Introduction
Non-small cell lung cancer (NSCLC) has high
morbidity and mortality rates globally. NSCLC is distinguished by
its rapid progression and asymptomatic nature, often leading to a
diagnosis at the advanced stages of disease, thereby impeding the
efficacy of treatment and prognosis for patients (1,2).
Consequently, there is a critical need to identify biomarkers for
early detection and screening, to investigate the fundamental
processes of lung cancer pathogenesis, and to identify novel
therapeutic targets.
One of the key features of cancer cells is an
abnormal cell cycle that leads to uncontrolled cell proliferation
(3-5). Furthermore, irregular expression of
cycle-related proteins enables cancer cells to invade, migrate and
evade apoptosis (6). Cytokinesis
cyclin 25 homologous protein C (CDC25C) is a novel tumor-associated
antigen that was identified by bioinformatics. In the study of lung
(7-9), liver (10) and colon (11) cancers, the expression of CDC25C
was found to be markedly elevated in tumor tissues compared with
normal tissues, indicating that CDC25C is involved in tumor
progression. Notably, CDC25C is significantly involved in the
transition from the G2 to M phase of the cell cycle (12).
The multilevel regulatory mechanisms of gene
expression in eukaryotic organisms occur during transcription,
post-transcription and translation as well as via subsequent
regulation. These mechanisms form the molecular basis for cell
differentiation, morphogenesis and individual development. In
particular, the regulation of gene transcription at the molecular
level is of great importance for understanding the molecular
mechanisms of related diseases and their prevention, diagnosis and
treatment. Signal transducer and activator of transcription 3
(STAT3) is a pivotal transcription factor involved in a myriad of
biological processes. STAT3 mediates the transmission of
extracellular signals to the nucleus, thereby initiating the
expression of specific target genes. In various human cancer types,
abnormal activation of STAT3 can induce tumor occurrence and
promote tumor development, is essential for enhancing the
proliferation and survival of cancer cells, making it an attractive
anticancer target.
N6-methyladenosine (m6A) is the most
prevalent form of RNA methylation modification, encompassing ~2/3
of all RNA modifications. Dysregulation of m6A
methylation influences tumorigenicity and tumor progression, and
its involvement in liver, breast and lung cancer has been proven
(3,4). m6A regulatory proteins
influence the biological behavior of NSCLC by regulating the mRNA
abundance of target genes and serve a key role in the occurrence
and prognosis of cancer (13).
The regulation of m6A involves various types of
regulators, including 'writer', 'eraser' and 'reader' proteins.
alkB homolog 5 RNA demethylase (ALKBH5), identified as an
m6A demethylase, demonstrates prognostic significance in
pancreatic (14) and colon
(15) cancers. The YTH
N6-methyladenosine RNA binding protein (YTHDF) family, comprising
YTHDF1, YTHDF2 and YTHDF3 are m6A 'reader' proteins.
YTHDF1 and YTHDF3 have been demonstrated to enhance mRNA stability
and translation through their interaction with target mRNAs
(16). YTHDF2, the initial
m6A binding protein to be identified and thoroughly
researched, has a role in controlling mRNA stability through the
identification and attachment to m6A modification sites
on mRNAs. YTHDF2 is significantly involved in the development of
different types of cancer such as lung (17,18), liver (19,20) and gastric (21) cancer, where it regulates the
biological processes of tumor cell proliferation and apoptosis,
influencing tumor progression and prognosis.
Despite notable progress in both fundamental and
clinical research pertaining to lung cancer treatment, as well as
continuous innovations in therapeutic strategies, the 5-year
survival rate of patients remains alarmingly low. This elevated
mortality is primarily attributed to the ambiguous mechanisms
underlying lung cancer, which present a significant challenge for
effective clinical management. A thorough understanding of the
transcriptional and post-transcriptional regulatory mechanisms of
oncogenes will assist the development of targeted drugs. In the
present study, the expression mechanism of CDC25C in NSCLC was
investigated at the transcriptional and post-transcriptional
regulatory level. Multi-omics analysis was used to compare the
differences between the low-expression and high-expression CDC25C
groups. The results of the present study revealed the
transcriptional and post-transcriptional regulation involved in
controlling CDC25C expression in NSCLC and suggested that CDC25C
may serve as an important prognostic marker for tumor progression
and metastasis in NSCLC.
Materials and methods
Cell culture and transfection
The human NSCLC cell lines, H838, A549 and H1299,
were cultured in RPMI-1640 (cat. no. C11875500BT; Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (cat. no.
FSP500; Shanghai ExCell Biology, Inc.). All transfections were
undertaken using Lipofectamine 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.). To prevent the off-target effects of small
interfering (si)RNA, an siRNA-pool was constructed by mixing three
types of siRNA for each gene. The final concentration for siRNA
transfection was set at 75 nM. The cells were infected for 12 h at
37°C. After transfection for 48 h, the cells were collected for
subsequent experiments. The siRNA oligonucleotides were synthesized
by Beijing Tsingke Biotech Co., Ltd., and were as follows: Control
siRNA forward, 5′-UUC UCC GAA CGU GUC ACG UTT-3′ and reverse,
5′-ACG UGA CAC GUU CGG AGA ATT-3′; CDC25C siRNA-1 forward, 5′-GAG
UUG CUG AGG UGU CGA ATT-3′ and reverse. 5′-UUC GAC ACC UCA GCA ACU
CTT-3′; CDC25C siRNA-2 forward, 5′-GAU GCA AUG UGU AGU UCA UTT-3′
and reverse, 5′-AUG AAC UAC ACA UUG CAU CTT-3′; CDC25C siRNA-3
forward, 5′-GGU GAU UCU GCA AAC CUA ATT-3′ and reverse. 5′-UUA GGU
UUG CAG AAU CAC CTT-3′; YTHDF2 siRNA-1 forward, 5′-CUG GAU AUA GUA
GCA AUU ATT-3′ and reverse, 5′-UAA UUG CUA CUA UAU CCA GTT-3′;
YTHDF2 siRNA-2 forward, 5′-CCA UUA CUA GUA ACA UCG UTT-3′ and
reverse, 5′-ACG AUG UUA CUA GUA AUG GTT-3′; YTHDF2 siRNA-3 forward,
5′-GAU AUU CAC CGU UCC AUU ATT-3′ and reverse, 5′-UAA UGG AAC GGU
GAA UAU CTT-3′; YTHDF3 siRNA-1 forward, 5′-CAC CAA UGU CAG AUC
CAUA-3′ and reverse, 5′-UAU GGA UCU GAC AUU GGU G-3′; YTHDF3
siRNA-2 forward, 5′-GGC CCA CUC UAU UUA CUC UTT-3′ and reverse,
5′-AGA GUA AAU AGA GUG GGC CTT-3′; YTHDF3 siRNA-3 forward, 5′-GCC
UCA GCC AUU AAU UCA ATT-3′ and reverse, 5′-UUG AAU UAA UGG CUG AGG
CTT-3′; ALKBH5 siRNA-1 forward, 5′-GAC UGU GCU CAG UGG AUA UTT-3′
and reverse, 5′-AUA UCC ACU GAG CAC AGU CTT-3′; ALKBH5 siRNA-2
forward, 5′-GAU AUG CUG CUG AUG AAA U-3′ and reverse, 5′-AUU UCA
UCA GCA GCA UAU C-3′; and ALKBH5 siRNA-3 forward, 5′-GCU UCA GCU
CUG AGA ACU A-3′ and reverse, 5′-UAG UUC UCA GAG CUG AAG C-3′.
The pMT170 vector (Sangon Biotech Co., Ltd.) was
used to construct the CDC25C-overexpressing lentivirus (Lv-CDC25C).
The generation system used was the second. Subsequently, pMT170
plasmid (100 µg) and virus packaging plasmids (pCMV-dR8.91,
65 µg; pCMV-VSV-G6363, 35 µg) were cotransfected into
293T cells (Sangon Biotech Co., Ltd.) using Lipofectamine™ 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 6 h.
Medium was replaced with fresh DMEM (Thermo Fisher Scientific,
Inc.) containing 10% FBS and incubated at 37°C for 48 h. The cell
supernatant was collected, then filtered through a 0.45-µm
filter (Pall Life Sciences). MOI for lentivirus transfection was
10. H1299 cells in the exponential growth phase were plated in
6-well plates and cultured for 24 h. Following 24 h incubation, the
cell medium was replaced with complete medium (RPMI-1640 medium
comprising 10% FBS; Gibco; Thermo Fisher Scientific, Inc.). The
cells were stably and continuously expressed after screening with
puromycin (2 µg/ml). The time interval between transduction
and follow-up experiment was 14 days. The puromycin concentration
for maintenance was 1 µg/ml. The pcDNA 3.1 vector (Sangon
Biotech Co., Ltd.) was employed to construct the YTHDF2, YTHDF3,
CDC25C and STAT3-overexpressing vectors. shYTHDF2 containing
YTHDF2-targeting shRNA was constructed by pMAGic7.1 lentiviral
vector. The target sequence of YTHDF2 was 5′-CCA TTA CTA GTA ACA
TCG T-3′. Empty vector was used as a negative control.
Cell proliferation assay
Cell viability was assessed by employing the Cell
Counting Kit-8 (CCK-8; cat. no. BMU106-CN; Abbkine Scientific Co.,
Ltd.). Cells were plated in 96-well plates at a density of 2,000
cells per well. For each well, 10 µl of reagent was added,
followed by incubation at 37°C for 2 h. The optical density (OD)
was measured at 450 nm. After cells adhered, their viability was
evaluated at 0, 24, 48, 72 and 96 h following the manufacturer's
protocol.
Colony formation assay
Colony formation assays were performed to assess the
extended-term proliferation of NSCLC cells. After 7-14 days, the
cells were fixed with methyl alcohol at room temperature for 15
min, and subsequently subjected to staining using crystal violet
(0.5% wt/vol) for an additional 15 min at room temperature. The
number of cells in a single clone exceeding 50 was considered to
form a colony. Colonies were manually counted.
Transwell (migration) and Matrigel
(invasion) assays
The migratory and invasive capabilities of cells
were assessed using the Transwell assay. Cells were suspended in
serum-free media, and then seeded at a density of
2.5×104 cells per well into the upper compartments of
chambers coated without (cat. no. 353097; Corning, Inc.) (for
migration) or with (cat. no. 354480; Corning, Inc.) (for invasion)
Matrigel, and complete medium with 20% FBS was added to the lower
chambers. Following 48 h of incubation at 37°C, the cells that
moved and penetrated to the other side of the membranes were fixed,
stained with crystal violet (0.5% wt/vol). Cells were fixed with 4%
paraformaldehyde at room temperature for 15 min, followed by
staining with crystal violet for an additional 15 min at room
temperature. Cells were observed under a light microscope at ×40
magnification, and images were captured from 3 randomly selected
fields of view. In the wound-healing assay, a single layer of cells
was then directly scratched using a toothpick. The migration
distance of cells was assessed at both 0 and 24 or 48 h.
Chromatin immunoprecipitation (ChIP)-PCR
and ChIP-qPCR
A total of 1×106 H1299 cells were
harvested and crosslinked by formaldehyde at a final concentration
of 1%. After stopping cross-link with glycine, chromatin was
sheared to 100-300 bp with sonication. The STAT3 antibody (cat. no.
9139s; Cell Signaling Technology, Inc.) and 100 µl protein G
magnetic beads were applied to pull down the target protein. Then
the protein was digested with proteinase K. DNA immunoprecipitated
by the target protein was harvested and purified, and detected by
PCR or qPCR. The sequences of the CDC25C primers are: forward,
5′-TAA CTC TGC TGC CCT CAA-3′ and reverse, 5′-CAT TCC CTG CTC CTC
ATA-3′. The GAPDH primers used for ChIP are the same as those used
in qPCR.
RNA extraction and reverse-transcription
quantitative PCR (RT-qPCR)
TRIzol reagent from Invitrogen (Thermo Fisher
Scientific, Inc.) was used to extract total RNA from samples, and
isopropanol was used to separate RNA from the reaction system. The
RNA was washed twice with 75% ethanol. RT-qPCR was used to evaluate
mRNA levels. The primers were synthesized by Sangon Biotech Co.,
Ltd. The One-Step RT-PCR Kit (Analytik Jena Gmb) was used with the
Analytik Jena qTOWER 2.2 thermal cycler. The One-Step RT-PCR Kit
was used according to the manufacturer's protocol. Actin was used
as the internal reference. The fluorescence intensity of SYBR Green
(Takara Biotechnology Co., Ltd.) was captured in real time. The
cycle threshold (CT) value (2−ΔΔCq method) was used to
calculate relative mRNA expression level (22). Thermocycling conditions were as
follows: Initial denaturation at 95°C for 10 min, followed by 40
cycles of 95°C for 15 sec and 60°C for 30 sec. The primer sequences
were as follows: CDC25C forward, 5′-GAA CCC CAA AAC GTT GCC TC-3′
and reverse, 5′-TCT ATG GC CAC GGT CCA AAC-3′; and GAPDH forward,
5′-CTC CTC CTG TTC GAC AGT CAG C-3′ and reverse, 5′-CCC AAT ACG ACC
AAA TCC GTT-3′.
