Introduction
The tyrosine kinase receptor c-MET, also called MET
or hepatocyte growth factor receptor (HGFR), is the only known
receptor for hepatocyte growth factor (HGF) (1). Aberrant MET signaling plays a pivotal
role in angiogenesis as well as in tumor cell proliferation,
survival and migration (2–4). Several studies suggest that HGF/c-MET
signaling promotes angio-genesis directly by stimulating
endothelial cells in response to VEGF in various tumor types
(5–8). Moreover, MET has been described as an
oncogene in different pathologies such as liver cancer (9) and MET gene amplification is reported
in 2–3% of various types of cancer (10).
In patients with neuroblastoma, a pediatric tumor of
the neural crest, high concentrations of HGF and elevated soluble
VEGF-A were found correlated with higher stage and poor outcome
(11). MET is expressed in most
neuroblastoma cell lines and HGF stimulated invasion of
neuroblastoma cells in vitro and in vivo, and
promoted the development of angiogenic neuroblastoma tumors in
vivo (12,13). Recent studies in adult cancers
suggest that tumor hypoxia induced by anti-angiogenic therapy such
as sorafenib leads to increased c-MET activation, cell migration
and aggressiveness (14,15) suggesting a combined inhibiting
approach to overcome this escape mechanism. Several preclinical
studies have explored the use of combined HGF/MET and VEGF/VEGFR
signaling inhibition in adult pathologies such as hepatocellular
cancer (16–18) or glioblastoma (15) with reduced tumor aggressiveness and
metastasis, encouraging the evaluation in neuroblastoma.
Cabozantinib (XL-184) is a multi-targeted tyrosine kinase inhibitor
which potently inhibits VEGFR and MET signaling (19). The principal targets of
cabozantinib are MET, VEGFR2, AXL and RET, but the compound is also
reported to inhibit other kinases including KIT, FLT-3 and TEK
(19). Cabozantinib has shown
effective inhibition of cell proliferation and migration/invasion
in vitro (18,20–22).
Cabozantinib exhibited inhibition of tumor growth which is mediated
by inhibition of angiogenesis and potent anti-metastatic effects in
different types of cancer (18,20,23,24).
Clinical phase I to III trials have shown antitumor activity in
advanced solid tumors such as prostate, lung, renal and thyroid
cancer, and the agent is approved for the treatment of progressive,
metastatic medullary thyroid cancer (25–32).
This study defined HGF/MET signaling activation as
an escape mechanism to selective VEGFR inhibition in neuroblastoma
and explored the antitumor activity of concomitant inhibition of
VEGFR2 and MET in comparison to specific pan-VEGFR inhibition in
preclinical neuroblastoma models.
Materials and methods
Drugs
Cabozantinib (XL-184; kindly provided by Dana Aftab
(Exelixis, Inc., San Francisco, CA, USA) was stored as a solid
powder. Axitinib (AG-013736, ref A-1107) was purchased from LC
Laboratories (Woburn, MA, USA). For in vitro experiments,
cabozantinib and axitinib were dissolved in 100% dimethyl sulfoxide
(DMSO) and diluted in complete cell culture. For in vivo
experiments, cabozantinib was dissolved daily in water/HCl to a
final concentration of 3 and 6 mg/ml; axitinib was suspended in 5%
carboxyl methylcellulose to a final concentration of 6 mg/ml.
Cell lines
Luciferase gene transfected IGR-N91 and IMR-32 cells
were derived from metastatic neuroblastoma as previously reported
(33). IMR-32-Luc and IGR-N91-Luc
were maintained in Roswell Park Memorial Institute (RPMI)-1640
medium and Dulbecco's modified Eagle's medium (DMEM), respectively,
containing 10% fetal calf serum (FCS; all from Life Technologies)
at 37°C and 5% CO2. Cell lines were regularly tested and
found to be free of mycoplasma.
The IncuCyte proliferation and migration
phase-contrast imaging assay
For cell proliferation assay, 15,000 IMR-32-Luc and
IGR-N91-Luc cells/well were seeded in 96-well plates and incubated
for 72 h with cabozantinib or axitinib at concentrations of 0–10
µmol/l or 0.5% DMSO. Cell confluence was imaged by phase
contrast using the IncuCyte HD system (IncuCyte™ live-cell). Frames
were captured at 4-h intervals from 2 separate regions/well using a
×10 objective. Cultures were run three times in at least
quadruplicates. Proliferation growth curves were constructed by
imaging plates using IncuCyte™ Zoom software, where growth curves
were built from confluence measurements acquired during
round-the-clock kinetic imaging. Cell migration was evaluated by
scratch assays. A scratch was made on confluent monolayers using a
96-pin WoundMaker™ and incubated with cabozantinib at 5
µmol/l for 48 h. Wound images were automatically acquired
and registered by IncuCyte Zoom software system. Data were
processed and analyzed using IncuCyte Zoom 96-Well Cell Invasion
Software Application Module (all from Essen BioScience, Inc., Ann
Arbor, MI, USA). Data are presented as the wound width closure. The
rate of wound closure was compared between treated and non-treated
conditions.
