Introduction
Methylation of islands has been shown to inhibit
transcription directly or stabilize chromatin in a conformation
that prevents transcription (1).
Hypermethylation of CpG islands is an important epigenetic
mechanism for silencing the transcription of many genes (2). DNA methylation inhibits gene
transcription by affecting the chromatin structure, in particular
via the protein complexes comprising methyl-binding domains,
transcriptional corepressors, and histone deacetylase in
hypermethylated regions of DNA (3–5). In
hematopoietic development, this was first demonstrated for
erythropoiesis, and later also for lymphoid and myeloid
differentiation (6–9). Aberrant methylation of
tumor-suppressor and growth-regulatory genes has been reported as
the most frequent alteration in both hematologic neoplasms and
solid tumors (10). Promoter
methylation is an increasingly recognized mechanism of
transcriptional silencing in human cancer. Downregulated expression
of target genes results from single transitional changes in these
important regulatory sequences. It has been demonstrated that
promoter methylation can be induced by viral agents or by
recruitment of the methyl-transferase enzymatic machinery (11,12).
Inhibins and activins, members of the transforming
growth factor-β (TGF-β) superfamily, are polypeptides that were
originally isolated from ovarian fluid, based on their effect on
pituitary follicle-stimulating hormone (FSH) production and
secretion. Inhibins are heterodimers that are composed of a common
α subunit and one of two homologous β subunits (βA and βB)
(13,14). More recently, both inhibins and
activins have been implicated in endocrine-related cancers
(15). The inhibin-α gene was
identified as a tumor suppressor gene in the gonads and adrenal
glands by functional studies using knockout mice (16,17).
This has raised the question of whether it plays a broader role as
a tumor suppressor outside the reproductive axis. Furthermore,
hypermethylation of the inhibin-α gene promoter and loss of
heterozygosity (LOH) at 2q32-36, the chromosome region harboring
the inhibin-α gene, has been reported in prostate carcinoma and
gastric cancer (18,19). Moreover, there was a positive
correlation between loss of inhibin expression and malignancy of
these human prostate carcinoma cells (20). Hypermethylation of the inhibin-α
promoter and LOH are frequently associated with silencing or loss
of expression of tumor suppressor genes, and the effects of
deletions involving the 2q33-36 regions in human leukemia cells are
unclear. Recently many studies have been made regulating promoter
methylation which is a useful biomarker for the diagnosis of cancer
patients (21,22).
We investigated the epigenetic modifications,
changes in LOH and mutations of the inhibin-α gene, and regulation
of transcriptional expression by inhibitors of DNA methylation
(5-aza-2′-deoxycytidine, 5-AzaC) in human lymphoid and myeloid
leukemia cells.
Materials and methods
Cell culture
Lymphoid (Jurkat, Molt-4, IM-9, and Raji) and
myeloid (HL-60, Kasumi-1, and K562) human leukemia cell lines were
purchased from the American Tissue Culture Collection (ATCC,
Rockville, MD, USA). Cells were cultured in RPMI-1640 medium
(Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum
(FBS), 100 U/ml penicillin and 100 μg/ml streptomycin at
37°C in a humidified atmosphere of 5% CO2 in 95%
air.
Bisulfite modification
The methylation status of the promoter CpG islands
of the inhibin-α subunit gene was analyzed by methylation specific
PCR (MSP) using sodium-bisulfite- converted DNA (23). Genomic DNA was extracted using the
Wizard Genomic DNA purification kit (Promega, Madison, WI, USA).
DNA (2 μg) in a volume of a 50 μl was denatured with
NaOH (final concentration, 0.2 M) and incubated at 37°C for 15 min.
Then, 30 μl of 10 mM hydroquinone and 520 μl of 3 M
sodium bisulfite (Sigma-Aldrich, St. Louis, MO, USA) at pH 5.0,
both freshly prepared, were added and mixed, and samples were
incubated under mineral oil at 55°C for 16 h. DNA was then desalted
using the Wizard DNA Clean-Up System (Promega), desulfonated by
addition of NaOH (final concentration, 0.3 M), and incubated at
37°C for 15 min. The solution was neutralized by addition of
ammonium acetate (final concentration, 3.0 M), and the DNA was
ethanol- precipitated, dried, re-suspended in 20 μl of water
and used immediately or stored at −20°C.
Determination of methylation status
Methylation was assessed by PCR and sequence
analysis of bisulfite-treated DNA. The bisulfite reaction converted
unmethylated cytosines to uracils, whereas methylated cytosines
were unchanged. The inhibin-α subunit 5′-UTR region was amplified
by nested PCR using primers for the bisulfite-converted sequence
(18). Primers 1
(5′-GATAAGAGTTTAGATTGGTTTTATTGGTT-3′) and 2
(5′-ACACCATAACTCACCTAACCCTACTAATAA-3′) were used for the first
round of PCR, and primers 3 (5′-ACCCCTTCTACCAAAATCTACCCAAAA-3′) and
4 (5′-GAAGGTGTTGTATGTTTGTATGTGTGAGTT-3′) were used for the second
round. The first round of PCR was performed in 25 μl
reactions with 2 μl of bisulfite-converted DNA, 1X PCR
buffer [10 mM Tris (pH 8.3), 50 mM KCl, 1.5 mM MgCl2],
200 μM dNTPs, 10 pmol of primers 1 and 2, and 1 U AmpliTaq
Gold DNA polymerase (Roche, Applied Biosystems, Foster City, CA,
USA). PCR cycles consisted of 95°C for 15 min, followed by 5 cycles
of 95°C for 1 min, 50°C for 2 min, and 72°C for 3 min, and then 30
cycles of 95°C for 1 min, 55°C for 2 min, and 72°C for 2 min, with
a final incubation step of 72°C for 10 min. A sample of 2 μl
from the first PCR was amplified in a 25-μl reaction as
above, except that primers 3 and 4 were used. PCR cycling
conditions were as for the first reaction, with the exception that
the annealing temperature was increased to 60°C. PCR products were
gel-purified, ligated into the pCR 2.1 vector, and cloned using the
TOPO TA Cloning kit according to the manufacturer's instructions
(Invitrogen, Carlsbad, CA, USA). For each PCR, 10 clones were
sequenced and methylation at each of the seven CpG sites was
determined. Overall percentage methylation of each sample was
determined as the mean of the percentage methylation at the seven
individual CpG sites.
