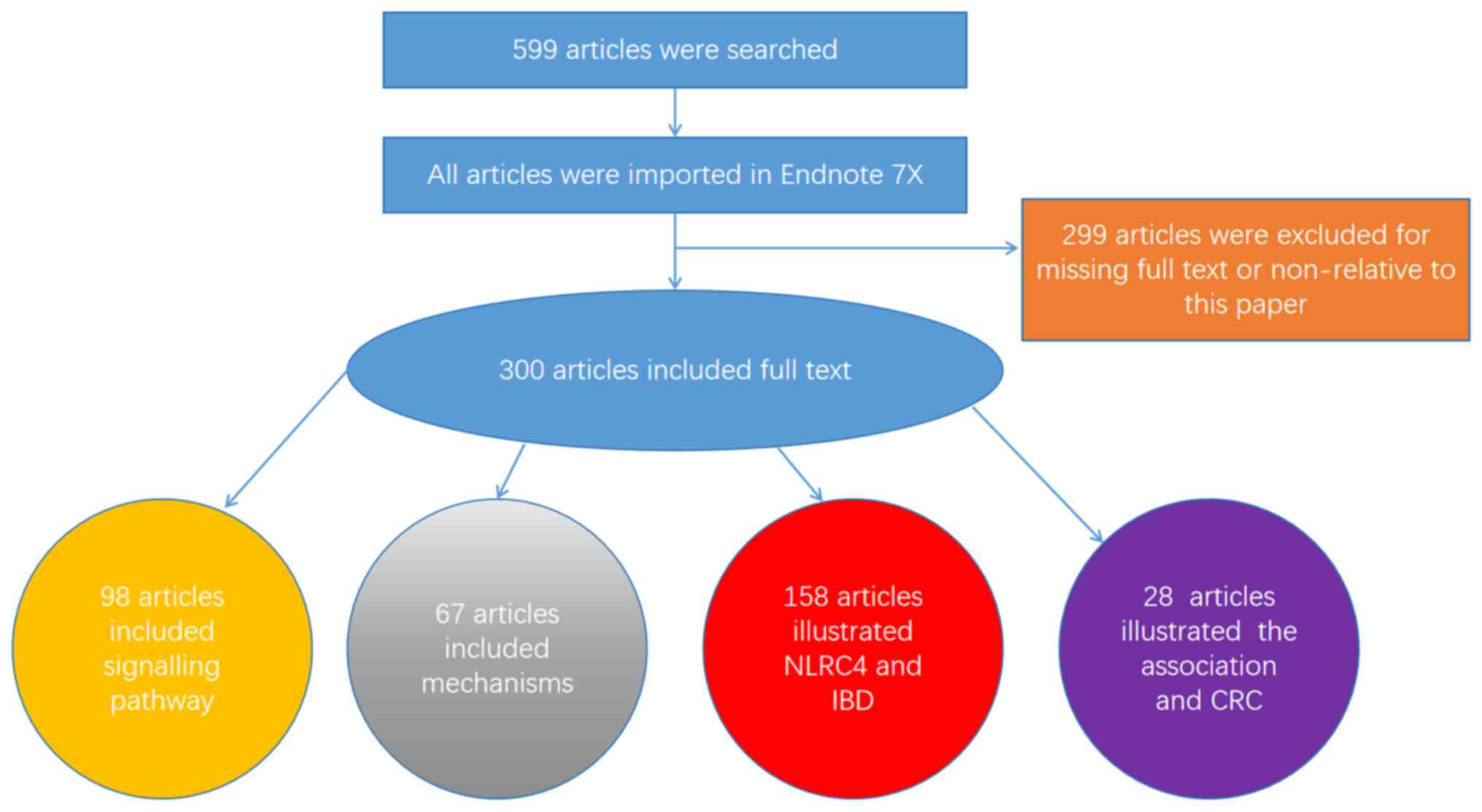
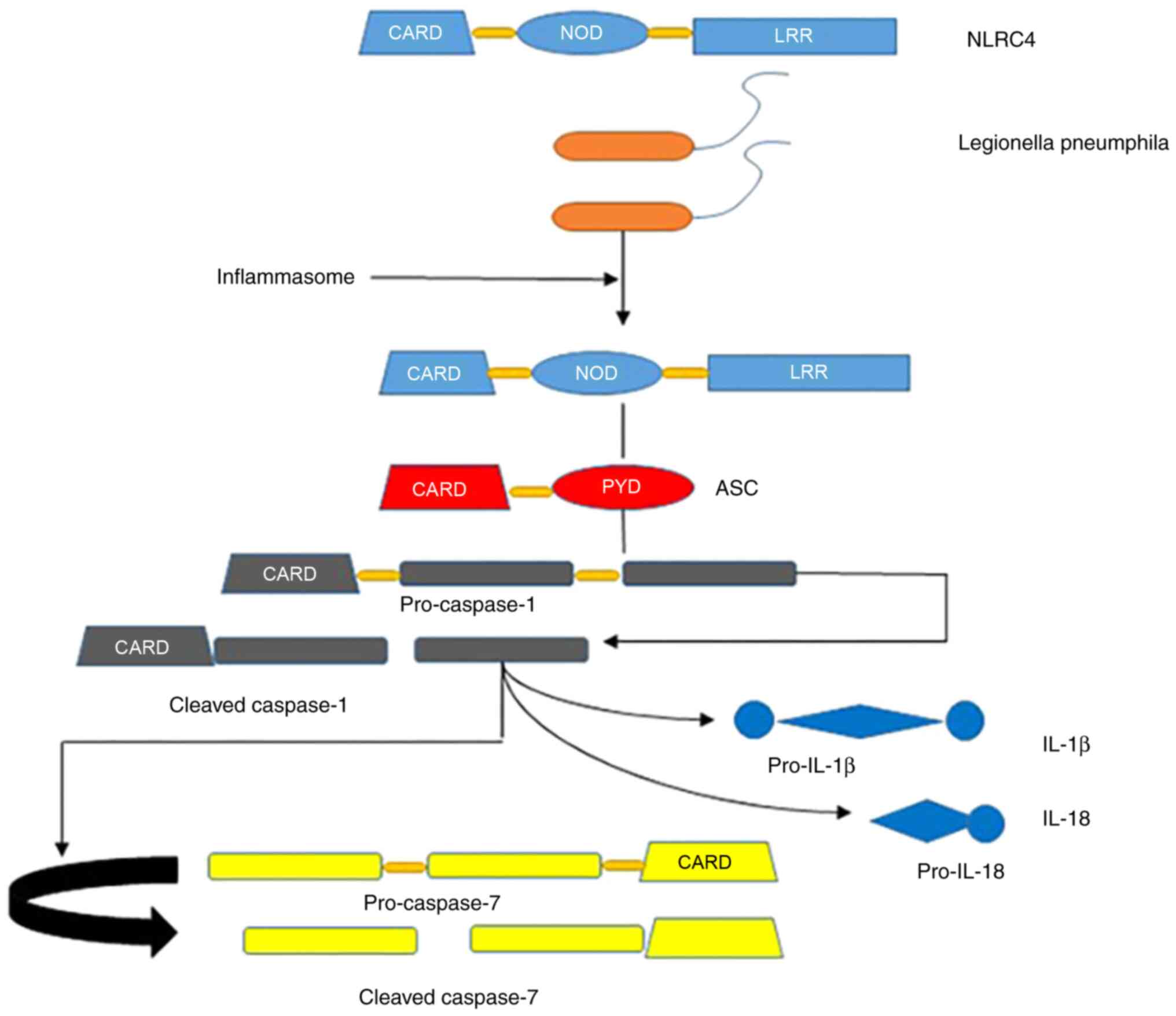
Unlike other NLR proteins, comprehensive structural
analyses of NLRC4 have significantly advanced the understanding of
its activation triggers (2).
NLRC4, as with other NLR family members, exhibits a tripartite
structural arrangement: An N-terminal domain for homotypic
interactions, a central domain for nucleotide binding and a series
of C-terminal leucine-rich repeats (LRRs). The stability of the
monomer is maintained through these interactions and the binding of
ADP to the nucleotide binding site (6). Two studies (7,8)
have shown that following co-expression of NLRC4 with NAIP2 and the
type III secretion system (T3SS) needle protein, PrgJ, NAIP
interacts with NLRC4 upon ligand binding, potentially forming
disc-like structures composed of unfolded NLRC4 monomers. These
monomers engage in extensive molecular interactions with adjacent
NLRC4 molecules on opposing faces (2). NLRC4 is identified as a crucial
component of the immune response against intestinal pathogens
(2,9-11).
