Introduction
Mantle cell lymphoma (MCL) is a highly aggressive
subtype of B-cell lymphoma, which accounts for 6–8% of all
non-Hodgkin lymphoma diagnoses (1). The malignant cells are present in the
mantle zone of the lymph node (2)
and are characterized by the molecular hallmark of translocation
t(11;14)(q13;q32), which is associated with cyclin D1
overexpression (3,4). More than 80% of patients with MCL are
diagnosed at stage III or IV of the disease, which is characterized
by lymphadenopathy, hepatosplenomegaly and bone marrow involvement
(5). Over the last decade,
high-dose cytarabine and autologous stem cell transplantation have
been used to treat younger patients, whereas rituximab and
bendamustine are used to treat older patients. Furthermore, the
proteasome inhibitor bortezomib, the immunomodulator lenalidomide,
and the Bruton's tyrosine kinase inhibitor ibrutinib have been
approved for use in novel therapeutic regimens (4). Although these therapeutic regimens
initially exhibit a high rate of complete response, the majority of
patients receiving these treatments experience short remission
duration with a continuous relapse pattern, leading to incurable
MCL with a median survival period of 4–5 years (6). Therefore, there is an urgent need to
explore novel molecular targeting agents for the treatment of
MCL.
Mammalian target of rapamycin (mTOR) is a highly
conserved 289 kDa serine/threonine kinase (7), which is involved in various signaling
pathways, including Ras, phosphoinositide 3-kinase (PI3K)/protein
kinase B (Akt), hypoxia-inducible factor-1 and nuclear factor
(NF)-κB. mTOR serves a critical role in the regulation of cell
differentiation, proliferation and survival (8), and is associated with tumorigenesis,
angiogenesis, tumor growth and metastasis (9–11).
In addition, mTOR is aberrantly activated in various malignancies
and is an indicator of more aggressive diseases, as well as poorer
disease prognosis (10–13). mTOR serves as the catalytic subunit
of two distinct protein complexes, known as mTOR complex (mTORC)1
and mTORC2. mTORC1 contains mTOR, Raptor and mTOR-associated
protein, LST8 homolog (mLST8), whereas mTORC2 is formed by mTOR,
Rictor and mLST8 (14). mTORC1
phosphorylates ribosomal protein S6 kinase (p70S6K) and eukaryotic
initiation factor 4E binding protein (4E-BP1), which results in
protein, nucleotide and lipid synthesis for cell growth and
proliferation. mTORC2 autophosphorylates mTOR on Ser2481, and also
phosphorylates Akt on Ser473 to control survival and apoptosis
(15,16). Numerous hematological malignancies
exhibit elevated or aberrant mTOR activation, and mTORC1 and mTORC2
are activated in primary MCL specimens (17). Therefore, numerous clinical trials
aimed at evaluating the potential of single agent mTOR-targeted
therapies have been launched. Weekly intravenous injection of the
mTOR inhibitor temsirolimus has been reported to result in an
overall response rate of 38 and 41% in two studies of patients with
relapsed MCL (18,19). Several other clinical trials have
indicated that mTOR inhibition can produce antitumor responses in
relapsed lymphoma. However, the use of an mTOR targeting agent as a
novel therapeutic regimen requires further exploration,
particularly for lymphoid malignancies such as MCL (12–14).
Celastrol is a triterpene purified from
Tripterygium wilfordii Hook, which possesses antioxidative,
ant-apoptotic, ant-inflammatory, anticarcinogenic and ant-obesity
properties (20,21). In previous studies, celastrol has
been reported to exert anticancer effects on leukemia (22), breast cancer (23), and head and neck cancer (24), as well as other types of tumor.
However, the precise mechanism of action of celastrol in lymphoma,
particularly in MCL, has not been fully elucidated.
The present study synthesized dihydrocelastrol
(DHCE) as a dihydro-analog of celastrol. DHCE exerted potent
anti-tumor activity in MCL cells, in vitro and in
vivo, with minimal cytotoxic effects on normal cells.
Furthermore, the molecular mechanisms of DHCE action were
investigated, and the results revealed that DHCE was an effective
inhibitor of mTORC1 and mTORC2. DHCE suppressed growth and
proliferation by inhibiting mTORC1-mediated phosphorylation of
p70S6K and 4E-BP1. Simultaneously, DHCE induced apoptosis and
inhibited cell survival by suppressing mTORC2-mediated
phosphorylation of Akt and NF-κB activity. Taken together, these
findings suggested that DHCE may have the potential to serve as a
novel therapeutic agent for the treatment of MCL.
Materials and methods
Cells and cell culture
The human MCL cell lines Jeko-1 and Granta519 were
purchased from American Type Culture Collection (Manassas, VA,
USA). The human MCL cell lines Z138, Mino and Rec-1 were provided
by Dr Xue Han of Tianjin Medical University Cancer Hospital
(Tianjin, China). Human peripheral blood mononuclear cells (PBMCs)
were acquired from three healthy normal donors (2 males, 1 female;
age, 24, 24 and 28 years, respectively) using Ficoll-Hypaque
density gradient centrifugation at 568 × g for 30 min at room
temperature. Informed consent was obtained from each healthy donor.
The present study was approved by the institutional review board of
Shanghai Tenth People's Hospital (Shanghai, China). The cells were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
All cells were maintained in a humidified atmosphere containing 5%
CO2 at 37°C.
Reagents and antibodies
DHCE was synthesized from celastrol using the method
previously described by Klaic et al (25). DHCE solution (2 mM) was dissolved
in dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany), stored at −20°C and diluted with cell culture medium to
obtain various concentrations. Antibodies against caspase-3 (cat.
no. 9665, 1:1,000), cleaved caspase-8 (cat. no. 9496, 1:1,000),
caspase-9 (cat. no. 9508, 1:1,000), B-cell lymphoma 2
(Bcl-2)-associated X protein (Bax) (cat. no. 2772, 1:1,000), Bcl-2
(cat. no. 2872, 1:1,000), Bcl-extra large (Bcl-xL) (cat. no. 2764,
1:1,000), Akt (cat. no. 2920, 1:2,000), phosphorylated (p)-Akt
(cat. no. 4060, 1:2,000), p-p70S6K (cat. no. 9205, 1:1,000), p70S6K
(cat. no. 2708, 1:1,000), p-4E-BP1 (cat. no. 9456, 1:1,000), 4E-BP1
(cat. no. 9644, 1:1,000), p-mTOR (Ser2481) (cat. no. 2974,
1:1,000), mTOR (cat. no. 2983, 1:1,000) and β-actin (cat. no. 3700,
1:1,000) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Cyclin D1 (cat. no. ab134175, 1:1,000),
cyclin-dependent kinase 4 (CDK4) (cat. no. ab108357, 1:1,000), CDK6
(cat. no. ab124821, 1:1,000), NF-κB inhibitor α (IκBα) (cat. no.
ab32518, 1:1,000), p-IκBα (cat. no. ab92700, 1:1,000), NF-κB-p65
(cat. no. ab32536, 1:50,000) and p-NF-κB-p65 (cat. no. ab76302,
1:10,000) were obtained from Abcam (Cambridge, UK). The pan-caspase
inhibitor Z-VAD-FMK was purchased from Selleck Chemicals (Houston,
TX, USA). Cell Counting kit-8 (CCK-8) was obtained from Shanghai
Yeasen Biotechnology Co., Ltd. (Shanghai, China) and the BD
Pharmingen™ Annexin V/propidium iodide (PI) Apoptosis Detection kit
was obtained from BD Biosciences (Franklin Lakes, NJ, USA).
