Contents
Introduction
The current concept of epithelial-mesenchymal
transition
EMT in development and in cancer
Epigenetic regulation of EMT
Therapeutic options targeting epigenetics and
EMT
Summary and future directions
Introduction
Epithelial-mesenchymal transition (EMT) as well the
reverse process of mesenchymal-epithelial transition (MET) is
essential for development and physiological response to injury
(such as wound healing) as well in carcinogenesis (1–3).
Under normal conditions, epithelial cells are linked
together as well as to the extracellular matrix environment by
different types of intercellular junctions (desmosomes, adherens
and tight junctions) enabling tissue maintenance and stability.
Epithelial cells can gain the potency to acquire a mesenchymal
phenotype to allow for physiological circadian tissue changes but
also of tissue loss or damage (4).
Interestingly, this process is also associated with an intermediate
stem cell phenotype, thus reflecting the highly conserved
mechanisms during embryogenesis (5–7).
Over the last few years, the interest in
understanding EMT and MET has significantly increased since we
further understand the essential role of EMT in cancer progression,
particularly during the complex initial processes of tissue
invasion and extravasation (8).
The regulatory mechanisms of EMT have been intensively investigated
and can be described by networks of activating/deactivating
signalling pathways. Furthermore, EMT is additionally influenced
and regulated by epigenetic mechanisms, such as DNA methylation and
histone modifications as well as microRNAs (miRNAs, see below).
This epigenetic regulation is particularly important as it accounts
for the observed reversibility of EMT-associated processes and the
plasticity of (cancer) cells to react upon various external and
internal stimuli.
Taken together, these data highlight the complex
nature of regulations involved in EMT and provide the basis for
development of a new types of drugs specifically targeting EMT in
human cancers. In this context, we provide a concise review of the
current concepts of EMT in human carcinogenesis and an outlook on
therapeutic anti-cancer approaches on the epigenetic level.
The current concept of
epithelial-mesenchymal transition
According to Kalluri and Weinberg (9) the biological process of EMT is
described as follows: i) epithelial cells are tightly integrated in
their cellular environment by tight junctions or desmosomes; ii)
under the influence of different EMT mediators (such as growth
factors or cytokines, discussed in detail below) epithelial cells
gain a mesenchymal status, which iii) is associated with different
biological properties, particularly the ability to invade and
metastasize. EMT refers to a collective series of transcriptional