Western blot analysis
Total tumor lysates were separated
electrophoretically and proteins were detected using monoclonal
mouse antibody anti-human β-actin (S125; diluted 1:1,000; Cell
Signaling Technology, Danvers, MA, USA), mouse polyclonal
anti-human PARP-1 (Ab-2, 1:600; Calbiochem, San Diego, CA, USA),
rabbit polyclonal anti-human p-ERK1/2 (Thr202/Tyr204), ERK1/2,
p-AKT (Ser473), AKT, p-VEGFR2 (Tyr1175) (19A10), VEGFR2, p-MET
(Tyr1234/1235), p-MET (Tyr1249), MET (1:1,000, all from Cell
Signaling Technology) as previously described (34).
Angiogenesis and MAPK proteome array
Protein lysates of parental IGR-N91-Luc and
axitinib-resistant AxiR cells were subjected to the human
angiogenesis and phospho-MAPK array kit (R&D Systems, Inc.,
Minneapolis, MN, USA) according to the manufacturer's
recommendations.
Experimental in vivo design
Animal experiments were carried out under conditions
established by the European Community (Directive 2010/63/UE) and
approved by the Comité d'Ethique en Expérimentation Animale n°26
(CEEA26) and the French Ministry (MENESR) (approval number:
00328.01). Antitumor activity was evaluated against orthotopic and
systemic neuroblastoma in female Swiss athymic mice, NSG or BalbC
RAGγc mice, established by injection of 106 cells into
the left adrenal or the tail vein, respectively, under isoflurane
anesthesia as previously reported (33,34)
and all efforts were made to minimize suffering. Briefly, treatment
started when positive bioluminescent signals were detected.
Cabozantinib was administered by oral gavage at 30 or 60 mg/kg/day
for a minimum of 14 days and axitinib at 30 mg/kg/twice daily (BID)
during 29 days; control animals received vehicle. Animals were
followed daily for clinical status, weekly for body weight.
Antitumor activity was determined using ultrasound and
bioluminescence, respectively. Statistical difference was
determined using the two-tailed non-parametric Mann-Whitney or
Kruskal-Wallis test and Prism® software version 6.00.
For pharmacodynamic analysis, primary tumors were harvested 2 h
after the end of treatment at day 29.
Histology and immunohistochemistry
Hematoxylin-eosin-safranin staining was performed on
paraformaldehyde fixed tumors for morphology; immunohistochemistry
for CD34 and caspase-3 expression was determined using rat
anti-mouse CD34 antibody (1:20; Hycult Technologies) and rat
anti-human cleaved caspase-3 antibody (1:100; Cell Signaling
Technology) as previously reported (34).
Results
Acquired resistance to selective VEGFR1-3
inhibition in the IGR-N91-Luc-AxiR cell line is associated with HGF
overexpression
In order to explore resistance mechanisms to
selective VEGFR inhibition we first developed in vitro a
resistant cell line, IGR-N91-Luc-AxiR, through chronic exposure of
IGR-N91-Luc cells in culture to the VEGFR1-3 tyrosine kinase
inhibitor axitinib. Axitinib at the 50% growth inhibiting
concentration (IC50 of 0.75 µmol/l) allowed
maintaining cell viability as well as selection of resistant clones
in the cell line. Constant resistant behavior was achieved after
five consecutive passages under continuous axitinib exposure. In
regard to cell proliferation inhibition, the IC50 of
axitinib in the parental IGR-N91-Luc cells was 0.75 µmol/l
when administered once as measured by phase-contrast imaging during
72 h. In contrast, proliferation of the resistant IGR-N91-Luc-AxiR
cells was less affected with an IC50 of 3 µM
(Fig. 1A). Indeed, growth of
IGR-N91-Luc-AxiR cells under axitinib treatment was identical to
that of the non-treated parental IGR-N91-Luc cells (data not
shown). We further explored the migration capacity of the parental
and axitinib-resistant IGR-N91-Luc cells under axitinib treatment.
Cells were seeded in monolayers into 96-well plates and a scratch
was done when they were at confluence. Axitinib at the
IC50 or 0.5% DMSO were added into each well and
migration was followed during 48 h when the wound of non-treated
parental IGR-N91-Luc control cells was nearly closed. For parental
IGR-N91-Luc cells wound repair inhibition of up to 54% was observed
under axitinib treatment whereas IGR-N91-Luc-AxiR cells showed no
reduction in cell migration under axitinib and had nearly closed
the scratch wound similar to non-treated cells (Fig. 1B). Thus, axitinib-resistant IGR-N91
neuroblastoma cells exhibit reduced sensitivity to inhibition of
both growth proliferation and cell migration to axitinib. To
explore potential escape mechanisms involved in this resistance we
subjected the cell lysates of parental IGR-N91-Luc and
IGR-N91-Luc-AxiR cells to proteome arrays for angiogenesis protein
and MAPK signaling expression which simultaneously detect the
relative levels of 55 angiogenesis-related proteins and the
relative phosphorylation of 24 kinases captured by 26 different
antibodies, respectively, spotted in duplicates on a nitrocellulose
membrane (Fig. 1C).