Cell viability assay
Jeko-1, Z138, Mino, Rec-1 and Granta519 cells were
plated into 96-well plates at a density of 2×105
cells/ml and were treated with 0.2, 0.4, 0.6, 0.8 and 1.2 µM
DHCE for 24, 48 and 72 h. PBMCs were plated into 96-well plates at
a density of 3×105 cells/ml and were treated with 1.6
µM DHCE for 48 h. After incubation at 37°C and 5%
CO2, 10 µl CCK-8 solution was added to each well
and the cells were incubated for an additional 2 h at 37°C.
Absorbance was then measured at 450 nm using a microplate
reader.
Cell apoptosis detection
Double-staining with the BD Pharmingen™ Annexin V/PI
Apoptosis Detection kit was used to detect cell apoptosis.
Following DHCE (0.4, 0.6 and 0.8 µM) and/or Z-VAD-FMK (50
µM) exposure for 48 h, 1×105 Jeko-1 and Z138
cells were resuspended in 95 µl binding buffer and 5
µl fluorescein isothiocyanate-conjugated Annexin V after
centrifugation at 142 × g and 4°C for 5 min. After incubation for
30 min at 4°C in the dark, 500 µl binding buffer and 20
µl PI solution were added to each cell suspension, and the
BD FASCanto II flow cytometer (BD Biosciences) was used for
analysis by FlowJo 10 (FlowJo LLC, Ashland, OR, USA). Apoptotic
cells were identified as Annexin V+/PI−
(early apoptosis) and Annexin V+/PI+ (late
apoptosis).
Cell cycle analysis
Following 0.4 and 0.8 µM DHCE exposure for 36
h, Jeko-1 and Z138 cells at a density of 2×105 cells/ml
were washed with cold PBS, and fixed in 70% ice-cold ethanol
overnight at −20°C. The ethanol-fixed samples were washed with PBS,
and incubated in 500 µl PI/RNase staining buffer (BD
Biosciences) for 15 min at room temperature in the dark prior to
flow cytometry. Results were analyzed by ModFit LT 3.2 (Verity
Software House, Inc., Topsham, ME, USA).
Clonogenic assay
Jeko-1 and Z138 cells (1×104 cells/well)
were mixed with RPMI-1640 media containing 10% FBS and 0.33% agar,
and were layered on top of a base layer of 0.5% agar in each well
of 6-well plates. After culturing for 2 weeks, cells were incubated
with 0.5% crystal violet for 1 h at room temperature. Subsequently,
images of cell colonies were captured using a digital camera after
staining with extra crystal violet. The number of cell colonies was
semi-quantified using ImageJ 1.51 software (National Institutes of
Health, Bethesda, MD, USA).
5-Ethynyl-2′-deoxyuridine (EdU) labeling
and immunofluo-rescence
Jeko-1 and Z138 cells were plated into a 6-well
plate at a density of 4×105 cells/well, and were
incubated with or without 0.8 µM DHCE at 37°C for 48 h.
Subsequently, all cells were incubated with 50 µM EdU
(Guangzhou RiboBio Co., Ltd., Guangzhou, China) at 37°C for 1 h.
After being fixed with 4% paraformaldehyde at room temperature for
30 min, the cells were treated with 0.5% Triton X-100 at room
temperature for 10 min and washed three times with PBS. Thereafter,
the cells were exposed to 100 µl 1X Apollo reaction cocktail
at 37°C for 30 min and incubated with DAPI at room temperature to
stain the cell nuclei for 5 min. A confocal laser-scanning
microscope was used to detect stained cells.
DAPI and terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) staining
Jeko-1 and Z138 cells were plated into a 6-well
plate at a density of 4×105 cells/well, and were
incubated with or without 0.8 µM DHCE at 37°C for 48 h.
After centrifugation at 142 × g and 37°C for 5 min and washing with
PBS, cells were fixed in 4% paraformaldehyde at 37°C for 30 min and
permeabilized with 0.1% Triton X-100 at 37°C for 10 min. After
washing, cells were incubated with TUNEL (Beyotime Institute of
Biotechnology, Shanghai, China) reaction mixture for 60 min at
37°C. Prior to plating on coverslips, cells were stained with DAPI
for 5 min at room temperature in the dark. The cells were imaged
under a fluorescence microscope (Zeiss Axiovert 25; Zeiss GmbH,
Jena, Germany).
Western blotting
Jeko-1 and Z138 cells were incubated in lysis buffer
[100 mM Tris-HCl (pH 6.8), 4% SDS, 20% glycerol] for protein
extraction. Protein concentration was determined using the
bicinchoninic acid method. Samples containing 30 µg protein
were separated by 10 or 12.5% SDS-PAGE and were then transferred to
polyvinylidene difluoride membranes. Membranes were blocked with 5%
non-fat milk or 5% bovine serum albumin [Anlite (Shanghai)
Pharmaceutical Technology Co., Ltd., Shanghai, China] at room
temperature for 1 h, and were then incubated overnight at 4°C with
primary antibodies. Subsequently, the membranes were probed with
secondary antibodies [goat ant-mouse immunoglobulin G (IgG), cat.
no. 926-32210, 1:1,000; goat ant-rabbit IgG, cat. no. 926-32211,
1:1,000; LI-COR Biosciences, Lincoln, NE, USA) for 60 min at room
temperature, and the blots were detected using the Odyssey
two-color infrared laser imaging system (LI-COR Biosciences).
Tumor xenograft model
A total of 8 male nude mice (athymic, BALB/C nu/nu;
age, 6 weeks; weight, 17–20 g) were purchased from the Shanghai
SLAC Laboratory Animal Co., Ltd. (Shanghai, China). All mice were
maintained in an air-conditioned room at 24°C with a 12-hour
light/dark cycle and 45% relative humidity. All mice had free
access to water and food. All animal studies were approved by the
institutional review board of the Shanghai Tenth People's Hospital
(ID: SYXK 2011–0111). Human MCL Jeko-1 cells (10×106)
were suspended in 100 µl serum-free culture medium and were
subcutaneously injected into the upper flank region of nude mice.