DNA analysis
DNA was isolated from cultured cells using standard
methods. Two regions of the inhibin-α subunit gene were amplified
from genomic DNA by PCR using specific oligonucleotide primers
(24). The first 240-bp region
(fragment A), which includes 140 bp of the 5′-UTR and 100 bp of
exon 1, was amplified using the primers AF
(5′-GACTGGGGAAGACTGGATGA-3′) and AR (5′-TCACCTTGGCCAGAACAAGT-3′).
The second 396-bp region (fragment B), which comprises part of exon
2, was amplified using the primers BF (AGCAGCCTCCAATAGCTCTG-3′) and
BR (5′-AGCTCCTGGAAGGAGATGTTC-3′). Genomic DNA (200 ng) was
amplified in a 50-μl volume reaction containing 1X PCR
buffer, 2 mM MgCl2, 2.5% DMSO, 0.2 mM dNTP, 20 pmol of
each specific primer and 1.5 U AmpliTaq Gold DNA polymerase. The
amplification conditions were as follows: 35 cycles comprising an
initial denaturation at 95°C for 14 min, then denaturation at 95°C
for 40 sec, annealing at 57°C for 30 sec, and extension at 72°C for
1 min, followed by a final extension at 72°C for 7 min.
Polymorphism −16C>T in 5′-UTR was screened in the samples by
restriction enzyme analysis using SpeI (New England Biolabs,
Ipswich, MA, USA). Briefly, fragment A was amplified by PCR and 5
μl of purified PCR product was digested overnight at 37°C
with 5 U of Spel, electrophoresed in 8% polyacrylamide gels,
stained with ethidium bromide and photographed. The presence of the
240-bp fragment indicated a variant homozygous for C, whereas the
presence of two fragments of 120 bp corresponded to a variant
homozygous for T. Substitution 769G>A of exon 2 was analyzed by
digestion of fragment B with appropriate restriction enzymes. Five
microliters of purified PCR product was digested overnight at 37°C
with 5 U of BsrFI and analyzed as described above. The
restriction site, which renders two fragments of 340 and 56 bp, is
abolished in the mutated allele. In addition, 5 μl of
purified PCR product was digested overnight at 37°C with 5 U of
Fnu4HI, electrophoresed in 15% polyacrylamide gels, stained
with ethidium bromide and visualized by image analysis. The 396-bp
fragment yields four fragments of 153, 107, 51 and 25 bp, among
others of lower molecular weight, in the wild-type allele, whereas
the allele with substitution 769G>A yields four fragments of
153, 107, 76, and 51 bp, among others of lower molecular
weight.
LOH analysis
LOH was determined using microsatellite markers on
2q32-q33 (D2S389) and 2q33-q36 (D2S128), as described previously
(18). The primers used were
D2S389 (5′-TAAAGCCTAGTGGAAGATCATC-3′,
5′-GCTGAGTTAACAGTTATCAACAATT-3′) and D2S128
(5′-AAACTGAGATTTGTCTAAGGGG-3′, 5′-AGCCAGGAATTTTTGCTATT-3′). PCR was
performed in 20 μl reactions consisting of 200 ng of DNA, 1X
PCR buffer, 0.2 mM dNTPs, 10 pmol of each primer, and 1 U AmpliTaq
Gold DNA polymerase. The amplification conditions were as follows:
35 cycles of an initial denaturation at 95°C for 14 min, a second
denaturation at 95°C for 1 min, annealing at 55°C for 1 min, and
extension at 72°C for 1 min, followed by a final extension at 72°C
for 10 min. Then, 10 μ1 of PCR products was mixed with 10
μ1 of stop solution containing 95% formamide, 10 mM NaOH,
0.25% bromophenol blue, and 0.25% xylene cyanol FF. The mixture was
denatured at 95°C for 5 min, placed on ice for 5 min,
electrophoresed in 12% polyacrylamide gels containing 10% glycerol
with 1X TBE buffer, and stained with ethidium bromide.
5-Aza-2′-deoxycytidine treatment
Cells were seeded at a density of
5×105/100 mm dish, allowed to attach for 24 h and then
treated with various concentration of 5-aza-2′-deoxycytidine
(5-AzaC, Sigma) for 5 days. The medium and drug were replaced every
2 days. At the end of the treatment period, the medium was removed
and the cell pellets were used for analysis.
RNA extraction and real-time PCR
Total RNA was extracted from cultured cells using
the TRIzol Reagent kit following the manufacturer's protocol
(Invitrogen). First-strand cDNA was synthesized from 1 μg of
DNase-treated RNA using a reverse transcription system (Promega)
according to the manufacturer's protocol with random hexamers. PCR
was performed with 2 μl cDNA in a 25-μl reaction
mixture of 1X PCR buffer, 0.2 mM of each dNTP, 10 pmol of primers
for inhibin-α (5′-AGGAAGAGGAGGATGTCTCC-3′ and
5′-GAGTAACCTCCATCCCGAGGT-3′; 823 bp), betaglycan
(5′-ACATGGATAAGAAGCGATTCAGC-3′ and 5′-AACGCAATGCCCATCACGGTTAG-3′,
331 bp), and β-actin (5′-CTTCTACAATGAGCTGCGTG-3′ and
5′-TCATGAGGTAGTCAGTCAGG-3′; 305 bp), and 1 U AmpliTaq Gold DNA
polymerase. The reactions were carried out in a thermal cycler with
an initial denaturation step at 95°C for 14 min, followed by 35
cycles (25 cycle for β-actin) of denaturation at 95°C for 1 min,
primer annealing at 50°C (inhibin-α) to 55°C (β-actin) for 1 min,
and a final extension at 72°C for 1 min. The reaction was
terminated at 72°C for 10 min; samples were stored at 4°C. Ten
microliters of PCR products were separated by electrophoresis in a
2% agarose gel containing ethidium bromide (0.5 μg/ml) and
visualized by image analysis (Gel Doc 1000 Gel Documentation
System; Bio-Rad, Hercules, CA, USA). Real-time PCR was performed on
a StepOnePlus Real-Time PCR System with Power SYBR Green PCR Master
Mix (Applied Biosystems). The gene-specific primer sequences were:
inhibin-α, 5′-CTCGGATGGAGGTTACTCTTTCAA-3′ and
5′-GAAGACCCCCCACCCTTAGA-3′ (88 bp); betaglycan,
5′-CAAAGCAGCAGAAGGGTGTGT-3′ and 5′-GGTGATTAGCTCGATGATGTGTACTT-3′
(73 bp); and β-actin, 5′-GCGAGAAGATGACCCAGATC-3′ and
5′-GGATAGCACAGCCTGGATAG-3′ (77 bp). PCR was performed with 1
μl of cDNA in a 20-μl reaction mixture containing 10
μl of Power SYBR Green PCR Master Mix, 2 μl of
primers, and 7 μl of PCR-grade water. The reaction
conditions were denaturation at 95°C for 10 min, followed by 40
cycles of 95°C for 15 sec and 60°C for 1 min. The crossing points
of the target genes with β-actin were calculated using the formula
2−(target gene-β-actin), and relative amounts were
quantified.