A gene mutation in NLRC4 is suspected to be the first identified
recessive mutation leading to the monogenic disorder, inflammatory
bowel disease (IBD), characterized by autoinflammation and immune
system irregularities triggered by environmental stimuli (12). Inflammation plays a key role in
tumorigenesis leading to oncogenic mutations, tumor promotion and
angiogenesis. Tumor promoting inflammation is driven by numerous
factors, including the presence of the proinflammatory cytokines,
interleukin (IL)-1β and IL-18. An important source of IL-1β and
IL-18 secretion is via activation of the inflammasome. The
inflammasome is a multiprotein complex that, upon activation, leads
to the processing and secretion of IL-1β and IL-18, which is
mediated by the cysteine protease, CASP1. Several inflammasomes,
including NLR family pyrin domain containing 3 (NLRP3), NLRC4 and
NLRP6, have been elucidated in tumorigenesis (12). Notably, inflammasomes play
different roles in different types of cancer, showing the
complexity of inflammation during tumorigenesis. Understanding
these roles will help to identify new therapeutic targets and
improve the treatment strategies of patients with cancer (12).
Inflammation plays a role in all stages of
tumorigenesis. The key signaling pathway that leads to acute and
chronic inflammation is via activation of the CASP1 inflammasome.
Inflammasome complexes assemble upon activation of certain
nucleotide-binding domains (NBDs), LRR proteins (NLRs), absent in
melanoma 2 (AIM2)-like receptors or pyridines (13). The activation of inflammasome and
IL-18 signaling pathways has a great protective effect in
colitis-associated CRC, while excessive inflammation driven by
inflammasome or IL-1 signaling pathways promotes breast cancer,
fibrosarcoma, gastric cancer and lung metastasis (13). Inflammasomes are multimeric
complexes consisting of NLRs, which react to a diverse array of
endogenous (damage-associated molecular patterns) and exogenous
(pathogen-associated molecular patterns) stimuli. Multiple lines of
evidence suggest that in cancer, inflammasomes are positively
correlated with features such as elevated IL-1β and IL-18 levels,
activation of NF-κB signaling, increased mitochondrial oxidative
stress and activation of the autophagy process (10,11,13,14). A number of NLRs, such as NLRP3 and
NLRC4, have also been emphasized in carcinogenesis and are closely
associated with activating inflammatory caspases (14). A particular genetic variant,
Ala160Thr, exhibits pathogenic properties in vitro,
amplifies NLRC4 signaling in response to stimuli (though with less
potency than dominant mutations in NLRC4) and results in a slight
increase in IL-18 in vivo. The variant form of NLRC4
(Ala160Thr) implicated in the recessive immune dysregulation was
identified from intestinal epithelial cells (IECs) and colon
tissue. Consequently, individuals carrying the NLRC4 (Ala160Thr)
mutation are at a higher risk for developing ulcerative colitis
(15). NLRC4 mutations can also
cause autoinflammatory disease (16). Additionally, a previous study has
found that NLRC4 expression is upregulated in tumor tissues
compared with normal tissues and is associated with prognosis in
lung adenocarcinoma (17). NLRC4
mediates the maturation and release of CASP1, further promoting the
release of inflammatory factors, such as IL-1β and IL-18, and plays
a critical role in various tumors. Depending on the type of cancer,
NLRC4 may act as a tumor promoter or suppressor (18). For instance, NLRC4 promotes tumor
progression in breast cancer (19), glioma (20) and liver cancer (21), but functions as a tumor suppressor
in melanoma (22) and CRC
(23). Another study demonstrated
that high expression of NLRC4 was associated with a favorable
prognosis in CRC (24). The
present review examines the relationship between NLRC4 and
inflammation, its function, signaling pathways and the mechanisms
by which NLRC4 is involved in CRC.
The NLR family comprises intracellular receptors
that detect bacterial molecules, with NLRC4 as one of its members
(25). These NLRs are proteins
characterized by LRR motifs that enable them to recognize bacterial
elements within the cytoplasm of eukaryotic cells. Upon detecting
these bacterial elements, the inflammasome complex, which
integrates certain NLRs, is triggered. This complex is crucial for
activating CASP1, an enzyme that processes pro-inflammatory
cytokines, such as IL-1β and IL-18, into their active forms
(26,27). Initially, NLRC4 was primarily
recognized as a sensor for flagellin in eukaryotic cells. However,
research involving multiple pathogenic organisms, including
Salmonella (18,28-30), Legionella (31-34), Shigella (25,35-38) and Pseudomonas (39-42), particularly at elevated pathogen
concentrations, has shown that NLRC4 can initiate CASP1 activity
regardless of the presence of bacterial flagellin (18,30). Additionally, emerging evidence
suggests that NLRC4 can also limit bacterial infections in a
CASP1-independent manner (43,44). These findings imply that NLRC4 may
detect a broader spectrum of bacterial molecules and participate in
multiple immune response pathways.
NLRC4 functions as a sensor within the innate immune
system. A 2004 study demonstrated that bone marrow-derived
macrophages (BMDMs) from NLRC4-deficient mice were unable to
initiate CASP1 activation, resulting in a failure in pyroptosis
when challenged with S. Typhimurium (5). Various pathogens possess virulence
factors similar to flagellin, which are crucial for activating the
NLRC4 inflammasome. These include Pseudomonas aeruginosa
(Pscl), Shigella flexneri (Mxil), Escherichia coli
(EprJ and Escl), Burkholderia pseudomallei (BsaK) and S.
Typhimurium (PrgJ) (47).
Notably, the NLRC4 inflammasome can also be activated in the
absence of flagellin during certain bacterial infections (48). Thus, NLRC4 is a key modulator of
the innate immune response, capable of recognizing a range of
bacterial virulence factors (18). The apoptosis inhibitory proteins
of the NLR family, known as NAIPs, act as essential sensors for
NLRC4 inflammasome activation. The murine genome encodes seven
distinct NAIPs, while the human genome contains only one variant,
hNAIP (49). Specifically, NAIP5
and NAIP6 have been shown to activate NLRC4 in response to
bacterial flagellin (50). NAIP2,
similar to NAIP5, serves as a receptor for the inflammasome,
recognizing T3SS rod proteins such as BsaK from B.
pseudomallei and PrgJ from S. Typhimurium (33). Furthermore, hNAIP can induce NLRC4
inflammasome activation in response to both flagellin and T3SS
components (51-53). During bacterial invasion, the
transcriptional regulation of NLRC4 and NAIPs is mediated by
interferon regulatory factor 8 (IRF8) (42).
Functional redundancies between NLRP3 and NLRC4 can
enhance host defense. Research indicates that mice with a dual
deficiency in NLRC4 and NLRP3 due to knockout exhibit increased
susceptibility to S. Typhimurium, with significantly higher
bacterial loads in the mesenteric lymph nodes, liver and spleen
compared with the healthy controls (54). Similarly, Citrobacter
rodentium, another intestinal pathogen, can induce heightened
pathology and increased susceptibility in mice lacking CASP1, NLRC4
and NLRP3, suggesting that NLRC4 is crucial for the defense against
C. rodentium (55). In
addition to its role in hematopoietic immune cell lineages during
infection, NLRC4 is also activated in non-hematopoietic cell
populations, notably within IECs (2). NLRC4 provides protection against
certain pathogens, including Salmonella (28,30), Citrobacter (11,56) and Legionella (33,57). However, its activation can also be
detrimental, potentially causing an excessive inflammatory response
during certain infections, such as those by Helicobacter
(18).
NLRC4 is a key factor in the pathogenesis of
autoinflammatory diseases. While NLRC4 activation is crucial for
initiating immune reactions and promoting inflammation in response
to bacterial invasion, excessive activation can lead to unnecessary
cellular death and cytokine release. Therefore, mutations causing
NLRC4 overactivation are likely to be detrimental, potentially
resulting in autoinflammatory diseases. Such genetic mutations can
lead to continuous CASP1 activation and increased production of
IL-1β and IL-18 in macrophages derived from patients expressing
mutated NLRC4 (58). A distinct
de novo gain-of-function (GOF) mutation in NLRC4, resulting
in a p.Val341Ala amino acid substitution within the helical domain
1, leads to inflammasome activation and is associated with
conditions such as near-fatal or fatal episodes of
autoinflammation, periodic fever and neonatal-onset enterocolitis
(15,59). Mice expressing this NLRC4 mutation
exhibit severe cold-induced exanthema, splenomegaly, increased
neutrophil infiltration, arthritis and dermatitis. In patients,
this mutation typically results in inflammatory arthritis, skin
erythema and recurrent fever (60,61). Given the significant role of
NLRC4-associated cytokine signaling in disease development,
therapeutic interventions targeting these pathways have been
explored. Previous research has suggested that a combined
therapeutic approach using rapamycin and anakinra may benefit
patients with NLRC4 mutations (62).