When tumors were measurable, mice were randomly divided into the
control and DHCE groups. In the DHCE group, mice were administered
3 mg/kg DHCE (DHCE was dissolved in 5% DMSO, 15% Tween-80 and
saline), whereas in the control group, mice received 100 µl
vehicle (5% DMSO, 15% Tween-80 and saline) daily for 19 consecutive
days. Tumor size and mouse weight were measured, and the tumor
volume was calculated as (length x width2) × 0.5. At the
end of treatment, mice were sacrificed by cervical dislocation
following intraperitoneal injection of 1% pentobarbital sodium (50
mg/kg). Hematoxylin-eosin (26)
and TUNEL staining of the tumor tissue sections were then
performed.
Drug combination study
Jeko-1 and Z138 cells were treated for 24 h with
DHCE (0.4, 0.6, 0.8 and 1.0 µM) and/or bort-ezomib (8, 12,
16 and 20 nM) (Sigma-Aldrich; Merck KGaA). Combination index (CI)
values were calculated using the Chou-Talalay equation (27): CI values <1 indicated synergism;
CI values equal to 1 indicated an additive effect; and CI values
>1 indicated antagonism.
Statistical analysis
Data are expressed as the means ± standard
deviation. Statistical analysis was conducted using an unpaired
Student's t-test or one-way analysis of variance followed by least
significant difference test for multiple comparisons. All
statistical analyses were performed using SPSS version 20.0
statistical analysis software (IBM Corp., Armonk, NY, USA). P≤0.05
was considered to indicate a statistically significant
difference.
Results
DHCE inhibits MCL cell growth and
proliferation
As shown in Fig.
1A, DHCE is a synthesized dihydro-analog compound with a
molecular weight of 452.6. In the present study, five MCL cell
lines (Jeko-1, Z138, Granta519, Mino and Rec-1) were treated with
various doses of DHCE. After 48 h of DHCE exposure, cell viability
was measured using a CCK-8 assay. MCL cell viability was decreased
in a dose-dependent manner (Fig.
1B), and the 48 h half maximal inhibitory concentration values
of DHCE in Jeko-1, Z138, Mino, Rec-1 and Granta519 cells were
0.65±0.10, 0.45±0.04, 0.42±0.01, 0.35±0.03 and 0.91±0.08 µM,
respectively. Jeko-1 and Z138 cells were selected for subsequent
analyses, as they have previously been used in numerous studies
regarding MCL (28,29). Furthermore, a time-course study of
DHCE further demonstrated that cell viability was inhibited in a
time-dependent manner (Fig. 1C).
Conversely, at concentrations as high as 1.6 µM, DHCE
conferred minimal cytotoxic effects on normal PBMCs obtained from
three healthy volunteers (Fig.
1D). To investigate the inhibitory effects of DHCE on
tumorigenic potential, anchorage-independent colony formation in
soft agar was compared between the control and treatment groups.
DHCE treatment reduced the ability of Jeko-1 and Z138 cells to form
tumorspheres in a dose-dependent manner, and colony formation was
almost completely inhibited among Z138 cells treated with 0.8
µM DHCE (Fig. 1E). To
further explore the effects of DHCE on cell proliferation, the
integration of EdU was measured, in order to detect the levels of
DNA synthesis; cell nuclei were stained with DAPI. The results
demonstrated that DHCE suppressed DNA synthesis in MCL cells, thus
indicating that DHCE inhibited proliferation of MCL cells (Fig. 1F). These findings suggested that
DHCE may decrease cell viability and inhibit cell proliferation in
a dose- and time-dependent manner; however, DHCE is not cytotoxic
to normal cells.
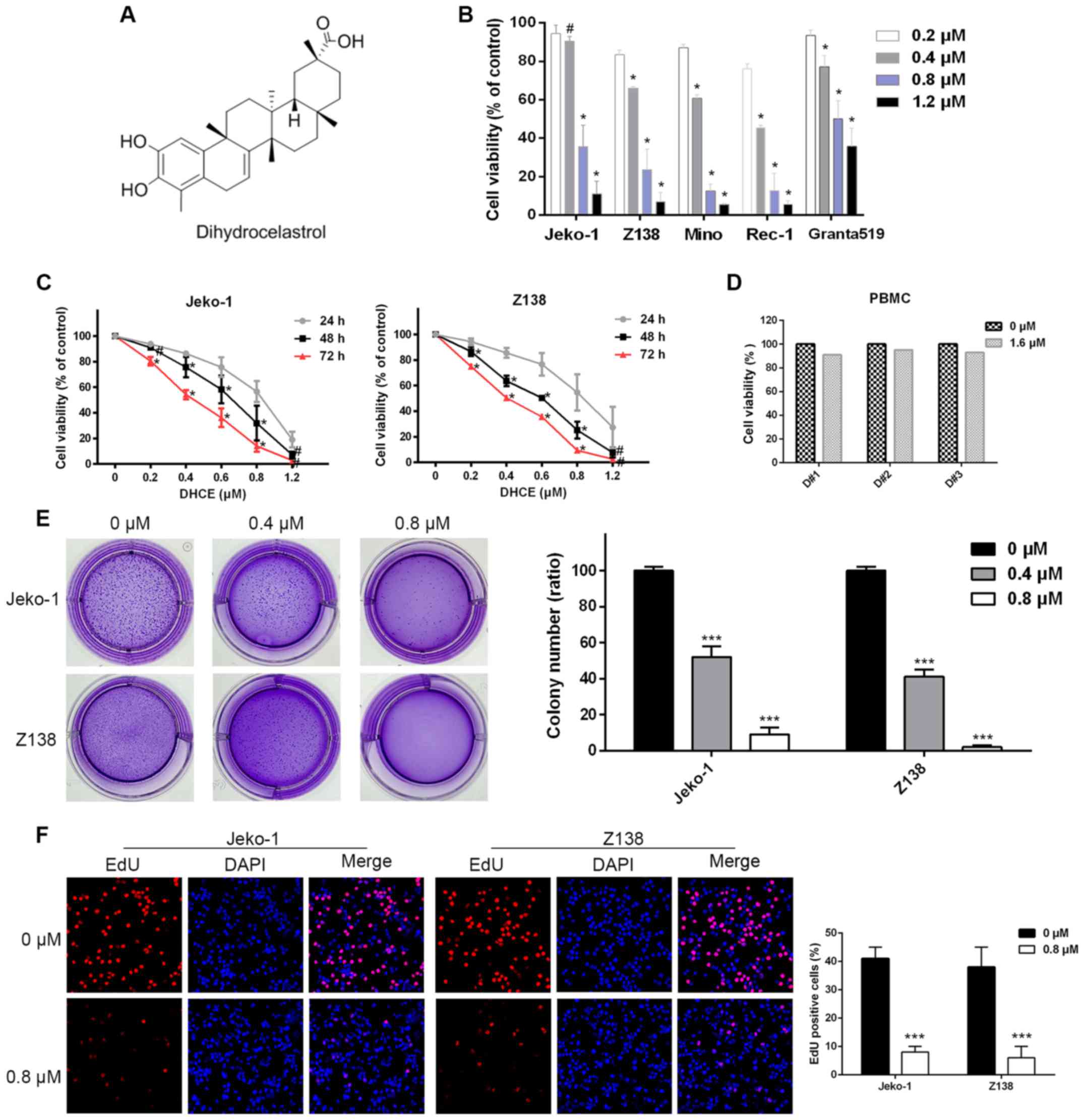 | Figure 1DHCE inhibits proliferation and
growth of MCL cells. (A) Chemical structure of DHCE. (B) MCL cell
lines were treated with DHCE for 48 h. Cell viability was analyzed
using a Cell Counting kit-8 assay. #P>0.05,
*P<0.05 compared with the 0.2 µM group. (C)
Jeko-1 and Z138 cells were treated with DHCE (0.2–1.2 µM)
for 24, 48 and 72 h, and viability was then analyzed.