Western blotting
The cells were disrupted using RIPA lysis buffer
(KGP702-100; Nanjing KeyGen Biotech Co., Ltd.) and then suspended
in loading buffer (LT101; Epizyme, Inc.). Protein concentration was
determined using a BCA Protein Assay Kit (Beyotime Institute of
Biotechnology). A total of 10 µg/lane protein was separated
by 10% SDS-PAGE, proteins were placed in rapid QuickBlock Blocking
Buffer for WB (Beyotime Institute of Biotechnology; cat. no. P0252)
and blocked for 15 min at room temperature, then transferred to
PVDF membrane (cat. no. 3010040001; Roche Diagnostics) incubated
with appropriate primary antibodies at 4°C overnight and secondary
antibodies at room temperature for 1 h (Table SI). The PVDF membrane was rinsed
with TBST (containing 0.1% Tween-20).
Dual-luciferase reporter assay
The 3′ untranslated region (UTR) sequence of CDC25C
was inserted into a pmiR-GLO dual luciferase expression vector
(Beijing Tsingke Biotech Co., Ltd.) containing firefly and Renilla
luciferase, resulting in the construction of a wild-type CDC25C
reporter plasmid. The CDC25C 3′ UTR mutant reporter plasmid was
constructed by substituting adenosine bases with cytosine within
the m6A consensus sites. The firefly and Renilla
luciferase activity ratios were evaluated 48 h after transfection
with the Dualucif Firefly Renilla Assay Kit (cat. no. F6075M;
Suzhou UE Landy Biotechnology Co., Ltd.). Lipofectamine 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) was used in the
transfection process.
The dual-luciferase reporter assay was performed to
explore the interaction between STAT3 and the CDC25C promoter.
Segments from the CDC25C promoter (P1, P2, P3, P4 and P5) were
incorporated into the pGL3-basic vector, whereas STAT3 was
integrated into the pcDNA3.1 vector. The CDC25C (P1, P2, P3, P4, P5
or Basic) firefly luciferase plasmids were co-transfected with
STAT3 vectors and the pRL-TK Renilla luciferase vector into
cells. The firefly and Renilla luciferase activity ratios
were evaluated 48 h after transfection with the Dualucif Firefly
Renilla Assay Kit. The dual-luciferase reporter assay was also
performed to explore the interaction between STAT3 and the YTHDF1
promoter or ALKBH5 promoter.
Total m6A measurement
The m6A levels in the Total RNA were
assessed with m6A methylation quantification kit (cat.
no. P-9005-48; EpiGentek Inc.). After isolating and purifying the
Total RNA, it was bound to the assay wells and cultured with the
capture antibody. The m6A level was determined based on
fluorescence after incubation with the fluoro-developer
solution.
RNA immunoprecipitation assay (RIP)
RIP was performed with the RNA-Binding Protein
Immunoprecipitation Kit (cat. no. Bes5101; Guangzhou Bersinbio Co.,
Ltd.). Briefly, the cells were collected at 4°C and 10,000 × g for
10 min, followed by lysis in a full RIPA buffer with a mix of
protease inhibitors and RNase inhibitor. Following this, the
cellular lysates were subjected to incubation with RIP buffer that
included 100 µl magnetic beads linked to the designated
antibody (cat. nos. 17479-1-AP, 24744-1-AP and 25537-1-AP;
Proteintech Group, Inc.) or IgG. The samples were subjected to
proteinase K cleavage in order to extract the immunoprecipitated
RNA. qPCR analysis was employed to illustrate the existence of
binding targets.
Subcutaneous and orthotopic implantation
in a NSCLC model
A total of 12 female nude mice (age, 4-6 weeks;
weight, 10-14 g) were allocated into negative control (6 mice) and
lentiviral transfection groups (6 mice) and reared under standard
environmental conditions (26-28°C; relative humidity, 40-60%;
12/12-h light/dark cycle, with commercial rat food and water
provided ad libitum). Control and Lv-CDC25C cells were
resuspended in PBS (1×106 per mouse) and subcutaneously
inoculated into the right flank of mice to establish NSCLC cell
xenograft models. The tumors sizes were observed on a weekly basis
starting from the second week. After 4 weeks, the tumors were
collected from the mice, dissected into 1-mm3 pieces and
subsequently transplanted into the right lungs of mice. Lung tissue
was collected from the mice 4 weeks later.
All animal care and experiments were approved
(approval no. SUSTech-JY2020016-1) by the Animal Care and Use
Committee of Southern University of Science and Technology
(Shenzhen, China), and the study complied with all relevant ethical
provisions on animal research. In the orthotopic tumor implantation
procedure, sodium pentobarbital (1%, 40 mg/kg, intraperitoneal
injection) was administered to anesthetize the nude mice. At our
laboratory Animal Centre, animal caretakers conduct daily
monitoring of the animals and promptly inform laboratory personnel
of any abnormalities. Euthanasia was performed when tumor volumes
exceeded 1,000 mm3 or when nude mice exhibited
significant appetite loss or abdominal distension. All animals used
in the experiments were sacrificed using pentobarbital (80 mg/kg,
intraperitoneal) followed by decapitation. The breathing and heart
rate of the mice were observed after decapitation to determine
whether they succumbed. No nude mice experienced unexpected
mortality during the experiment. All mice were euthanized at the
planned end of the experiment.
Human specimens
All human lung specimens were obtained from Southern
University of Science and Technology Hospital (Shenzhen, China)
with written informed consent from the patients, between August
2020 and December 2021. The age of the patients ranged from 40-80
years (4 men and 2 women). The inclusion criteria were as follows:
i) Patients with NSCLC; ii) no other disease. All experiments were
approved (approval no. 008 of 2020) by the Southern University of
Science and Technology Hospital, and the study complied with all
relevant ethical provisions on human lung specimen research.
Immunohistochemistry (IHC)
All tissues were fixed with 4% paraformaldehyde at
room temperature for 24 h, embedded in paraffin, and sectioned into
6-µm slices. Following deparaffinization with xylene (45
min) and rehydration through a graded ethanol series (75, 85, 95
and 100% ethanol, 5 min each), sections were subjected to antigen
retrieval by treatment with 0.01 mol/l citrate buffer for 15 min at
100°C. Sections were then incubated in 3% hydrogen peroxide
solution for 30 min at 37°C to block endogenous peroxidase
activity. Next, the sections were blocked with 10% goat serum (cat.
no. G1208-5ML; Wuhan Servicebio Technology Co., Ltd.) for 10 min at
room temperature. The sections were subsequently incubated at 4°C
for 16 h with primary antibodies against CDC25C (1:500; cat. no.
16485-1-AP; Proteintech Group, Inc.) or STAT3 (1:500; cat. no.
10253-2-AP; Proteintech Group, Inc.). Then, the sections were
washed in PBST (containing 0.1% Tween-20). After washing, the
tissue sections were treated with streptavidin-horseradish
peroxidase-conjugated secondary antibody from the
Immunohistochemistry kit (1:200; cat. no G1215-200T; Wuhan
Servicebio Technology Co., Ltd.) for 2 h at room temperature.
Diaminobenzidine substrate was used for colorimetric development
after washing with PBST. These results were observed under light
microscope.
Hematoxylin and eosin (H&E)
staining
The tissues were deparaffinized with xylene and
immersed in an EDTA antigen retrieval buffer. Then, the tissues
were stained with H&E. The sections were stained with
hematoxylin dye for 2 min at room temperature, treated with 1%
hydrochloric acid alcohol for 45 sec at room temperature, stained
with eosin dye for 2 min at room temperature, dehydrated with
ethanol (75, 85, 95, 100%), washed with xylene, and finally sealed
with neutral resin. The results were observed under light
microscope.
Fluorescence in situ hybridization (FISH)
and immunofluorescence (IF) analysis
Cells were fixed in 4% paraformaldehyde for 20 min
at room temperature, followed by permeabilization with 0.1% Triton
X-100 for 20 min. The cells were then immersed in pre-hybridization
buffer enriched with 10% formamide. Hybridization was conducted
using a comparable buffer, supplemented with competitor RNA and BSA
(Invitrogen; Thermo Fisher Scientific, Inc.) to minimize background
interference. The CDC25C FISH probe was as follows (all 5′-3′): CTG
GGC TAC ATT TCA TTA GGT GCT GGT; CCA AAC CAT TCG GAG TGC TACA AAGA;
CTA CAC ATT GCA TCT CTC TTT CTA TGG C; CAC CAA GTT TCC ATT GTC ATT
TTC TTT ATT and ACT TTA TCT GGT ATT GTG TTG TCC TTG AAT.
The CDC25C probe, mixed in 200 µl
hybridization buffer, was applied to a surface within a humid and
dark chamber. Subsequently, a glass coverslip was inverted and
placed onto the droplet, followed by incubation at 37°C for 16 h.
Following incubation with the CDC25C probe, the cells were
subjected to primary antibody treatment at 37°C for 2 h, then
incubated with an anti-mouse fluorescent-labeled secondary antibody
for 2 h at room temperature. Next, several cycles of washing were
conducted, which involved a DAPI (1 µg/ml) staining process.
Afterward, the coverslip was affixed to a microscope slide using an
anti-fade mounting solution.
Methylated RNA immunoprecipitation
The m6A modifications on CDC25C were
detected through the utilization of the MeRIP m6A Kit
(cat. no. Bes5203-2; Guangzhou Bersinbio Co., Ltd.). In short, 300
nt RNA fragments generated by ultrasonication were subjected to
overnight incubation at 4°C with m6A antibody or
IgG-conjugated beads. RNA-antibody complex was incubated with 100
µl protein A/G magnetic beads at a temperature of 4°C for a
duration of 3 h, followed by elution using an elution buffer (cat.
no. Bes5203-2; Guangzhou Bersinbio Co., Ltd.). qPCR analysis was
employed to illustrate the existence of binding targets.
RNA sequencing
VAHTS Stranded mRNA-seq Library Prep Kit for
Illumina V2 (NR612-02; Co., Ltd.) was used for library preparation
according to the instructions. Reads were aligned to the human
Ensemble genome GRCh38 using Hisat2 aligner (v2.1.0) under
parameters: '--rna-strandness RF'. The reads mapped the genome were
calculated using featureCounts (v1.6.3). Differential gene
expression analysis was performed using the DESeq2 R-package
(Guangzhou Epibiotek Co., Ltd.).
MeRIP-seq library preparation
Total RNA was extracted using TRIzol™ Reagent (cat.
no. 15596018; Invitrogen; Thermo Fisher Scientific, Inc.). The
concentration of total RNA was measured by Qubit RNA HS assay kit
(cat. no. Q32852; Invitrogen; Thermo Fisher Scientific, Inc.).
Total RNA (100 µg) was fragmented into 100-200 nt RNA
fragments using 10X RNA Fragmentation Buffer (100 mM Tris-HCl, 100
mM ZnCl2 in nuclease-free H2O). The reaction was stopped
by adding 10X EDTA (0.5 M EDTA). Methylated RNA immunoprecipitation
was performed using EpiTM m6A immunoprecipitation kit
(cat. no. R1804; Guangzhou Epibiotek Co., Ltd.). The
m6A-enriched RNA was purified using TRIzol™ Reagent
(cat. no. 15596018; Invitrogen; Thermo Fisher Scientific, Inc.).
The library was prepared by smart-seq method. Both the input
samples without IP and the m6A IP samples were subjected
to 150-bp, paired-end sequencing on an Illumina NovaSeq 6000
sequencer (Guangzhou Epibiotek Co., Ltd.).
The RNA-seq data and relevant clinical data across
tumor types and normal tissues were downloaded from The Cancer
Genome Atlas (TCGA) database (https://www.cancer.gov/ccg/research/genome-sequencing/tcga)
and the Genotype-Tissue Expression (GTEx) database by UCSC XENA1
(https://xenabrowser.net/). R software v3.6.3
(https://cran.r-project.org/) was used
for statistical analysis, and the ggplot2 package was used for
visualization. The Wilcoxon rank sum test detected two sets of
data, and P<0.05 was considered statistically significant.
BP-1-102 treatment
H1299 cells were treated with 5 µg/ml
BP-1-102 (cat. no. HY-100493; MedChemExpress) at 37°C for 0, 24,
36, 48, 72 and 96 h. Then, the cells were collected for subsequent
experiments.
Actinomycin D treatment
H1299 cells were treated with 5 µg/ml
actinomycin D (cat. no. 15021S, Cell Signaling Technology, Inc.) at
37°C for 0, 2 and 4 h. Then, total cell RNA was extracted, followed
by RT-qPCR to measure the stability of mRNAs. The gene expression
at 0 h was considered the baseline.
Bioinformatic analysis
CDC25C mRNA m6A modification was
predicted by SRAMP (http://www.cuilab.cn/sramp). The default database
settings were used. The binding sites of STAT3 on the CDC25C
promoter were predicted in the human transcription factor database
(HumanTFDB; http://bioinfo.life.hust.edu.cn/HumanTFDB/). Gene
Ontology (https://geneon-tology.org/) and Kyoto
Encyclopedia of Genes and Genomes analyses (https://www.kegg.jp/kegg/) were used for the analysis
of sequencing data.
