Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies occurring around the world (1,2).
Although there are various clinical therapeutic strategies for HCC,
metastasis and recurrence often develop (3). A number of molecules involved in HCC
development have been identified (4); however, our understanding of their
underlying mechanisms is limited.
The hepatocellular carcinoma-related protein (HCRP1)
gene, also known as the human vacuolar protein sorting 37 homologue
A (hVps37A) gene, is located on chromosome 8p22, and is frequently
deleted in HCC. HCRP1 was first described as a growth inhibitory
protein in HCC cells (5).
Thereafter, additional physiologic functions of HCRP1 were explored
in other tumors such as ovarian, oral and oropharyngeal, breast,
and non-small cell lung cancer (6–10).
HCRP1 is a member of the endosomal sorting complex required for
transport (ESCRT)-I complex and affects lysosomal sorting of EGFR
(11). To date, the regulatory
mechanism of HCRP1 in HCC remains unclear.
The role of epithelial-mesenchymal transition (EMT)
processes as a central factor governing cancer invasion and
metastasis has been established in the last decade and involves the
activity of a core set of transcription factors that are activated
by signal transduction pathways in neoplastic cells (12). Transforming growth factor-β (TGF-β)
is a potent pleiotropic cytokine that regulates mammalian
development, differentiation, and homeostasis in essentially all
cell types and tissues, and plays an important role in the
induction of EMT in various cancers (13). Strong inducers of EMT such as TGF-β
are capable of orchestrating both fibrogenesis and carcinogenesis
(14). The aim of the present
study was to investigate the role of HCRP1 in HCC and elucidate its
underlying mechanisms.
Materials and methods
Cell culture
Two human HCC cell lines, HepG2 and SMMC-7721, were
obtained from the Chinese Academy of Sciences (Shanghai, China).
The HepG2 cells were cultured in DMEM (cat. no. 11995-065, Gibco,
Grand Island, NY, USA) and supplemented with 10% fetal bovine serum
(FBS, cat. no. S1810, Biowest, Nuaillé, France), whereas the
SMMC-7721 cells were cultured in RPMI-1640 (cat. no. 11875-093,
Gibco) with 10% FBS; both cell lines were incubated at 37°C in 5%
CO2 atmosphere. The TGF-β receptor kinase inhibitor
LY364947 (cat. no. S2805, Selleck) was diluted to 10 µM with
Opti-MEM® (cat. no. 31985062, Gibco). In addition, 10
cases of paraffin-embedded tissues samples (containing HCC and the
corresponding non-tumorous liver tissues) were collected from
Huashan Hospital of Fudan University. Ethics approval was obtained
from the Clinical Research Ethics Committee of Fudan
University.
Immunohistochemistry
Four-µm-thick sections were prepared from
formalin-fixed, paraffin-embedded blocks, deparaffinized in xylene,
and rehydrated using a series of graded ethanol washes. After
inhibition of endogenous peroxidase and antigen retrieval
(microwave irradiation in 0.01 M citrate buffer at pH 6.0), the
sections were respectively incubated with the primary antibodies
anti-HCRP1 (cat. no. A1505, ABclonal, College Park, MD, USA) and
anti-E-cadherin (cat. no. 3195, Cell Signaling, Beverly, MA, USA)
at 4°C overnight, followed by horseradish peroxidase conjugated
secondary antibodies (Dako, cat. no. K406511, Glostrup, Denmark).
PBS was used as negative control instead of the primary antibody.
Slides were then developed for 5 min with the chromogen,
3,3′-diaminobenzidine (DAB), and counterstained with hematoxylin to
distinguish the nucleus from the cytoplasm.
Western blot analysis
Total protein was extracted from cells using a RIPA
lysis buffer (cat. no. sc-24948, Santa Cruz, Dallas, TX, USA).