FITC-flow cytometric analysis of
inhibin-α protein
Cultured cells were detached with 0.05% trypsin-EDTA
solution. After washing with cold PBS, cells were incubated with a
1:50 dilution of anti-inhibin-α goat polyclonal antibody (Santa
Cruz Biotechnology, Santa Cruz, CA, USA) or normal goat serum as a
negative control for 30 min at 4°C. After washing three times with
cold PBS, cells were stained with a fluorescein isothiocyanate
(FITC)-labeled donkey antibody (1:50 dilution) to rabbit
immunoglobulin for 30 min at 4°C. Washing was repeated in the same
manner and cell-surface immunofluorescence was analyzed using a
FACSCalibur instrument together with CellQuest software
(Becton-Dickinson, San Jose, CA, USA).
Determination of cell doubling time
Cells were treated with 5 μM 5-AzaC for 5
days and washed with PBS. Cells were seeded at 2×104/ml
in 12-well plates containing culture medium, and cell number/dish
was determined by trypan blue assay daily for 5 consecutive days.
Untreated cells were analyzed under similar conditions as a
control. The average cell number from two plates was determined,
and the mean cell numbers were plotted to calculate the doubling
times. The cell population doubling time was calibrated using the
Kuchler formula (25).
Cell cycle analysis
Cells (5×105) were treated with 5
μM 5-AzaC for 5 days. At the end of the treatment period,
cells were harvested, washed with PBS, fixed with 70% ethanol for 1
h, treated with RNAsin (20 μg/ml) at 37°C for 1 h, and
stained with propidium iodide (50 μg/ml; Sigma). DNA content
at each cell cycle stage was analyzed using a FACSCalibur
instrument together with CellQuest software (Becton-Dickinson).
Statistical analysis
Values are expressed as means ± SD. Student's t-test
was used to evaluate differences among the samples. Statistical
analyses were performed using GraphPad Prism 5 software (GraphPad
Software Inc., San Diego, CA, USA). *P<0.05 and **P<0.01 were
considered to indicate statistical significance.
Results
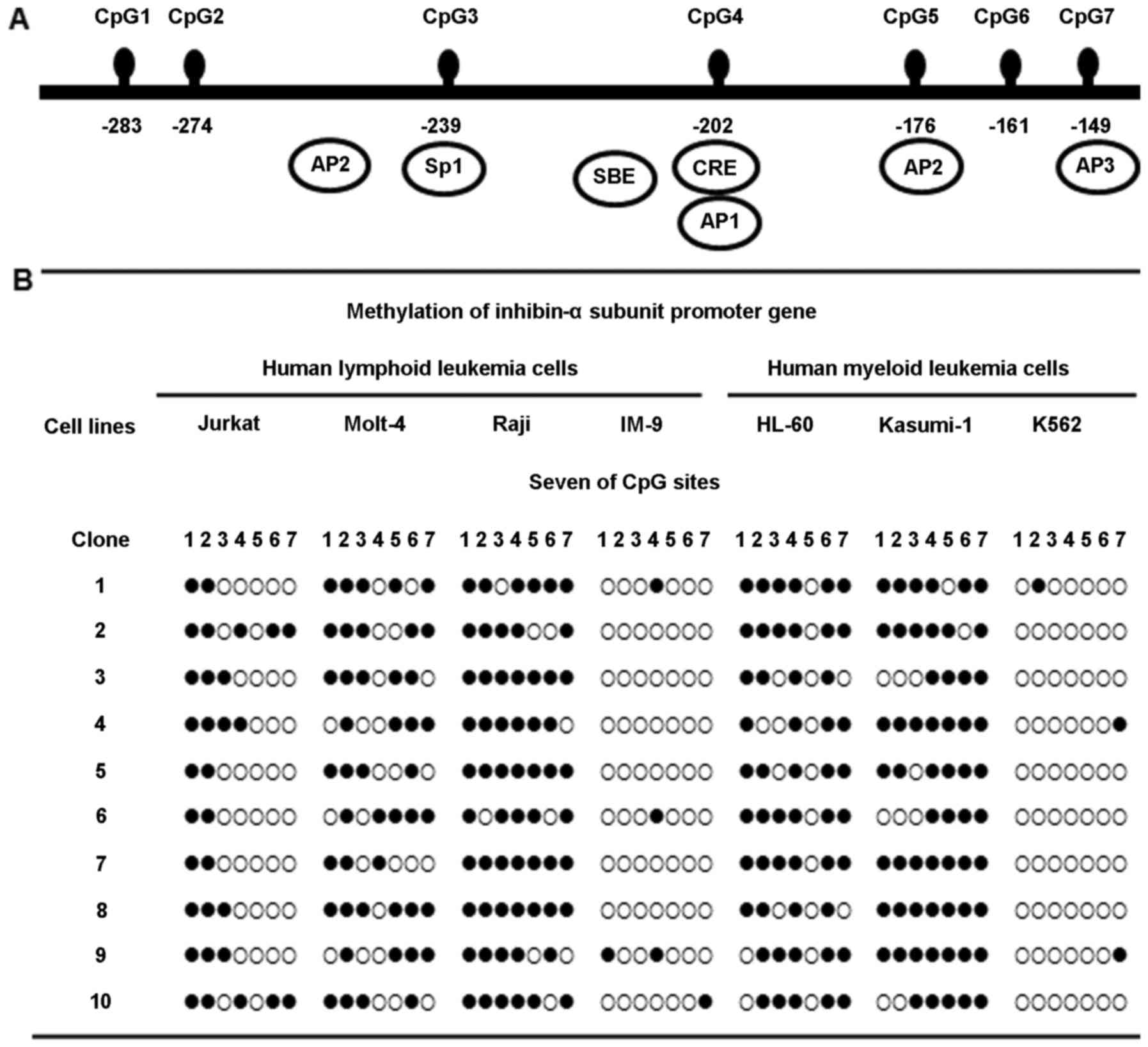
Methylation status of the inhibin-α gene
promoter in human leukemia cells
Methylation at the seven CpG sites, in the 135 bp
region from –149 to –284 of the ATG site in the human inhibin-α
gene promoter, was investigated by bisulfite genomic sequencing
(Fig. 1A). Molt-4, Raji, HL-60,
and Kasumi-1 cells showed marked hypermethylation of the inhibin-α
subunit gene promoter; in contrast, Jurkat cells exhibited
hypomethylation. This region was almost unmethylated in IM-9 and
K562 cells (Fig. 1B).