and post-translational events that cause epithelial cells to take
on mesenchymal features, thus allowing the cells to separate from
the tissue context, lose baso-apical polarity and gain motility
(3,10–12)
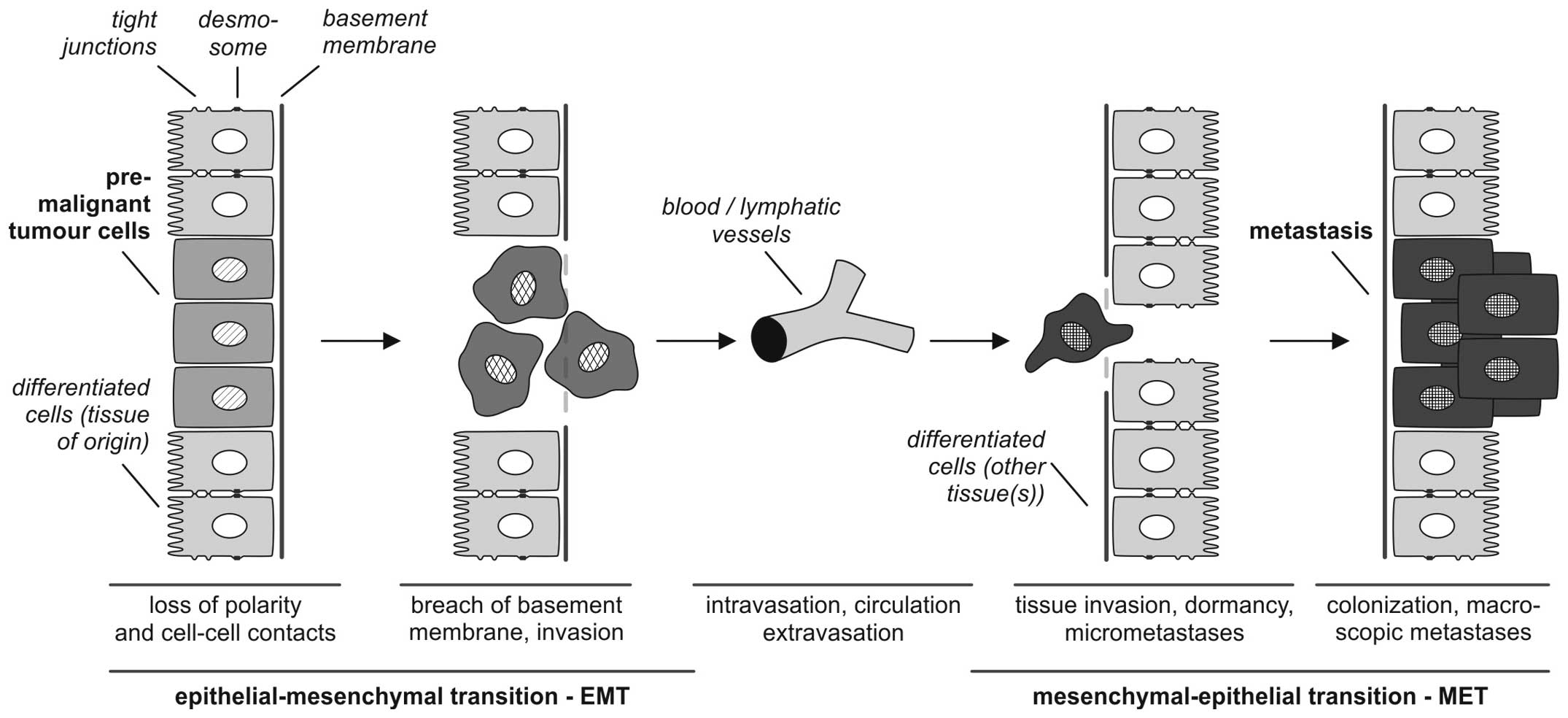
(Fig. 1).
It is of central importance, that EMT processes are
reversible so that mesenchymal cells can undergo MET to
differentiate back to epithelial phenotypes. This reverse
transition plays a key role in the formation of macroscopic
metastases in different organs (13).
For experimental approaches it is important to
characterize the EMT or MET status of tumor cells to investigate
the influence of agonistic or antagonistic acting drugs. Different
markers of extracellular (fibronectin, vitronectin) and cellular
localization (vimentin, E-cadherin) are suitable to identify the
EMT-MET-related differentiation status (1,12,14,15)
(Table I). Cellular markers are
either cytoplasmic membrane proteins (such as E-cadherin, claudins,
occludin, desmoplakin) or cytoplasmic proteins (cytokeratins,
vimentin or mucins). In particular, the epithelial phenotype is
typically characterized by cytokeratin expression which stabilizes
the cytoskeleton of epithelial cells. Additionally, these
cytokeratins hierarchically classify the epithelial differentiation
status depending on the tissue/organ context as described by Moll
et al(16).
 | Table IEMT-related changes in protein
expression pattern (12,14,15). |
Table I
EMT-related changes in protein
expression pattern (12,14,15).
| Upregulation
(mesenchymal markers) | Upregulation
nuclear localisation | Downregulation
(epithelial markers) |
|---|
|
↑N-cadherin | β-catenin |
↓E-cadherin |
|
↑Vimentin | Smad-2/3 | ↓Desmoplakin |
| ↑Fibronectin | NF-κB | ↓Cytokeratin |
| ↑MMPs | Snai1/2, twist | ↓Occludin |
One of the fundamental molecular aspects of EMT in
converting differentiated epithelial tumor cells into
de-differentiated, migratory mesenchymal cells is the repression of
epithelial genes, such as E-cadherin, which results in the loss of
epithelial cell-cell contacts. For tumor progression, the loss of
E-cadherin is a central feature in the early stages of metastasis
(17–21), further supporting the involvement
of an EMT-like process in metastatic tissue invasion. During EMT,
E-cadherin is replaced by N-cadherin, a process referred to as
‘cadherin switching’ (22,23).
Additionally, intermediary filaments, such as
vimentin or smooth muscle cells are used as mesenchymal markers.
The intercellular connection status described by the expression
pattern of E-cadherin, claudins, occludins or desmoplakin indicate
a tissue-integrative epithelial status, whereas the linkage to the
extracellular matrix is mediated by glycoproteins, such as
fibronectin and vitronectins connecting the extracellular matrix
with cellular integrins.