Total protein concentration was determined by using a BCA protein
assay kit (cat. no. P0012S, Beyotime, Beijing, China). Equal
amounts of proteins were separated by SDS-PAGE and
electrophoretically transferred to polyvinylidene fluoride
membranes (cat. no. IPFL00010, Merck Millipore, Darmstadt,
Germany). The membranes were blocked with 5% milk in Tris-HCl
buffered solution for 2 h, and then subsequently incubated
overnight at 4°C with the respective primary and secondary
antibodies anti-mouse lgG (H+L) (cat. no. SA0001-1, Proteintech)
and anti-rabbit lgG (H+L) (cat. no. SA0001-2, Proteintech) at room
temperature for 1 h. To confirm equal protein loading, the
membranes were incubated with GAPDH (cat. no. G9545, Sigma-Aldrich,
St. Louis, MO, USA) or β-actin (cat. no. 8457S, Cell Signaling) as
internal controls. The signals were visualized using an enhanced
chemiluminescent substrate and detected by using a FluorChem Q
imaging system (Protein Simple, Santa Clara, CA, USA). Images from
the western blot assay were quantified using the Quantity
One® software (Bio-Rad, Hercules, CA, USA). The
expression level was normalized with respect to that of the
internal control. The following antibodies were used: anti-HCRP1
(cat. no. A1505, ABclonal), anti-E-cadherin (cat. no. 3195, Cell
Signaling, Beverly, MA, USA), anti-N-cadherin (cat. no. 4061, Cell
Signaling), anti-vimentin (cat. no. 5741, Cell Signaling),
anti-MMP9 (cat. no. 2270, Cell Signaling), anti-β-catenin (cat. no.
8480, Cell Signaling), anti-Snail (cat. no. 3879, Cell Signaling),
Smad2 (phospho-Thr220) antibody (cat. no. 11323, SAB, College Park,
MD, USA), anti-Smad2 (cat. no. 21322, SAB).
Real-time quantitative PCR (qPCR)
Total RNA was extracted using RNAiso Plus (cat. no.
9108, Takara, Dalian, China) according to the manufacturer's
instructions, and then reversed to cDNA using
PrimeScript® RT Master Mix (cat. no. DRR036A, Takara) at
37°C for 15 min, 85°C for 5 sec, and then at 4°C. QPCR for HCRP1
and GAPDH was performed in a 10-µl reaction volume using the
SYBR® PremixEx Taq™ (cat. no. RR420A, Takara) and
ABI7900HT real-time PCR system (Life, Singapore). The thermal cycle
conditions consisted of one cycle at 95°C for 10 min, followed by
40 cycles of amplification at 95°C for 15 sec, and then 60°C for 1
min. The expression level of HCRP1 mRNA was normalized to the
geometric mean of the GAPDH mRNA, which was obtained by calculating
for the 2−ΔΔCt value, whereas Ct value represented the
threshold cycle for each transcript (HCRP1 primers: forward,
5′-CAACAAGUCAUACCACAGCTT-3′; and reverse,
5′-GCUGUGGUAUGACUUGUUGTT-3′. GAPDH primers: forward,
5′′-CTGACTTCAACAGCGACACC-3′; and reverse,
5′-TGCTGTAGCCAAATTCGTTGT-3′).
Lentiviral-vector infections
Lentiviral vectors expressing a constitutively
active form of HCRP1 (HanBio, Shanghai, China) were respectively
introduced into SMMC-7721 cells via standard infections at a
multiplicity of infection (MOI) of 1:5 for 24 h. To knock down
HCRP1 expression, small interfering RNAs (siRNAs) (sequences: 1,
5′-GAAAGUAGCUGCACAUGAAdTdT-3′, 5′-UUCAUGUGCAGCUACUUUCdTdT-3′; 2,
5′-CACCAUAAACAACCUGACAdTdT-3′, 5′-UGUCAGGUUGUUUAUGGUGdTdT-3′; and
3, 5′-CUCUCAGAACUAAGUGUGUdTdT-3′, 5′-ACACACUUAGUUCUGAGAGdTdT-3′)
were delivered into the cells. The silencing efficiency was
validated by qPCR and the third sequence was selected for
recombination with a HCRP1-siRNA lentivirus (shHCRP1) (HanBio) and
used in infecting the HepG2 cells. An empty lentiviral vector was
used as negative control. The stable clones were selected in media
containing puromycin dihydrochloride (cat. no. P9620,
Sigma-Aldrich, Shanghai, China) for two weeks.
Wound healing assays
HepG2 and SMMC-7721 cells (density: 5×105
cells/ml) were seeded onto 6-well plates in triplicate and
incubated overnight. Wound healing assays were performed with a
1,000-µl sterile pipette tip, which was used in making a
scratch across the confluent monolayer. Marked fields were observed
at 0, 12, 36 and 48 h to assess the rate of gap closure. Only the
time-points at which the effect was clearly observed were selected.