Statistical analysis
The data were analyzed using GraphPad Prism 8.0
(Dotmatics). The data are presented as the mean ± standard
deviation from a minimum of three independent biological
replicates. One-way ANOVA was performed to evaluate the
differences. Paired Student's t test was performed to evaluate the
differences between two groups. The survival probability was
determined using the Kaplan-Meier plotter (https://kmplot.com/analysis/) followed by the log-rank
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Overexpression of CDC25C regulates the
proliferation, invasion and migration of NSCLC cells
To examine the impact of CDC25C on NSCLC
advancement, the expression levels of CDC25C in different types of
cancer were analyzed using TCGA datasets (Fig. 1A and Table SII). The results indicated a
significant increase in the expression of CDC25C in lung
adenocarcinoma samples (Fig. 1B).
The RNA-seq data of 539 tumors and 59 normal tissues from patients
with NSCLC from TCGA datasets were also reanalyzed. Kaplan-Meier
curves indicated a significant association between CDC25C
expression and the survival of patients with NSCLC. Compared with
the CDC25C low expression group, the high expression group
exhibited a significantly poorer prognosis (Fig. 1C). Next, the CDC25C levels in 30
paraffin-embedded specimens from patients with NSCLC were measured
using IHC (Fig. 1D).
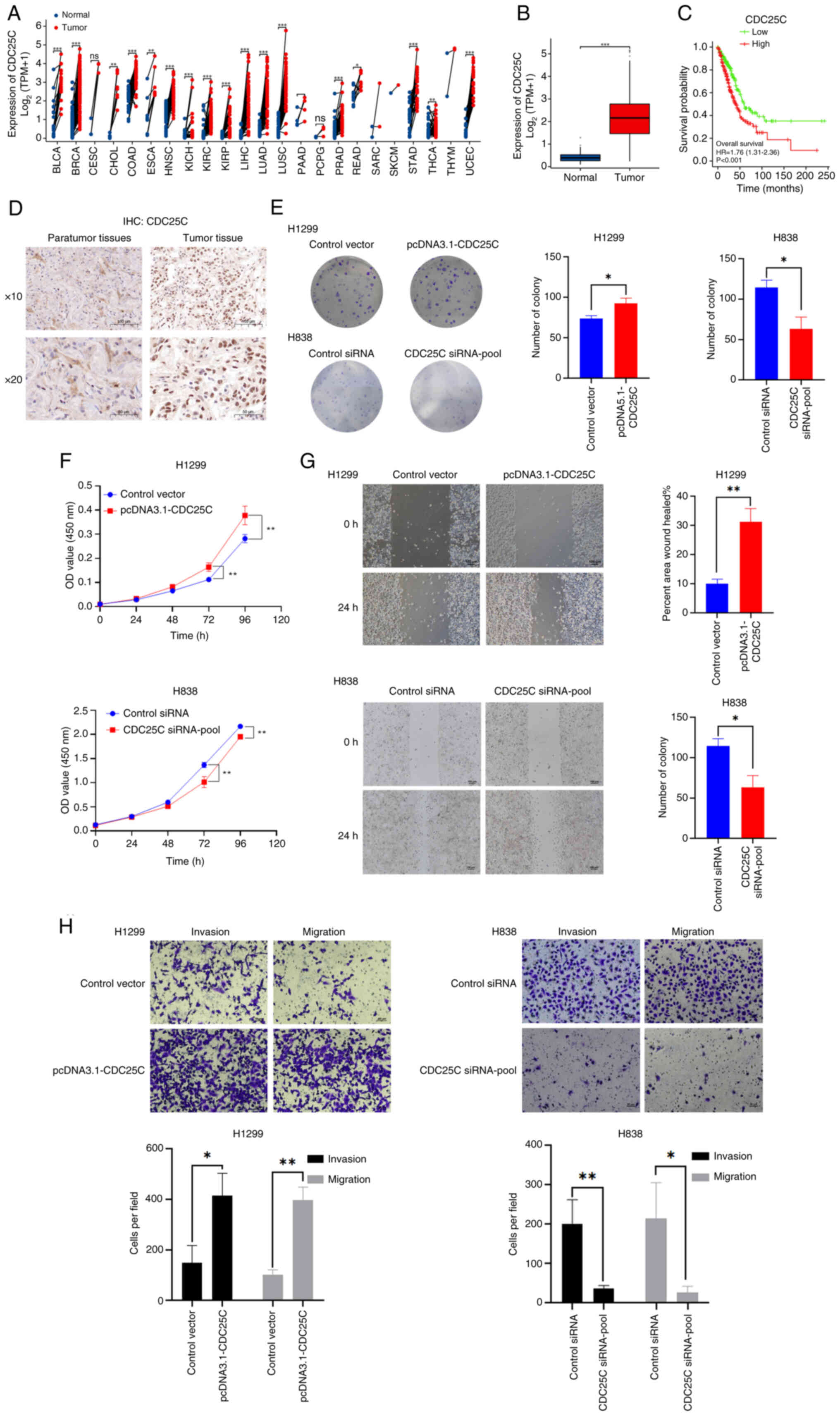 | Figure 1Upregulation of CDC25C is a
distinctive feature in NSCLC. (A) CDC25C expression in various
cancer types using TCGA datasets. (B) TCGA database indicated that
CDC25C was elevated in tumor tissues (n=539) compared with normal
tissues (n=59). (C) High expression of CDC25C was linked to a
poorer prognosis for patients with NSCLC. (D) Representative images
of immunohistochemistry images of tumor and para-tumor tissues. (E)
Colony formation and (F) Cell Counting Kit-8 assays were utilized
to assess the proliferative capacities of NSCLC cell lines. (G)
Wound healing assays were performed to assess the migratory
potential of NSCLC cell lines following the modulation of CDC25C
expression. The change in cell boundary from 0 to 24 h depicted the
trajectory of cellular movement. (H) Transwell and Matrigel assays
were conducted to assess the migratory and invasive capacities of
NSCLC cell lines. Cell counting was performed at ×40 magnification.
The results are presented as the mean ± standard deviation of three
independent experiments. *P<0.05,
**P<0.01 and ***P<0.001. CDC25C,
cytokinesis cyclin 25 homologous protein C; NSCLC, non-small cell
lung cancer; TCGA, The Cancer Genome Atlas; siRNA, small
interfering RNA; BLCA, bladder urothelial carcinoma; BRCA, breast
invasive carcinoma; CESC, cervical squamous cell carcinoma and
endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon
adenocarcinoma; ESCA, esophageal carcinoma; HNSC, head and neck
squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney
renal clear cell carcinoma; KIRP, kidney renal papillary cell
carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung
adenocarcinoma; LUSC, lung squamous cell carcinoma; PAAD,
pancreatic adenocarcinoma; PCPG, pheochromocytoma and
paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum
adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD,
stomach adenocarcinoma; THCA, thyroid carcinoma; THYM, thymoma;
UCEC, uterine corpus endometrial carcinoma. |
To interrogate the role of CDC25C in NSCLC cells,
siRNAs (CDC25C siRNA1-3) were employed to knockdown CDC25C
expression, or CDC25C was overexpressed by transfecting H1299 cells
with a plasmid encoding CDC25C (Fig.
S1A). As demonstrated by CCK-8 and colony formation assays, the
downregulation of CDC25C was found to impair the proliferative
capacity of H838 cells (Fig. 1E and
F), whereas the upregulation of CDC25C exhibited an opposite
effect. Subsequently, a wound healing assay also revealed that
knockdown of CDC25C tended to attenuate the migratory ability of
H838 cells (Fig. 1G).
Additionally, Transwell assays were performed and it was found that
the knockdown of CDC25C expression inhibited cell migration and
invasion (Fig. 1H). The
overexpression of CDC25C resulted in the opposite outcome. These
findings suggested that ectopic expression of CDC25C modulates the
proliferation, invasion and migration of NSCLC cells.
CDC25C promotes NSCLC growth and
metastasis
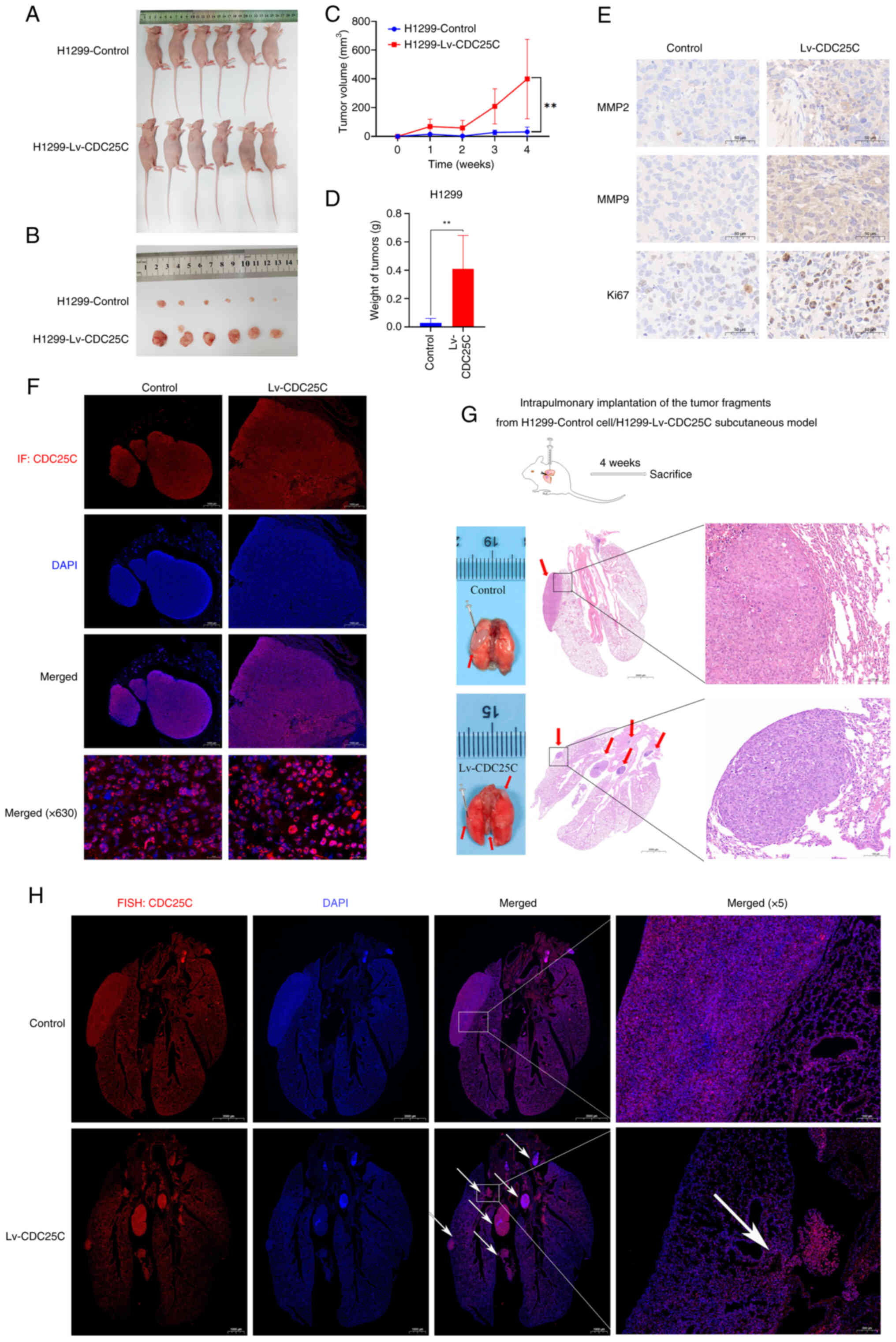
To assess the impact of CDC25C on NSCLC, H1299 cells
(control and Lv-CDC25C groups) were implanted subcutaneously into
nude mice (Figs. 2A and S1A). The NSCLC xenograft model derived
from the Lv-CDC25C group showed a significant increase in tumor
mass compared with the control group. A significant rise in tumor
volume was also noted within the Lv-CDC25C group (Fig. 2B-D). The MMP-2 and MMP-9
expression levels in the tumors were assessed to indicate the
invasive and metastatic capabilities of the tumor cells. Ki67 is a
marker found in cells undergoing proliferation, and its role is
intricately connected to mitosis, playing a crucial role in cell
multiplication. The control group showed markedly lower expression
levels of MMP-2, MMP-9 and Ki67 compared with the Lv-CDC25C group
(Fig. 2E). The IF analysis
results indicated that the tumor tissues of the Lv-CDC25C group
exhibited higher expression of CDC25C protein compared with the
control group (Fig. 2F).