#P>0.05, *P<0.05, 48 h group compared
with the 72 and 24 h groups; 72 h group compared with the 24 and 48
h groups. (D) PBMCs obtained from three healthy volunteers were
treated with DHCE (1.6 µM) for 48 h, and cell viability was
then measured. (E) Representative images of overall colony
formation of Jeko-1 and Z138 cells treated with DHCE. The number of
colonies formed in each well was quantified. (F) Representative
photomicrographs (original magnification, ×50) exhibited decreased
proliferation, as determined by EdU immunofluorescence (red),
following treatment with 0.8 µM DHCE for 48 h. DAPI was used
to counterstain nuclei (blue). The number of EdU-positive cells was
quantified. Data are presented as the means ± standard error of the
mean (n=3). ***P<0.001 compared with the 0 µM
group. DHCE, dihydrocelastrol; EdU, 5-ethynyl-2′-deoxyuridine; MCL,
mantle cell lymphoma; PBMCs, peripheral blood mononuclear
cells. |
DHCE promotes cell cycle arrest in MCL
cells
The cyclin D1-CDK4 and cyclin D1-CDK6 complexes are
critical for the G1/S checkpoint, and regulate the
progression of cells from G1 phase to S phase (30). To evaluate the effects of DHCE on
cell cycle progression, Jeko-1 and Z138 cells were treated with
DHCE and analyzed by flow cytometry. The results demonstrated that
following treatment with DHCE, the percentage of Jeko-1 and Z138
cells in the G0/G1 phase of the cell cycle
was significantly increased (Fig,
2A). The present study also analyzed the expression levels of
cell cycle regulatory proteins, and indicated that DHCE suppressed
the expression of cyclin D1, CDK4 and CDK6 (Fig. 2B). Taken together, these data
suggested that DHCE may induce G0/G1 phase
arrest in a dose-dependent manner by inhibiting the expression of
cell cycle regulatory proteins.
DHCE induces apoptosis of MCL cells
Evasion of apoptosis has a key role in tumorigenesis
and chemotherapeutic resistance (31). To investigate whether apoptosis is
associated with the mechanism underlying the cytotoxic effects of
DHCE, Jeko-1 and Z138 cells were incubated with various
concentrations of DHCE for 48 h. DHCE markedly increased the
percentage of apoptotic cells in a dose-dependent manner, compared
with in the control group (Fig.
3A). Furthermore, Jeko-1 and Z138 cells were simultaneously
treated with DHCE and a pan-caspase inhibitor, Z-VAD-FMK. The
results demonstrated that Z-VAD-FMK induced a significant reduction
in DHCE-induced cell apoptosis (Fig.
3B; data of Z138 cells not shown), thus indicating that
DHCE-mediated apoptosis is dependent upon caspase activity. These
findings demonstrated that DHCE may induce MCL cell apoptosis in a
dose-dependent manner.
 | Figure 3DHCE induces apoptosis of mantle cell
lymphoma cells. (A) Cells were treated with DHCE for 48 h and
analyzed by FACS using Annexin V/PI staining. Statistical analysis
of apoptosis is presented in the right panel. (B) Cells were
incubated with or without the pan-caspase inhibitor Z-VAD-FMK, and
were then treated with 0.8 µM DHCE for 48 h, stained with
Annexin V/PI and analyzed by FACS. Statistical analysis of
apoptosis is presented in the right panel. (C) Representative
fluorescent images (original magnification, ×50) detected increased
numbers of apoptotic cells, as evaluated by TUNEL staining (green),
following treatment with 0.8 µM DHCE for 48 h. DAPI was used
as a nuclear stain (blue). The overall number of TUNEL-positive
cells was determined and analyzed in the right panel. (D)
Expression levels of apoptosis-associated proteins were analyzed
using western blot analysis, with β-actin used as an internal
control. Data are expressed as the means ± standard deviation
(n=3). *P<0.05, **P<0.01,
***P<0.001 compared with the 0 µM group, or as
indicated. Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma
2; Bcl-xL, Bcl-extra large; DHCE, dihydrocelastrol; FACS,
fluorescence-activated cell sorting; PI, propidium iodide; TUNEL,
terminal deoxynucleotidyl-transferase-mediated dUTP nick end
labeling. |
DHCE induces DNA strand breaks due to
apoptosis
A series of morphological alterations are triggered
when apoptosis is activated, including cell shrinkage, aberrant
chromatin compaction, DNA hydrolysis, nuclear fragmentation and the
formation of apoptotic bodies (32). To directly confirm that DHCE
promoted the apoptosis of MCL cells, a confocal laser-scanning
microscope was used to detect the binding of TUNEL reagent with DNA
fragments. Compared with in the control group, MCL cells treated
with DHCE for 48 h exhibited chromatin fragmentation (Fig. 3C), which is a typical morphological
characteristic of apoptotic cells. These cytological observations
further confirmed that DHCE promotes the apoptosis of MCL
cells.
DHCE induces caspase-dependent apoptosis
through the extrinsic and intrinsic pathways
It is widely known that apoptosis signaling pathways
consist of extrinsic and intrinsic pathways. Upon receiving
specific signals instructing the cells to undergo apoptosis,
caspase proteases lead to the cleaving of cellular components
including structural proteins in the cytoskeleton and nuclear
proteins, such as DNA repair enzymes, through extrinsic and
intrinsic pathways (31). DHCE
markedly activated caspase-8 and caspase-3 (Fig. 3D), which are associated with the
extrinsic pathway of apoptosis. In addition, DHCE suppressed the
expression of the ant-apoptotic proteins Bcl-2 and Bcl-xL, and
upregulated the expression of Bax, which is a proapoptotic protein
of the intrinsic pathway (Fig. 3D)
(33). Increased procaspase-9
substrate cleavage was also detected (Fig. 3D). These findings suggested that
DHCE may activate the extrinsic and intrinsic pathways of apoptosis
in a dose-dependent manner.