Mutations of the inhibin-α gene in human
leukemia cells
A mutation study of the inhibin-α gene in human
leukemia cells was carried out. The PCR product (fragment A)
including nucleotide −16 was digested with SpeI (Fig. 2A). Polymorphisms were identified
within the 5′-UTR and exon 1 and used to divide the cell lines into
the following two groups: i) CC genotype (Molt-4, Raji, and HL-60
cells) and ii) CT genotype (Jurkat and IM-9 cells) + TT genotype
(Kasumi-1 and K562 cells). Interestingly, inhibin-α gene mutation
patterns differed between lymphoid leukemia cells (CT,
heterozygote) and myeloid leukemia cells (TT, homozygote).
Substitution 769G>A of exon 2 in human leukemia cells was
analyzed by restriction enzyme digestion. A PCR product comprising
nucleotide 769, fragment B, was digested with BsrFI
(Fig. 2B) and/or Fnu4HI
(Fig. 2C). The single base change
at 769G>A of exon 2 was not found in the seven human leukemia
cell lines.
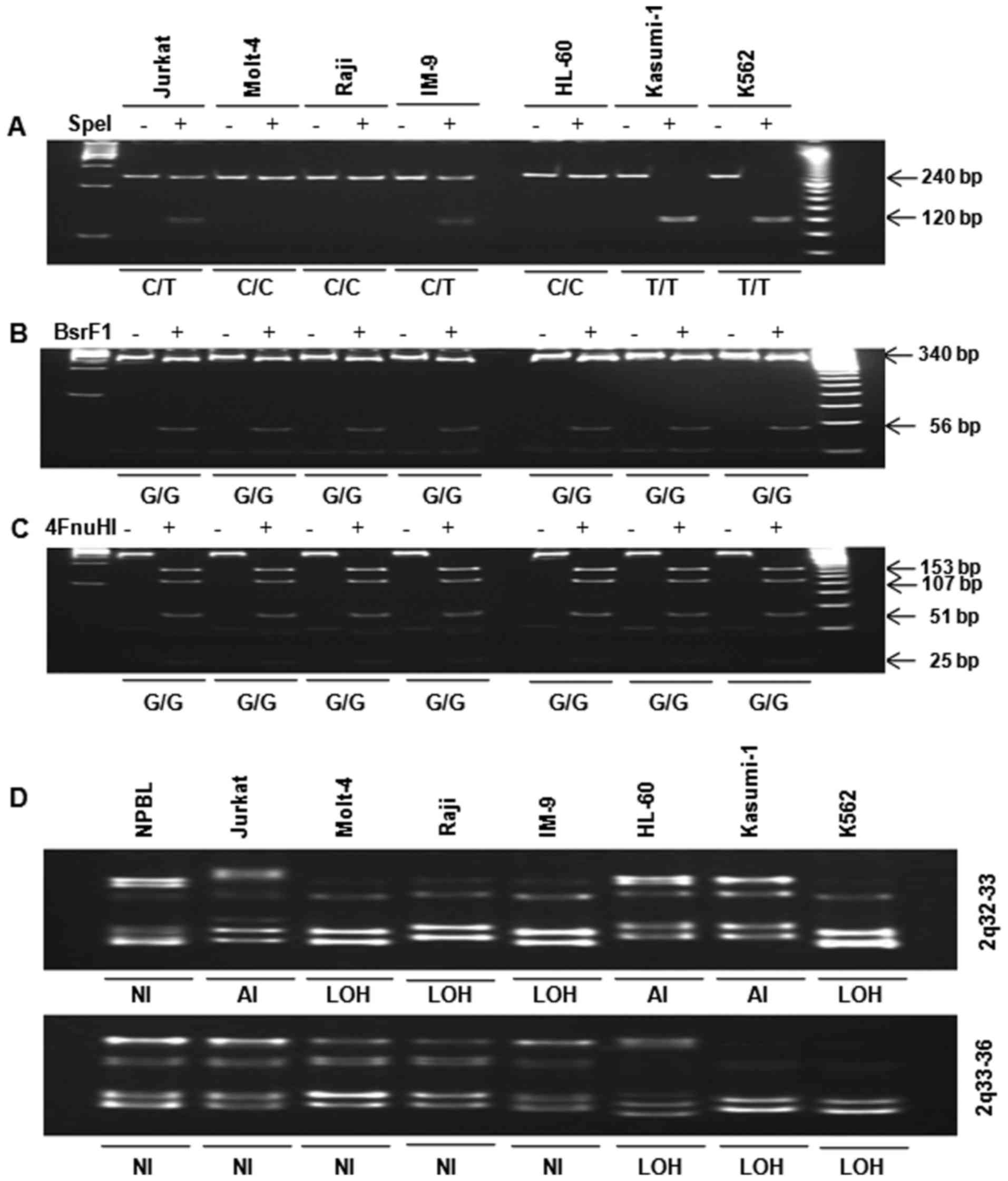 | Figure 2Analysis of polymorphism −16C>T in
the 5′-UTR and substitution 769G>A in exon 2 by restriction
enzyme digestion. (A) Fragment A was digested with SpeI. The
presence of a 240-bp fragment indicated a variant homozygous for C,
whereas the presence of two fragments of 120 bp corresponded to a
variant homozygous for T. (B) Fragment B was digested with
BsrFI. Digestion of the 396-bp PCR product yielded two
fragments of 340 and 56 bp in the wild-type allele (G), while the
mutated allele remained uncleaved (A). (C) Fragment C was digested
with FnuHI. Digestion of the 396-bp PCR product yielded four
fragments of 153, 107, 51 and 25 bp (among others of lower
molecular weight) in the wild-type allele, whereas the allele with
the 769G>A substitution yielded four fragments of 153, 107, 76
and 51 bp (among others of lower molecular weight). PCR products
were incubated overnight with (+E) and without (−E) restriction
enzyme, separated in 8 and 15% polyacrylamide gels, stained with
ethidium bromide and photographed. LOH analysis of 2q in human
leukemia cells. Genomic DNA was amplified by PCR for analysis of
the 2q chromosomal region. PCR products were resolved in 12%
polyacrylamide gels, stained with ethidium bromide and
photographed. (D) 2q32-33 and 2q33-36. LOH was considered in the
presence of a 50% decrease in band intensity. NI, not informative;
LOH, loss of heterozygosity; AI, allelic imbalance. |
LOH at 2q in human leukemia cells
LOH was determined by PCR of genomic DNA. Analysis
of the 2q chromosome arm revealed that LOH with at least one
microsatellite marker occurred at 2q32-36 in Jurkat, Molt-4, Raji,
IM-9, HL-60, Kasumi-1, and K562 cells (Fig. 2D). LOH at 2q32-33 was observed in
human lymphoid (Molt-4, Raji, IM-9) and myeloid (K562) leukemia
cells. LOH at 2q33-36 was observed in human myeloid (HL-60,
Kasumi-1 and K562) leukemia cells, but not in human lymphoid