IGR-N91-Luc-AxiR cells exhibited overexpression of HGF and
phosphorylation of downstream effector ERK1 as compared to the
parental cells as well as reduced expression of PEDF, a
physiological negative regulator of angiogenesis. In response to
axitinib treatment at 0.75 µmol/l, the parental IGR-N91-Luc
cells showed inhibition of VEGFR2 phosphorylation up to 120 min
with an activation of p-ERK at the latest time-point explored. In
contrast, IGR-N91-Luc-AxiR cells exhibited activated/phosphorylated
ERK at baseline, consistent with the MAPK array, which diminished
at the latest time-point and no reduction of VEGFR2 phosphorylation
was observed to axitinib exposure (Fig. 1D). Thus, activation of the HGF/MET
and MAPK signaling pathway is involved in resistance to VEGFR
inhibition in neuroblastoma.
HGF stimulates the MET signaling pathway
in neuroblastoma cells and cabozantinib inhibits proliferation and
migration
We next explored the effects of HGF stimulation on
the two parental IGR-N91-Luc and IMR-32-Luc neuroblastoma cell
lines. Cells cultured under serum-free medium conditions overnight
were incubated with 60 ng/ml HGF and harvested at indicated
time-points up to 120 min for western blot analyses (Fig. 2A). In the IMR-32-Luc cells HGF
exposure resulted in significant activation of its receptor MET
with an increase of phosphorylation in sites Tyr1234/1235 and to a
lesser extent at Tyr1349 whereas expression levels of the total
receptor remained stable. The effects were less prominent in the
IGR-N91-Luc cells. Based on the hypothesis raised above that HGF
signaling is involved in promoting cell survival under selective
VEGFR inhibition, we subjected both parental cell lines to the
potent VEGFR2 and MET tyrosine kinase inhibitor cabozantinib to the
proliferation assay for 72 h. Cabozantinib inhibited cell growth of
IGR-N91-Luc and IMR-32-Luc cells in a concentration-dependent
manner, with IC50 doses of 1.4 and 2.8 µM,
respectively (Fig. 2B). Despite
the similar IC50 of the cells, treatment with
cabozantinib at 5 and 10 µM for 2 and 24 h resulted in
inhibition of phosphorylation of MET and downstream signaling
effectors AKT and MAPK in IMR-32-Luc cells, whereas the inhibition
was found only at higher concentrations and at 2 h in the
IGR-N91-Luc cells (Fig. 2C).
Effects of selective HGF stimulation on cell proliferation capacity
were impossible to evaluate due to the lack of growth of these
neuroblastoma cells in the absence of serum. When exploring the
effects on migration in both cell lines we found no significant
increase in cell migration to HGF selective medium as compared to
10% FCS growth conditions although IMR-32-Luc cells seemed
stimulated under HGF (ns). Cabozantinib at 5 µM reduced
significantly migratory capacity in both cell lines, under both
selective HGF and serum conditions, and migration was completely
inhibited in IMR-32-Luc (Fig. 2D).
Thus, activation of HGF-mediated MET signaling is involved in
neuroblastoma cell migration and inhibition of MET results in
reduced tumor cell migration capacity (1,8,9).
Specific inhibition of VEGFR1-3 signaling
pathway is associated with increased metastases of IGR-N91-Luc
neuroblastoma tumors
In vivo we first explored pan-VEGFR
inhibition using axitinib against the orthotopic IGR-N91-Luc
neuroblastoma xenograft model in BalbC RAGγC mice in order to
extend our previous data in the subcutaneous and orthotopic IGR-N91
xenografts that had shown growth inhibition to axitinib (35). Axitinib treatment was initiated 2
days after injection of cells into the left adrenal administered by
oral gavage twice daily at 30 mg/kg/dose and was given for a longer
time period (i.e. 29 days) than the previous experiments in order
to allow potential development of metastases in this model. At day
29, several animals in the axitinib treated group had reduced tumor
load although the median tumor volume of primary adrenal tumors was
not significantly reduced compared to controls as determined by
ultrasound (Fig. 3A). At autopsy
at day 29, macroscopically visible metastases were detected in 6
out of 16 animals treated with axitinib, located in the liver,
whereas this was the case in 1 out of 8 control animals (Fig. 3B). Bioluminescence imaging ex
vivo detected a signal increase in tumor burden in the livers
of treated animals as compared to the treated animals (ns; Fig. 3C). Western blot analysis of adrenal
tumors of 7 control and 14 treated animals revealed enhanced MET
and ERK phosphorylation in some tumors, independent of tumor size
and the occurrence of metastasis. AKT phosphorylation was inhibited
in smaller samples in comparison to controls (Fig. 3D). In addition 10 out of the 15
analyzed tumors treated showed SRC phosphorylation which was
present only in 2 out of 7 controls. Thus, selective VEGFR1-3
inhibition may result in enhanced metastatic spread and SRC
activation in neuroblastoma tumors.
Cabozantinib inhibits tumor growth of
orthotopic IGR-N91-Luc neuroblastoma in a dose-dependent
manner
Based on our observation in vitro that
HGF-mediated MET upregulation could be involved in the invasiveness
of neuroblastoma cells, we investigated the dual inhibition of
VEGFR and MET against the adrenal IGR-N91-Luc model in NSG mice.
Cabozantinib at 30 and 60 mg/kg resulted in a dose-dependent tumor
growth inhibition of 60 and 87%, respectively, compared to the
control group (P=0.005; Kruskal-Wallis test) with 7 animals in each
group (Fig. 4A–C). Protracted
administration of cabozantinib at both doses was well tolerated
with minor body weight loss in treated animals. To gain insight
into the mechanisms by which cabozantinib inhibits tumor growth, we
explored anti-angiogenic and pro-apoptotic effects in adrenal
tumors using immunohistochemistry and western blot analysis.