Next, it was determined whether CDC25C
overexpression promoted metastasis of lung cancer cells. For this
purpose, tumor fragments from the H1299 cell (control and Lv-CDC25C
groups) subcutaneous model were cut into 1-mm3 pieces
and embedded in the lungs of nude mice (Fig. 2G). Compared with the control
group, different sites of lung cancer tissue metastasis occurred in
the Lv-CDC25C group. The H&E staining results revealed that the
tumor tissues in the Lv-CDC25C group exhibited higher levels of
invasiveness. To further investigate the difference in CDC25C
expression in lung cancer and normal lung tissues, FISH technology
was employed to assess the CDC25C mRNA expression levels. In both
the control and Lv-CDC25C groups, it was observed that the CDC25C
mRNA expression level was higher in the tumor tissue than in the
normal lung tissue. Furthermore, multiple high-expression CDC25C
mRNA metastatic tumors were observed in the Lv-CDC25C group.
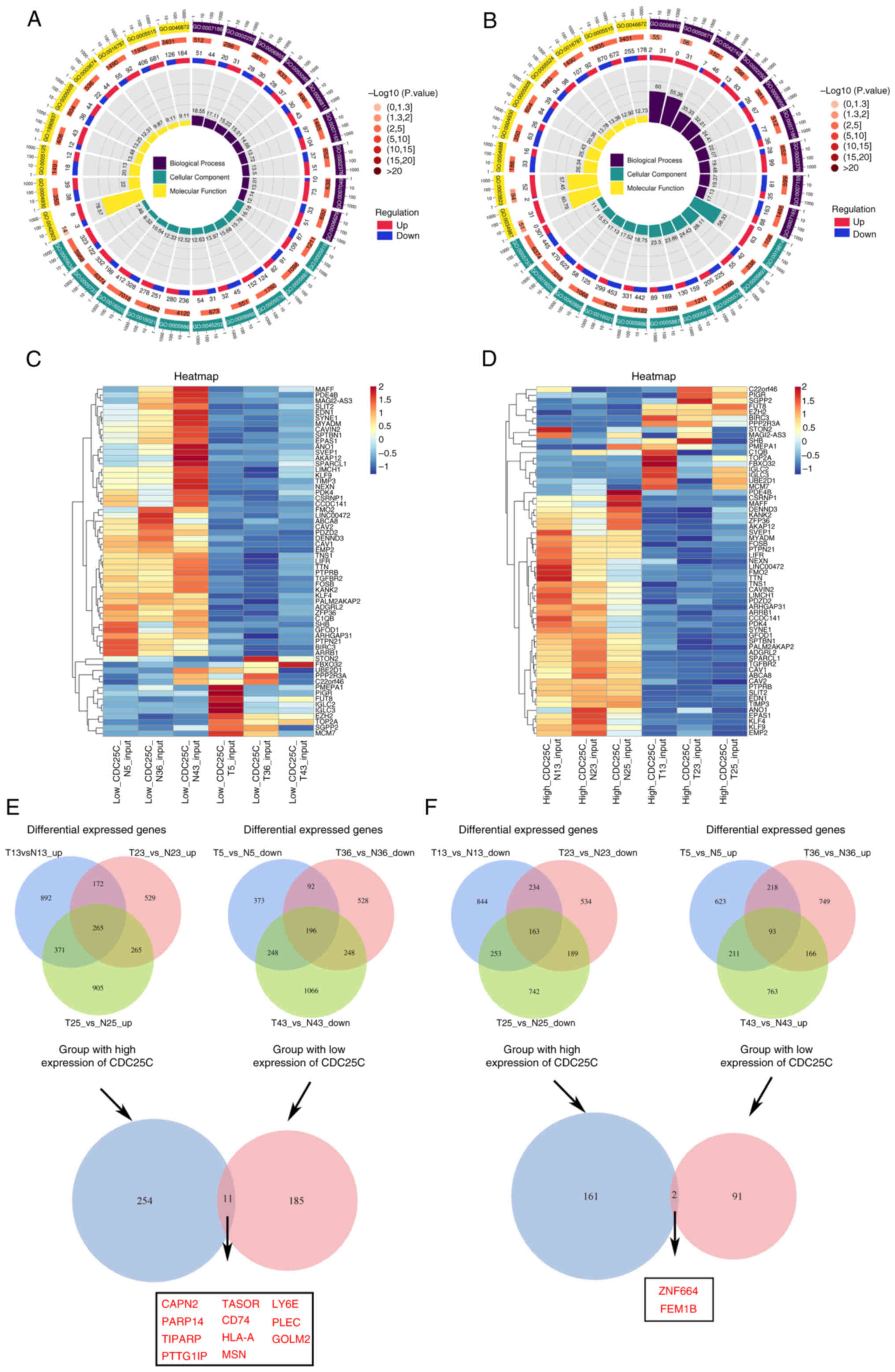
Characterization of CDC25C in NSCLC
NSCLC is a multifaceted disease characterized by
alterations in gene expression within tumor cells throughout its
initiation and progression. Advances in sequencing technology have
enabled the comprehensive analysis of intricate genetic
modifications associated with cancer. To elucidate the role of
CDC25C in NSCLC, three tissues samples (N5/T5, N36/T36 and N43/T43)
exhibiting low CDC25C gene expression levels and three tissues
samples (N13/T13, N23/T23 and N25/T25) demonstrating high
expression were selected from a cohort of 30 patients with lung
cancer for RNA-seq. For each patient, both para-tumor and tumor
tissues were collected for comparative analysis. In the CDC25C low
expression group, 1,863 genes were found to be upregulated and
1,391 genes downregulated in the tumor tissues compared with the
adjacent normal tissues (Fig. S2A
and C). In the CDC25C high expression group, 2,020 genes were
upregulated while 2,593 genes were downregulated in the tumor
tissues relative to the adjacent normal tissues (Fig. S2B and C). Both Gene Ontology and
Kyoto Encyclopedia of Genes and Genomes analyses (KEGG) revealed
that the pathways enriched in the differentially expressed genes
(DEGs) differed significantly between the CDC25C low expression
(Figs. 3A and S2D) and high expression (Figs. 3B and S2E) groups. The DEGs in the tumor and
para-tumor tissues from the CDC25C low expression (Fig. 3C) and high expression (Fig. 3D) groups were separately analyzed.
To identify specific molecules that interact with CDC25C, 11 genes
that were significantly upregulated in the tumor tissues with high
CDC25C expression levels and significantly downregulated in tumor
tissues with low CDC25C expression levels (Fig. 3E) were screened. Additionally, 2
genes that were significantly downregulated in the tumor tissues
with high CDC25C expression and significantly upregulated in the
tumor tissues with low CDC25C expression were also identified
(Fig. 3F).
BP-1-102 deactivates the transcriptional
function of STAT3 and inhibits the transcription of CDC25C
To elucidate the fundamental mechanisms responsible
for the heightened expression of CDC25C in tumor tissues, research
on both transcriptional and post-transcriptional regulation was
conducted. The HumanTFDB was utilized to predict the presence of a
STAT3 binding site in the CDC25C promoter. By IHC, elevated STAT3
levels were detected in tumor tissues compared with the para-tumor
tissues (Fig. 4A). Subsequently,
five step-deletion distal sequences of the CDC25C promoter were
inserted into the pGL3-Basic vector (Fig. 4B). Compared with the pGL3-Basic +
pcDNA3.1 group, the luciferase activity of the pGL3-Basic +
pcDNA3.1-STAT3 group was not significantly different. Compared with
the pGL3-Basic + pcDNA3.1-STAT3 group, the luciferase activity of
the pGL3-P1 (Full) + pcDNA3.1-STAT3, pGL3-P2 + pcDNA3.1-STAT3 and
pGL3-P3 + pcDNA3.1-STAT3 groups were significantly elevated
(Figs. 4B and S1B). For further confirmation, ChIP-PCR
and ChIP-qPCR were performed to detect the enrichment of STAT3 on
the CDC25C promoter. The results demonstrated that STAT3 was
enriched at the promoter region of CDC25C (Fig. 4C). These results suggested that
STAT3 could bind to the CDC25C promoter and regulate CDC25C
transcription.
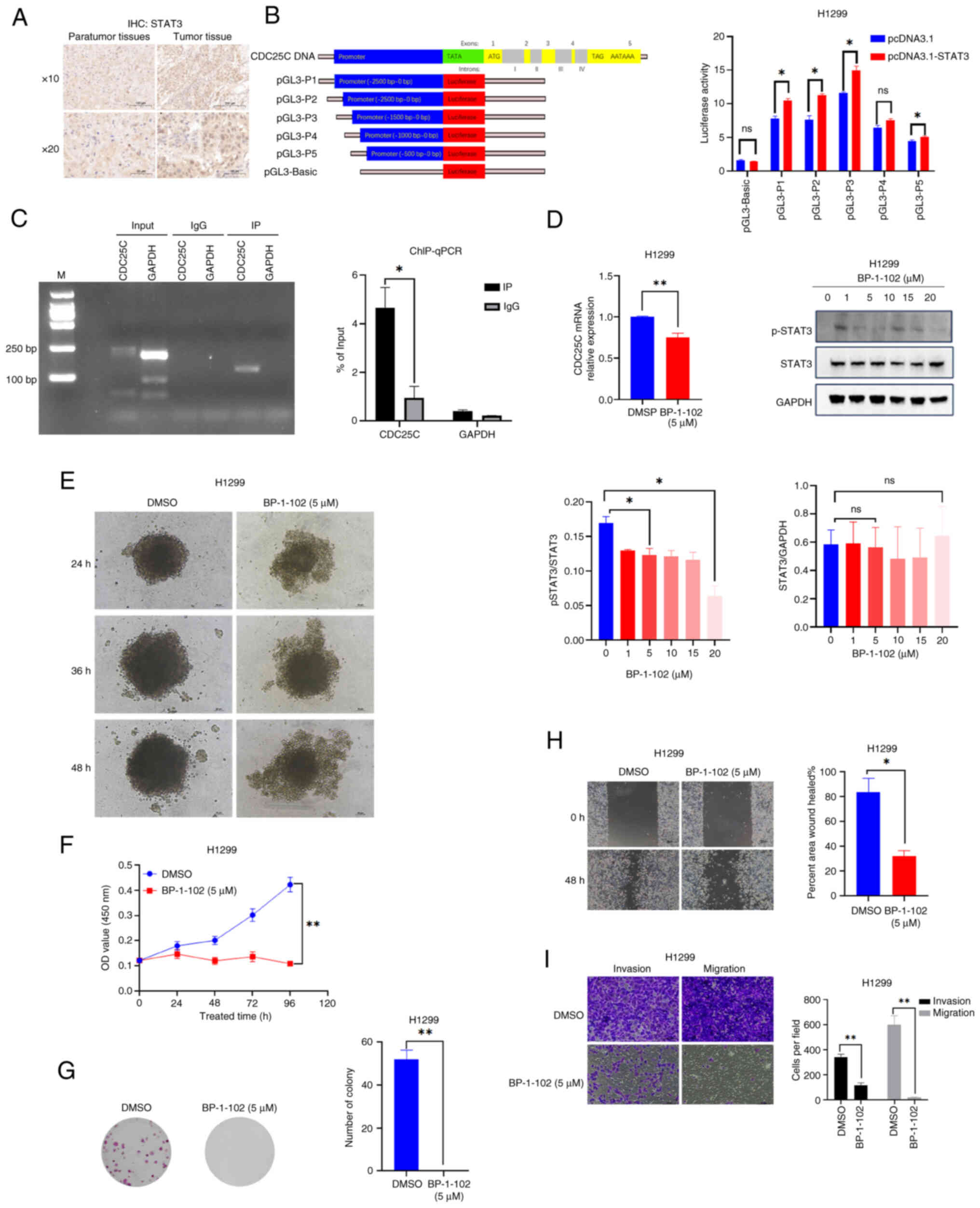 | Figure 4BP-1-102 deactivates the
transcriptional function of STAT3 and inhibits tumor growth. (A)
Immunohistochemistry revealed the presence of clusters of cells
with positive STAT3 staining in tumor tissues, whereas para-tumor
tissues exhibited negative staining. (B) Diagram showing the
conserved STAT3 response elements in the CDC25C promoter. CDC25C
promoter (pGL3-Basic, P1, P2, P3, P4 and P5) luciferase activity
was detected by dual-luciferase assays. (C) Chromatin
immunoprecipitation-qPCR was performed to identify the enrichment
of STAT3 on the CDC25C promoter region in H1299 cells; IgG served
as an antibody control. (D) Western blot analysis of p-STAT3 and
STAT3 in H1299 cells treated with BP-1-102 for 24 h; the CDC25C
mRNA level in H1299 cells treated with BP-1-102 (5 µM) was
analyzed by qPCR. (E) H1299 cell pellet analysis after treatment
with BP-1-102 (5 µM). (F-I) BP-1-102 (5 µM) causes
the viability and the migration and invasion abilities of cells to
decrease after STAT3 was inhibited; cell counting was performed at
a ×40 magnification. The results are presented as the mean ±
standard deviation of three independent experiments.