The functions of NLRC4 in various forms of
programmed cell death, including PANoptosis, necroptosis, apoptosis
and pyroptosis, have been extensively studied. During infection,
activating specific programmed cell death pathways is essential for
eliminating invading pathogens from the host. Necroptosis and
pyroptosis are typically characterized by their lytic nature and
ability to elicit an immune response, whereas apoptosis was
traditionally considered immunologically silent (2). However, one previous study has
suggested that apoptosis may not always be silent (63). Emerging research has demonstrated
that some infectious pathogens and non-microbial stressors can
induce an inflammatory form of cell death known as PANoptosis
(64). PANoptosis is a distinct
and physiologically relevant pathway triggered by specific stimuli
and regulated by the PANoptosome, a coordinating structure that
facilitates the simultaneous activation of key components
associated with necroptosis, apoptosis and pyroptosis (65-74). The involvement of NLRC4 in
pyroptosis has been linked to retinal ganglion cell death and has
provided new insights into the pyroptosis of microglia. This
finding highlights potential therapeutic approaches for mitigating
irreversible vision loss caused by glaucoma by targeting pyroptosis
(75). Apoptosis is a programmed
cell death process that enables the systematic and effective
removal of damaged cells resulting from development or DNA damage.
Apoptosis can be initiated by external signals, such as ligands
binding to death receptors on the cell surface, or by internal
factors such as genotoxic stress (76). The relationship between apoptosis
and NLRC4 has been well established, with poly (ADP-ribose)
polymerase 1 cleavage by CASPs serving as a definitive indicator of
apoptosis (76). Necroptosis, a
regulated form of necrotic cell death, is also crucial for the
organism's defense against certain pathogens (77). During Salmonella infection,
a recent study observed activation of mixed lineage kinase
domain-like pseudokinase (necroptosis), CASP8, CASP7 and CASP3
(apoptosis), as well as CASP1 and GSDMD (pyroptosis) (62).
During various phases of cancer progression,
including metastasis, angiogenesis, proliferation and
immunosuppression, aberrant activation of the inflammasome plays a
crucial role. Conversely, inflammasome activation can maintain the
intestinal barrier and initiate tumor suppression, highlighting its
complex role in tumorigenesis (78). Animal experiments involving
deficiencies in key inflammasome factors such as NLRC4, Nlrp3,
CASP1 and PYCARD have identified the pivotal role of NLR
inflammasomes in the pathogenesis of colitis-associated cancer
(CAC) (79-81). No significant differences in CAC
disease outcomes or pathology were observed when comparing
NLRC4−/− animals with controls (wild-type animals)
(82). Additionally, another
study found that treating NLRC4−/− mice with dextran
sulfate sodium (DSS) and azoxymethane (AOM), which induce DNA
damage, resulted in increased colonic epithelial cell
proliferation, reduced apoptosis and larger tumor volumes (23). Additionally, NLRC4-deficient mice
exhibited increased sensitivity to colitis induced by DSS compared
with wild-type mice (82).
Chemokines and cytokines play a crucial role in eliminating cancer
cells, and NLRC4 activation is essential for the synthesis of these
molecules in tumor-associated macrophages. In the B16F10 melanoma
mouse model, NLRC4 is indispensable for the generation of IFN-γ by
CD8+ and CD4+ T cells (83). However, a previous investigation
indicated that NLRC4 does not contribute to melanoma progression,
as no difference in tumor incidence was observed between
NLRC4-deficient mice and wild-type littermates (84,85). Inflammation is believed to
influence several phases of tumorigenesis, contributing to the
host's resistance to harmful microbial infections and maintaining
tissue balance. Disruptions in this process could potentially
trigger inflammatory disorders and malignancies (86). NLRC4 is a pivotal component of the
inflammasome complex, and its dysregulation is closely associated
with the development of CRC associated with colitis (82). The role of NLRC4 in carcinogenesis
varies by malignancy; it can act as either a suppressor or promoter
(24). NLRC4 acts as a suppressor
in CRC (23). Peng et al
(24) found that high NLRC4
expression was associated with a favorable prognosis in CRC.
However, it was not determined whether NLRC4 was an independent
prognostic factor for CRC. Therefore, further investigation is
needed to clarify this. The function of NLRC4 is summarized in
Table I.
Mutations in genes encoding inflammasome components
often result in increased susceptibility to autoinflammatory
diseases, infections or cancer in humans (83). NLRC4 may protect against CRC
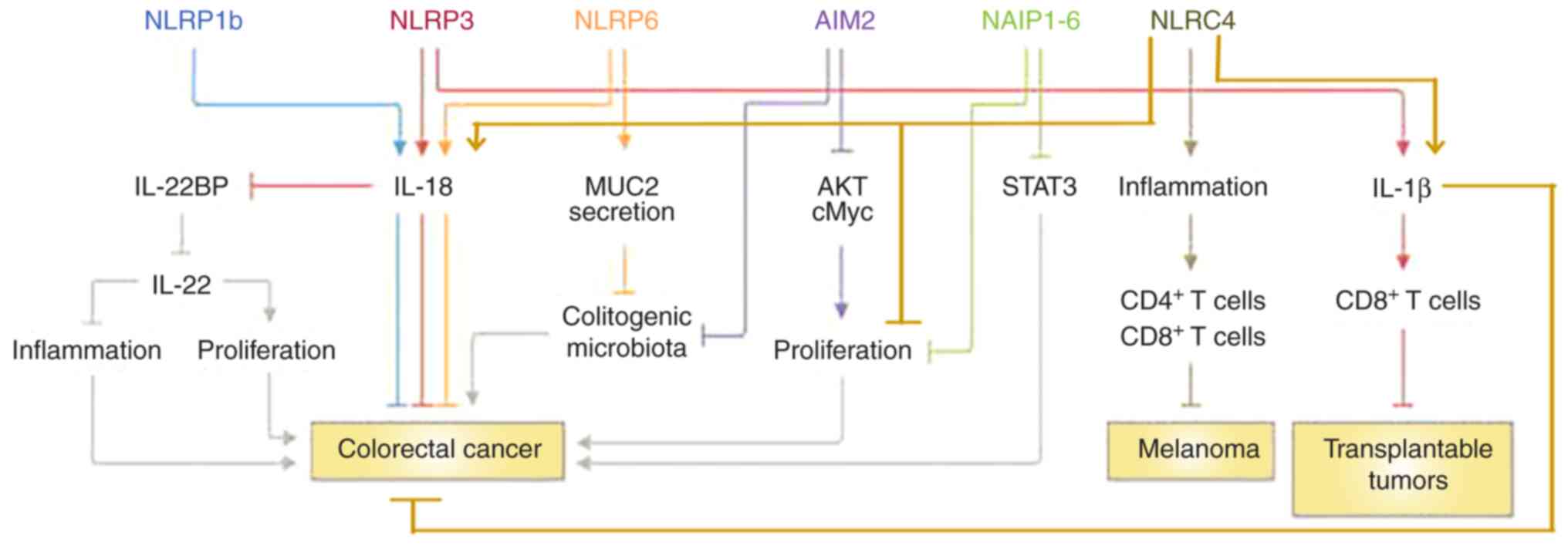
through the CASP1 signaling pathway (87). It is noteworthy that deficiencies
in CASP1 and ASC render mice vulnerable to DSS-induced colitis and
associated CRC (13,79-81,83,88), demonstrating that inflammasomes
have a protective function in a CRC inflammatory model. NLRC4 may
also protect against CRC through the NLRP3/IL-8 signaling pathway
(83) (Fig. 3). NLRC4 and NLRP3 are two
different inflammasomes with different expression levels that
typically change consistently (75,89-91), although the precise mechanism
remains unclear. One possibility is that inflammasome sensors such
as NLRP3 facilitate the release of IL-18, an immune signaling
protein that aids in restoring the epithelial barrier against
injury. This function could explain how IL-18 exerts a protective
effect against CRC associated with colitis (13,92-99).
Additionally, NLRC4 may play an anticancer role
(suppressing transplantable tumors) through the IL-1β/IL-1 receptor
signaling axis in dendritic cells (100). This pathway may stimulate an
effective CD8+ T-cell response toward transplantable
tumor cells (83) (Fig. 3). NLRC4 could potentially inhibit
CRC implantation through this signaling pathway, although no
literature currently shows that NLRC4 directly kills CRC cells via
this axis.