Cabozantinib treated xenografts showed significantly reduced
microvessel density compared to control tumors as determined by
CD34 staining (P=0.0116 for 30 mg/kg and P=0.0025 for 60 mg/kg
treated tumors; Kruskal-Wallis test; Fig. 4D). A dose-dependent increase in
cleaved caspase-3-positive nuclei was noted for cabozantinib at the
higher dose as compared to controls (P=0.0091 for 60 mg/kg;
Fig. 4E). When investigating the
effects on the VEGF and HGF signaling pathways by exploring known
key downstream effectors (Fig.
4F), we observed the appearance of ERK1/2 phosphorylation in
tumor samples to cabozantinib treatment as compared to controls. No
modification was observed in regard to AKT activation levels. In
contrast, SRC phosphorylation was found inhibited at both
concentrations in comparison to control tumors.
 | Figure 4Cabozantinib exhibits antitumor
activity against IGR-N91-Luc orthotopic neuroblastoma mediated by
anti-angiogenic effects and induction of cell death. Animals
bearing orthotopic adrenal IGR-N91-Luc tumors were treated orally
with cabozantinib at 30 mg/kg/day (light blue line), 60 mg/kg/day
(dark blue line) or vehicle (grey line). (A) Graphs show growth
curves of individual mice (light grey) and arithmetic means (±
standard error of mean). Tumor volumes were detected by
bioluminescence (B) and ultrasonography (C). Images depict
representative adrenal tumors of control and treated animals at day
22. Paraffin-embedded sections of IGR-N91-Luc orthotopic primary
tumors treated with vehicle (light grey, n=7), cabozantinib at 30
mg/kg (dark grey, n=7) or 60 mg/kg (black, n=6), harvested at day
29 post treatment initiation, were stained immunohistochemically
with anti-CD34 and anti-cleaved caspase-3 antibodies. (D) The
percentage of CD34-positive area was determined and microvessel
density was quantified using CaloPix software and statistically
evaluated by Kruskal-Wallis test. (E) The apoptosis index was
determined as number of cleaved caspase-3 positive cells per
mm2. Representative examples of histological stainings
are shown at ×100 magnification. Positive staining appears as brown
color. Graphs represent means (± standard error of mean,
Kruskal-Wallis test; *P<0.05,
**P<0.01). (F) Total lysates from three individual
tumors were subjected to western blotting for expression analyses
of phosphorylated (p-) and non-phosphorylated AKT, ERK1/2, SRC and
β-actin. |
Thus, cabozantinib acts by inhibition of
angiogenesis at both dose levels whereas induction of cell death
demands higher concentrations; inhibition of SRC appears associated
with antitumor activity.
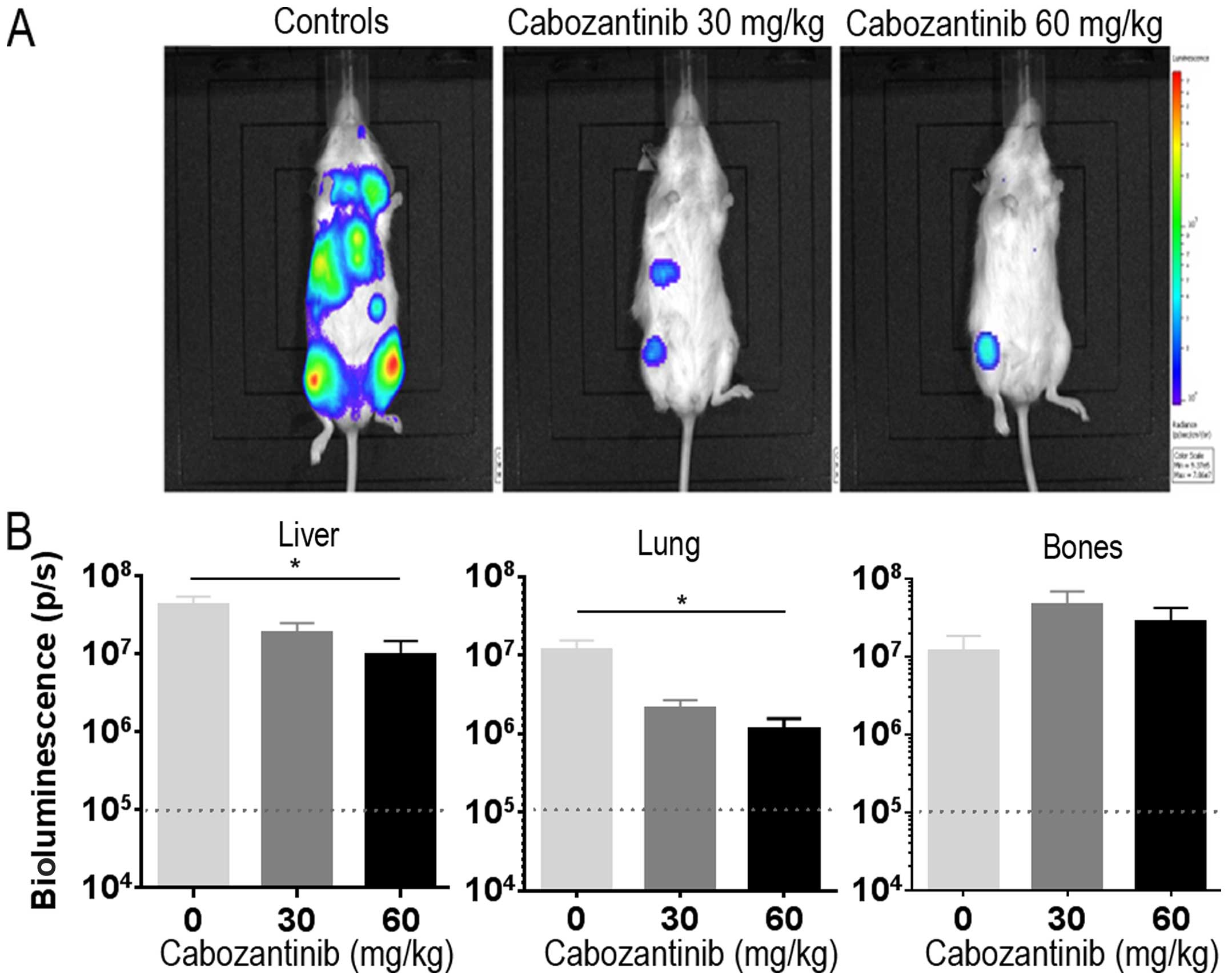
Cabozantinib reduces metastatic spread in
the systemic IMR-32-Luc neuroblastoma model
To be able to best explore the inhibition of
metastatic spread of neuroblastoma, we used our systemic IMR-32-Luc
model in female NSG mice which develop metastases preferably in
lungs, liver and bones (33).
Cabozantinib orally at 30 and 60 mg/kg/day was initiated at day 10
post tail vein injection, when bioluminescence signals were
detectable in all animals and was given for 34 days when the
animals were sacrificed. Bioluminescence signals in vivo in