Each assay was performed in triplicate and repeated thrice.
Cell migration and invasion assay
The cell migration study was performed using 24-well
tissue culture plates with Transwell® chambers (cat. no.
3422, Corning, NY, USA) according to the manufacturer's
instructions. The cell invasion study was performed using a
polycarbonate membrane that was precoated with 30 µl of
Matrigel® (cat. no. 354234, BD, Shanghai, China) at 37°C
for 4 h. The cells were detached with trypsin and washed with
serum-free medium. HepG2 cells and SMMC-7721 cells (both at
densities of 5×104 cells/ml) were added to the upper
chamber that contained a total volume of 100 µl of
serum-free medium. The lower chamber contained 600 µl of
conditioned medium (DMEM with 10% FBS or RPIM-1640 with 10% FBS).
After 24 h, the cells that adhered to the bottom side of the
membrane were fixed in methanol, stained with 0.1% crystal violet,
and the average cell number per high power field was calculated.
Each assay was conducted in triplicate and repeated thrice.
Statistical analysis
Statistical analysis was performed using Graphpad
Prism® 5.0 (La Jolla, CA, USA). Numeration data were
analyzed using the chi-square test, and the exact probability
method was used when there was a theoretical frequency of <5.
The Student's t-test was used to compare the difference between the
two groups. P-values <0.05 were considered statistically