Bacterial flagellin, a classic pathogen-associated
molecular pattern, interacts with Toll-like receptor 5 and the
NAIP5 receptor (integral to the NLRC4 inflammasome), stimulating
immune responses in mammals. However, the role of flagellin
receptors in lower animal species is less understood (107). NLRC4 inflammasome may induce
colitis inflammation and CRC via p53 signaling pathway. p53
activation is instrumental in promoting apoptosis in coelomocytes
(107). Numerous studies have
shown that the p53 signaling pathway is closely related to tumor
cell apoptosis, thereby inhibiting tumor development (108-115). However, one study indicated that
NLRC4 is not associated with the p53 signaling pathway in
protecting against colonic tumorigenesis (101). Elevated pyroptosis, an indicator
of a 'hot' tumor environment characterized by CD8+ T
cells and various T cell subtypes, is influenced by oncogenic
pathways including PI3K/AKT/mTOR signaling, angiogenesis,
IL-2/STAT5 signaling, IL-6/Janus kinase/STAT3 signaling,
epithelial-mesenchymal transition, KRAS signaling, DNA repair and
the p53 pathway (116).
Therefore, we suggest that NLRC4 is involved in the p53 signaling
pathway to inhibit tumor development. Mechanistically, NLRC4 is
considered to play a significant role in this pathway in CRC. These
potential signaling pathways are summarized in Table II.
From the above, it can be inferred that NLRC4
suppresses and eliminates CRC cells through pyroptosis, apoptosis,
necroptosis and PANoptosis. The assembly of the inflammasome
complex triggers the activation of CASP1, which is responsible for
the maturation of IL-1β and IL-18 into their active forms and the
cleavage of GSDMD, thereby inducing pyroptosis, a type of
inflammatory cell death (117,118). Components such as NAIP-NLRC4,
NLRP6, NLRP9, AIM2 and Pyrin can assemble into inflammasomes,
playing a role in modulating the host's immune and inflammatory
responses (119,120). NLRC4 protects against CRC as a
cytosolic sensor by regulating the immune response. NLRP6, NLRP7,
NLRP9, NLRP12, the DNA sensor IFNγ-inducible protein 16 and the RNA
sensor RIG-I have been associated with promoting CASP1 activation,
though confirmation of their capacity to assemble into an
inflammasome complex is still needed (121).
NLRC4 protects IECs to prevent CRC. IECs form a
crucial barrier against pathogen invasion. The intestinal immune
system's defense and its disease potential are significantly
influenced by a coordinated IEC-specific response involving the
CASP1 and CASP8 inflammasomes (10). Irak et al (122) suggested that serum levels of
monocyte chemoattractant protein 2/chemokine (C-C motif) ligand 8
and NLRC4 could contribute to the development of Crohn's disease
and play a protective role in maintaining intestinal homeostasis
and mitigating inflammation. Another study indicated that the
prompt and targeted elimination of infected enterocytes by the
epithelium-autonomous NAIP/NLRC4 system is crucial to prevent an
excessive TNF-induced inflammatory response that could otherwise
damage the epithelial barrier (123). NAIPs protect against colonic
tumors by facilitating the clearance of epithelial cells stimulated
by carcinogens, likely independent of the NLRC4 inflammasome
(101). The administration of
AOM, a DNA-damaging substance, along with repeated DSS injections,
induces CRC progression associated with colitis (124,125). In mice, oxazolone, a haptenating
agent, can also trigger hemorrhagic colonic inflammation and severe
submucosal edema, in addition to DSS (126). Although NLRC4 does not
contribute to the rapid genetic reconfiguration of the intestine in
response to flagellin, its inflammasome activation generates IL-1β
and IL-18, which protect mice from both mucosal and systemic
inflammation (82). In summary,
NLRC4 protects the intestinal mucosa from pathogen attack through
various pathways and inflammatory protective factors, thereby
preventing the occurrence of CRC.
Mutations in NLRC4 resulting in GOF have been
associated with several conditions, including early-onset recurrent
fever, recurrent macrophage activation syndrome, enterocolitis and
even cancer (15,16,18,60,120,127). The NLRC4 (Ala160Thr) variant can
cause recessive immune dysregulation and autoinflammation, or act
as a heterozygous risk factor for the development of ulcerative
colitis. This variant often affects epithelial cells and colon
tissue (15).
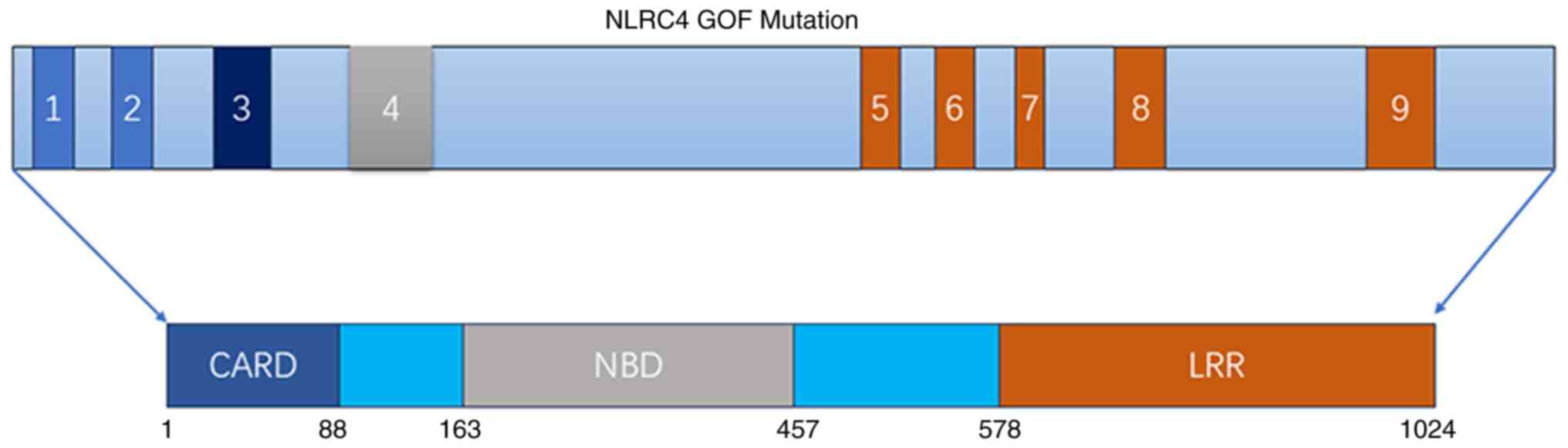
The NLRC4 protein features a CARD at its N-terminus,
a central NBD and a LRR domain (128). Mutations frequently occur in the
NBD and LRR domains. Bardet et al (128) identified two mutations in the
NBD: p.Arg207Lys and p.Thr337Asn (Fig. 4). Romberg et al (59) reported a p.Val341Ala mutation in
the NBD, while Barsalou et al (62) identified a p.Val341Leu mutation.
Additionally, the p.Ser445Pro mutation at the NBD was described by
Volker-Touw et al (129).
Other mutations in the NBD include p.Val341Ala, p.Thr337Ser,
p.His443Pro and p.Ser445Pro, as noted in the literature (1,2,58,60,129). In the LRR domain, mutations such
as p.Gln657Leu (61) and
p.Trp655Cys (130) have been
observed. Some mutations induce clinical symptoms, while others do
not (16,128,131). Mice expressing a murine NLRC4
(Val341Ala) mutant showed elevated systemic IL-18 levels,
indicating that the mechanisms by which this mutant induces
elevated IL-18 production are conserved between humans and mice.
However, while experiments that are germfree or with infections
argue against a role for commensal or pathogenic bacteria,
identifying the triggers and mechanisms that synergize with IL-18
to drive NLRC4 (Val341Ala)-associated pathologies requires further
research using this NLRC4 (Val341Ala) mouse model (132). The NLRC4 Val341Ala mutation is
closely related to colitis, increasing IL-18 levels and potentially
raising the risk of CRC from a mechanistic perspective (132). Research on the elevated
expression level of Val431Ala in the tissues of patients with CRC
is lacking, as is a direct link between this factor and CRC. To
date, few studies have reported an association between NLRC4 GOF
mutations and CRC, with only 1 study indicating that NLRC4
mutations were present in 4% of CRC cases (24). Therefore, the strategies for
treating CRC that rely on NLRC4 are currently in the basic research
stage, and there are not yet many clinically relevant studies. This
is also one of the purposes of writing the present review.
NLRC4 is a significant inflammasome that protects
against CRC through various signaling pathways and mechanisms.
