH, Spiess P and Shukla A: Asbestos modulates
thioredoxin-thioredoxin interacting protein interaction to regulate
inflammasome activation. Part Fibre Toxicol. 11:242014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Murthy S, Adamcakova-Dodd A, Perry SS,
Tephly LA, Keller RM, Metwali N, Meyerholz DK, Wang Y, Glogauer M,
Thorne PS, et al: Modulation of reactive oxygen species by Rac1 or
catalase prevents asbestos-induced pulmonary fibrosis. Am J Physiol
Lung Cell Mol Physiol. 297:L846–L855. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Powis G and Kirkpatrick DL: Thioredoxin
signaling as a target for cancer therapy. Curr Opin Pharmacol.
7:392–397. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Yang H, Li W, Xu H, Yang X and Gan
L: Thioredoxin 1 upregulates FOXO1 transcriptional activity in drug
resistance in ovarian cancer cells. Biochim Biophys Acta.
1852:395–405. 2015. View Article : Google Scholar
|
|
17
|
Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee
KH, Kim TY, Han SW, Oh DY, Kim TY, et al: Histone deacetylase
inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances
anti-tumor effects of the poly (ADP-ribose) polymerase (PARP)
inhibitor olaparib in triple-negative breast cancer cells. Breast
Cancer Res. 17:332015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding L, Zhang Z, Liang G, Yao Z, Wu H,
Wang B, Zhang J, Tariq M, Ying M and Yang B: SAHA triggered MET
activation contributes to SAHA tolerance in solid cancer cells.
Cancer Lett. 356:828–836. 2015. View Article : Google Scholar
|
|
19
|
Hurwitz JL, Stasik I, Kerr EM, Holohan C,
Redmond KM, McLaughlin KM, Busacca S, Barbone D, Broaddus VC, Gray
SG, et al: Vorinostat/SAHA-induced apoptosis in malignant
mesothelioma is FLIP/caspase 8-dependent and HR23B-independent. Eur
J Cancer. 48:1096–1107. 2012. View Article : Google Scholar
|
|
20
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of lung cancer Calu-6 cells via
caspase-dependent apoptosis. Chem Biol Interact. 177:107–114. 2009.
View Article : Google Scholar
|
|
21
|
You BR and Park WH: Gallic acid-induced
lung cancer cell death is related to glutathione depletion as well
as reactive oxygen species increase. Toxicol In Vitro.
24:1356–1362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
You BR, Kim SH and Park WH: Reactive
oxygen species, glutathione, and thioredoxin influence suberoyl
bishydroxamic acid-induced apoptosis in A549 lung cancer cells.
Tumour Biol. 36:3429–3439. 2015. View Article : Google Scholar
|
|
23
|
You BR, Shin HR, Han BR and Park WH: PX-12
induces apoptosis in Calu-6 cells in an oxidative stress-dependent
manner. Tumour Biol. 36:2087–2095. 2015. View Article : Google Scholar
|
|
24
|
Butler LM, Zhou X, Xu WS, Scher HI,
Rifkind RA, Marks PA and Richon VM: The histone deacetylase
inhibitor SAHA arrests cancer cell growth, up-regulates
thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc
Natl Acad Sci USA. 99:11700–11705. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Crisanti MC, Wallace AF, Kapoor V,
Vandermeers F, Dowling ML, Pereira LP, Coleman K, Campling BG,
Fridlender ZG, Kao GD, et al: The HDAC inhibitor panobinostat
(LBH589) inhibits mesothelioma and lung cancer cells in vitro and
in vivo with particular efficacy for small cell lung cancer. Mol
Cancer Ther. 8:2221–2231. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie HJ, Noh JH, Kim JK, Jung KH, Eun JW,
Bae HJ, Kim MG, Chang YG, Lee JY, Park H, et al: HDAC1 inactivation
induces mitotic defect and caspase-independent autophagic cell
death in liver cancer. PLoS One. 7:e342652012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cacan E, Ali MW, Boyd NH, Hooks SB and
Greer SF: Inhibition of HDAC1 and DNMT1 modulate RGS10 expression
and decrease ovarian cancer chemoresistance. PLoS One.
9:e874552014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chae JS, Gil Hwang S, Lim DS and Choi EJ:
Thioredoxin-1 functions as a molecular switch regulating the
oxidative stress-induced activation of MST1. Free Radic Biol Med.
53:2335–2343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ungerstedt J, Du Y, Zhang H, Nair D and
Holmgren A: In vivo redox state of human thioredoxin and redox
shift by the histone deacetylase inhibitor suberoylanilide
hydroxamic acid (SAHA). Free Radic Biol Med. 53:2002–2007. 2012.
View Article : Google Scholar : PubMed/NCBI
|