*P<0.05 and **P<0.01. STAT3, signal
transducer and activator of transcription 3; CDC25C, cytokinesis
cyclin 25 homologous protein C; qPCR, quantitative PCR; p-,
phosphorylated; ns, not significant. |
The ability of BP-1-102 to inhibit STAT3
phosphorylation in H1299 cells was examined and it was found that
CDC25C mRNA expression decreased following BP-1-102 treatment
(Fig. 4D). It was also observed
that treatment with BP-1-102 (5 µM) decreased the ability of
H1299 cancer cells to form pellets (Fig. 4E). Furthermore, as demonstrated by
CCK-8 and colony formation assays, treatment with BP-1-102 (5
µM) weakened the proliferation capability of H1299 cells
(Fig. 4F and G). Comparable
impacts on the migration and invasion of H1299 cells were observed,
as demonstrated by Transwell and scratch assays (Fig. 4H and I). These findings indicate
that STAT3 has the capacity to directly interact with the CDC25C
promoter and contribute to the transcriptional regulation of
CDC25C. It was also found that BP-1-102 deactivated the
transcriptional function of STAT3 and inhibited the transcription
of CDC25C.
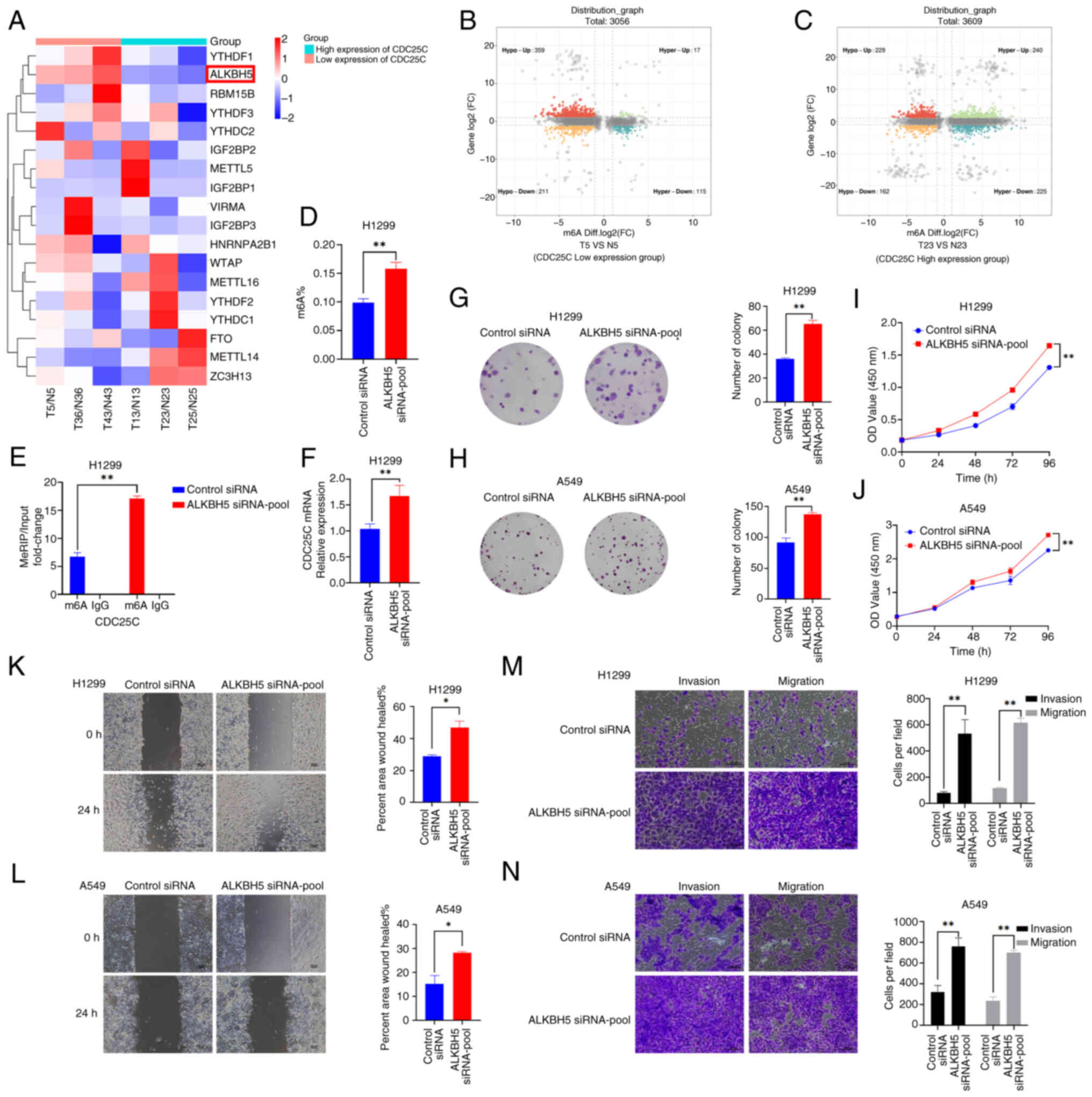
ALKBH5 regulates the expression of CDC25C
by changing the m6A modification status
m6A modification precisely modulates gene
expression at the post-transcriptional level by influencing RNA
stability, translation efficiency and interactions with other
proteins, thereby impacting various cellular biological processes.
To investigate the regulatory mechanism of m6A
modification on CDC25C expression in NSCLC, the expression of
m6A-related genes in the high and low CDC25C expression
groups were compared following RNA-seq (Fig. 5A). Subsequently, MeRIP sequencing
of the tumor and para-tumor tissues from both groups (low and high
CDC25C expression groups) were conducted. The analyses revealed
that the number of genes exhibiting hypermethylation in the low
CDC25C expression group (Fig. 5B;
and Fig. S3A and B) was
significantly lower than that observed in the high CDC25C
expression group (Fig. 5C; and
Fig. S3C and D). To explore
whether the expression of CDC25C is subject to the crosstalk
between transcriptional and post-transcriptional regulation,
MeRIP-seq and mRNA-seq of the cancer tissues with differential
expression levels were conducted to select the differentially
expressed m6A-related proteins, YTHDF1 and ALKBH5; and
the HumanTFDB was utilized to predict the binding sites of STAT3.
The binding of STAT3 to YTHDF1 or ALKBH5 was verified through
dual-luciferase assays. Nevertheless, it was discovered that the
fluorescence expression levels of the wild-type plasmid and the
site-mutated plasmid were not significantly different in H1299
cells (Fig. S4A and B). Hence,
it was hypothesized that other m6A-related proteins
might be transcriptionally regulated by STAT3. The RNA-seq results
identified that ALKBH5 was significantly downregulated in the high
CDC25C expression group compared with the low CDC25C expression
group and was therefore selected as the focus of subsequent
research.
siRNAs were used to knock down the expression of
ALKBH5 (ALKBH5 siRNA1-3; Fig.
S1C). The ALKBH5 knockdown group showed a higher overall
m6A level compared with the control group (Fig. 5D). Furthermore, ALKBH5 knockdown
increased the levels of m6A modification of CDC25C mRNA
(Fig. 5E) and the expression of
CDC25C mRNA (Fig. 5F).
Downregulation of ALKBH5 in H1299 and A549 cells led to a
significant increase in the cell proliferation rate compared with
the control groups (Fig. 5G-J).
The result of wound healing and Transwell assays demonstrated that
decreased ALKBH5 expression led to the enhanced migration and
invasion of NSCLC cells (Fig.
5K-N). Therefore, these findings suggested that aberrant
expression of ALKBH5 may be associated with the development and
advancement of NSCLC. These results also suggested that ALKBH5
activity may decrease the m6A level of CDC25C mRNA in
NSCLC, potentially resulting in a decrease in the expression level
of CDC25C mRNA.
YTHDF2 and YTHDF3 compete for binding
with CDC25C in an m6A-dependent manner to regulate
CDC25C expression
Recognition of m6A by different YTHDF
proteins promotes mRNA decay or translation. YTHDF1 and YTHDF3
promote the stability and translation of target mRNA, while YTHDF2
facilitates mRNA decay. Initially, endogenous Co-IP experiments
were conducted and a reciprocal interaction between YTHDF1, YTHDF2
and YTHDF3 was observed (Fig.
6A). Additionally, it was noted that YTHDF1, YTHDF2 and YTHDF3
exhibited binding to CDC25C mRNA within H1299 cells (Fig. 6B). Furthermore, YTHDF3 exhibited a
high binding ability to CDC25C. Notably, the overexpression of
YTHDF3 in H1299 cells promoted the expression of CDC25C (Fig. 6C). In addition, reducing the
expression of YTHDF3 led to a significant increase in the
association between YTHDF2 and CDC25C mRNA, suggesting that YTHDF2
and YTHDF3 may compete for binding with CDC25C mRNA in NSCLC cells
(Figs. 6D and S1D). The m6A profile showed
that m6A peaks were highly enriched around 3′-UTR and
the stop codon. Furthermore, SRAMP database prediction was
conducted, revealing multiple highly reliable m6A
modification sites in CDC25C mRNA (Table SIII). Subsequently, a cytosine
substitution for adenosine in the CDC25C 3′ UTR sequence was
implemented to generate a mutant form of CDC25C that is resistant
to m6A modification (Fig.
6E). The subsequent dual-luciferase reporter assay provided
confirmation that CDC25C mRNA is a direct target of YTHDF2
(Figs. 6F and S1D). Furthermore, in H1299 cells
treated with actinomycin D and transfected with YTHDF2 siRNA, the
degradation of CDC25C mRNA was observed to be reduced compared with
the control (Fig. 6G). It was
therefore hypothesized that CDC25C mRNA carrying the m6A
modification, was competitively bound by YTHDF2 or YTHDF3. However,
in NSCLC, YTHDF3 exhibited a preference for binding to CDC25C mRNA
and facilitating CDC25C translation. Conversely, when YTHDF3 was
downregulated, YTHDF2 interacted with CDC25C and accelerated CDC25C
mRNA decay.