significant.
Results
HCRP1 is downregulated in HCC tissues and
cell lines
Immunohistochemistry analysis was performed to
examine the expression of HCRP1 in tissue samples from 10 cases of
HCC. HCRP1 was downregulated in HCC tissues (Fig. 1A). Then, we assessed the expression
of HCRP1 in various HCC cell lines (i.e., HepG2, Hep3B, hun-7,
BEL-7404, SMMC-7721, and MHCC97-H), which was also downregulated
compared to that of the normal liver cell line, LO2 (Fig. 1B). The expression of HCRP1 in HepG2
cells (with low invasion capability) was relatively high, whereas
that in SMMC-7721 cells (with high invasion capability) was
comparatively low. Thus, we performed a knockdown experiment using
siRNAs that were transfected into HepG2 cells, as well as
functional studies using lentiviral vector-mediated
HCRP1-overexpressing SMMC-7721 cells.
HCRP1 affects the migration and invasion
of HCC cells
The transfection efficiency of the lentiviral vector
was validated by RT-PCR and western blot analysis (Fig. 2A), and the biological functions of
HCRP1 were also investigated. We performed the wound healing at
several time-points (0, 12, 24, 36 and 48 h). Only the time-points
at which the effect was obviously apparent were selected. Compared
to the negative controls, HCRP1-deletion cells (HepG2/shHCRP1
cells) underwent rapid wound closure after 36 h (Fig. 2B), whereas HCRP1-overexpressing