leukemia cells. However, K562 cells exhibited LOH at both 2q32-33
and 2q33-36.
Effect of 5-AzaC treatment on inhibin-α
and betaglycan mRNA levels in human leukemia cells
Basal expression of inhibin-α mRNA was not detected
in human leukemia cells, whereas betaglycan mRNA was expressed in
the majority of cells (Fig. 3A).
To evaluate the role of methylation in the inactivation of the
inhibin-α gene promoter in human leukemia cells, a DNA
methyltransferase inhibitor, 5-AzaC, was used. Human leukemia cells
were treated with 2 and 5 μM 5-AzaC, and inhibin-α and
betaglycan mRNA levels were measured by real-time PCR (Fig. 3B). 5-AzaC treatment resulted in
increased inhibin-α and betaglycan mRNA levels in all seven human
leukemia cell lines. The magnitude of the increase in inhibin-α and
betaglycan mRNA levels caused by 5-AzaC treatment was greater in
lymphoid than in myeloid leukemia cells.
Effect of 5-AzaC treatment on inhibin-α
protein levels in human leukemia cells
Human leukemia cells were treated with 5 μM
5-AzaC, and inhibin-α subunit protein levels were measured by flow
cytometry and FITC staining (Fig.
4). Treatment with 5-AzaC resulted in 11.3–32.3- and 14.0–27.2-
fold increases in inhibin-α protein levels in human lymphoid and
myeloid leukemia cells. Fluorescence intensities after 5-AzaC
treatment were higher in human lymphoid compared to myeloid
leukemia cells.
Effect of 5-AzaC on the growth and
doubling time of human leukemia cells
Cells exposed to 0.5, 2, and 5 μM 5-AzaC
exhibited significant growth inhibition in a dose-dependent manner,
and the population doubling time of human leukemia cells was
increased by 1.3–2.6-fold (Table
I).
 | Table ISuppression by 5-AzaC of the growth
of human leukemia cells. |
Table I
Suppression by 5-AzaC of the growth
of human leukemia cells.
| Human leukemia cell
lines | Viability (%)
| Doubling time (h)
| Fold growth
suppression |
|---|
| 0 μM | 5-AzaC
| 5.0 μM | 5-AzaC
|
|---|
| 0.5 μM | 2.0 μM | 0 μM | 5.0 μM |
|---|
| Jurkat | 100 | 28.8 | 28.8 | 28.1 | 20 | 50 | 2.5 |
| Molt-4 | 100 | 42.1 | 40.6 | 39.5 | 36 | 68 | 1.9 |
| Raji | 100 | 24.9 | 23.2 | 21.9 | 26 | 57 | 2.2 |
| IM-9 | 100 | 38.6 | 35.6 | 33.2 | 34 | 55 | 1.6 |
| HL-60 | 100 | 82.4 | 73.6 | 66.5 | 27 | 69 | 2.6 |
| Kasumi-1 | 100 | 76.5 | 71.1 | 67.7 | 67 | 83 | 1.3 |
| K562 | 100 | 88.2 | 82.8 | 81.1 | 47 | 84 | 1.8 |
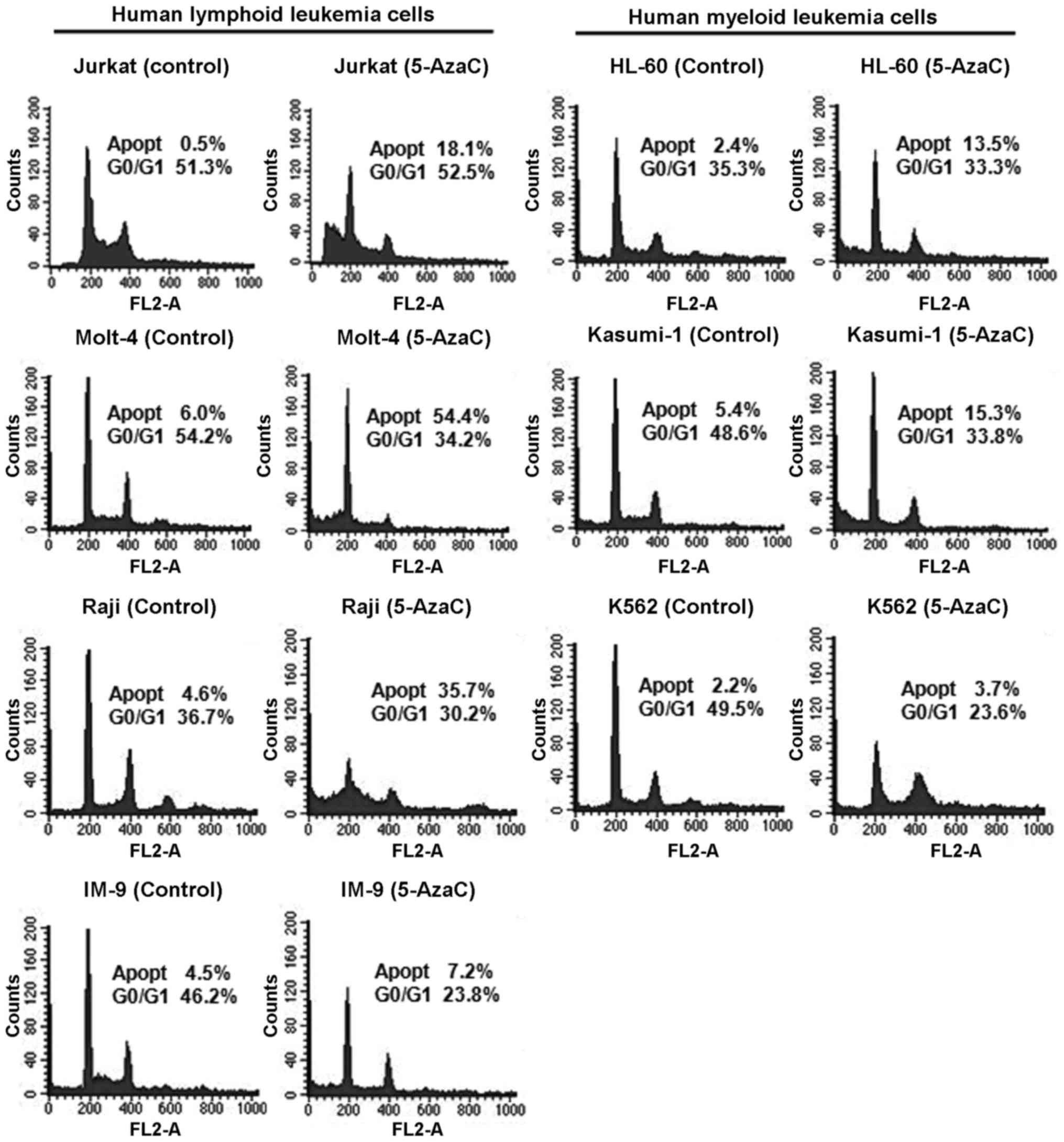
Effect of 5-AzaC on the cell cycle in
human leukemia cells
The cell cycle profiles of human leukemia cells
treated with 5 μM 5-AzaC were analyzed by flow cytometry
(Fig. 5). The results suggested
changes in the cell cycle and induction of apoptosis in
5-AzaC-treated cells. Treatment of Jurkat, Molt-4, Raji, IM-9,
HL-60, Kasumi-1, and K562 cells with 5-AzaC resulted in a
1.7–36.2-fold increase in the proportion of apoptotic cells.