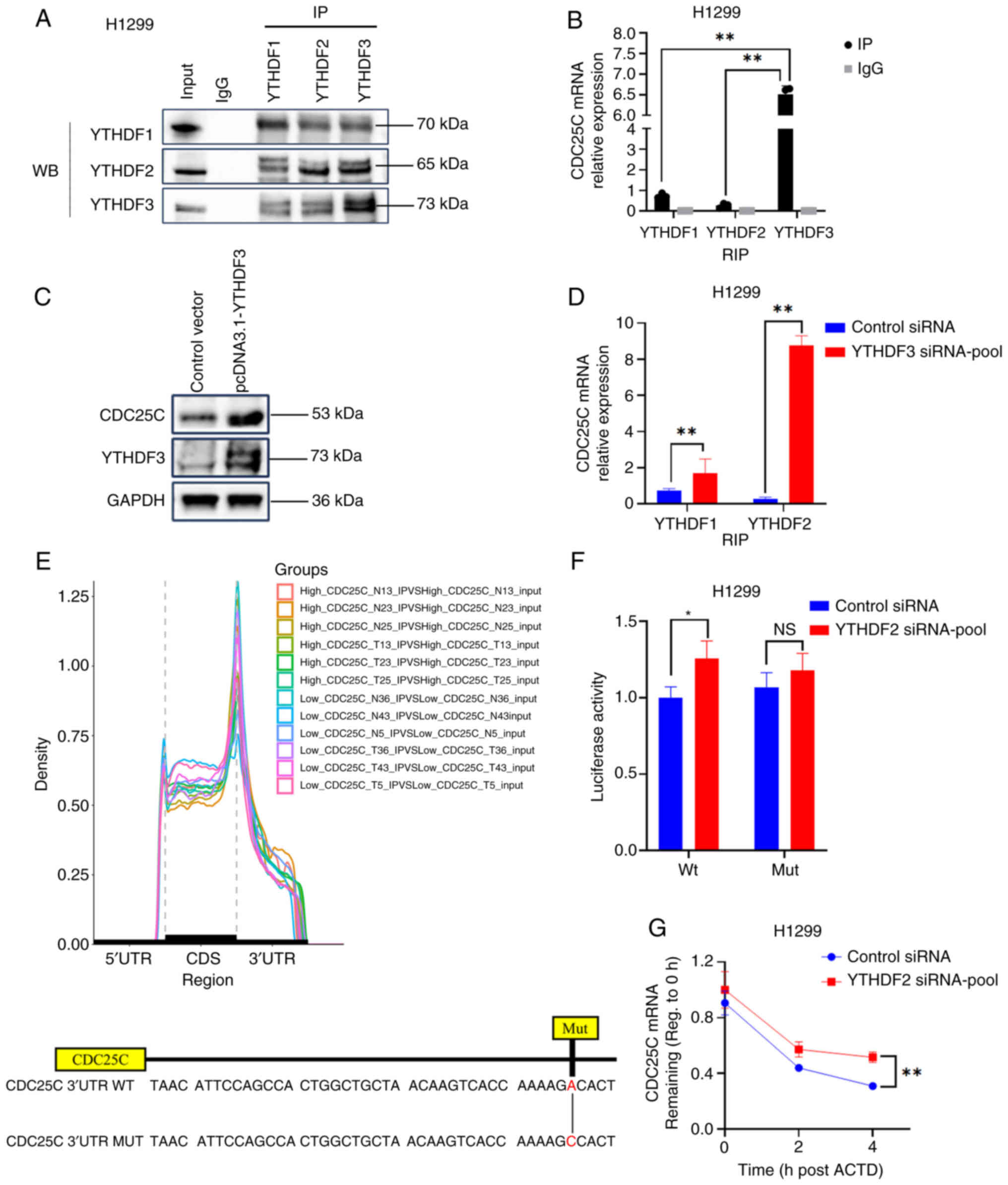 | Figure 6YTHDF2 and YTHDF3 competitively bind
to CDC25C mRNA via m6A modification. (A) Co-IP
experiments were conducted using lysates obtained from H1299 cells,
with IP performed using antibodies targeting YTHDF1, YTHDF2 or
YTHDF3. (B) RIP assay was used to evaluate the binding of
YTHDF1/2/3 to CDC25C mRNA in H1299 cells. (C) WB indicated that
overexpression of YTHDF3 promoted the translation of CDC25C. (D)
RIP experiment showed that knocking down YTHDF3 promoted the
binding of YTHDF2 to CDC25C mRNA in H1299 cells. (E) The
distribution of m6A peaks in the CDC25C high expression
and low expression groups. The diagram illustrates the potential
m6A modification site and its corresponding mutants in
the 3′UTR of CDC25C mRNA. (F) Luciferase assay. (G) The mRNA level
of CDC25C in H1299 cells treated with 5 µg/ml ACTD and
transfected with YTHDF2 siRNA was analyzed by quantitative PCR; The
results are presented as the mean ± standard deviation of three
independent experiments. *P<0.05 and
**P<0.01. YTHDF, YTH N6-methyladenosine RNA binding
protein; CDC25C, cytokinesis cyclin 25 homologous protein C;
m6A, N6-methyladenosine; IP, immunoprecipitation; RIP,
RNA IP; WB, western blot analysis; siRNA, small interfering RNA;
UTR, untranslated region; ACTD, actinomycin D; Wt, wild-type; Mut,
mutant. |
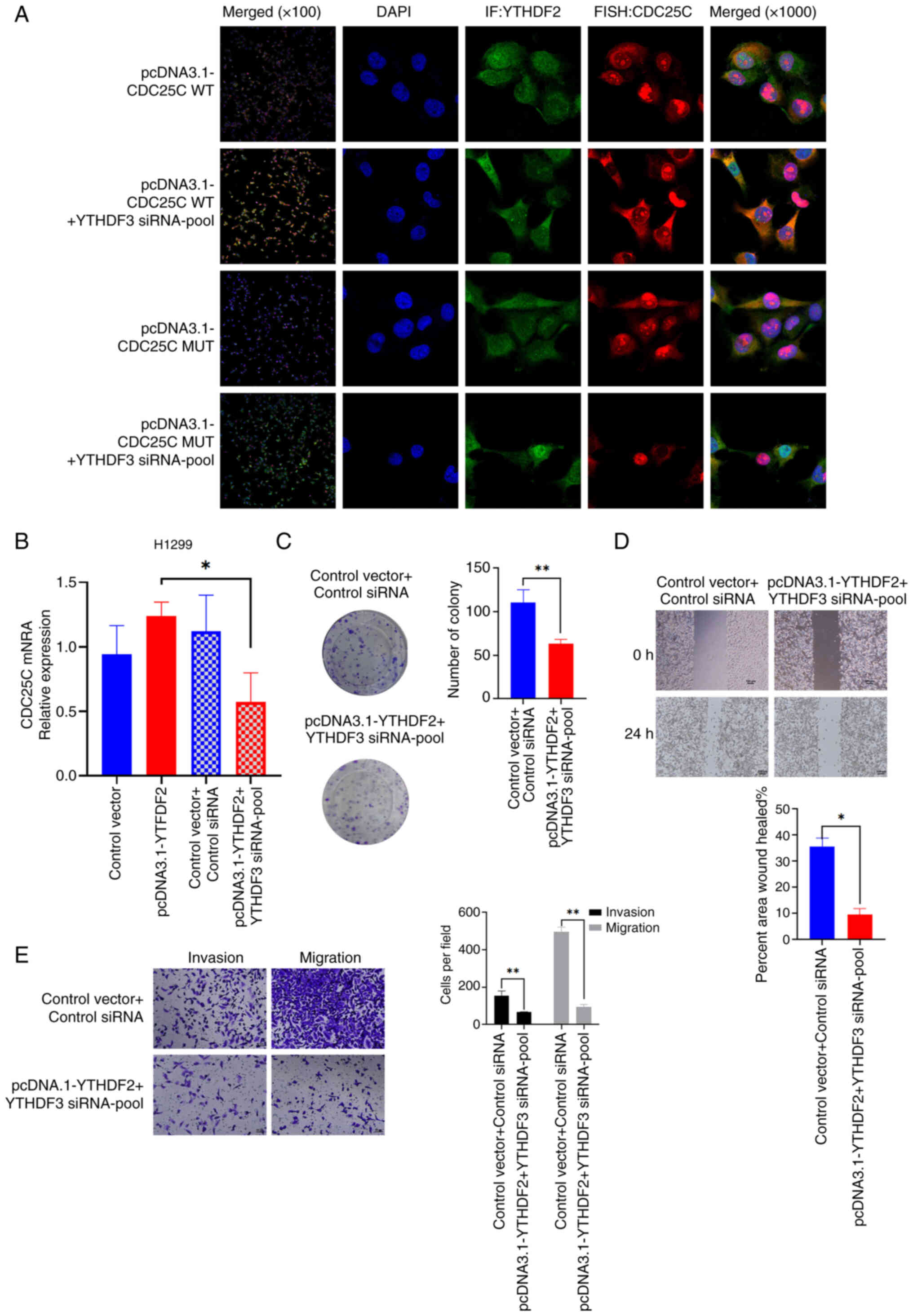
The colocalization of CDC25C mRNA and YTHDF2 protein
in H1299 cells was confirmed using RNA FISH technology combined
with IF analysis. The colocalization of YTHDF2 and CDC25C mRNA was
found to be significantly increased upon the knockdown of YTHDF3
expression. In addition, colocalization with mutant CDC25C mRNA was
weakened (Fig. 7A). Next, it was
explored whether the expression of CDC25C was dependent on the
competitive relationship between YTHDF2 and YTHDF3. It was found
that the CDC25C mRNA level was decreased in H1299 cells
co-transfected with the YTHDF3 siRNA-pool and pcDNA3.1-YTHDF2
(Figs. 7B and S1E). As indicated by colony formation
assays, co-transfection of YTHDF3 siRNA and pcDNA3.1-YTHDF2
weakened the proliferation capability of NSCLC cells (Fig. 7C). Additionally, wound healing and
Transwell assays also indicated that co-transfected with YTHDF3
siRNA and pcDNA3.1-YTHDF2 tended to attenuate the migration and
invasion of NSCLC cells (Fig. 7D and
E). In summary, the results suggested that YTHDF2 and YTHDF3
competitively bind to CDC25C mRNA in an m6A-dependent
manner to regulate CDC25C expression.
Validating the regulation of CDC25C by YTHDF2 and
YTHDF3 in vivo to achieve inhibition of tumor growth would
be useful to understand these interactions. However, overexpression
of YTHDF2 and knockdown of YTHDF3 inhibited the proliferation of
cells. Since the previous experimental results demonstrated that
knockdown of YTHDF3 alone promoted the mRNA expression of CDC25C
and thereby promoted cell proliferation, to further determine
whether it is the overexpression of YTHDF2 that inhibits cell
proliferation and leads to the inability to obtain a stable
Lv-YTHDF2 cell line, a stable shYTHDF2 cell line was constructed
for mRNA sequencing (Figs. S1F and
S5A). Through KEGG enrichment analysis, it was found that genes
related to the TGF-β signaling pathway were upregulated compared
with the control group (Fig.
S5B). In the KEGG enrichment analysis results, it was also
observed that genes related to the NOD-like receptor signaling
pathway were downregulated (Fig.
S5C). Hence, it was hypothesized that overexpression of YTHDF2
might suppress cell proliferation via the TGF-β and NOD-like
receptor pathways.
After uncovering the transcriptional regulation of
CDC25C by STAT3 and the targeted degradation of CDC25C by YTHDF2,
it was also hypothesized that STAT3 might interact with
m6A-modified proteins. If it had been possible to
disclose the crosstalk between STAT3 and m6A-related
proteins in regulating CDC25C, it would constitute a highlight of
the present study. Hence, MeRIP-seq and RNA-seq of shYTHDF2 cells
were conducted. Regrettably, no significant differences were
observed in STAT3 m6A modification and mRNA expression
in the sequencing results. Nevertheless, our sequencing outcomes
indicated that knockdown of YTHDF2 influenced the mRNA expression
of STAT1, a member of the STAT family, and the MeRIP results
demonstrated that STAT1 underwent hypermethylation after YTHDF2
expression was knocked down (Table
SIV).
Discussion
Significant progress has been made in basic and
clinical research regarding NSCLC treatment, and treatment methods
have been continuously innovated. However, the 5-year mortality
rate of patients remains high due to the unclear pathogenesis of
lung cancer, which has become a bottleneck in clinical treatment.
Transcriptional regulation and post-transcriptional modification
are crucial mechanisms governing gene expression. Certain studies
have indicated a connection between m6A modification and
the progression of lung cancer (23,24). Aberrations in these processes have
been increasingly recognized as pivotal factors contributing to
cancer pathogenesis. The results of the present study indicated
that the transcriptional activator, STAT3, directly interacts with
the CDC25C promoter, thereby regulating its expression.
Furthermore, through multi-omics analysis, the genes related to the
post-transcriptional modification (m6A methylation) of
CDC25C were identified. The findings suggested that the
downregulation of ALKBH5 in NSCLC results in a significant increase
in the m6A modification of CDC25C mRNA. The results of
the present study also indicated that YTHDF3 and YTHDF2 compete to
bind to CDC25C mRNA, thereby facilitating or inhibiting its
expression. Thus, the present study discloses that dysregulated
expression of the CDC25C gene in NSCLC is influenced by multiple
regulatory layers encompassing both transcriptional and
post-transcriptional mechanisms.
STAT3 functions as a transcription factor with a
role in numerous biological processes. STAT3 is capable of relaying
signals from outside the cell to the nucleus, subsequently
initiating the transcription of specific target genes (25). Although STAT3 serves various roles
in cancer, increasing evidence indicates that its prolonged
activation promotes the proliferation of cancer cells and is
associated with tumor malignancy (26). Nan et al (26) consider that STAT3 has the
potential to influence the expression of interleukin-6 and
cyclooxygenase-2 in NSCLC with EGFR mutations. The progression of
NSCLC can be further inhibited by pharmacological and genetic
approaches to inhibit the activity of STAT3. In triple-negative
breast cancer, it has been found that STAT3 directly interacts with
a regulatory region of the TNFRSF1A gene, and the expression levels
of TNFRSF1A rely on the activity of STAT3 both in models of
constitutive and cytokine-induced activation (27). In light of the crucial role of
STAT3 in tumorigenesis and development, targeting STAT3 for drug
development has emerged as one of the primary drug research focuses
over the past two decades. Numerous compounds have been reported to
possess inhibitory activity on STAT3-related pathways and have
entered the clinical research phase, with indications encompassing
liver, colorectal and lung cancer. Owing to the complex mechanisms
of tumorigenesis, the simultaneous inhibition of multiple targets
through drug combination constitutes a notable anticancer strategy.
Histone deacetylases (HDACs) represent a class of epigenetic
enzymes closely associated with tumorigenesis. A previous
investigation demonstrated that the inhibition of HDAC will result
in the compensatory activation of the related drug target, STAT3,
in breast cancer via a cascade of reactions (28). Hence, designing STAT3-HDAC
dual-target inhibitors can enhance the therapeutic efficacy against
tumors. A STAT3-HDAC dual-target inhibitor termed 'Compound 14' has
exhibited high anti-proliferative capacity and favorable antitumor
activity in both in vitro and in vivo experiments.
Another study has indicated that the selective STAT3 inhibitor,
WB436B, can significantly restrain the growth and metastasis of
pancreatic cancer and prolong the survival time of tumor-bearing
mice, furnishing a solid experimental foundation for STAT3 as a
drug target for the treatment of pancreatic cancer (29). It was shown in the present study
that STAT3, as a transcriptional activator of CDC25C, affects
CDC25C mRNA expression. It was also demonstrated that BP-1-102 (a
specific inhibitor of STAT3 activity) effectively suppressed the
proliferation, invasion and migration of NSCLC cells. Therefore,
the results of the present study indicated that STAT3 promotes
NSCLC development by regulating CDC25C expression.