cells (SMMC-7721/HCRP1 cells) exhibited slow wound closure after 48
h (Fig. 2C). Moreover, the
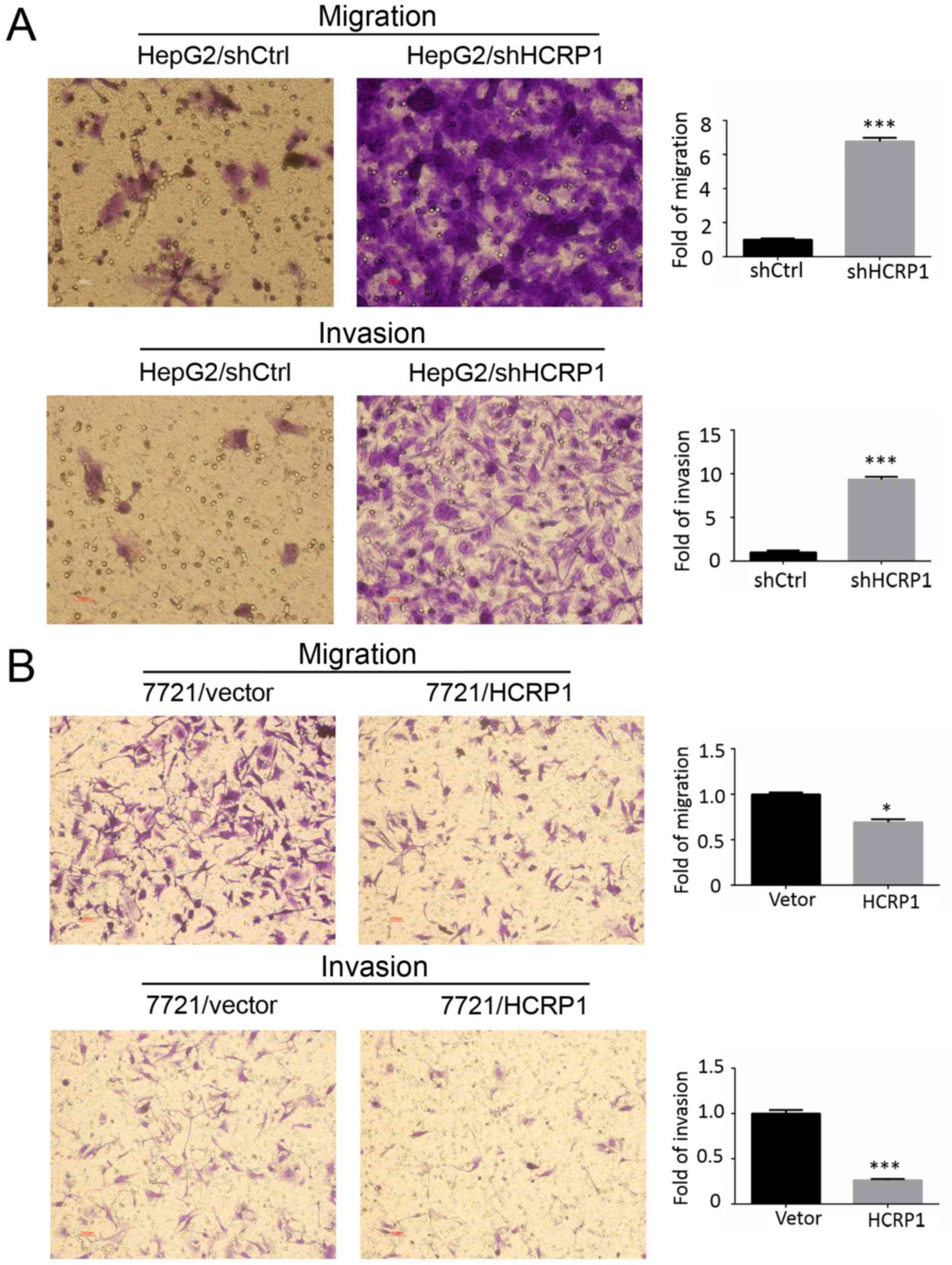
Transwell assay showed that knocking down HCRP1 resulted in a
significant increase in the migration and invasion of HepG2 cells
(Fig. 3A), whereas HCRP1
overexpression decreased the migration and invasion of SMMC-7721
cells (Fig. 3B). Based on these
findings, we confirm that HCRP1 inhibits the migration and invasion
of HCC cells.
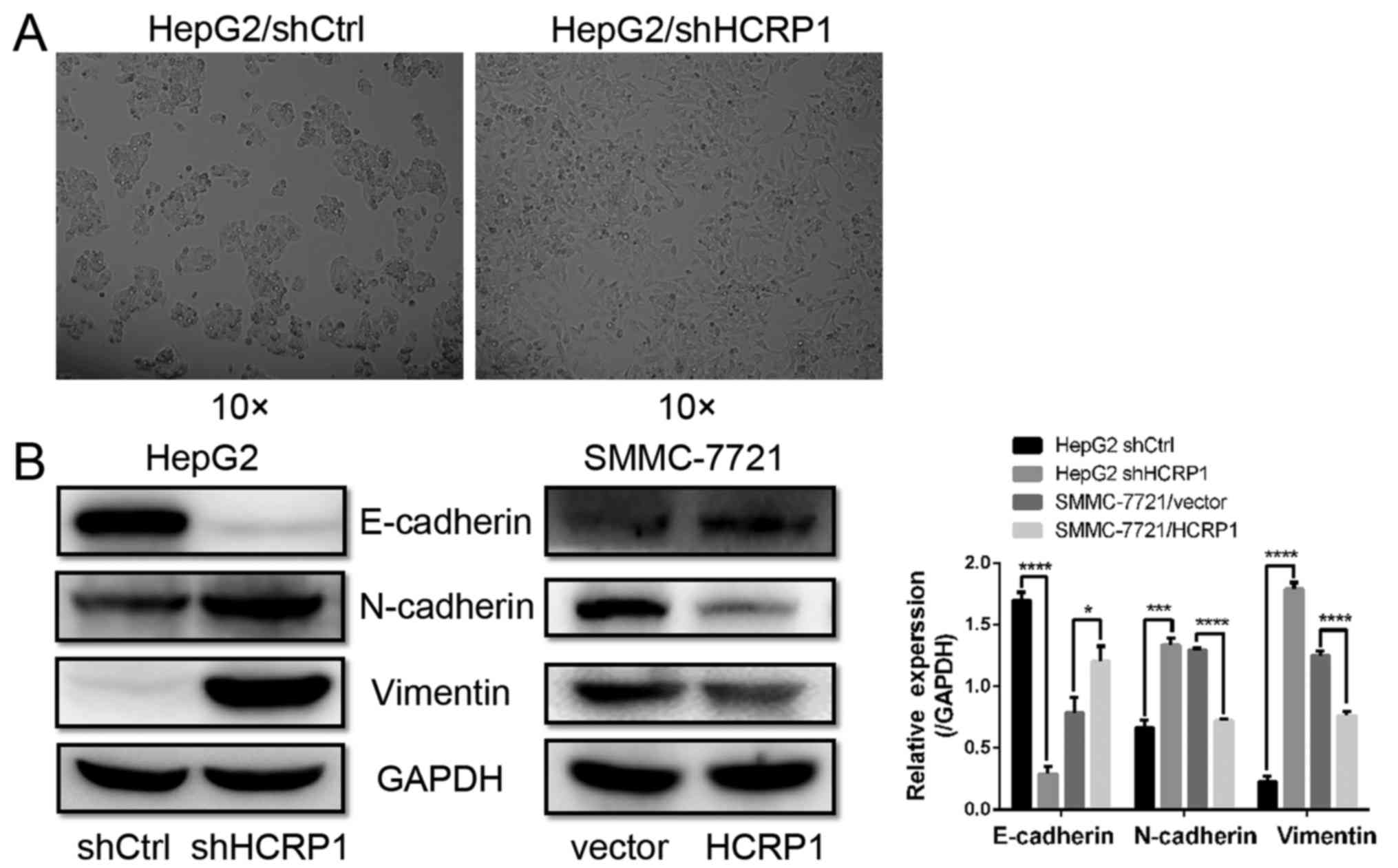
HCRP1 deletion induces HepG2 cells to
enter the EMT
The present study observed alterations in the
morphology of HepG2/shHCRP1 cells, wherein these became
spindle-shaped, which is an indication of a mesenchymal phenotype
(Fig. 4A). To determine whether
HCRP1 is involved in EMT, we investigated a panel of EMT-related
markers after HCRP1 knockdown. We observed a significant reduction
of E-cadherin expression (epithelial phenotype marker), whereas
that of N-cadherin and vimentin (mesenchymal phenotype markers)
were upregulated following lentiviral vector-shHCRP1 transfection
in HepG2 cells (Fig. 4B). We also
detected a significant downregulation of N-cadherin and vimentin
and an upregulation of E-cadherin in SMMC-7721/HCRP1 cells
(Fig. 4B). These results indicated
that HCRP1 may be involved in the process of EMT in HCC cells.