control animals were distributed over the liver region, thorax, and
bilateral lower extremities consistent with the previous metastatic
homing pattern in this model (33). Cabozantinib treated animals in
treatment groups exhibited reduced bioluminescence signals
(Fig. 5A). Luciferase activity of
tumor cells ex vivo was significantly reduced in liver (n=6,
P=0.0238; Kruskal-Wallis test) and lung (n=6, P=0.0141) in the 60
mg/kg treated animals as compared with the control group. This was
not the case in bones (Fig. 5B).
Thus, cabozantinib reduces metastatic spread to visceral organs in
IMR-32 neuroblastoma.
Discussion
Inhibition of angiogenesis is significantly hampered
by the induction of pro-angiogenic factors resulting in secondary
tumor escape from treatment. In the present study, we explored the
escape mechanisms to selective pan-VEGFR inhibition in preclinical
neuroblastoma models. We found HGF mediated MET, MAPK and SRC
pathway signaling involved in neuroblastoma escape to VEGFR1-3
inhibition which resulted in enhanced cell migration, suggesting
that dual inhibition of VEGFR and MET may be a rational approach
for further investigation in neuroblastoma treatment.
Through a new resistant cell line that was developed
in vitro by consistent exposure to axitinib we could show
that the loss of sensitivity to selective VEGFR1-3 inhibition was
associated with the induction of HGF and subsequent signaling
through downstream effector MAPK pathway. The HGF/MET pathway is
not constitutively upregulated in neuroblastoma but most
neuroblastoma cell lines tested exhibited low MET expression (data
not shown) which could be stimulated by its physiological ligand
HGF, as shown here for the two cell lines IGR-N91-Luc and
IMR-32-Luc. MET signaling is involved in cell proliferation as well
as cellular migration (36). Hecht
et al (12,13) provided the first evidence that the
HGF/c-Met pathway is essential for invasiveness and malignant
progression of human neuroblastomas in vitro and in chick
chorioallantoic membranes. Elevated concentrations of HGF and
soluble VEGF-A have been reported in patients with neuroblastoma
and were correlated with higher stage disease (11). Both of our luciferase transfected
cell lines are sensitive to proliferation and migration inhibiting
effects of cabozantinib, an inhibitor of VEGFR2 and MET kinases,
with IC50s in the low micromolar range. Our findings
were consistent with the recent results described by Zhang et
al (37) where IMR-32 was one
of the most sensitive neuroblastoma cell lines tested. In addition,
we demonstrated that wound repair capacity was significantly
reduced in IGR-N91-Luc cells and more importantly in the IMR-32-Luc
cell line. This cell line also showed enhanced MET
activation/phosphorylation when stimulated with HGF as compared to
IGR-N91-Luc suggesting differences in the capacity of neuroblastoma
cells to activate this survival pathway. The in vitro
effects of cabozantinib were associated with inhibition of MET.
In vivo we confirmed our previous data on
tumor growth inhibition with the pan-VEGFR inhibitor axitinib in
IGR-N91-Luc neuroblastoma (35).