DHCE suppresses mTORC1/p70S6K/4E-BP1
mTORC1 phosphorylates p70S6K and 4E-BP1, which are
the two best-characterized downstream targets of mTORC1, thus
regulating protein translation and contributing to cell growth and
proliferation. To address the potential involvement of mTORC1 in
DHCE-mediated inhibition of MCL cell proliferation, the present
study measured the protein expression levels of p70S6K and 4E-BP1.
Western blot analysis of Jeko-1 and Z138 cells demonstrated that
phosphorylation of p70S6K and 4E-BP1 was inhibited by DHCE in a
dose-dependent manner (Fig. 4A).
Although DHCE treatment decreased the total protein expression of
4E-BP1 after 24 h, phosphorylation of 4E-BP1 was markedly inhibited
compared with the control. These results indicated that
DHCE-mediated inhibition of cell proliferation and growth may
result from the suppression of mTORC1/p70S6K/4E-BP1 activity
(Fig. 4B).
 | Figure 4Mechanisms underlying the antitumor
activity of DHCE. (A) DHCE suppressed mTORC1/p70S6K/4E-BP1
signaling. Cells were treated with 0.4 or 0.8 µM DHCE for 24
h. Expression levels of mTORC1 kinase activity-related proteins
were analyzed using western blot analysis, and phosphorylated
protein levels were normalized to the corresponding total protein
levels. (B) Diagram illustrating the effects of DHCE on mTORC1 and
mTORC2. (C) DHCE suppressed mTORC2/Akt/NF-κB activity. Cells were
treated with 0.4 or 0.8 µM DHCE for 24 h. Expression levels
of mTORC2 kinase activity-related proteins were analyzed using
western blot analysis, and phosphorylated protein levels were
normalized to the corresponding total protein levels. Data are
expressed as the means ± standard deviation (n=3).
*P<0.05 compared with the 0 µM group. 4E-BP1,
eukaryotic initiation factor 4E binding protein; Akt, protein
kinase B; DHCE, dihydrocelastrol; eIF4E, eukaryotic translation
initiation factor 4E; IκBα, NF-κB inhibitor α; MCL, mantle cell
lymphoma; mTOR, mammalian target of rapamycin; mTORC, mTOR complex;
NF-κB, nuclear factor-κB; p-, phosphorylated; p70S6K, ribosomal
protein S6 kinase. |
DHCE blocks mTORC2/Akt/NF-κB activity
signaling
In contrast to mTORC1, mTORC2 modulates cell
survival and apoptosis. Unlike rapamycin, a highly potent and
selective inhibitor of mTORC1, which has little or no apoptotic
effects on MCL cells (34), DHCE
strongly induced apoptosis of Jeko-1 and Z138 cells (Fig. 3). Previous studies demonstrated
that Akt regulated the activation of NF-κB, and that NF-κB can be
stimulated downstream of mTORC2 by either Akt-dependent or
Akt-independent mechanisms (35–38).
The present western blot analysis results demonstrated that
treatment of MCL cells with DHCE downregulated the protein
expression levels of NF-κB target genes, including Bcl-2, Bcl-xL
and cyclin D1. Therefore, it was hypothesized that DHCE may induce
apoptosis of MCL cells by suppressing mTORC2/Akt/NF-κB signaling.
Treatment of cells with DHCE resulted in a marked, dose-dependent
inhibition of Akt (Ser473) and mTOR (Ser2481) phosphorylation in
Jeko-1 and Z138 cells compared with in the control group (Fig. 4C). DHCE treatment exerted only a
minimal effect on total mTOR and total Akt protein expression.
Subsequently, the effects of DHCE on NF-κB activity were assessed
and demonstrated that DHCE treatment resulted in decreased IκBα and
NF-κB-p65 phosphorylation. These findings suggested that DHCE may
block the activity of mTORC2/Akt/NF-κB, thus leading to apoptosis
and inhibition of cell survival (Fig.
4B).
DHCE inhibits tumor growth in a xenograft
mouse model
The present study further examined the therapeutic
efficacy of DHCE using a xenograft mouse model. Nude mice bearing
subcutaneous Jeko-1 cell xenograft tumors were administered daily
intraperitoneal injections of either DHCE (3 mg/kg) or a vehicle
control. The diameter and volume of the largest tumor in these mice
was 1.98 cm and 3.20 cm3, respectively. DHCE treatment
markedly decreased tumor size (Fig.
5A) and significantly decreased tumor volume (Fig. 5B); however, body weight was not
significantly altered between the DHCE-treated and vehicle-treated
mice (Fig. 5C). The results of
H&E staining demonstrated that necrosis in the tumor samples of
the DHCE-treated group was increased compared with in the vehicle
control group. TUNEL staining revealed that DHCE treatment
increased apoptosis compared with in the vehicle control group
(Fig. 5D). DHCE treatment was well
tolerated; no symptoms of poor health or abnormal behaviors were
observed in DHCE-treated or vehicle-treated mice, and microscopic
examination of individual organs revealed no evidence of tissue
damage in either treatment group (data not shown). Consistent with
the in vitro observations, these in vivo findings
demonstrated that DHCE may inhibit MCL tumor growth in a xenograft
model without exerting lethal toxicity.
DHCE acts synergistically with bortezomib
in MCL cells
Combination chemotherapy is a rational strategy for
the treatment of MCL (4).
Combination treatment with DHCE and bortezomib inhibited growth in
both cell lines to a greater degree than treatment with DHCE or
bortezomib alone. In the present study, CI values were <1.0,
indicating synergism of DHCE and bortezomib (Fig. 6). Combined treatment of MCL cells
with DHCE and bortezomib exhibited clear synergistic effects on the
inhibition of cell proliferation.
Discussion
Despite recent developments resulting in the
generation of novel treatment regimens for MCL, the disease remains
incurable, with many patients experiencing short periods of
remission followed by continuous relapse. Novel drugs are required
to further improve the outcome of patients with MCL. Numerous
reports have confirmed that the triterpenoid celastrol is an
effective antitumor analog in leukemia, breast cancer, gastric
cancer, head and neck cancer, and multiple myeloma (22–24).
However, little is currently known regarding the use of this
compound in lymphoma, and to the best of our knowledge, the present
study is the first to directly assess the use of a triterpenoid in
MCL. DHCE is a novel triterpene, and the present study explored the
effects and relevant mechanisms of DHCE in MCL cells.