Interestingly, human lymphoid leukemia cells exhibited a greater
increase in the proportion of apoptotic cells than myeloid leukemia
cells after treatment with 5-AzaC.
Discussion
Methylation of CpG sites within the regulatory
regions of tumor-suppressor genes is a common aberration in human
cancers that is frequently associated with gene silencing. In this
study, the degree of methylation varied among the seven CpG sites
in the inhibin-α gene promoter in four human lymphoid, and three
human myeloid leukemia cells. Seven CpG sites were significantly
hypermethylated in human lymphoid (Molt-4 and Raji) leukemia cells
and human myeloid (HL-60 and Kasumi-1) leukemia cells, while
lymphoid (Jurkat) leukemia cells exhibited hypomethylation. In
contrast, this region was not methylated in lymphoid (IM-9) or
myeloid (K562) leukemia cells. The inhibin-α subunit PC3 prostate
cancer cell lines. The methylation pattern ranges from dense to
sparse methylation, with CpG sites 0–3 being undermethylated in the
DU145 and PC3 cell lines compared to the LNCaP cells (20). Our findings suggested that the
methylation pattern at CpG sites did not differ significantly
between human lymphoid and myeloid leukemia cells. Germline cells
of chronic lymphocytic leukemia (CLL) patients with allele-specific
expression (ASE) showed increased levels of DNA methylation in the
promoter region (26).
Transcriptional silencing of tumor-suppressor genes
can be caused by mutations. Two polymorphic sites were identified:
−16>T in the 5-UTR and 769G>A in exon 2. Mutation at the
−16-bp site of the 5′-UTR was heterozygotic in lymphoid (Jurkat and
IM-9) cells and homozygotic in myeloid (K562) cells. The −16-bp
mutation in the 5′-UTR differed significantly between human
lymphoid and myeloid leukemia cells, suggesting that human leukemia
cells may be affected by the −16>T allele variant and supporting
the concept that the 5′-UTR allele variant. In this study, Molt-4,
Raji, IM-9, HL-60, and Kasumi-1 cells showed LOH at chromosome
2q32-36, but Jurkat cells did not. In contrast, K562 cells
exhibited LOH at both 2q32-36 and 2q33-36. Changes at chromosome 2q
occur in prostate carcinoma and pediatric adrenocortical tumors
(27–29). In bladder carcinoma and
head-and-neck squamous cell carcinoma (30,31),
2q deletion is correlated with advanced disease and a poor
prognosis. Taken together, our results suggest that low expression
of the inhibin-α subunit gene is related to hypermethylation,
mutation and LOH.
Induction of inhibin-α subunit mRNA expression in
the human gastric cancer cell lines by treatment with 5-AzaC
demonstrates the presence of all necessary transcription factors
(19). In the human leukemia cell
lines analyzed in this study, expression of inhibin-α subunit mRNA
after 5-AzaC treatment was not correlated with methylation status.