However, in the more immunocompromized BalbC RAGγC mice, we
observed a limited growth inhibiting effect on the primary adrenal
tumor whereas animals treated with axitinib presented metastases
more frequently at study end, mainly to the liver, suggesting
actually the induction of a more aggressive disease pattern under
axitinib treatment. When exploring tumor samples, we found, beside
the induction of MET, SRC activation/phosphorylation in most
axitinib treated tumors as compared to controls. Both, activation
of MET and SRC, have been reported to increase aggressiveness of
tumor cells and promote metastases in response to anti-VEGF
antibody and multi-tyrosine kinase VEGFR inhibitors such as
sorafenib, sunitinib and bevacizumab (15,24,38,39).
Moreover, the HGF/MET signaling has been described to induce
angiogenesis independently of VEGF (40). We further detected in our resistant
cell line the reduction of expression levels of PEDF, which is a
physiological negative regulator of angiogenesis. Loss of PEDF
could be another mechanism of resistance in neuroblastoma and its
role needs to be explored further.
Consistent with the in vitro evaluation,
cabozantinib resulted in significant dose-dependent inhibition of
tumor growth of adrenal IGR-N91-Luc tumors compared to controls.
These effects appeared more prominent as those observed in the
previous experiments to axitinib (35) although the results in sensu
stricto cannot be compared directly as they were performed in
independent experiments. Orthotopic primary IGR-N91-Luc tumors
showed reduced microvessel density for the 30 and the 60 mg/kg dose
levels, however, at the higher treatment dose also significant
induction of apoptotic cell death was observed. Prolonged exposure
of cabozantinib resulted in a reduction of SRC phosphorylation in
treated tumors. SRC activation was detected in tumors of mice
following prolonged axinitinib treatment and, thus, its inhibition
may be additionally involved in the superior effects of
cabozantinib albeit it is not inhibiting SRC directly. In our
systemic neuroblastoma model, cabozantinib treatment resulted in
significant reduction of metastasis formation in the IMR-32-Luc,
mainly to the liver and the lungs. However, we observed no
reduction of bone metastases as it had been reported in patients
with metastatic prostate cancer and the difference of the mouse
environment to the human microenvironment could underlie these
distinct results in an organ which is very much influenced by the
microenvironment (26,28). Recently, it has been shown that
cabozantinib could affect bone microenvironment in normal condition
highlighting its potential role in mediating treatment responses,
supporting the observations in patients with bone metastases in
prostate cancer. In addition, other factors of the tumor
microenvironment (including myeloid cells or microphages) are
likely critical in promoting metastasis and a potential effect of
cabozantinib on the tumor microenvironment needs further
investigations (26,41).
Our results with pan-VEGFR inhibition in
vitro and in vivo support previously raised concerns
that a more aggressive pattern of tumor cells may occur with
anti-angiogenic therapies (42–44)
through adaptive mechanisms such as activation of alternative
pro-angiogenic signaling pathways such as HGF/c-MET and SRC
signaling that may result in enhanced invasiveness and metastases.
Dual VEGFR2 and MET inhibiting antitumor activity was mediated by
anti-angiogenic, anti-migratory and pro-apoptotic effects, and
associated with reduction of metastatic spread supporting the
hypothesis that inhibition of a larger spectrum of targets beyond
the VEGF pathway may help to circumvent the resistance to more
selective angiogenesis blockade. The enhanced aggressiveness
associated with HGF signaling may be considered in the further
development of anti-angiogenic treatments in neuroblastoma.
Acknowledgments
We are grateful to the Platform of Preclinical
Evaluation (PFEP) and Dr Patrick Gonin for providing
immunocompromized mice and health care of animals as well as Dr
Valérie Rouffiac of the Platform of Cell Imaging (PFIC) for their
advice in bioluminescence imaging and Dr Ingrid Leguerney for
ultrasound imaging. We thank Carole Lecinse for critical reading of
the manuscript. The present study was supported by a grant from
'Fédération Enfants et Santé' and the 'Société Française des
Cancers de l'Enfant.
References
|
1
|
Birchmeier C and Gherardi E: Developmental
roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends
Cell Biol. 8:404–410. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grant DS, Kleinman HK, Goldberg ID,
Bhargava MM, Nickoloff BJ, Kinsella JL, Polverini P and Rosen EM:
Scatter factor induces blood vessel formation in vivo. Proc Natl
Acad Sci USA. 90:1937–1941. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang L, Yang N, Park JW, Katsaros D,
Fracchioli S, Cao G, O'Brien-Jenkins A, Randall TC, Rubin SC and
Coukos G: Tumor-derived vascular endothelial growth factor
up-regulates angiopoietin-2 in host endothelium and destabilizes
host vasculature, supporting angiogenesis in ovarian cancer. Cancer
Res. 63:3403–3412. 2003.PubMed/NCBI
|
|
4
|
Sulpice E, Ding S, Muscatelli-Groux B,
Bergé M, Han ZC, Plouet J, Tobelem G and Merkulova-Rainon T:
Cross-talk between the VEGF-A and HGF signalling pathways in
endothe-lial cells. Biol Cell. 101:525–539. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC
and Van Waes C: Hepatocyte growth factor/scatter factor-induced
activation of MEK and PI3K signal pathways contributes to
expression of proangiogenic cytokines interleukin-8 and vascular
endothelial growth factor in head and neck squamous cell carcinoma.