The initiation of the complex process of EMT is
triggered by multiple cellular signaling mechanisms including
‘classical’ developmental pathways, such as Hedgehog, Wnt and
Notch, as well as signaling by growth factors including
transforming growth factor β (TGFβ), fibroblast growth factor
(FGF), epidermal growth factor (EGF), and, platelet-derived growth
factor (PDGF) (1,11,12)
(Table II). Additionally,
epigenetic mechanisms (discussed below) as well as miRNA-based
regulation have been reported (24,25).
Insight into the underlying mechanism of the transcriptional
regulation of EMT came from the initial identification of the
transcription factor, Snai1 (Snail), as a target of the
above-mentioned EMT-promoting signaling pathways, which acts as a
direct transcriptional repressor of the E-cadherin gene (26,27).
In recent years, additional transcription factors have been
identified which repress E-cadherin and mediate the transcriptional
initiation of EMT: zinc finger protein Snai2 (Slug) (28), the two-handed zinc
finger/homeodomain proteins ZEB1 (δEF1 or ZFHX1A) (29) and ZEB2 (SIP1 or ZFHX1B) (30), the basic helix-loop-helix protein
E12/E47 (Tcf3) (31), and Twist
(although it is not clear whether the latter directly binds the
E-boxes within the E-cadherin promoter).
| Table II‘Classical’ contextual EMT-inducing
pathways in human carcinogenesis. |
Table II
‘Classical’ contextual EMT-inducing
pathways in human carcinogenesis.
| Pathway | Cancer
association | Refs. |
|---|
| Bile acids | Hepatobiliary
carcinoma cells | (103,104) |
| Bone morphogenetic
protein | Tumor cells | (105) |
| Environmental and
social factors (Nicotine, ultraviolet light) | Tumor cells | (106,107) |
| Epidermal growth
factor | Tumor cells | (108) |
| Estrogens | Breast and ovarian
cancer | (109,110) |
| Fibroblast growth
factor | Tumor cells | (111) |
| Hepatocyte growth
factor | Liver tumor
cells | (112) |
| Hypoxia/autocrine
motility factor | Ovarian, pancreatic
cancer | (113–115) |
| Integrins | Tumor cells | (116,117) |
| Interleukin-related
protein/Interleukin-6 | Tumor cells | (118,119) |
| Notch | Tumor cells | (105) |
| Platelet-derived
growth factor | Liver and colon
cancer | (120) |
| Prostaglandine
(2)/cyclooxygenase 2 | NSCLC cells | (121,122) |
| Scatter factor | Malignant
mesothelioma | (123,124) |
| Sonic hedgehog | Tumor cells | (105) |
| Transforming growth
factor-β | Tumor cells | (116) |
| Vascular
endothelial growth factor | Tumor cells | |
| WNT | Tumor cells | (125) |
EMT in development and in cancer
EMT represents the intersection of different aspects
of human development which are sequential rather than parallel
processes (2). During the early
phase of human development, EMT is involved in morphogenesis and
stem cell plasticity required for correct implantation,
gastrulation and organogenesis (32,33).
In the adult organism, subsequent processes relying on regulated
EMT or MET are tissue maintenance allowing for reconstruction or
maintenance of tissue, as well as cell homeostasis after
inflammatory or degenerative insults. In the case of chronic
inflammatory and degenerative diseases, such as organ fibrosis, the
EMT/MET system is over-regulated which may lead to organ
insufficiency or failure (34).
Finally, another cancer-related function of EMT was ascribed to
cancer stem cells which are centrally involved in tumor
progression, metastasis and recurrence after therapy (35,36).
The involved molecular mechanisms of EMT are
summarized in the following paragraphs for i) morphogenesis, ii)
chronic diseases and finally for iii) cancer.