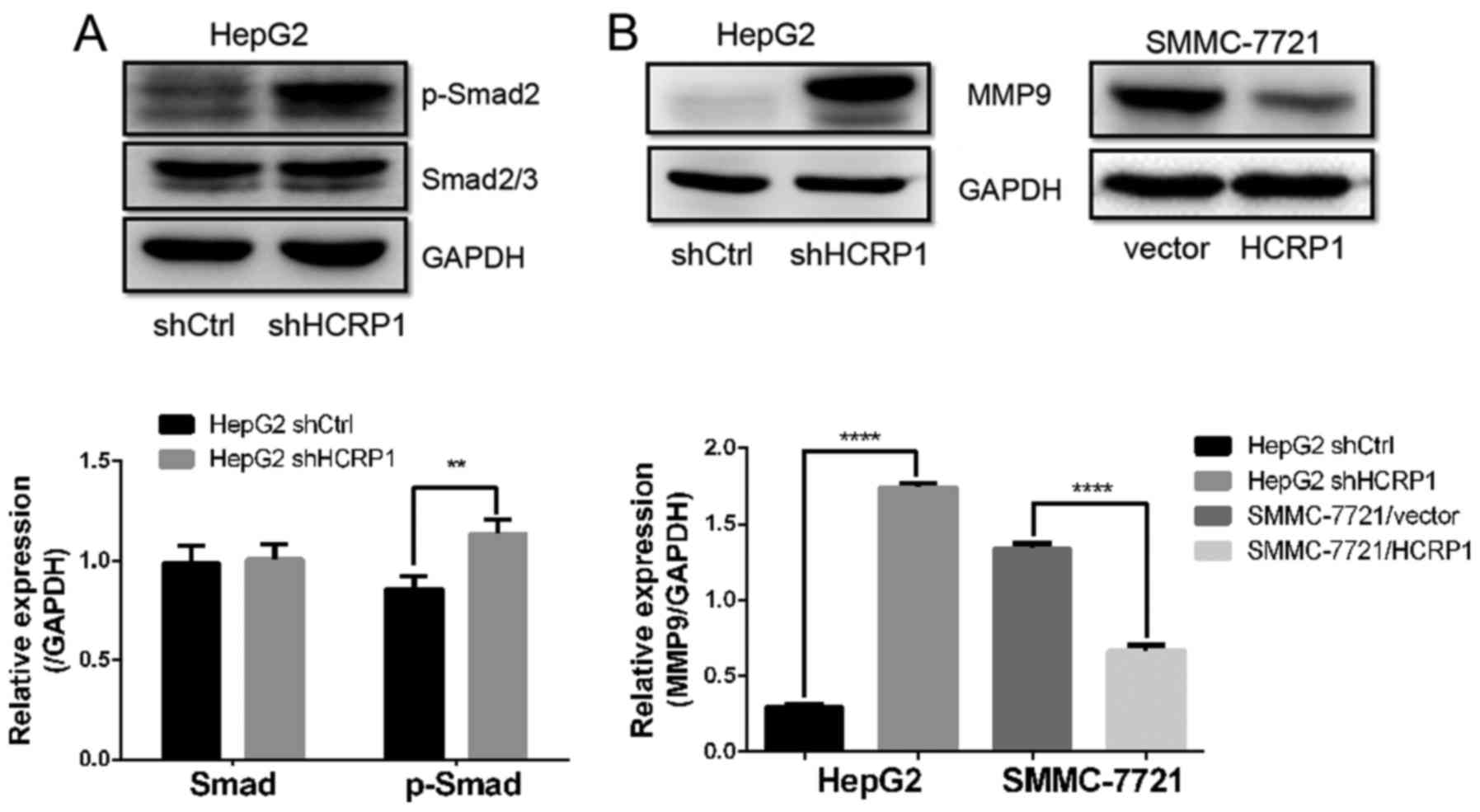
HCRP1 deletion induces EMT through the
TGF-β pathway
TGF-β is considered as an important inducer of EMT
in the progression of various cancers. To investigate whether the
TGF-β signaling pathway is involved in HCRP1 deletion-induced EMT,
we detected the phosphorylation status of Smad2, a downstream
effector of the TGF-β signaling pathway. We observed a significant
increase in the expression of p-Smad2 in HepG2/shHCRP1 cells, which
indicated that HCRP1 downregulation also activated the TGF-β
signaling pathway (Fig. 5A).
Furthermore, the expression of MMP9, which is a target of TGF-β
signaling during EMT, increased after HCRP1 downregulation and
decreased after HCRP1 overexpression (Fig. 5B). To further investigate the
interaction between HCRP1 and TGF-β, we used a specific TGF-β
receptor kinase inhibitor, LY364947, to block the TGF-β signaling
pathway. The inhibitor counteracted the effects of HCRP1 deletion
in HepG2 cells, as indicated by a decrease in the expression of
p-Smad2 and mesenchymal cell markers N-cadherin and vimentin, as
well as the transcription factor Snail. In addition, the inhibitor
restored the expressions of epithelial cell markers E-cadherin and
β-catenin, which were diminished by HCRP1 deletion (Fig. 6).