Mutations in NLRC4 may contribute to CRC development and could be
associated with a poor prognosis. There is a lack of original
studies on NLRC4 mutations and their prognosis in CRC due to some
limited conditions in our institution (such as no funding support
and lack of suitable patients and samples). However, the present
review highlights the need to explore the relationship between
NLRC4 mutations and CRC further. The clinical utility of detecting
NLRC4 in the serum and tissue of patients with CRC requires
additional investigation by researchers and clinicians. NLRC4
contributes to the suppression of CRC, which is the conclusion from
numerous experiments and partial clinical study. However, the
development of a NLRC4-dependent novel strategy to treat patients
with CRC also requires further study. For instance, investigations
into how to prevent NLRC4 mutations, how to block their induction
of CRC and how to improve the treatment of CRC with serum or tissue
NLRC4 mutations are needed.
Not applicable.
GT, YS, HL, HQ and ZT participated in collecting the
literature; GT wrote the paper and drew the figures; YS summarized
the tables. HL, HQ and ZT participated in revising the review. All
authors read and approved the final version of the manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
The authors thank Mrs. Liqing Li (Central
Laboratory, Huzhou Central Hospital, Huzhou, Zhejiang 313003, P.R.
China) for her valuable suggestions on the manuscript.
This study was supported by the Science and Technology Project
of Zhejiang Province (grant no. 2018C37090).
|
1
|
Poyet JL, Srinivasula SM, Tnani M, Razmara
M, Fernandes-Alnemri T and Alnemri ES: Identification of Ipaf, a
human caspase-1-activating protein related to Apaf-1. J Biol Chem.
276:28309–28313. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Duncan JA and Canna SW: The NLRC4
inflammasome. Immunol Rev. 281:115–123. 2018. View Article : Google Scholar :
|
|
3
|
Gutierrez O, Pipaon C and Fernandez-Luna
JL: Ipaf is upregulated by tumor necrosis factor-alpha in human
leukemia cells. FEBS Lett. 568:79–82. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sadasivam S, Gupta S, Radha V, Batta K,
Kundu TK and Swarup G: Caspase-1 activator Ipaf is a p53-inducible
gene involved in apoptosis. Oncogene. 24:627–636. 2005. View Article : Google Scholar
|
|
5
|
Mariathasan S, Newton K, Monack DM, Vucic
D, French DM, Lee WP, Roose-Girma M, Erickson S and Dixit VM:
Differential activation of the inflammasome by caspase-1 adaptors
ASC and Ipaf. Nature. 430:213–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang
C, Wang R, Zhang Y, Martinon F, Miao D, et al: Crystal structure of
NLRC4 reveals its autoinhibition mechanism. Science. 341:172–175.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang X, Shaw DK, Hammond HL, Sutterwala
FS, Rayamajhi M, Shirey KA, Perkins DJ, Bonventre JV, Velayutham
TS, Evans SM, et al: The prostaglandin E2-EP3 receptor axis
regulates anaplasma phagocytophilum-mediated NLRC4 inflammasome
activation. PLoS Pathog. 12:e10058032016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang L, Chen S, Ruan J, Wu J, Tong AB,
Yin Q, Li Y, David L, Lu A, Wang WL, et al: Cryo-EM structure of
the activated NAIP2-NLRC4 inflammasome reveals nucleated
polymerization. Science. 350:404–409. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sellin ME, Müller AA, Felmy B, Dolowschiak
T, Diard M, Tardivel A, Maslowski KM and Hardt WD:
Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected
enterocyte expulsion to restrict Salmonella replication in the
intestinal mucosa. Cell Host Microbe. 16:237–248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rauch I, Deets KA, Ji DX, von Moltke J,
Tenthorey JL, Lee AY, Philip NH, Ayres JS, Brodsky IE, Gronert K
and Vance RE: NAIP-NLRC4 inflammasomes coordinate intestinal
epithelial cell expulsion with eicosanoid and IL-18 release via
activation of caspase-1 and -8. Immunity. 46:649–659. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nordlander S, Pott J and Maloy KJ: NLRC4
expression in intestinal epithelial cells mediates protection
against an enteric pathogen. Mucosal Immunol. 7:775–785. 2014.
View Article : Google Scholar :
|
|
12
|
Janowski AM, Kolb R, Zhang W and
Sutterwala FS: Beneficial and detrimental roles of NLRs in
carcinogenesis. Front Immunol. 4:3702013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dupaul-Chicoine J, Yeretssian G, Doiron K,
Bergstrom KSB, McIntire CR, LeBlanc PM, Meunier C, Turbide C, Gros
P, Beauchemin N, et al: Control of intestinal homeostasis, colitis,
and colitis-associated colorectal cancer by the inflammatory
caspases. Immunity. 32:367–378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhiyu W, Wang N, Wang Q, Peng C, Zhang J,
Liu P, Ou A, Zhong S, Cordero MD and Lin Y: The inflammasome: An
emerging therapeutic oncotarget for cancer prevention. Oncotarget.
7:50766–50780. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Steiner A, Reygaerts T, Pontillo A,
Ceccherini I, Moecking J, Moghaddas F, Davidson S, Caroli F, Grossi
A, Castro FFM, et al: Recessive NLRC4-autoinflammatory disease
reveals an ulcerative colitis locus. J Clin Immunol. 42:325–335.
2022. View Article : Google Scholar :
|
|
16
|
Wang J, Ye Q, Zheng W, Yu X, Luo F, Fang
R, Shangguan Y, Du Z, Lee PY, Jin T and Zhou Q: Low-ratio somatic
NLRC4 mutation causes late-onset autoinflammatory disease. Ann
Rheum Dis. 81:1173–1178. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu C, Zhao J, Wang X, Wang Y, Zhang W and
Zhu G: A novel pyroptosis related genes signature for predicting
prognosis and estimating tumor immune microenvironment in lung
adenocarcinoma. Transl Cancer Res. 11:2647–2659. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sundaram B and Kanneganti TD: Advances in
understanding activation and function of the NLRC4 inflammasome.
Int J Mol Sci. 22:10482021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin H and Kim HJ: NLRC4, ASC and caspase-1
are inflammasome components that are mediated by P2Y2R
activation in breast cancer cells. Int J Mol Sci. 21:33372020.
View Article : Google Scholar
|
|
20
|
Lim J, Kim MJ, Park Y, Ahn JW, Hwang SJ,
Moon JS, Cho KG and Kwack K: Upregulation of the NLRC4 inflammasome
contributes to poor prognosis in glioma patients. Sci Rep.
9:78952019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sonohara F, Inokawa Y, Kanda M, Nishikawa
Y, Yamada S, Fujii T, Sugimoto H, Kodera Y and Nomoto S:
Association of inflammasome components in background liver with
poor prognosis after curatively-resected hepatocellular carcinoma.
Anticancer Res. 37:293–300. 2017. View Article : Google Scholar
|
|
22
|
Janowski AM, Colegio OR, Hornick EE,
McNiff JM, Martin MD, Badovinac VP, Norian LA, Zhang W, Cassel SL
and Sutterwala FS: NLRC4 suppresses melanoma tumor progression
independently of inflammasome activation. J Clin Invest.
126:3917–3928. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu B, Elinav E, Huber S, Booth CJ, Strowig
T, Jin C, Eisenbarth SC and Flavell RA: Inflammation-induced
tumorigenesis in the colon is regulated by caspase-1 and NLRC4.
Proc Natl Acad Sci USA. 107:21635–21640. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peng L, Zhu N, Wang D, Zhou Y and Liu Y:
Comprehensive analysis of prognostic value and immune infiltration
of NLRC4 and CASP1 in colorectal cancer. Int J Gen Med.
15:5425–5440. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abdelaziz DH, Amr K and Amer AO:
Nlrc4/Ipaf/CLAN/CARD12: More than a flagellin sensor. Int J Biochem
Cell Biol. 42:789–791. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun Q and Scott MJ: Caspase-1 as a
multifunctional inflammatory mediator: Noncytokine maturation
roles. J Leukoc Biol. 100:961–967. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lamkanfi M, Kanneganti TD, Franchi L and
Núñez G: Caspase-1 inflammasomes in infection and inflammation. J
Leukoc Biol. 82:220–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Naseer N, Zhang J, Bauer R, Constant DA,
Nice TJ, Brodsky IE, Rauch I and Shin S: Salmonella enterica
Serovar typhimurium induces NAIP/NLRC4- and NLRP3/ASC-independent,
caspase-4-dependent inflammasome activation in human intestinal
epithelial cells. Infect Immun. 90:e00663212022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Naseer N, Egan MS, Reyes Ruiz VM, Scott
WP, Hunter EN, Demissie T, Rauch I, Brodsky IE and Shin S: Human
NAIP/NLRC4 and NLRP3 inflammasomes detect Salmonella type III
secretion system activities to restrict intracellular bacterial
replication. PLoS Pathog. 18:e10097182022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gram AM, Wright JA, Pickering RJ, Lam NL,
Booty LM, Webster SJ and Bryant CE: Salmonella flagellin activates
NAIP/NLRC4 and canonical NLRP3 inflammasomes in human macrophages.