Cell cycle dysregulation and evasion of apoptosis,
both of which are hallmarks of tumor cells, contribute to
tumorigenesis and chemical resistance (39). In general, when confronted with
stress or damage, cells activate DNA damage checkpoints and
initiate DNA repair; however, with persistent cellular impairments,
DNA damage cannot be appropriately repaired and the DNA damage
checkpoint pathway is activated to eliminate potentially harmful
DNA-damaged cells via apoptosis (40). Targeting the apoptosis pathways is
a promising opportunity to eradicate cancer (31). Cyclin D1, along with CDK4 and CDK6,
regulates G1/S progression through phosphorylation of
retinoblastoma protein (39).
Treatment of MCL cells with DHCE resulted in a downregulation of
cyclin D1, CDK4 and CDK6 expression, which then led to
G0/G1 phase cell cycle arrest.
Fluorescence-activated cell sorting analysis and TUNEL/DAPI double
staining revealed that DHCE induced apoptosis of MCL cells in a
dose-dependent manner, which was suppressed by the pan-caspase
inhibitor Z-VAD-FMK. The ability of DHCE to induce apoptosis was
associated with the activity of apoptotic proteins. Proteins of the
Bcl-2 family are key regulators of the intrinsic apoptotic pathway,
including Bcl-2, Bcl-xL and Bax, which ultimately activate
caspase-9 at the apoptosome (41).
Caspase-8 is a key initiator caspase that can directly activate
caspase-3, thus promoting apoptosis via the extrinsic pathway
(31). DHCE treatment resulted in
activation of caspase-3, caspase-8 and caspase-9. Furthermore, DHCE
treatment decreased the expression of the ant-apoptotic
mitochondrial proteins Bcl-2 and Bcl-xL, and increased the
expression of the proapoptotic mitochondrial protein Bax. These
findings suggested that DHCE treatment may induce the
caspase-dependent apoptosis of MCL cells via extrinsic and
intrinsic apoptotic pathways.
The mTOR pathway is considered a promising target
for cancer therapeutics (42,43).
mTORC1 activity affects cell growth and metabolism by
phosphorylating p70S6K and 4E-BP1, which regulate protein
synthesis, as well as lipid, nucleotide and glucose metabolism.
Upon phosphorylation, p70S6K becomes activated, which is critical
for lipid and ribosome biogenesis pathways and protein translation.
The phosphorylation of 4E-BP1 causes its dissociation from the
translation initiation factor eukaryotic translation initiation
factor 4E, thus triggering 5′cap-dependent mRNA translation
(44). mTORC2 controls apoptosis
by autophosphorylating mTOR on Ser2481 with Rictor, and by
phosphorylating Akt on Ser473 (7).
Rapamycin, which is a well-known mTORC1 inhibitor, and analogs of
rapamycin, incompletely inhibit mTORC1 and only inhibit mTORC2 when
administered at high doses for a prolonged duration (34,44),
due to the presence of a negative feedback loop between mTORC1 and
the insulin/PI3K signaling pathway. mTORC1 phosphorylates growth
factor receptor bound protein 10 to inhibit insulin/insulin-like
growth factor 1 receptor signaling, which is an upstream activator
of Akt and mTORC2. Therefore, inhibition of mTORC1 alone removes
the negative feedback on insulin/PI3K/Akt signaling, allowing Akt
to become activated, which ultimately promotes cell survival and
prevents apoptosis (45). Other
similar feedback mechanisms also exist, which limit the
effectiveness of mTORC1 inhibitors. Treatment of established cell
lines or primary MCL cultures with rapamycin induces little or no
apoptotic response (34).
Inhibition of mTORC1 alone has negligible effects on apoptosis in
several cell lines; however, suppressing both mTORC1 and mTORC2
presents a more effective strategy. In the present study, it was
demonstrated that treatment of MCL cells with DHCE reduced p70S6K
and 4E-BP1 phosphorylation, indicating that DHCE suppressed mTORC1
activity. Therefore, it may be concluded that DHCE inhibits the
growth and proliferation of MCL cells through suppression of the
mTORC1/p70S6K/4E-BP1 pathway.
Although the activity of mTORC1 can be assessed by
determining the phosphorylation status of its downstream
substrates, a specific downstream marker of mTORC2 activity has not
been identified to date. Previous studies have demonstrated that
knockdown of the mTORC2 protein Rictor, but not the mTORC1 protein
Raptor, significantly inhibits activity of Akt as well as that of
NF-κB (46,47), which controls cell survival and
apoptosis (48,49). Once phosphorylated by mTORC2, Akt
regulates transcriptional activity of NF-κB by inducing
phosphorylation and subsequent degradation of IκB (36), or by phosphorylating IκB kinase,
which increases the activity of NF-κB and stimulates the
transcription of prosurvival genes (42). In addition, mTORC2 directly
promotes NF-κB activation through the
serum/glucocorticoid-regulated kinase 1-dependent pathway or via
the phosphorylation of protein kinase C (35,37,38).
In the present study, it was demonstrated that phosphorylation of
mTOR (Ser2481) and Akt (Ser473) were significantly decreased in MCL
cells following treatment with DHCE, thus indicating that DHCE
inhibited mTORC2. Consistent with these findings, a reduction in
the phosphorylation of IκBα and NF-κB-p65 was observed, thus
indicating the reduction of NF-κB activity. Due to the suppression
of NF-κB activity, the expression levels of NF-κB targets,
including Bcl-2, Bcl-xL and cyclin D1, were also decreased.
Therefore, DHCE may exhibit a potent apoptotic effect on MCL cells
due to the suppression of mTORC2/Akt/NF-κB activity. These findings
are in agreement with previous studies (35,37,38).
However, a recent report indicated that mTORC2 activity suppresses
NF-κB signaling (50). Further
studies are required to determine how mTORC1 and mTORC2 regulate
NF-κB activity.
In the present study, DHCE was able to suppress
tumor growth in vivo without causing toxicity by inducing
the apop-tosis of MCL cells. Intraperitoneal administration of 3
mg/kg DHCE for 19 days resulted in a significant inhibition of
tumor growth, with no clear indications of toxicity. Consistent
with the in vitro results, immunohistochemical analysis of
the tumor samples confirmed that DHCE administration induced
apoptosis.
Combined treatment of MCL cells with DHCE and
bortezomib, a proteasome inhibitor, enhanced the inhibition of cell
proliferation that was achieved with DHCE or bortezomib alone. To
further evaluate the synergy between the two drugs, the CI values
were calculated; the results demonstrated that values were <1.0,
thus indicating that DHCE and bortezomib acted synergistically to
induce cytotoxicity in MCL cells. Bortezomib is approved for use by
the Food and Drug Administration of United States of America and is
currently used in MCL treatment regimens. Combination therapies are
often used as a strategy to minimize cytotoxicity and resistance
(4). The present results indicated
that combined therapy with DHCE and bortezomib may be a promising
strategy to overcome cytotoxicity and resistance in patients with
MCL. However, further validation and exploration is required to
fully determine the cooperative mechanism and synergism between
these drugs.