In prostate cancer cell lines, expression of inhibin-α subunit mRNA
was correlated with methylation status after treatment with 5-AzaC
and trichostatin A (TSA). A reciprocal relationship between the
degree of methylation and re-expression of inhibin-α subunit was
evident after treatment with 5-AzaC. PC3 cells, which exhibited the
lowest degree of methylation, were easily demethylated and
expressed high levels of inhibin-α subunit mRNA; in contrast, LNCaP
cells, which were the most highly methylated, showed lower
expression of inhibin-α subunit mRNA (20). These results suggest that the
expression level is not dependent on the degree of methylation
within the promoter region.
The pattern of methylation of the inhibin-α gene
reflected the level of the encoded protein in human leukemia cells.
Immunostaining of 5-AzaC-treated-cells was performed to evaluate
inhibin-α protein levels. Human myeloid leukemia (HL-60 and
Kasumi-1) cells treated with 5-AzaC showed lower inhibin-α protein
levels than the other cell lines. Interestingly, the increase in
inhibin-α protein levels was greater in human lymphoid than in
myeloid leukemia cells. However, the inhibin-α subunit protein
level was not correlated with the methylation status of those cell
lines after 5-AzaC treatment. The percentage of positively stained
demethylated LNCaP and DU145 cells was lower compared with that of
demethylated PC3 cells (20).
Sequential gene expression changed in cancer cell lines after
treatment with 5-AzaC and then focused on the genes with expression
levels that changed gradually, because the effect of
hypomethylation by 5-AzaC would gradually occur. Monitoring of
changes in mRNA levels after 5-AzaC treatment enables
identification of genes whose expression levels changed gradually
(32). 5-AzaC is a DNA
demethylating and anti-cancer agent resulting in the induction of
genes suppressed via DNA hypermethylation (33). Some of the genes upregulated by
5-AzaC treatment may be transcriptionally repressed by promoter
hypermethylation in gastric cancer (34). We found a correlation between
inhibin-α mRNA and protein levels in human leukemia cells after
treatment with 5-AzaC. Also, betaglycan mRNA levels were influenced
by 5-AzaC treatment. However, pattern of increases of inhibin-α and
betaglycan mRNA showed correlation in gene expression between human
lymphoid and myeloid leukemia cells.
Our data show that treatment with 5-AzaC led to a
substantial increase in the doubling times of surviving leukemia
cells, and an increased proportion of apoptotic cells due to
nonspecific suppression of cell growth. 5-AzaC-induced growth
inhibition results from the release of methylation silencing of
cell cycle regulatory genes, such as p16 (35). Moreover, 5-AzaC affects the levels
of several proteins involved in cell cycle regulation, apoptosis,
and survival (36,37). Our results suggest that inhibin-α
has a critical function in cells. 5-AzaC exerts a cytotoxic
effect.
In this study, epigenetically mediated aberrant
transcriptional silencing of the inhibin-α gene in human leukemia
cells was characterized. The results suggested that this gene
likely plays an important role in leukemia tumorigenesis as a
putative tumor suppressor. Methylation of the inhibin-α gene
promoter was evident in some human leukemia cell lines, but whether
this is a cause or consequence of tumorigenesis remains to be
determined. Moreover, functional studies of the inhibin-α gene may
provide insight into the development of leukemia treatment. Our
results suggest that the inhibin-α gene promoter is poised for
activation in the cell lines tested and that the effects on
transcription are primarily indirect and mediated by activation of
transcription factors. Induction of the expression of other genes,
either alone or in combination with inhibin-α, may explain the
observed growth suppression in human leukemia cells.
References
|
1
|
Antequera F, Boyes J and Bird A: High
levels of de novo methylation and altered chromatin structure at
CpG islands in cell lines. Cell. 62:503–514. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Esteller M: CpG island hypermethylation
and tumor suppressor genes: A booming present, a brighter future.
Oncogene. 21:5427–5440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Okano M, Xie S and Li E: Cloning and
characterization of a family of novel mammalian DNA (cytosine-5)
methyltransferases. Nat Genet. 19:219–220. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jones PL, Veenstra GJ, Wade PA, Vermaak D,
Kass SU, Landsberger N, Strouboulis J and Wolffe AP: Methylated DNA
and MeCP2 recruit histone deacetylase to repress transcription. Nat
Genet. 19:187–191. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nan X, Ng HH, Johnson CA, Laherty CD,
Turner BM, Eisenman RN and Bird A: Transcriptional repression by
the methyl-CpG-binding protein MeCP2 involves a histone deacetylase
complex. Nature. 393:386–389. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
van der Ploeg LH and Flavell RA: DNA
methylation in the human gamma delta beta-globin locus in erythroid
and nonerythroid tissues. Cell. 19:947–958. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lichtenstein M, Keini G, Cedar H and
Bergman Y: B cell-specific demethylation: A novel role for the
intronic κ chain enhancer sequence. Cell. 76:913–923. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lübbert M, Miller CW and Koef fler HP:
Changes of DNA methylation and chromatin structure in the human
myeloperoxidase gene during myeloid differentiation. Blood.
78:345–356. 1991.PubMed/NCBI
|
|
9
|
Felgner J, Kreipe H, Heidorn K, Jaquet K,
Heuss R, Zschunke F, Radzun HJ and Parwaresch MR: Lineage-specific
methylation of the c-fms gene in blood cells and macrophages.
Leukemia. 6:420–425. 1992.PubMed/NCBI
|
|
10
|
Baylin SB, Herman JG, Graff JR, Vertino PM
and Issa JP: Alterations in DNA methylation: A fundamental aspect
of neoplasia. Adv Cancer Res. 72:141–196. 1998. View Article : Google Scholar
|
|
11
|
Toyooka S, Carbone M, Toyooka KO,
Bocchetta M, Shivapurkar N, Minna JD and Gazdar AF: Progressive
aberrant methylation of the RASSF1A gene in simian virus 40
infected human mesothelial cells. Oncogene. 21:4340–4344. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsai CN, Tsai CL, Tse KP, Chang HY and
Chang YS: The Epstein- Barr virus oncogene product, latent membrane
protein 1, induces the downregulation of E-cadherin gene expression
via activation of DNA methyltransferases. Proc Natl Acad Sci USA.
99:10084–10089. 2002. View Article : Google Scholar
|
|
13
|
Mathews LS: Activin receptors and cellular
signaling by the receptor serine kinase family. Endocr Rev.