J Immunol. 206:631–640. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schell U, Simon S and Hilbi H:
Inflammasome recognition and regulation of the Legionella
flagellum. Curr Top Microbiol Immunol. 397:161–181. 2016.PubMed/NCBI
|
|
32
|
Cerqueira DM, Pereira MS, Silva AL, Cunha
LD and Zamboni DS: Caspase-1 but not caspase-11 is required for
NLRC4-mediated pyroptosis and restriction of infection by
flagellated Legionella species in mouse macrophages and in vivo. J
Immunol. 195:2303–2311. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu
H, Liu L and Shao F: The NLRC4 inflammasome receptors for bacterial
flagellin and type III secretion apparatus. Nature. 477:596–600.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pereira MSF, Morgantetti GF, Massis LM,
Horta CV, Hori JI and Zamboni DS: Activation of NLRC4 by
flagellated bacteria triggers caspase-1-dependent and -independent
responses to restrict Legionella pneumophila replication in
macrophages and in vivo. J Immunol. 187:6447–6455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luchetti G, Roncaioli JL, Chavez RA,
Schubert AF, Kofoed EM, Reja R, Cheung TK, Liang Y, Webster JD,
Lehoux I, et al: Shigella ubiquitin ligase IpaH7.8 targets
gasdermin D for degradation to prevent pyroptosis and enable
infection. Cell Host Microbe. 29:1521–1530.e10. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mitchell PS, Roncaioli JL, Turcotte EA,
Goers L, Chavez RA, Lee AY, Lesser CF, Rauch I and Vance RE:
NAIP-NLRC4-deficient mice are susceptible to shigellosis. Elife.
9:e590222020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hermansson AK, Paciello I and Bernardini
ML: The orchestra and its maestro: Shigella's fine-tuning of the
inflammasome platforms. Curr Top Microbiol Immunol. 397:91–115.
2016.PubMed/NCBI
|
|
38
|
Suzuki S, Mimuro H, Kim M, Ogawa M, Ashida
H, Toyotome T, Franchi L, Suzuki M, Sanada T, Suzuki T, et al:
Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates
inflammasomes to demolish macrophages. Proc Natl Acad Sci USA.
111:E4254–E4263. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Santoni K, Pericat D, Gorse L, Buyck J,
Pinilla M, Prouvensier L, Bagayoko S, Hessel A, Leon-Icaza SA,
Bellard E, et al: Caspase-1-driven neutrophil pyroptosis and its
role in host susceptibility to Pseudomonas aeruginosa. PLoS Pathog.
18:e10103052022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mohamed MF, Gupta K, Goldufsky JW, Roy R,
Callaghan LT, Wetzel DM, Kuzel TM, Reiser J and Shafikhani SH:
CrkII/Abl phosphorylation cascade is critical for NLRC4
inflammasome activity and is blocked by Pseudomonas aeruginosa
ExoT. Nat Commun. 13:12952022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Graustein AD, Berrington WR, Buckingham
KJ, Nguyen FK, Joudeh LL, Rosenfeld M, Bamshad MJ, Gibson RL, Hawn
TR and Emond MJ: Inflammasome genetic variants, macrophage
function, and clinical outcomes in cystic fibrosis. Am J Respir
Cell Mol Biol. 65:157–166. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Karki R, Lee E, Place D, Samir P, Mavuluri
J, Sharma BR, Balakrishnan A, Malireddi RKS, Geiger R, Zhu Q, et
al: IRF8 regulates transcription of Naips for NLRC4 inflammasome
activation. Cell. 173:920–933.e13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mascarenhas DPA, Cerqueira DM, Pereira
MSF, Castanheira FVS, Fernandes TD, Manin GZ, Cunha LD and Zamboni
DS: Inhibition of caspase-1 or gasdermin-D enable caspase-8
activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog.
13:e10065022017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Furuoka M, Ozaki K, Sadatomi D, Mamiya S,
Yonezawa T, Tanimura S and Takeda K: TNF-α induces caspase-1
activation independently of simultaneously induced NLRP3 in 3T3-L1
cells. J Cell Physiol. 231:2761–2767. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hua L, Liang S, Zhou Y, Wu X, Cai H, Liu
Z, Ou Y, Chen Y, Chen X, Yan Y, et al: Artemisinin-derived
artemisitene blocks ROS-mediated NLRP3 inflammasome and alleviates
ulcerative colitis. Int Immunopharmacol. 113:1094312022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Taman H, Fenton CG, Anderssen E,
Florholmen J and Paulssen RH: DNA hypo-methylation facilitates
anti-inflammatory responses in severe ulcerative colitis. PLoS One.
16:e02489052021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Miao EA, Mao DP, Yudkovsky N, Bonneau R,
Lorang CG, Warren SE, Leaf IA and Aderem A: Innate immune detection
of the type III secretion apparatus through the NLRC4 inflammasome.
Proc Natl Acad Sci USA. 107:3076–3080. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Miao EA, Alpuche-Aranda CM, Dors M, Clark
AE, Bader MW, Miller SI and Aderem A: Cytoplasmic flagellin
activates caspase-1 and secretion of interleukin 1beta via Ipaf.
Nat Immunol. 7:569–575. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
49
|
Endrizzi MG, Hadinoto V, Growney JD,
Miller W and Dietrich WF: Genomic sequence analysis of the mouse
Naip gene array. Genome Res. 10:1095–1102. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kofoed EM and Vance RE: Innate immune
recognition of bacterial ligands by NAIPs determines inflammasome
specificity. Nature. 477:592–595. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rayamajhi M, Zak DE, Chavarria-Smith J,
Vance RE and Miao EA: Cutting edge: Mouse NAIP1 detects the type
III secretion system needle protein. J Immunol. 191:3986–3989.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yang J, Zhao Y, Shi J and Shao F: Human
NAIP and mouse NAIP1 recognize bacterial type III secretion needle
protein for inflammasome activation. Proc Natl Acad Sci USA.
110:14408–14413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kortmann J, Brubaker SW and Monack DM:
Cutting edge: Inflammasome activation in primary human macrophages
is dependent on flagellin. J Immunol. 195:815–819. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Broz P, Newton K, Lamkanfi M, Mariathasan
S, Dixit VM and Monack DM: Redundant roles for inflammasome
receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp
Med. 207:1745–1755. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu Z, Zaki MH, Vogel P, Gurung P, Finlay
BB, Deng W, Lamkanfi M and Kanneganti TD: Role of inflammasomes in
host defense against Citrobacter rodentium infection. J Biol Chem.
287:16955–16964. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Man SM, Karki R, Briard B, Burton A,
Gingras S, Pelletier S and Kanneganti TD: Differential roles of
caspase-1 and caspase-11 in infection and inflammation. Sci Rep.
7:451262017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gonçalves AV, Margolis SR, Quirino GFS,
Mascarenhas DPA, Rauch I, Nichols RD, Ansaldo E, Fontana MF, Vance
RE and Zamboni DS: Gasdermin-D and caspase-7 are the key
caspase-1/8 substrates downstream of the NAIP5/NLRC4 inflammasome
required for restriction of Legionella pneumophila. PLoS Pathog.
15:e10078862019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Canna SW, de Jesus AA, Gouni S, Brooks SR,
Marrero B, Liu Y, DiMattia MA, Zaal KJ, Sanchez GA, Kim H, et al:
An activating NLRC4 inflammasome mutation causes autoinflammation
with recurrent macrophage activation syndrome. Nat Genet.
46:1140–1146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Romberg N, Al Moussawi K, Nelson-Williams
C, Stiegler AL, Loring E, Choi M, Overton J, Meffre E, Khokha MK,
Huttner AJ, et al: Mutation of NLRC4 causes a syndrome of
enterocolitis and autoinflammation. Nat Genet. 46:1135–1139. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kitamura A, Sasaki Y, Abe T, Kano H and
Yasutomo K: An inherited mutation in NLRC4 causes autoinflammation
in human and mice. J Exp Med. 211:2385–2396. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chear CT, Nallusamy R, Canna SW, Chan KC,
Baharin MF, Hishamshah M, Ghani H, Ripen AM and Mohamad SB: A novel
de novo NLRC4 mutation reinforces the likely pathogenicity of
specific LRR domain mutation. Clin Immunol. 211:1083282020.