The relationship between HCRP1 and EMT
markers in HCC tissues
To further explore the relationship between HCRP1
and EMT, immunohistochemical analysis was performed to detect the
expression of HCRP1 and E-cadherin in tissues from 10 cases of HCC
tissues. The results revealed that HCRP1 and E-cadherin were not
expressed in HCC tissues, whereas highly expressed in non-tumorous
liver tissues (Fig. 7).
Discussion
HCRP1 is a novel HCC-related protein whose gene is
located at a frequently deleted region such as a loss of
heterozygosity (LOH) region in HCC (4). However, HCRP1 as a novel tumor
suppressor gene in HCC, and its biological functions and underlying
mechanisms remain elusive. In the present study, we first examined
the expression of HCRP1 in 10 pairs of HCC tissues and the
corresponding non-cancerous tissues, which showed that HCRP1 was
downregulated in HCC tissues. Then, we examined the expression
level of HCRP1 in LO2 normal liver cells and various other HCC cell
lines, which revealed that HCRP1 was also downregulated in HCC cell
lines. In addition, loss/gain-of-function studies (HCRP1 deletion
in HepG2 cells and HCRP1 overexpression in SMMC-7721 cells by
lentiviral vector infections) showed that HCRP1 is a regulator of
cell migration and invasion. Moreover, we demonstrated that HCRP1
downregulation in HCC contributes to EMT, and TGF-β participates in
this process. Our study provides a novel mechanism underlying HCC
metastasis.
The wound healing assay and Transwell assay
demonstrated that HCRP1 deletion enhanced the ability for migration
and invasion in HepG2 cells, whereas overexpression of HCRP1
inhibited these two activities in SMMC-7721 cells. These results
confirmed the role of HCRP1 on migration and invasion in HCC cells.
Interestingly, HCRP1 knockdown induced an alteration in cell
morphology, which indicated that HCRP1 is involved in the EMT of
HCC cells.
EMT plays a crucial role in the early steps of
metastasis in cancer progression, including HCC (15). During EMT development, epithelial
cells lose their polarity and acquire a mesenchymal phenotype,
thereby becoming migratory and invasive (16). The pivotal role of EMT in HCC has
been increasingly recognized and various molecular mechanisms
underlying hepatocellular EMT have been identified such as TGF-β
signaling (17). TGF-β signaling
commences by its binding to three high-affinity receptors, namely,
TβR-I, TβR-II, TβR-III, and stimulates the latent transcription
factors, Smad2 and Smad3 (13).
Changes in cell behavior regulated by the activation of Smad2/3 are
referred to as 'canonical TGF-β signaling'. Therefore, to
investigate whether HCRP1 deletion induces EMT via the TGF-β
signaling pathway, we detected the protein expression level of
phosphorylated Smad2. We observed a significant increase in the
expression of p-Smad2 in HepG2/shHCRP1 cells. We also detected the
expression of MMP9, which is well established as a functional
target of TGF-β signaling during EMT (18). In addition, we also determined that
the TGF-β receptor kinase inhibitor LY364947 decreased the
expression of p-Smad2 and mesenchymal cell markers N-cadherin and
vimentin, as well as the transcription factor Snail, whose levels
of expression increased due to the downregulation of HCRP1. All
this evidence support that the TGF-β pathway is activated during
HCRP1 deletion-induced EMT.
The expression of HCRP1 is a predictor of
disease-free survival in HCC and could be a helpful predictor that
might influence medical decisions in treating HCC (19). A more recent report found that the
TGF-β inhibitor LY2157299 appears to modulate EMT and has a
clinically meaningful benefit to patients (20). Given that TGF-β is a downstream
signal of HCRP1, we consider TGF-β inhibitor as an effective
reagent for HCC patients with low HCRP1 expression.
Acknowledgments
This study was supported by The National Natural
Science Foundation of China (grant nos. 81272387, 81470857 and
81502272). The authors would like to thank LetPub (www.letpub.com) for its linguistic assistance during
the preparation of this manuscript.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao P, Wang JG, Gao P, Li X and Brewer R:
Sudden unexpected death from natural diseases: Fifteen years'
experience with 484 cases in Seychelles. J Forensic Leg Med.