15:310–325. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mather JP, Moore A and Li RH: Activins,
inhibins, and follistatins: Further thoughts on a growing family of
regulators. Proc Soc Exp Biol Med. 215:209–222. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Risbridger GP, Schmitt JF and Robertson
DM: Activins and inhibins in endocrine and other tumors. Endocr
Rev. 22:836–858. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matzuk MM, Finegold MJ, Su JG, Hsueh AJ
and Bradley A: α-inhibin is a tumour-suppressor gene with gonadal
specificity in mice. Nature. 360:313–319. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matzuk MM and Bradley A: Identification
and analysis of tumor suppressor genes using transgenic mouse
models. Semin Cancer Biol. 5:37–45. 1994.PubMed/NCBI
|
|
18
|
Schmitt JF, Millar DS, Pedersen JS, Clark
SL, Venter DJ, Frydenberg M, Molloy PL and Risbridger GP:
Hypermethylation of the inhibin α-subunit gene in prostate
carcinoma. Mol Endocrinol. 16:213–220. 2002.PubMed/NCBI
|
|
19
|
Kim YI, Shim J, Kim BH, Lee SJ, Lee HK,
Cho C and Cho BN: Transcriptional silencing of the inhibin-α gene
in human gastric carcinoma cells. Int J Oncol. 41:690–700.
2012.PubMed/NCBI
|
|
20
|
Balanathan P, Ball EM, Wang H, Harris SE,
Shelling AN and Risbridger GP: Epigenetic regulation of inhibin
α-subunit gene in prostate cancer cell lines. J Mol Endocrinol.
32:55–67. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chatterton Z, Burke D, Emslie KR, Craig
JM, Ng J, Ashley DM, Mechinaud F, Saffery R and Wong NC: Validation
of DNA methylation biomarkers for diagnosis of acute lymphoblastic
leukemia. Clin Chem. 60:995–1003. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu P, Shen JK, Xu J, Trahan CA, Hornicek
FJ and Duan Z: Aberrant DNA methylations in chondrosarcoma.
Epigenomics. 8:1519–1525. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Clark SJ, Harrison J, Paul CL and Frommer
M: High sensitivity mapping of methylated cytosines. Nucleic Acids
Res. 22:2990–2997. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sundblad V, Chiauzzi VA, Andreone L, Campo
S, Charreau EH and Dain L: Controversial role of inhibin α-subunit
gene in the aetiology of premature ovarian failure. Hum Reprod.
21:1154–1160. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuchler RJ: Development of animal cell
populations in vitro. Biochemical Methods in Cell Culture and
Virology. Kuchler RJ: Dowden, Hutchinson and Ross Inc. Press;
Stroudsburg, PA: pp. 90–113. 1977
|
|
26
|
Wei QX, Claus R, Hielscher T, Mertens D,
Raval A, Oakes CC, Tanner SM, de la Chapelle A, Byrd JC,
Stilgenbauer S, et al: Germline allele-specific expression of DAPK1
in chronic lymphocytic leukemia. PLoS One. 8:e552612013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alers JC, Rochat J, Krijtenburg PJ, Hop
WC, Kranse R, Rosenberg C, Tanke HJ, Schröder FH and van Dekken H:
Identification of genetic markers for prostatic cancer progression.
Lab Invest. 80:931–942. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Suarez BK, Lin J, Burmester JK, Broman KW,
Weber JL, Banerjee TK, Goddard KA, Witte JS, Elston RC and Catalona
WJ: A genome screen of multiplex sibships with prostate cancer. Am
J Hum Genet. 66:933–944. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Longui CA, Lemos-Marini SH, Figueiredo B,
Mendonca BB, Castro M, Liberatore R Jr, Watanabe C, Lancellotti CL,
Rocha MN, Melo MB, et al: Inhibin α-subunit (INHA) gene and locus
changes in paediatric adrenocortical tumours from TP53 R337H
mutation heterozygote carriers. J Med Genet. 41:354–359. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao J, Richter J, Wagner U, Roth B,
Schraml P, Zellweger T, Ackermann D, Schmid U, Moch H, Mihatsch MJ,
et al: Chromosomal imbalances in noninvasive papillary bladder
neoplasms (pTa). Cancer Res. 59:4658–4661. 1999.PubMed/NCBI
|
|
31
|
Ransom DT, Barnett TC, Bot J, de Boer B,
Metcalf C, Davidson JA and Turbett GR: Loss of heterozygosity on
chromosome 2q: Possibly a poor prognostic factor in head and neck
cancer. Head Neck. 20:404–410. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Arai M, Yokosuka O, Hirasawa Y, Fukai K,
Chiba T, Imazeki F, Kanda T, Yatomi M, Takiguchi Y, Seki N, et al:
Sequential gene expression changes in cancer cell lines after
treatment with the demethylation agent 5-Aza-2′-deoxycytidine.
Cancer. 106:2514–2525. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sayar N, Karahan G, Konu O, Bozkurt B,
Bozdogan O and Yulug IG: Transgelin gene is frequently
downregulated by promoter DNA hypermethylation in breast cancer.
Clin Epigenetics. 7:1042015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mikata R, Yokosuka O, Fukai K, Imazeki F,
Arai M, Tada M, Kurihara T, Zhang K, Kanda T and Saisho H: Analysis
of genes upregulated by the demethylating agent
5-aza-2′-deoxycytidine in gastric cancer cell lines. Int J Cancer.
119:1616–1622. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bender CM, Pao MM and Jones PA: Inhibition
of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth
of human tumor cell lines. Cancer Res. 58:95–101. 1998.PubMed/NCBI
|
|
36
|
Song SH, Jong HS, Choi HH, Inoue H, Tanabe
T, Kim NK and Bang YJ: Transcriptional silencing of
Cyclooxygenase-2 by hyper-methylation of the 5′ CpG island in human
gastric carcinoma cells. Cancer Res. 61:4628–4635. 2001.PubMed/NCBI
|
|
37
|
Valdez BC, Li Y, Murray D, Corn P,
Champlin RE and Andersson BS: 5-Aza-2′-deoxycytidine sensitizes
busulfan- resistant myeloid leukemia cells by regulating expression
of genes involved in cell cycle checkpoint and apoptosis. Leuk Res.
34:364–372. 2010. View Article : Google Scholar
|