View Article : Google Scholar
|
|
62
|
Barsalou J, Blincoe A, Fernandez I,
Dal-Soglio D, Marchitto L, Selleri S, Haddad E, Benyoucef A and
Touzot F: Rapamycin as an adjunctive therapy for NLRC4 associated
macrophage activation syndrome. Front Immunol. 9:21622018.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang Y, Gao W, Shi X, Ding J, Liu W, He H,
Wang K and Shao F: Chemotherapy drugs induce pyroptosis through
caspase-3 cleavage of a gasdermin. Nature. 547:99–103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Christgen S, Zheng M, Kesavardhana S,
Karki R, Malireddi RKS, Banoth B, Place DE, Briard B, Sharma BR,
Tuladhar S, et al: Identification of the PANoptosome: A molecular
platform triggering pyroptosis, apoptosis, and necroptosis
(PANoptosis). Front Cell Infect Microbiol. 10:2372020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pandian N and Kanneganti TD: PANoptosis: A
unique innate immune inflammatory cell death modality. J Immunol.
209:1625–1633. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Pan H, Pan J, Li P and Gao J:
Characterization of PANoptosis patterns predicts survival and
immunotherapy response in gastric cancer. Clin Immunol.
238:1090192022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lin JF, Hu PS, Wang YY, Tan YT, Yu K, Liao
K, Wu QN, Li T, Meng Q, Lin JZ, et al: Phosphorylated NFS1 weakens
oxaliplatin-based chemosensitivity of colorectal cancer by
preventing PANoptosis. Signal Transduct Target Ther. 7:542022.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wang Y and Kanneganti TD: From pyroptosis,
apoptosis and necroptosis to PANoptosis: A mechanistic compendium
of programmed cell death pathways. Comput Struct Biotechnol J.
19:4641–4657. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Place DE, Lee S and Kanneganti TD:
PANoptosis in microbial infection. Curr Opin Microbiol. 59:42–49.
2021. View Article : Google Scholar
|
|
70
|
Lee S, Karki R, Wang Y, Nguyen LN,
Kalathur RC and Kanneganti TD: AIM2 forms a complex with pyrin and
ZBP1 to drive PANoptosis and host defence. Nature. 597:415–419.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Karki R, Sundaram B, Sharma BR, Lee S,
Malireddi RKS, Nguyen LN, Christgen S, Zheng M, Wang Y, Samir P, et
al: ADAR1 restricts ZBP1-mediated immune response and PANoptosis to
promote tumorigenesis. Cell Rep. 37:1098582021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jiang W, Deng Z, Dai X and Zhao W:
PANoptosis: A new insight into oral infectious diseases. Front
Immunol. 12:7896102021. View Article : Google Scholar :
|
|
73
|
Zheng M and Kanneganti TD: The regulation
of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis,
apoptosis, and necroptosis (PANoptosis). Immunol Rev. 297:26–38.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Samir P, Malireddi RKS and Kanneganti TD:
The PANoptosome: A deadly protein complex driving pyroptosis,
apoptosis, and necroptosis (PANoptosis). Front Cell Infect
Microbiol. 10:2382020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chen H, Deng Y, Gan X, Li Y, Huang W, Lu
L, Wei L, Su L, Luo J, Zou B, et al: NLRP12 collaborates with NLRP3
and NLRC4 to promote pyroptosis inducing ganglion cell death of
acute glaucoma. Mol Neurodegener. 15:262020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Pistritto G, Trisciuoglio D, Ceci C,
Garufi A and D'Orazi G: Apoptosis as anticancer mechanism: Function
and dysfunction of its modulators and targeted therapeutic
strategies. Aging (Albany NY). 8:603–619. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yan J, Wan P, Choksi S and Liu ZG:
Necroptosis and tumor progression. Trends Cancer. 8:21–27. 2022.
View Article : Google Scholar
|
|
78
|
Karki R and Kanneganti TD: Diverging
inflammasome signals in tumorigenesis and potential targeting. Nat
Rev Cancer. 19:197–214. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Allen IC, TeKippe EM, Woodford RM, Uronis
JM, Holl EK, Rogers AB, Herfarth HH, Jobin C and Ting JP: The NLRP3
inflammasome functions as a negative regulator of tumorigenesis
during colitis-associated cancer. J Exp Med. 207:1045–1056. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zaki MH, Boyd KL, Vogel P, Kastan MB,
Lamkanfi M and Kanneganti TD: The NLRP3 inflammasome protects
against loss of epithelial integrity and mortality during
experimental colitis. Immunity. 32:379–391. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zaki MH, Vogel P, Body-Malapel M, Lamkanfi
M and Kanneganti TD: IL-18 production downstream of the Nlrp3
inflammasome confers protection against colorectal tumor formation.
J Immunol. 185:4912–4920. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Carvalho FA, Nalbantoglu I, Aitken JD,
Uchiyama R, Su Y, Doho GH, Vijay-Kumar M and Gewirtz AT: Cytosolic
flagellin receptor NLRC4 protects mice against mucosal and systemic
challenges. Mucosal Immunol. 5:288–298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Karki R, Man SM and Kanneganti TD:
Inflammasomes and cancer. Cancer Immunol Res. 5:94–99. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tenthorey JL, Chavez RA, Thompson TW,
Deets KA, Vance RE and Rauch I: NLRC4 inflammasome activation is
NLRP3- and phosphorylation-independent during infection and does
not protect from melanoma. J Exp Med. 217:e201917362020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ohashi K, Wang Z, Yang YM, Billet S, Tu W,
Pimienta M, Cassel SL, Pandol SJ, Lu SC, Sutterwala FS, et al:
NOD-like receptor C4 inflammasome regulates the growth of colon
cancer liver metastasis in NAFLD. Hepatology. 70:1582–1599. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chen GY and Núñez G: Inflammasomes in
intestinal inflammation and cancer. Gastroenterology.
141:1986–1999. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Peng L, Youwei R and Yanghong Z: Research
progress of NLRC4 and colorectal cancer. J Hubei Univ Sci Technol
(Med Sci). 36:176–179. 2022. View Article : Google Scholar
|
|
88
|
Bast A, Krause K, Schmidt IHE, Pudla M,
Brakopp S, Hopf V, Breitbach K and Steinmetz I: Caspase-1-dependent
and -independent cell death pathways in Burkholderia pseudomallei
infection of macrophages. PLoS Pathog. 10:e10039862014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Freeman L, Guo H, David CN, Brickey WJ,
Jha S and Ting JPY: NLR members NLRC4 and NLRP3 mediate sterile
inflammasome activation in microglia and astrocytes. J Exp Med.
214:1351–1370. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Guo Q, Wu Y, Hou Y, Liu Y, Liu T, Zhang H,
Fan C, Guan H, Li Y, Shan Z and Teng W: Cytokine secretion and
pyroptosis of thyroid follicular cells mediated by enhanced NLRP3,
NLRP1, NLRC4, and AIM2 inflammasomes are associated with autoimmune
thyroiditis. Front Immunol. 9:11972018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Chiarini A, Armato U, Gui L and Dal Prà I:
'Other than NLRP3' inflammasomes: Multiple roles in brain disease.
Neuroscientist. 30:23–48. 2024. View Article : Google Scholar
|
|
92
|
Salcedo R, Worschech A, Cardone M, Jones
Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O'HUigin C, et al:
MyD88-mediated signaling prevents development of adenocarcinomas of
the colon: Role of interleukin 18. J Exp Med. 207:1625–1636. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Takagi H, Kanai T, Okazawa A, Kishi Y,
Sato T, Takaishi H, Inoue N, Ogata H, Iwao Y, Hoshino K, et al:
Contrasting action of IL-12 and IL-18 in the development of dextran
sodium sulphate colitis in mice. Scand J Gastroenterol. 38:837–844.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Chen GY, Liu M, Wang F, Bertin J and Núñez
G: A functional role for Nlrp6 in intestinal inflammation and
tumorigenesis. J Immunol. 186:7187–7194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wilson JE, Petrucelli AS, Chen L,
Koblansky AA, Truax AD, Oyama Y, Rogers AB, Brickey WJ, Wang Y,
Schneider M, et al: Inflammasome-independent role of AIM2 in
suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med.
21:906–913. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Bakhshi S and Shamsi S: MCC950 in the
treatment of NLRP3-mediated inflammatory diseases: Latest evidence
and therapeutic outcomes. Int Immunopharmacol. 106:1085952022.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Cai Y, Chen J, Liu J, Zhu K, Xu Z, Shen J,
Wang D and Chu L: Identification of six hub genes and two key
pathways in two rat renal fibrosis models based on bioinformatics
and RNA-seq transcriptome analyses. Front Mol Biosci.