Cancer Res. 61:5911–5918. 2001.PubMed/NCBI
|
|
6
|
Dong G, Lee TL, Yeh NT, Geoghegan J, Van
Waes C and Chen Z: Metastatic squamous cell carcinoma cells that
overexpress c-Met exhibit enhanced angiogenesis factor expression,
scattering and metastasis in response to hepatocyte growth factor.
Oncogene. 23:6199–6208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abounader R, Lal B, Luddy C, Koe G,
Davidson B, Rosen EM and Laterra J: In vivo targeting of SF/HGF and
c-met expression via U1snRNA/ribozymes inhibits glioma growth and
angiogenesis and promotes apoptosis. FASEB J. 16:108–110. 2002.
|
|
8
|
Abounader R and Laterra J: Scatter
factor/hepatocyte growth factor in brain tumor growth and
angiogenesis. Neuro Oncol. 7:436–451. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Goyal L, Muzumdar MD and Zhu AX: Targeting
the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res.
19:2310–2318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kawakami H, Okamoto I, Okamoto W, Tanizaki
J, Nakagawa K and Nishio K: Targeting MET amplification as a new
oncogenic driver. Cancers (Basel). 6:1540–1552. 2014. View Article : Google Scholar
|
|
11
|
Sköldenberg EG, Larsson A, Jakobson A,
Hedborg F, Kogner P, Christofferson RH and Azarbayjani F: The
angiogenic growth factors HGF and VEGF in serum and plasma from
neuroblastoma patients. Anticancer Res. 29:3311–3319.
2009.PubMed/NCBI
|
|
12
|
Hecht M, Schulte JH, Eggert A, Wilting J
and Schweigerer L: The neurotrophin receptor TrkB cooperates with
c-Met in enhancing neuroblastoma invasiveness. Carcinogenesis.
26:2105–2115. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hecht M, Papoutsi M, Tran HD, Wilting J
and Schweigerer L: Hepatocyte growth factor/c-Met signaling
promotes the progression of experimental human neuroblastomas.
Cancer Res. 64:6109–6118. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pennacchietti S, Michieli P, Galluzzo M,
Mazzone M, Giordano S and Comoglio PM: Hypoxia promotes invasive
growth by transcriptional activation of the met protooncogene.
Cancer Cell. 3:347–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sennino B, Ishiguro-Oonuma T, Wei Y,
Naylor RM, Williamson CW, Bhagwandin V, Tabruyn SP, You WK, Chapman
HA, Christensen JG, et al: Suppression of tumor invasion and
metastasis by concurrent inhibition of c-Met and VEGF signaling in
pancreatic neuroendocrine tumors. Cancer Discov. 2:270–287. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qian F, Engst S, Yamaguchi K, Yu P, Won
KA, Mock L, Lou T, Tan J, Li C, Tam D, et al: Inhibition of tumor
cell growth, invasion, and metastasis by EXEL-2880 (XL880,
GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine
kinases. Cancer Res. 69:8009–8016. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huynh H, Ong R and Soo KC: Foretinib
demonstrates anti-tumor activity and improves overall survival in
preclinical models of hepatocellular carcinoma. Angiogenesis.
15:59–70. 2012. View Article : Google Scholar
|
|
18
|
Yakes FM, Chen J, Tan J, Yamaguchi K, Shi
Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al: Cabozantinib
(XL184), a novel MET and VEGFR2 inhibitor, simultaneously
suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer
Ther. 10:2298–2308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Guessous F, Kofman A, Schiff D
and Abounader R: XL-184, a MET, VEGFR-2 and RET kinase inhibitor
for the treatment of thyroid cancer, glioblastoma multiforme and
NSCLC. IDrugs. 13:112–121. 2010.PubMed/NCBI
|
|
20
|
Torres KE, Zhu QS, Bill K, Lopez G,
Ghadimi MP, Xie X, Young ED, Liu J, Nguyen T, Bolshakov S, et al:
Activated MET is a molecular prognosticator and potential
therapeutic target for malignant peripheral nerve sheath tumors.
Clin Cancer Res. 17:3943–3955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Navis AC, Bourgonje A, Wesseling P, Wright
A, Hendriks W, Verrijp K, van der Laak JA, Heerschap A and Leenders
WP: Effects of dual targeting of tumor cells and stroma in human
glioblastoma xenografts with a tyrosine kinase inhibitor against
c-MET and VEGFR2. PLoS One. 8:e582622013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bentzien F, Zuzow M, Heald N, Gibson A,
Shi Y, Goon L, Yu P, Engst S, Zhang W, Huang D, Zhao L, et al: In
vitro and in vivo activity of cabozantinib (XL184), an inhibitor of
RET, MET, and VEGFR2, in a model of medullary thyroid cancer.