In conclusion, DHCE exhibited potent antitumor
activity in MCL cell lines and in an MCL xenograft model, at doses
that have little effect on normal cells and are well tolerated in
mice. The results demonstrated that DHCE suppressed cell growth and
proliferation by inhibiting mTORC1-mediated phosphorylation of
p70S6K and 4E-BP1. Simultaneously, DHCE induced apoptosis and
inhibited cell survival by suppressing mTORC2-mediated
phosphorylation of Akt and NF-κB activity. Furthermore, combined
treatment with DHCE and bortezomib resulted in synergistic
cytotoxic effects on MCL cells. The present study therefore
supported the important role of the mTOR pathway in the
tumorigenesis of MCL, thus suggesting the necessity of dually
inhibiting mTORC1/p70S6K/4E-BP1 and mTORC2/Akt/NF-κB activity in
MCL therapy. With further investigation into the associated
molecular mechanisms and clinical implications of DHCE
administration, DHCE has the potential to serve as a novel
therapeutic regimen to improve the outcomes of patients with
MCL.
Acknowledgments
The authors would like to thank Dr. Xue Han of
Tianjin Medical University Cancer Hospital (Tianjin, China) for
providing the human MCL cell lines.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81570190,
81529001, 81670194 and 81602515).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JS, WZ, YX, and BL designed the research; JS, WZ,
YX, and BL organized, analyzed, and interpreted the data; YX, BL,
WB, LG, YoZ, XL, JH, ZX, SC, DY, BX, YW, HW and XW performed the
experiments; JS, WZ, YX and BL drafted the manuscript; JH, HW and
YiZ contributed to clinical sample collection. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Informed consent was obtained from each healthy
donor. The present study was approved by the institutional review
board of Shanghai Tenth People's Hospital (Shanghai, China). All
animal studies were approved by the institutional review board of
the Shanghai Tenth People's Hospital (ID: SYXK 2011-0111).
Patient consent for publication
All volunteers consent to the publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Spurgeon SE, Till BG, Martin P, Goy AH,
Dreyling MP, Gopal AK, LeBlanc M, Leonard JP, Friedberg JW, Baizer
L, et al: Recommendations for clinical trial development in mantle
cell lymphoma. J Natl Cancer Inst. 109:1092016.
|
|
2
|
Zhou Y, Wang H, Fang W, Romaguer JE, Zhang
Y, Delasalle KB, Kwak L, Yi Q, Du XL and Wang M: Incidence trends
of mantle cell lymphoma in the United States between 1992 and 2004.
Cancer. 113:791–798. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bosch F, Jares P, Campo E, Lopez-Guillermo
A, Piris MA, Villamor N, Tassies D, Jaffe ES, Montserrat E, Rozman
C, et al: PRAD-1/cyclin D1 gene overexpression in chronic
lymphoproliferative disorders: A highly specific marker of mantle
cell lymphoma. Blood. 84:2726–2732. 1994.PubMed/NCBI
|
|
4
|
Cheah CY, Seymour JF and Wang ML: Mantle
cell lymphoma. J Clin Oncol. 34:1256–1269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hamad N, Armytage T, McIlroy K, Singh N
and Ward C: Primary cutaneous mantle-cell lymphoma: A case report
and literature review. J Clin Oncol. 33:e104–e108. 2015. View Article : Google Scholar
|
|
6
|
Campo E and Rule S: Mantle cell lymphoma:
Evolving management strategies. Blood. 125:48–55. 2015. View Article : Google Scholar
|
|
7
|
Lee JS, Vo TT and Fruman DA: Targeting
mTOR for the treatment of B cell malignancies. Br J Clin Pharmacol.
82:1213–1228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Strimpakos AS, Karapanagiotou EM, Saif MW
and Syrigos KN: The role of mTOR in the management of solid tumors:
An overview. Cancer Treat Rev. 35:148–159. 2009. View Article : Google Scholar
|
|
9
|
Baldo P, Cecco S, Giacomin E, Lazzarini R,
Ros B and Marastoni S: mTOR pathway and mTOR inhibitors as agents
for cancer therapy. Curr Cancer Drug Targets. 8:647–665. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Inoki K, Corradetti MN and Guan KL:
Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet.
37:19–24. 2005. View
Article : Google Scholar
|
|
11
|
Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK,
Wei Y, Sun HL, Li LY, Ping B, Huang WC, et al: IKK beta suppression
of TSC1 links inflammation and tumor angiogenesis via the mTOR
pathway. Cell. 130:440–455. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eyre TA, Collins GP, Goldstone AH and
Cwynarski K: Time now to TORC the TORC? New developments in mTOR
pathway inhibition in lymphoid malignancies. Br J Haematol.
166:336–351. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peponi E, Drakos E, Reyes G, Leventaki V,
Rassidakis GZ and Medeiros LJ: Activation of mammalian target of
rapamycin signaling promotes cell cycle progression and protects
cells from apoptosis in mantle cell lymphoma. Am J Pathol.
169:2171–2180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dancey J: mTOR signaling and drug
development in cancer. Nat Rev Clin Oncol. 7:209–219. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gupta M, Hendrickson AE, Yun SS, Han JJ,
Schneider PA, Koh BD, Stenson MJ, Wellik LE, Shing JC, Peterson KL,
et al: Dual mTORC1/mTORC2 inhibition diminishes Akt activation and
induces Puma-dependent apoptosis in lymphoid malignancies. Blood.
119:476–487. 2012. View Article : Google Scholar :
|
|
18
|
Witzig TE, Geyer SM, Ghobrial I, Inwards
DJ, Fonseca R, Kurtin P, Ansell SM, Luyun R, Flynn PJ, Morton RF,
et al: Phase II trial of single-agent temsirolimus (CCI-779) for
relapsed mantle cell lymphoma. J Clin Oncol. 23:5347–5356. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ansell SM, Inwards DJ, Rowland KM Jr,
Flynn PJ, Morton RF, Moore DF Jr, Kaufmann SH, Ghobrial I, Kurtin
PJ, Maurer M, et al: Low-dose, single-agent temsirolimus for
relapsed mantle cell lymphoma: A phase 2 trial in the North Central
Cancer Treatment Group. Cancer. 113:508–514. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu J, Lee J, Salazar Hernandez MA,
Mazitschek R and Ozcan U: Treatment of obesity with celastrol.