In the present study, the transcriptional regulation
of CDC25C was first explored. Subsequently, the
post-transcriptional regulation of CDC25C expression by
m6A modification was further investigated. Studies have
demonstrated that the onset of lung cancer is attributed to
multiple factors, among which epigenetics is a significant
component. In recent years, the in-depth research on epigenetics in
the occurrence and development of cancer has offered novel
perspectives for clinical prevention, diagnosis, treatment, and the
development of new drugs. Regarding the early diagnosis, prognosis
evaluation and treatment monitoring of lung cancer, m6A
modification can be regarded as a potential and valuable tumor
diagnostic biomarker. Research has revealed that multiple
m6A regulatory proteins have crucial roles in lung
cancer. For instance, methyltransferases such as
methyltransferase-like (METTL3) (30) and METTL14 (31), along with methylation-reading
proteins such as YTHDF2 and insulin like growth factor 2 mRNA
binding protein (32), all
participate in processes such as the proliferation, migration,
invasion and drug resistance of lung cancer cells. By targeting
these m6A regulatory proteins, specific inhibitors or
agonists can be developed to regulate the m6A
modification levels to inhibit the growth and metastasis of lung
cancer cells, or to enhance their sensitivity to chemotherapy and
immunotherapy. With the successful clinical application of
epigenetic therapeutic drugs such as methyltransferase inhibitors
and HDAC inhibitors, m6A therapy has demonstrated
notable potential in the treatment of lung cancer (33). Moreover, m6A therapy
can be combined with other lung cancer treatment approaches (such
as surgery, radiotherapy, chemotherapy and immunotherapy) to
enhance the therapeutic effect and the survival rate of patients.
For instance, incorporating METTL3-targeted inhibitors with
immunotherapy can render patients with NSCLC resistant to
anti-programmed cell death protein-1 immunotherapy sensitive again
and reduce the side effects and drug resistance (34). Although m6A
modification has exhibited immense potential in the diagnosis and
treatment of NSCLC, challenges still persist in clinical
applications. The heterogeneity of NSCLC results in variations in
m6A modification among individual patients, augmenting
the complexity of individualized treatment. Furthermore, the
majority of current studies are conducted at the cellular and
animal levels, and clinical trials require longer follow-up periods
and larger research cohorts for further validation. The
transformation of basic research achievements into clinical
application also requires strict clinical trials to verify the
safety and efficacy of the treatment strategies.
ALKBH5 is among the earliest discovered
m6A demethylases and plays a pivotal role in a range of
human diseases, particularly cancer development, through its
demethylation function (35-37). ALKBH5 has been shown to have a
m6A demethylase function, but its target and mechanism
of action vary among different tumor types. In bladder cancer,
ALKBH5 regulates the level of m6A modification of target
mRNA and thus affects the epithelial-mesenchymal transition of
tumor cells, interfering with tumor invasion and metastasis
(38). In glioblastoma and breast
cancer, ALKBH5 promotes the maintenance of tumor stem cells by
removing m6A modifications from FOXM1 and NANOG
(39,40). In the present study, to further
elucidate the effects of ALKBH5 on the proliferation and invasion
activities of NSCLC cells, CCK-8, clonogenic and Transwell assays
were conducted. The research findings indicated that low expression
of ALKBH5 in NSCLC cells promoted cell proliferation, invasion and
migration. Furthermore, the present study demonstrated that ALKBH5
regulates the m6A level of CDC25C.
As m6A reader proteins, the YTHDF
protein family, which includes YTHDF1, YTHDF2 and YTHDF3, fulfill
distinct functional roles. YTHDF1 and YTHDF3 are involved in the
regulation of translation, whereas YTHDF2 primarily promotes the
degradation of m6A-methylated transcripts (41). Certain studies have indicated that
the targeting of YTHDF3 to programmed death-ligand 1 facilitates
immune evasion in NSCLC (42). In
triple-negative breast cancer, high expression of YTHDF3 is
associated with a poorer disease-free survival and overall survival
in patients (43). The findings
of the present study suggest that YTHDF3 increases the translation
of CDC25C mRNA in a manner dependent on m6A, further
promoting the progression of NSCLC. In pancreatic cancer cells,
there is a phenomenon of 'migration-proliferation dichotomy' of
YTHDF2, meaning that it promotes the proliferation of pancreatic
cancer cells and inhibits migration and invasion (44). In cervical cancer, knockdown of
YTHDF2 can significantly enhance the stability of GLI family zinc
finger 2 mRNA, further inducing the apoptosis of cervical cancer
cells (45). In prostate cancer,
YTHDF2 binds to the serine protease 8 mRNA and promotes its
degradation in a m6A-dependent manner, thereby promoting
prostate cancer cell proliferation (46). These findings demonstrate that
YTHDF2 exhibits distinct roles across various cancer tissues. The
results of the present study indicated that the downregulation of
ALKBH5 in NSCLC results in an increased m6A modification
of CDC25C mRNA. Moreover, heightened expression of YTHDF3 was more
likely to recognize CDC25C mRNA, thereby promoting its translation
and consequently facilitating the progression of NSCLC. Conversely,
knocking down YTHDF3 expression led to a higher likelihood of
YTHDF2 binding to CDC25C, which may promote its degradation and
inhibit the proliferation and migration of NSCLC cells. In summary,
while YTHDF2 may be capable of mediating the degradation of CDC25C
mRNA, it is more probable that the m6A site on CDC25C
mRNA is preferentially recognized by YTHDF3, thereby facilitating
its translation.
The present study uncovered the molecular process
of m6A alteration and the role of CDC25C, which may be
governed by ALKBH5 and YTHDF proteins, in managing NSCLC tumor
progression. Validating the regulation of CDC25C by YTHDF2 and
YTHDF3 in vivo to achieve inhibition of tumor growth would
be useful to understand these interactions. However, overexpression
of YTHDF2 and knockdown of YTHDF3 inhibited the proliferation of
cells. As a result, it was arduous to obtain a sufficient number of
stably transfected cell lines for subcutaneous xenograft tumor
experiments. In the tumor microenvironment, TGF-β is capable of
regulating the growth and metastasis of tumors through multiple
pathways, such as maintaining the homeostasis of tumor stem cells
(47), inhibiting immune
responses (48) as well as
inducing epithelial-mesenchymal transition and metastasis (49). Therefore, it was hypothesized that
overexpression of YTHDF2 inhibits TGF-β, thereby suppressing tumor
growth and metastasis. Studies have shown that the TGF-β signaling
pathway plays different roles in early-stage and late-stage tumors.
In early-stage tumors, TGF-β exerts an antitumor effect by
facilitating cell cycle arrest and apoptosis, whereas in advanced
tumors, it promotes the survival of tumor cells by creating an
immunosuppressive milieu (50,51). A stable shYTHDF2 cell line was
constructed for mRNA sequencing. Through KEGG enrichment analysis,
it was found that genes related to the TGF-β signaling pathway were
upregulated compared with the control group. It was also noticed
that genes related to the NOD-like receptor signaling pathway were
downregulated. The relationship between NOD-like receptors, as one
of the representatives of inflammatory immune receptors, and tumors
merits exploration. Correia et al (52) verified that NOD1 promotes the
apoptosis of breast cancer cells via the TNF signaling pathway.
Couturier-Maillard et al (53) demonstrated that in mice, a
deficiency in NOD2 causes intestinal ecological imbalance,
increasing the chances of transmissible colitis and colitis-related
carcinogenesis. Antibiotics or anti-IL-6 neutralizing antibodies
can ameliorate the aforementioned conditions. The transplantation
of feces from NOD2 knockout mice to other mice and subsequent
treatment with DSS-AOM resulted in higher body weight loss and
histological scores compared with mice in the other groups.
Therefore, it is inferred that NOD2 exerts a protective effect. It
was conjectured that overexpression of YTHDF2 might suppress cell
proliferation via the TGF-β and NOD-like receptor pathways, but
this requires further experimental validation.
The present study clarified the regulatory
mechanisms of CDC25C high expression in NSCLC from both the
post-transcriptional modification and transcriptional regulation
perspectives. Our next study will focus on the interactions between
the STAT family and the YTHDF family to reveal the potential role
of transcriptional and post-transcriptional regulation. ChIP-seq
analysis for STAT3 binding would be useful to provide direct
molecular evidence. ChIP-seq can be employed using samples from all
species with known genomic sequences to study the interaction
between any DNA-related protein and its target DNA and thus acquire
precise sequence information for each fragment. One of the most
significant advantages of ChIP-seq technology is its capacity to
achieve single-base pair resolution. Investigating whether
transcriptional regulation and post-transcriptional modification
are mutually influenced will be the authors' focus in the next
stage of research.
In conclusion, the results of the present study
indicated that BP-1-102 deactivated the transcriptional function of
STAT3 to downregulate CDC25C expression. ALKBH5 inhibited invasion
and metastasis by regulating CDC25C in a m6A-dependent
manner during NSCLC progression. Promoting the degradation of
CDC25C by YTHDF2 and inhibiting the transcription of CDC25C by
STAT3 may be potential treatment avenues for NSCLC.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. The data generated in the
present study may be found in the National Center for Biotechnology
Information under the accession numbers SRP553805 and SRP553846 or
at the following URL: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1203003;
https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1203088.
Authors' contributions
YZ and YW designed the study. YZ drafted the
manuscript. YZ, WM and KW performed the experiments. YZ, KW, HL, GZ
and XH participated in data analysis. YZ and YW were involved in
the amendment of the manuscript. GZ and XH provided funding
support. All authors read and approved the final version of the
manuscript. YZ and YW confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
All the human lung cancer and normal lung specimens
were collected at Southern University of Science and Technology
Hospital (Shenzhen, China) with written consent from the patients
(including for the utilization of their tissue for research) and
approval from the Institute Research Ethics Committee (approval no.
008 of 2020). The nude mouse tumorigenesis experiment was conducted
in accordance with the guidelines and approved (approval no.
SUSTech-JY202 0016-1) by the Ethics Committee of Southern
University of Science and Technology (Shenzhen, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present was supported by the Stability Support Plan for
Higher Education Institutions in Shenzhen (grant no.
20200925160201001), the Shenzhen Science and Technology Planning
Project (grant no. JCYJ202205301152 03008), the Dean project of
Southern University of Science and Technology Hospital (grant no.
2020-A2) and the Technology Major Project of Nanshan District
Health System (grant no. NSZD2023-063).
References
|
1
|
Del Campo JM, Maroto S, Sebastian L,
Sebastian L, Vaillo X, Bolufer S, Lirio F, Sesma J and Galvez C:
Uniportal VATS for diagnosis and staging in non-small cell lung
cancer (NSCLC). Diagnostics (Basel). 13:8262023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mathieu LN, Larkins E, Sinha AK,
Mishra-Kalyani PS, Jafri S, Kalavar S, Ghosh S, Goldberg KB, Pazdur
R, Beaver JA and Singh H: FDA approval summary: Atezolizumab as
adjuvant treatment following surgical resection and platinum-based
chemotherapy for stage II TO IIIA NSCLC. Clin Cancer Res.
29:2973–2978. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen Y, Li H, Zhang W, Qi W, Lu C, Huang
H, Yang Z, Liu B and Zhang L: Sesamin suppresses NSCLC cell
proliferation through cyclin D1 inhibition-dependent cell cycle
arrest via Akt/p53 pathway [Toxicology and applied pharmacology,
387 (2020) 114848]. Toxicol Appl Pharm. 400:1150482020. View Article : Google Scholar
|
|
4
|
Yang T, Xiao Y, Liu S, Luo F, Tang D, Yu Y
and Xie Y: Isorhamnetin induces cell cycle arrest and apoptosis by
triggering DNA damage and regulating the AMPK/mTOR/p70S6K signaling
pathway in doxorubicin-resistant breast cancer. Phytomedicine.
114:1547802023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li R, Zheng C, Shiu PH, Rangsinth P, Wang
W, Kwan YW, Wong ES, Zhang Y, Li J and Leung GP: Garcinone E
triggers apoptosis and cell cycle arrest in human colorectal cancer
cells by mediating a reactive oxygen species-dependent JNK
signaling pathway. Biomed Pharmacother. 162:1146172023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bazzaz R, Bijanpour H, Pirouzpanah SMB,
Yaghmaei P and Rashtchizadeh N: Adjuvant therapy with
γ-tocopherol-induce apoptosis in HT-29 colon cancer via
cyclin-dependent cell cycle arrest mechanism. J Biochem Mol Toxic.
33:e223992019. View Article : Google Scholar
|
|
7
|
Tao L, Cao YZ, Wei ZH, Jia Q, Yu S, Zhong
J, Wang A, Woodgett JR and Lu Y: Xanthatin triggers Chk1-mediated
DNA damage response and destabilizes Cdc25C lysosomal degradation
in lung cancer cells. Toxicol Appl Pharm. 337:85–94. 2017.