Thyroid. 23:1569–1577. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
You WK, Sennino B, Williamson CW, Falcón
B, Hashizume H, Yao LC, Aftab DT and McDonald DM: VEGF and c-Met
blockade amplify angiogenesis inhibition in pancreatic islet
cancer. Cancer Res. 71:4758–4768. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiang Q, Chen W, Ren M, Wang J, Zhang H,
Deng DY, Zhang L, Shang C and Chen Y: Cabozantinib suppresses tumor
growth and metastasis in hepatocellular carcinoma by a dual
blockade of VEGFR2 and MET. Clin Cancer Res. 20:2959–2970. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kurzrock R, Sherman SI, Ball DW,
Forastiere AA, Cohen RB, Mehra R, Pfister DG, Cohen EE, Janisch L,
Nauling F, et al: Activity of XL184 (Cabozantinib), an oral
tyrosine kinase inhibitor, in patients with medullary thyroid
cancer. J Clin Oncol. 29:2660–2666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smith DC, Smith MR, Sweeney C, Elfiky AA,
Logothetis C, Corn PG, Vogelzang NJ, Small EJ, Harzstark AL, Gordon
MS, et al: Cabozantinib in patients with advanced prostate cancer:
Results of a phase II randomized discontinuation trial. J Clin
Oncol. 31:412–419. 2013. View Article : Google Scholar
|
|
27
|
Zhu K, Kong X, Zhao D, Liang Z and Luo C:
c-MET kinase inhibitors: A patent review (2011–2013). Expert Opin
Ther Pat. 24:217–230. 2014. View Article : Google Scholar
|
|
28
|
Saylor PJ, Lee RJ and Smith MR: Emerging
therapies to prevent skeletal morbidity in men with prostate
cancer. J Clin Oncol. 29:3705–3714. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Agarwal N, Sonpavde G and Sternberg CN:
Novel molecular targets for the therapy of castration-resistant
prostate cancer. Eur Urol. 61:950–960. 2011. View Article : Google Scholar
|
|
30
|
Drilon A, Wang L, Hasanovic A, Suehara Y,
Lipson D, Stephens P, Ross J, Miller V, Ginsberg M, Zakowski MF, et
al: Response to Cabozantinib in patients with RET fusion-positive
lung adenocarcinomas. Cancer Discov. 3:630–635. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Choueiri TK, Pal SK, McDermott DF,
Morrissey S, Ferguson KC, Holland J, Kaelin WG and Dutcher JP: A
phase I study of cabozantinib (XL184) in patients with renal cell
cancer. Ann Oncol. 25:1603–1608. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Elisei R, Schlumberger MJ, Müller SP,
Schöffski P, Brose MS, Shah MH, Licitra L, Jarzab B, Medvedev V,
Kreissl MC, et al: Cabozantinib in progressive medullary thyroid
cancer. J Clin Oncol. 31:3639–3646. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Daudigeos-Dubus E, LE Dret L, Rouffiac V,
Bawa O, Leguerney I, Opolon P, Vassal G and Geoerger B:
Establishment and characterization of new orthotopic and metastatic
neuroblastoma models. In Vivo. 28:425–434. 2014.PubMed/NCBI
|
|
34
|
Daudigeos-Dubus E, Le Dret L,
Lanvers-Kaminsky C, Bawa O, Opolon P, Vievard A, Villa I, Pagès M,
Bosq J, Vassal G, et al: Regorafenib: Antitumor activity upon mono
and combination therapy in preclinical pediatric malignancy models.
PLoS One. 10:e01426122015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rössler J, Monnet Y, Farace F, Opolon P,
Daudigeos-Dubus E, Bourredjem A, Vassal G and Geoerger B: The
selective VEGFR1-3 inhibitor axitinib (AG-013736) shows antitumor
activity in human neuroblastoma xenografts. Int J Cancer.
128:2748–2758. 2011. View Article : Google Scholar
|
|
36
|
Sierra JR and Tsao MS: c-MET as a
potential therapeutic target and biomarker in cancer. Ther Adv Med
Oncol. 3(Suppl): S21–S35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang L, Scorsone K, Woodfield SE and Zage
PE: Sensitivity of neuroblastoma to the novel kinase inhibitor
cabozantinib is mediated by ERK inhibition. Cancer Chemother
Pharmacol. 76:977–987. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shojaei F, Simmons BH, Lee JH, Lappin PB
and Christensen JG: HGF/c-Met pathway is one of the mediators of
sunitinib-induced tumor cell type-dependent metastasis. Cancer
Lett. 320:48–55. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huveldt D, Lewis-Tuffin LJ, Carlson BL,
Schroeder MA, Rodriguez F, Giannini C, Galanis E, Sarkaria JN and
Anastasiadis PZ: Targeting Src family kinases inhibits
bevacizumab-induced glioma cell invasion. PLoS One. 8:e565052013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sengupta S, Gherardi E, Sellers LA, Wood
JM, Sasisekharan R and Fan TP: Hepatocyte growth factor/scatter
factor can induce angiogenesis independently of vascular
endothelial growth factor. Arterioscler Thromb Vasc Biol. 23:69–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Haider MT, Hunter KD, Robinson SP, Graham
TJ, Corey E, Dear TN, Hughes R, Brown NJ and Holen I: Rapid
modification of the bone microenvironment following short-term
treatment with Cabozantinib in vivo. Bone. 81:581–592. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bergers G and Hanahan D: Modes of
resistance to anti-angiogenic therapy. Nat Rev Cancer. 8:592–603.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bottsford-Miller JN, Coleman RL and Sood
AK: Resistance and escape from antiangiogenesis therapy: Clinical
implications and future strategies. J Clin Oncol. 30:4026–4034.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Giuliano S and Pagès G: Mechanisms of
resistance to anti-angiogenesis therapies. Biochimie. 95:1110–1119.
2013. View Article : Google Scholar : PubMed/NCBI
|