Cell. 161:999–1011. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kannaiyan R, Shanmugam MK and Sethi G:
Molecular targets of celastrol derived from Thunder of God Vine:
Potential role in the treatment of inflammatory disorders and
cancer. Cancer Lett. 303:9–20. 2011. View Article : Google Scholar
|
|
22
|
Sethi G, Ahn KS, Pandey MK and Aggarwal
BB: Celastrol, a novel triterpene, potentiates TNF-induced
apoptosis and suppresses invasion of tumor cells by inhibiting
NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB
activation. Blood. 109:2727–2735. 2007.
|
|
23
|
Jang SY, Jang SW and Ko J: Celastrol
inhibits the growth of estrogen positive human breast cancer cells
through modulation of estrogen receptor α. Cancer Lett. 300:57–65.
2011. View Article : Google Scholar
|
|
24
|
Fribley AM, Miller JR, Brownell AL,
Garshott DM, Zeng Q, Reist TE, Narula N, Cai P, Xi Y, Callaghan MU,
et al: Celastrol induces unfolded protein response-dependent cell
death in head and neck cancer. Exp Cell Res. 330:412–422. 2015.
View Article : Google Scholar
|
|
25
|
Klaić L, Trippier PC, Mishra RK, Morimoto
RI and Silverman RB: Remarkable stereospecific conjugate additions
to the Hsp90 inhibitor celastrol. J Am Chem Soc. 133:19634–19637.
2011. View Article : Google Scholar
|
|
26
|
Hu L, Wu H, Li B, Song D, Yang G, Chen G,
Xie B, Xu Z, Zhang Y, Yu D, et al: Dihydrocelastrol inhibits
multiple myeloma cell proliferation and promotes apoptosis through
ERK1/2 and IL-6/STAT3 pathways in vitro and in vivo. Acta Biochim
Biophys Sin (Shanghai). 49:420–427. 2017. View Article : Google Scholar
|
|
27
|
Ashton JC: Drug combination studies and
their synergy quantification using the Chou-Talalay method -
letter. Cancer Res. 75:24002015. View Article : Google Scholar
|
|
28
|
Rudelius M, Rosenfeldt MT, Leich E,
Rauert-Wunderlich H, Solimando AG, Beilhack A, Ott G and Rosenwald
A: Inhibition of focal adhesion kinase overcomes resistance of
mantle cell lymphoma to ibrutinib in the bone marrow
microenvironment. Haematologica. 103:116–125. 2018. View Article : Google Scholar :
|
|
29
|
Zhou J, Tiemann K, Chomchan P, Alluin J,
Swiderski P, Burnett J, Zhang X, Forman S, Chen R and Rossi J: Dual
functional BAFF receptor aptamers inhibit ligand-induced
proliferation and deliver siRNAs to NHL cells. Nucleic Acids Res.
41:4266–4283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hassan M, Watari H, AbuAlmaaty A, Ohba Y
and Sakuragi N: Apoptosis and molecular targeting therapy in
cancer. BioMed Res Int. 2014:1508452014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Taylor RC, Cullen SP and Martin SJ:
Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol
Cell Biol. 9:231–241. 2008. View Article : Google Scholar
|
|
33
|
Davids MS: Targeting BCL-2 in B-cell
lymphomas. Blood. 130:1081–1088. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dal Col J, Zancai P, Terrin L, Guidoboni
M, Ponzoni M, Pavan A, Spina M, Bergamin S, Rizzo S, Tirelli U, et
al: Distinct functional significance of Akt and mTOR constitutive
activation in mantle cell lymphoma. Blood. 111:5142–5151. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Madrid LV, Mayo MW, Reuther JY and Baldwin
AS Jr: Akt stimulates the transactivation potential of the RelA/p65
Subunit of NF-kappa B through utilization of the Ikappa B kinase
and activation of the mitogen-activated protein kinase p38. J Biol
Chem. 276:18934–18940. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ahmad A, Biersack B, Li Y, Kong D, Bao B,
Schobert R, Padhye SB and Sarkar FH: Targeted regulation of
PI3K/Akt/mTOR/NF-κB signaling by indole compounds and their
derivatives: Mechanistic details and biological implications for
cancer therapy. Anticancer Agents Med Chem. 13:1002–1013. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tanaka K, Babic I, Nathanson D, Akhavan D,
Guo D, Gini B, Dang J, Zhu S, Yang H, De Jesus J, et al: Oncogenic
EGFR signaling activates an mTORC2-NF-κB pathway that promotes
chemotherapy resistance. Cancer Discov. 1:524–538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee K, Gudapati P, Dragovic S, Spencer C,
Joyce S, Killeen N, Magnuson MA and Boothby M: Mammalian target of
rapamycin protein complex 2 regulates differentiation of Th1 and
Th2 cell subsets via distinct signaling pathways. Immunity.
32:743–753. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stewart ZA, Westfall MD and Pietenpol JA:
Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol
Sci. 24:139–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Halazonetis TD, Gorgoulis VG and Bartek J:
An oncogene-induced DNA damage model for cancer development.
Science. 319:1352–1355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Riedl SJ and Salvesen GS: The apoptosome:
Signalling platform of cell death. Nat Rev Mol Cell Biol.
8:405–413. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
LoPiccolo J, Blumenthal GM, Bernstein WB
and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: Effective
combinations and clinical considerations. Drug Resist Updat.
11:32–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sabatini DM: mTOR and cancer: Insights
into a complex relationship. Nat Rev Cancer. 6:729–734. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gingras AC, Gygi SP, Raught B, Polakiewicz
RD, Abraham RT, Hoekstra MF, Aebersold R and Sonenberg N:
Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism.
Genes Dev. 13:1422–1437. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hsu PP, Kang SA, Rameseder J, Zhang Y,
Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al:
The mTOR-regulated phosphoproteome reveals a mechanism of
mTORC1-mediated inhibition of growth factor signaling. Science.
332:1317–1322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang HT, Wang WW, Ren LH, Zhao XX, Wang
ZH, Zhuang DL and Bai YN: The mTORC2/Akt/NFκB pathway-mediated
activation of TRPC6 participates in adriamycin-induced podocyte
apoptosis. Cell Physiol Biochem. 40:1079–1093. 2016. View Article : Google Scholar
|
|
47
|
Lee K, Nam KT, Cho SH, Gudapati P, Hwang
Y, Park DS, Potter R, Chen J, Volanakis E and Boothby M: Vital
roles of mTOR complex 2 in Notch-driven thymocyte differentiation
and leukemia. J Exp Med. 209:713–728. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bassères DS and Baldwin AS: Nuclear
factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic
initiation and progression. Oncogene. 25:6817–6830. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yun S, Vincelette ND, Knorr KL, Almada LL,
Schneider PA, Peterson KL, Flatten KS, Dai H, Pratz KW, Hess AD, et
al: 4EBP1/c-MYC/PUMA and NF-κB/EGR1/BIM pathways underlie
cytotoxicity of mTOR dual inhibitors in malignant lymphoid cells.
Blood. 127:2711–2722. 2016. View Article : Google Scholar : PubMed/NCBI
|

