37:33–38. 2016. View Article : Google Scholar
|
|
3
|
Meguro M, Mizuguchi T, Kawamoto M and
Hirata K: The molecular pathogenesis and clinical implications of
hepatocellular carcinoma. Int J Hepatol. 2011:8186722011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellular carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu Z, Liang L, Wang H, Li T and Zhao M:
HCRP1, a novel gene that is downregulated in hepatocellular
carcinoma, encodes a growth-inhibitory protein. Biochem Biophys Res
Commun. 311:1057–1066. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wittinger M, Vanhara P, El-Gazzar A,
Savarese-Brenner B, Pils D, Anees M, Grunt TW, Sibilia M, Holcmann
M, Horvat R, et al: hVps37A Status affects prognosis and cetuximab
sensitivity in ovarian cancer. Clin Cancer Res. 17:7816–7827. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Perisanidis C, Savarese-Brenner B, Würger
T, Wrba F, Huynh A, Schopper C, Kornek G, Selzer E, Ewers R, Psyrri
A, et al: HCRP1 expression status is a significant prognostic
marker in oral and oropharyngeal cancer. Oral Dis. 19:206–211.
2013. View Article : Google Scholar
|
|
8
|
Xu J, Yang W, Wang Q, Zhang Q, Li X, Lin
X, Liu X and Qin Y: Decreased HCRP1 expression is associated with
poor prognosis in breast cancer patients. Int J Clin Exp Pathol.
7:7915–7922. 2014.
|
|
9
|
Yang W, Wang JG, Wang Q, Qin Y, Lin X,
Zhou D, Ren K, Hou C, Xu J and Liu X: Decreased HCRP1 promotes
breast cancer metastasis by enhancing EGFR phosphorylation. Biochem
Biophys Res Commun. 477:222–228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Du Y, Wang P, Sun H, Yang J, Lang X, Wang
Z, Zang S, Chen L, Ma J and Sun D: HCRP1 is downregulated in
non-small cell lung cancer and regulates proliferation, invasion,
and drug resistance. Tumour. 37:15893–15901. 2016. View Article : Google Scholar
|
|
11
|
Bache KG, Slagsvold T, Cabezas A, Rosendal
KR, Raiborg C and Stenmark H: The growth-regulatory protein
HCRP1/hVps37A is a subunit of mammalian ESCRT-I and mediates
receptor down-regulation. Mol Biol Cell. 15:4337–4346. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Voutsadakis IA: Epithelial-mesenchymal
transition (EMT) and regulation of EMT factors by steroid nuclear
receptors in breast cancer: A review and in silico investigation. J
Clin Med. 5:E112016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wendt MK, Tian M and Schiemann WP:
Deconstructing the mechanisms and consequences of TGF-β-induced EMT
during cancer progression. Cell Tissue Res. 347:85–101. 2012.
View Article : Google Scholar
|
|
14
|
Giannelli G, Koudelkova P, Dituri F and
Mikulits W: Role of epithelial to mesenchymal transition in
hepatocellular carcinoma. J Hepatol. 65:798–808. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van Zijl F, Mall S, Machat G, Pirker C,
Zeillinger R, Weinhaeusel A, Bilban M, Berger W and Mikulits W: A
human model of epithelial to mesenchymal transition to monitor drug
efficacy in hepatocellular carcinoma progression. Mol Cancer Ther.
10:850–860. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Zijl F, Zulehner G, Petz M, Schneller
D, Kornauth C, Hau M, Machat G, Grubinger M, Huber H and Mikulits
W: Epithelial-mesenchymal transition in hepatocellular carcinoma.
Future Oncol. 5:1169–1179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Duivenvoorden WC, Hirte HW and Singh G:
Transforming growth factor beta1 acts as an inducer of matrix
metalloproteinase expression and activity in human
bone-metastasizing cancer cells. Clin Exp Metastasis. 17:27–34.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lai MW, Huang SF, Lin SM, Chen TC, Lin CY,
Yeh CN, Yeh TS, Chen MF and Yeh CT: Expression of the HCRP1 mRNA in
HCC as an independent predictor of disease-free survival after
surgical resection. Hepatol Res. 39:164–176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giannelli G, Villa E and Lahn M:
Transforming growth factor-β as a therapeutic target in
hepatocellular carcinoma. Cancer Res. 74:1890–1894. 2014.
View Article : Google Scholar : PubMed/NCBI
|