View Article : Google Scholar
|
|
8
|
Wang JN, Zhang ZR, Che Y, Yuan ZY, Lu ZL,
Li Y, Li N, Wan J, Sun HD, Sun N, et al: Acetyl-macrocalin B, an
-kaurane diterpenoid, initiates apoptosis through the
ROS-p38-caspase 9-dependent pathway and induces G2/M phase arrest
via the Chk1/2-Cdc25C-Cdc2/cyclin B axis in non-small cell lung
cancer. Cancer Biol Ther. 19:609–621. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Senju M, Sueoka N, Sato A, Iwanaga K,
Sakao Y, Tomimitsu S, Tominaga M, Irie K, Hayashi S and Sueoka E:
Hsp90 inhibitors cause G2/M arrest associated with the reduction of
Cdc25C and Cdc2 in lung cancer cell lines. J Cancer Res Clin.
132:150–158. 2006. View Article : Google Scholar
|
|
10
|
Zhou Q and Chen T: BI6727, a polo-like
kinase 1 inhibitor, synergizes with gefitinib to suppress
hepatocellular carcinoma cells via a G2/M arrest mechanism.
Pharmazie. 77:230–235. 2022.PubMed/NCBI
|
|
11
|
Zhao Y, Wu Z, Zhang Y and Zhu L: HY-1
induces G2/M cell cycle arrest in human colon cancer cells through
the ATR-Chk1-Cdc25C and weel pathways. Cancer Sci. 104:1062–1066.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu K, Zheng MY, Lu R, Du J, Zhao Q, Li Z,
Li Y and Zhang S: The role of CDC25C in cell cycle regulation and
clinical cancer therapy: A systematic review. Cancer Cell Int.
20:2132020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang M, Hu X, Tang B and Deng FM:
Exploring the interplay between methylation patterns and non-coding
RNAs in non-small cell lung cancer: Implications for pathogenesis
and therapeutic targets. Heliyon. 10:e248112024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He Y, Hu H, Wang YD, Yuan H, Lu Z, Wu P,
Liu D, Tian L, Yin J, Jiang K and Miao Y: ALKBH5 inhibits
pancreatic cancer motility by decreasing long non-coding RNA
KCNK15-AS1 methylation. Cell Physiol Biochem. 48:838–846. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang P, Wang Q, Liu A, Zhu J and Feng J:
ALKBH5 holds prognostic values and inhibits the metastasis of colon
cancer. Pathol Oncol Res. 26:1615–1623. 2020. View Article : Google Scholar
|
|
16
|
Gao Y, Pei G, Li D, Li R, Shao Y, Zhang QC
and Li P: Multivalent m6A motifs promote phase
separation of YTHDF proteins. Cell Res. 29:767–769. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sheikhshabani SH, Modarres P,
Ghafouri-Fard S, Amini-Farsani Z, Khodaee L, Shaygan N,
Amini-Farsani Z and Omrani MD: Meta-analysis of microarray data to
determine gene indicators involved in cisplatin resistance in
non-small cell lung cancer. Cancer Rep (Hoboken). 7:e19702024.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Koh YW, Han JH, Haam S and Lee HW:
Prognostic and predictive value of YTHDF1 and YTHDF2 and their
correlation with tumor-infiltrating immune cells in non-small cell
carcinoma. Front Oncol. 12:9966342022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wen J, Xue L, Wei Y, Liang J, Jia W, Yong
T, Chu T, Li H, Han S, Liao J, et al: YTHDF2 is a therapeutic
target for HCC by suppressing immune evasion and angiogenesis
through ETV5/PD-L1/VEGFA Axis. Adv Sci (Weinh). 11:e23072422024.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun S, Liu Y, Zhou M, Wen J, Xue L, Han S,
Liang J, Wang Y, Wei Y, Yu J, et al: PA2G4 promotes the metastasis
of hepatocellular carcinoma by stabilizing FYN mRNA in a
YTHDF2-dependent manner. Cell Biosci. 12:552022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fang Y, Wu X, Gu Y, Shi R, Yu T, Pan Y,
Zhang J, Jing X, Ma P and Shu Y: LINC00659 cooperated with ALKBH5
to accelerate gastric cancer progression by stabilising JAK1 mRNA
in an m6A-YTHDF2-dependent manner. Clin Transl Med.
13:e12052023. View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Wang M, Liu Z, Fang X, Cong X and Hu Y:
The emerging role of m6A modification of non-coding RNA
in gastrointestinal cancers: A comprehensive review. Front Cell Dev
Biol. 11:12645522023. View Article : Google Scholar
|
|
24
|
Jin Z, Sheng J, Hu Y, Zhang Y, Wang X and
Huang Y: Shining a spotlight on m6A and the vital role
of RNA modification in endometrial cancer: A review. Front Genet.
14:12473092023. View Article : Google Scholar
|
|
25
|
Devarajan E and Huang S: STAT3 as a
central regulator of tumor metastases. Curr Mol Med. 9:626–633.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee H, Jeong AJ and Ye SK: Highlighted
STAT3 as a potential drug target for cancer therapy. BMB Rep.
52:415–423. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Egusquiaguirre SP, Yeh JE, Walker SR, Liu
S and Frank DA: The STAT3 target gene TNFRSF1A modulates the NF-κB
pathway in breast cancer cells. Neoplasia. 20:489–498. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ren Y, Li S, Zhu R, Wan C, Song D, Zhu J,
Cai G, Long S, Kong L and Yu L: Discovery of STAT3 and histone
deacetylase (HDAC) dual-pathway inhibitors for the treatment of
solid cancer. J Med Chem. 64:7468–7482. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen H, Zhou W, Bian A, Zhang Q, Miao Y,
Yin X, Ye J, Xu S, Ti C, Sun Z, et al: Selectively targeting STAT3
using a small molecule inhibitor is a potential therapeutic
strategy for pancreatic cancer. Clin Cancer Res. 29:815–830. 2023.
View Article : Google Scholar
|
|
30
|
Bai W, Xiao G, Xie G, Chen Z, Xu X, Zeng J
and Xie J: METTL3/IGF2BP1 influences the development of
non-small-cell lung cancer by mediating m6A methylation
modification of TRPV1. Thorac Cancer. 15:1871–1881. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ji X, Wan X, Sun H, Deng Q, Meng S, Xie B
and Zhou S: METTL14 enhances the m6A modification level
of lncRNA MSTRG.292666.16 to promote the progression of non-small
cell lung cancer. Cancer Cell Int. 24:612024. View Article : Google Scholar
|
|
32
|
Lu H, Ai J, Zheng Y, Zhou W, Zhang L, Zhu
Z, Zhang H and Wang S: IGFBP2/ITGA5 promotes gefitinib resistance
via activating STAT3/CXCL1 axis in non-small cell lung cancer. Cell
Death Dis. 15:4472024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang KL, Yeh TY, Hsu PC, Wong TH, Liu JR,
Chern JW, Lin MH and Yu CW: Discovery of novel anaplastic lymphoma
kinase (ALK) and histone deacetylase (HDAC) dual inhibitors
exhibiting antiproliferative activity against non-small cell lung
cancer. J Enzym Inhib Med Chem. 39:23186452024. View Article : Google Scholar
|
|
34
|
Yu HS, Liu J, Bu X, Ma Z, Yao Y, Li J,
Zhang T, Song W, Xiao X, Sun Y, et al: Targeting METTL3 reprograms
the tumor microenvironment to improve cancer immunotherapy. Cell
Chem Biol. 31:776–791.e7. 2024. View Article : Google Scholar
|
|
35
|
Wang Q, Huang YF, Jiang M, Tang Y, Wang Q,
Bai L, Yu C, Yang X, Ding K, Wang W, et al: The demethylase ALKBH5
mediates ZKSCAN3 expression through the m6A modification
to activate VEGFA transcription and thus participates in
MNNG-induced gastric cancer progression. J Hazard Mater.
473:1346902024. View Article : Google Scholar
|
|
36
|
Song K, Cao Q, Yang Y, Zuo Y and Wu X:
ALKBH5 modulates bone cancer pain in a rat model by suppressing
NR2B expression. Biotechnol Appl Biochem. 71:1105–1115. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang S, Zhu X, Hao Y, Su TT and Shi W:
ALKBH5-mediated m6A modification of circFOXP1 promotes
gastric cancer progression by regulating SOX4 expression and
sponging miR-338-3p. Commun Biol. 7:5652024. View Article : Google Scholar
|
|
38
|
Tang B, Yang Y, Kang M, Wang Y, Wang Y, Bi
Y, He S and Shimamoto F: m6A demethylase ALKBH5 inhibits
pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation
and mediating Wnt signaling. Mol Cancer. 19:32020. View Article : Google Scholar
|
|
39
|
Zhang C, Samanta D, Lu H, Bullen JW, Zhang
H, Chen I, He X and Semenza GL: Hypoxia induces the breast cancer
stem cell phenotype by HIF-dependent and ALKBH5-mediated
m6 A-demethylation of NANOG mRNA. P Natl Acad Sci USA.
113:E2047–E2056. 2016. View Article : Google Scholar
|
|
40
|
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S,
Lu Z, Chen Y, Sulman EP, Xie K, Bögler O, et al: The m6A
hallmark of cancer: RNA demethylase ALKBH5 maintains tumorigenicity
of glioblastoma stem-like cells by sustaining FOXM1 expression and
cell proliferation. Cancer Res. 31:591–606.e6. 2017.
|
|
41
|
Jin D, Guo J, Wu Y, Yang L, Wang X, Du J,
Dai J, Chen W, Gong K, Miao S, et al: m6A demethylase
ALKBH5 inhibits tumor growth and metastasis by reducing
YTHDFs-mediated YAP expression and inhibiting
miR-107/LATS2-mediated YAP activity in NSCLC. Mol Cancer.
19:402020. View Article : Google Scholar
|
|
42
|
Luo Y, Zeng C, Ouyang Z, Zhu W, Wang J,
Chen Z, Xiao C, Wu G, Li L, Qian Y, et al: YTH domain family
protein 3 accelerates non-small cell lung cancer immune evasion
through targeting CD8+ T lymphocytes. Cell Death Discov.
10:3202024. View Article : Google Scholar
|
|
43
|
Lin Y, Jin X, Nie Q, Chen M, Guo W, Chen
L, Li Y, Chen X, Zhang W, Chen H, et al: YTHDF3 facilitates
triple-negative breast cancer progression and metastasis by
stabilizing ZEB1 mRNA in an m6 A-dependent manner. Ann
Transl Med. 10:832022. View Article : Google Scholar
|
|
44
|
Chen J, Sun Y, Xu X, Wang D, He J, Zhou H,
Lu Y, Zeng J, Du F, Gong A and Xu M: YTH domain family 2
orchestrates epithelial-mesenchymal transition/proliferation
dichotomy in pancreatic cancer cells. Cell Cycle. 16:2259–2271.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wan F, Qiu F, Deng Y, Hu H, Zhang Y, Zhang
JY, Kuang P, Tian H, Wu D, Min H, et al: Knockdown of YTHDF2
initiates ERS induced apoptosis and cancer stemness suppression by
sustaining GLI2 stability in cervical cancer. Trans Oncol.
46:1019942024. View Article : Google Scholar
|
|
46
|
Zhao X, Lv S, Li N, Zou Q, Sun L and Song
T: YTHDF2 protein stabilization by the deubiquitinase OTUB1
promotes prostate cancer cell proliferation via PRSS8 mRNA
degradation. J Biol Chem. 300:107–152. 2024. View Article : Google Scholar
|
|
47
|
Yoon SH, Lee S, Kim HS, Song J, Baek M,
Ryu S, Lee HB, Moon HG, Noh DY, Jon S and Han W: NSDHL contributes
to breast cancer stem-like cell maintenance and tumor-initiating
capacity through TGF-β/Smad signaling pathway in MCF-7 tumor
spheroid. BMC Cancer. 24:13702024. View Article : Google Scholar
|
|
48
|
Qian J, Jiao Y, Wang G, Liu H, Cao X and
Yang H: Mechanism of TGF-β1 inhibiting Kupffer cell immune
responses in cholestatic cirrhosis. Exp Ther Med. 20:1541–1549.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zeng Q, Shi W, Xie L, Dai Y and Wei C:
TGF-β1/miR-30d-5p axis regulating the expression of EMT key factors
to induce apoptosis of lung cancer cells. Heliyon. 10:e378012024.
View Article : Google Scholar
|
|
50
|
Naik A and Thakur N: Epigenetic regulation
of TGF-βand vice versa in cancers-A review on recent developments.
Biochem Biophys Acta Rev Cancer. 1879:1892192024. View Article : Google Scholar
|
|
51
|
Hu Z, Liu Y, Liu M, Zhang Y and Wang C:
Roles of TGF-β signalling pathway-related lncRNAs in cancer
(Review). Oncol Lett. 25:1072023. View Article : Google Scholar
|
|
52
|
da Silva Correia J, Miranda Y,
Austin-Brown N, Hsu J, Mathison J, Xiang R, Zhou H, Li Q, Han J and
Ulevitch RJ: Nod1-dependent control of tumor growth. Proc Natl Acad
Sci USA. 103:1840–1845. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Couturier-Maillard A, Secher T, Rehman A,
Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T,
Bressenot A, Delanoye-Crespin A, et al: NOD2-mediated dysbiosis
predisposes mice to transmissible colitis and colorectal cancer. J
Clin Invest. 123:700–711. 2013.PubMed/NCBI
|