9:10357722022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Di Q, Zhao X, Tang H, Li X, Xiao Y, Wu H,
Wu Z, Quan J and Chen W: USP22 suppresses the NLRP3 inflammasome by
degrading NLRP3 via ATG5-dependent autophagy. Autophagy.
19:873–885. 2023. View Article : Google Scholar :
|
|
99
|
Kolb R, Phan L, Borcherding N, Liu Y, Yuan
F, Janowski AM, Xie Q, Markan KR, Li W, Potthoff MJ, et al:
Obesity-associated NLRC4 inflammasome activation drives breast
cancer progression. Nat Commun. 7:130072016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ghiringhelli F, Apetoh L, Tesniere A,
Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G,
Ullrich E, et al: Activation of the NLRP3 inflammasome in dendritic
cells induces IL-1beta-dependent adaptive immunity against tumors.
Nat Med. 15:1170–1178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Allam R, Maillard MH, Tardivel A,
Chennupati V, Bega H, Yu CW, Velin D, Schneider P and Maslowski KM:
Epithelial NAIPs protect against colonic tumorigenesis. J Exp Med.
212:369–383. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Güllülü Ö, Hehlgans S, Rödel C, Fokas E
and Rödel F: Tumor suppressor protein p53 and inhibitor of
apoptosis proteins in colorectal cancer-A promising signaling
network for therapeutic interventions. Cancers (Basel). 13:6242021.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Lee C, Do HTT, Her J, Kim Y, Seo D and
Rhee I: Inflammasome as a promising therapeutic target for cancer.
Life Sci. 231:1165932019. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Naqishbandi AM: Cytotoxic and apoptotic
potential of gemini-chrysophanol nanoparticles against human
colorectal cancer HCT-116 cell lines. BMC Pharmacol Toxicol.
23:562022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Moazzendizaji S, Sevbitov A, Ezzatifar F,
Jalili HR, Aalii M, Hemmatzadeh M, Aslani S, Gholizadeh Navashenaq
J, Safari R, Hosseinzadeh R, et al: microRNAs: Small molecules with
a large impact on colorectal cancer. Biotechnol Appl Biochem.
69:1893–1908. 2022. View Article : Google Scholar
|
|
106
|
Elrebehy MA, Al-Saeed S, Gamal S, El-Sayed
A, Ahmed AA, Waheed O, Ismail A, El-Mahdy HAM, Sallam AM and
Doghish AS: miRNAs as cornerstones in colorectal cancer
pathogenesis and resistance to therapy: A spotlight on signaling
pathways interplay-a review. Int J Biol Macromol. 214:583–600.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Dai F, Guo M, Shao Y and Li C: Vibrio
splendidus flagellin C binds tropomodulin to induce p38
MAPK-mediated p53-dependent coelomocyte apoptosis in Echinodermata.
J Biol Chem. 298:1020912022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Mello SS and Attardi LD: Deciphering p53
signaling in tumor suppression. Curr Opin Cell Biol. 51:65–72.
2018. View Article : Google Scholar :
|
|
109
|
Raghu D and Karunagaran D: Plumbagin
downregulates Wnt signaling independent of p53 in human colorectal
cancer cells. J Nat Prod. 77:1130–1134. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Golubovskaya VM and Cance WG: Targeting
the p53 pathway. Surg Oncol Clin N Am. 22:747–764. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Stegh AH: Targeting the p53 signaling
pathway in cancer therapy-the promises, challenges and perils.
Expert Opin Ther Targets. 16:67–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Morandell S and Yaffe MB: Exploiting
synthetic lethal interactions between DNA damage signaling,
checkpoint control, and p53 for targeted cancer therapy. Prog Mol
Biol Transl Sci. 110:289–314. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Golubovskaya VM and Cance WG: Focal
adhesion kinase and p53 signaling in cancer cells. Int Rev Cytol.
263:103–153. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
El-Deiry WS: Insights into cancer
therapeutic design based on p53 and TRAIL receptor signaling. Cell
Death Differ. 8:1066–1075. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Bates S and Vousden KH: p53 in signaling
checkpoint arrest or apoptosis. Curr Opin Genet Dev. 6:12–18. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Khan M, Ai M, Du K, Song J, Wang B, Lin J,
Ren A, Chen C, Huang Z, Qiu W, et al: Pyroptosis relates to tumor
microenvironment remodeling and prognosis: A pan-cancer
perspective. Front Immunol. 13:10622252022. View Article : Google Scholar
|
|
117
|
Ding J, Wang K, Liu W, She Y, Sun Q, Shi
J, Sun H, Wang DC and Shao F: Pore-forming activity and structural
autoinhibition of the gasdermin family. Nature. 535:111–116. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli
VG, Wu H and Lieberman J: Inflammasome-activated gasdermin D causes
pyroptosis by forming membrane pores. Nature. 535:153–158. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Broz P and Dixit VM: Inflammasomes:
Mechanism of assembly, regulation and signalling. Nat Rev Immunol.
16:407–420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Kay C, Wang R, Kirkby M and Man SM:
Molecular mechanisms activating the NAIP-NLRC4 inflammasome:
Implications in infectious disease, autoinflammation, and cancer.
Immunol Rev. 297:67–82. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Man SM: Inflammasomes in the
gastrointestinal tract: Infection, cancer and gut microbiota
homeostasis. Nat Rev Gastroenterol Hepatol. 15:721–737. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Irak K, Bayram M, Cifci S and Sener G:
Serum levels of NLRC4 and MCP-2/CCL8 in patients with active
Crohn's disease. PLoS One. 16:e02600342021. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Fattinger SA, Geiser P, Samperio Ventayol
P, Di Martino ML, Furter M, Felmy B, Bakkeren E, Hausmann A,
Barthel-Scherrer M, Gül E, et al: Epithelium-autonomous NAIP/NLRC4
prevents TNF-driven inflammatory destruction of the gut epithelial
barrier in Salmonella-infected mice. Mucosal Immunol. 14:615–629.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Mizoguchi A: Animal models of inflammatory
bowel disease. Prog Mol Biol Transl Sci. 105:263–320. 2012.
View Article : Google Scholar
|
|
125
|
Saleh M and Trinchieri G: Innate immune
mechanisms of colitis and colitis-associated colorectal cancer. Nat
Rev Immunol. 11:9–20. 2011. View Article : Google Scholar
|
|
126
|
Kiesler P, Fuss IJ and Strober W:
Experimental models of inflammatory bowel diseases. Cell Mol
Gastroenterol Hepatol. 1:154–170. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Henderson LA and Cron RQ: Macrophage
activation syndrome and secondary hemophagocytic
lymphohistiocytosis in childhood inflammatory disorders: Diagnosis
and management. Paediatric drugs. 22:29–44. 2020. View Article : Google Scholar :
|
|
128
|
Bardet J, Laverdure N, Fusaro M, Picard C,
Garnier L, Viel S, Collardeau-Frachon S, De Guillebon JM, Durieu I,
Casari-Thery C, et al: NLRC4 GOF mutations, a challenging diagnosis
from neonatal age to adulthood. J Clin Med. 10:43692021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Volker-Touw CM, de Koning HD, Giltay JC,
de Kovel CGF, van Kempen TS, Oberndorff KMEJ, Boes ML, van Steensel
MAM, van Well GTJ, Blokx WAM, et al: Erythematous nodes, urticarial
rash and arthralgias in a large pedigree with NLRC4-related
autoinflammatory disease, expansion of the phenotype. Br J
Dermatol. 176:244–248. 2017. View Article : Google Scholar
|
|
130
|
Moghaddas F, Zeng P, Zhang Y, Schützle H,
Brenner S, Hofmann SR, Berner R, Zhao Y, Lu B, Chen X, et al:
Autoinflammatory mutation in NLRC4 reveals a leucine-rich repeat
(LRR)-LRR oligomerization interface. J Allergy Clin Immunol.
142:1956–1967.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Trifiletti R, Lachman HM, Manusama O,
Zheng D, Spalice A, Chiurazzi P, Schornagel A, Serban AM, van Wijck
R, Cunningham JL, et al: Identification of ultra-rare genetic
variants in pediatric acute onset neuropsychiatric syndrome (PANS)
by exome and whole genome sequencing. Sci Rep. 12:111062022.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Eeckhout E, Asaoka T, Van Gorp H, Demon D,
Girard-Guyonvarc'h C, Andries V, Vereecke L, Gabay C, Lamkanfi M,
van Loo G and Wullaert A: The autoinflammation-associated
NLRC4V341A mutation increases microbiota-independent
IL-18 production but does not recapitulate human autoinflammatory
symptoms in mice. Front Immunol. 14:12726392023. View Article : Google Scholar
|