EMT in development
In order to enable cells to move to new localities,
EMT (and the opposite process, MET) is a central aspect in the
developing embryo and has been shown to contribute initially to
implantation, gastrulation and subsequently to the development of
somites, chondrocytes, cardiac valves, and to nephrogenesis
(37,38). The associated molecular steps
regulating EMT are highly conserved: as mentioned above, the key
players of EMT are the transcription factors, Snail, Twist and ZEB
and their important repressor target, E-cadherin. The primary goals
of all these EMT-related processes are loss of cell-cell adhesion
and polarity and changes in the cell shape, as well as enhanced
cell motility and ‘invasiveness’ during embryonic development for
organ maturation as reviewed in detail by Thiery et
al(2). Although the upstream
regulatory inputs seem heterogeneous at a first glance, some of the
master pathways [such as Hedgehog, Wnt, TGF-β/bone morphogenetic
protein (BMP), FGF and EGF] involved in cancer associated-EMT
(Table II) also govern EMT during
the different phases of embryogenesis. This emphasizes the
biological robustness of these pathways (7) and supports the theory that cancer may
be viewed as a deregulated program of development (39). Therefore, it is important to
further investigate the role of EMT/MET-related processes in
development as this knowledge may be transferred to
pathophysiological states, such as chronic disease and
carcinogenesis which may subsequently aid in the development of new
therapeutic approaches.
EMT in chronic diseases
Our knowledge regarding EMT in chronic diseases has
increased over the last few years, leading to a new comprehension
of chronic diseases. Again, since physiological regeneration and
reparation share the same molecular mechanism as EMT/MET in
development, it is fascinating to hypothesize that EMT/MET may also
play a role in chronic disease caused by over-regulated
regeneration and inflammation. This idea is supported by different
cellular tracing studies indicating that chronic disease-related
interstitial fibrosis is produced by myofibroblasts derived not
only from orthotopic fibroblasts, but from epithelial cells via EMT
(2). For example, hepatocytes or
alveolar epithelial cells differentiate into myofibroblastic cells
during carbon tetrachloride (CCl4)-induced liver fibrosis or TGF-β
treatment, respectively (40,41).
Furthermore, endothelial and mesothelial cells have the potency to
trans-differentiate into mesenchymal cells relevant for cardiac
(42), renal (43) or peritoneal fibrosis (44). As another example, we have
previously demonstrated that vascular smooth muscle cells exhibit a
reverse molecular epithelial phenotype in human atherosclerosis
associated with progressive atherosclerotic lesions (45). The major driving force behind such
a type of chronic disease-related fibrosis is TGF-β signaling and
the Snail cascade which may be inhibited by Smad7 gene transfer
(46), as well as a systemic
vitamin D analogue (47) or BMP-7
application in vivo(48).
This observation may be useful for future therapeutic approaches
aiming at protection from progressive organ fibrosis and the
associated end stage organ failure.
EMT in cancer
As recently reviewed in depth by Brabletz (8), the de-differentiation processes
mediated by EMT are now accepted as a hallmark of cancer. EMT plays
a key role in the initial steps of tumor cell dissemination and
metastasis. In this context, EMT is related to a current concept of
cancer stem cell, i.e. ‘migrating cancer stem cells’ [as termed by
Brabletz et al(5) and Jung
et al(49)]. In these
models, EMT enables cancer cells to trans-differentiate to
mesenchymal cancer cells accompanied by the induction of stem
cell-like properties.
Typically, EMT is found locally at the tumor front
with a characteristically increased expression of vimentin
paralleled by a loss of E-cadherin (50,51).
Since EMT is not always obvious in tumor specimens due to the
enhanced stromal cellularity at the tumor margin, the relevance of
EMT is still under debate (2).
Nevertheless, experimental and clinical data on solid tumors, such
as breast, colorectal and ovarian carcinoma have revealed that the
overexpression of the classical transcription markers, SNAIL1 and
SNAIL2, is associated with a worse outcome in terms of relapse or
survival (52–54). Additionally, the inhibition of EMT
signaling pathways can enhance the efficiency of ‘classical’
targeted therapy regimes in the experimental setting of hepatic,
pancreatic or lung cancer cells (2,55,56).
Therefore, detailed topographic analysis of the distribution of EMT
markers within the tumor specimen should be carried out for a
better prognostic and predictive stratification of cancer
patients.
As reviewed by Thiery et al(2) and Brabletz (8), the molecular EMT ‘machinery’ is
synergistically and reciprocally regulated together with other
control instances, such as the EMT-inhibiting miRNA-200 and
miRNA-34 families influencing differentiation, stemness,
proliferation and drug sensitivity. Additionally, the expression of
these EMT/MET inducers or inhibitors is under the contextual
control of the environment as summarized in Table II.
Taken together, the triggering pathways mentioned in
Table II induce Snail gene
expression, in turn leading to the repression of E-cadherin by the
phosphatidylinositol-3 kinase (PI3K)/mitogen-activated protein
kinase (MAPK), Smad, RTK, Notch, β-catenin and glioma-associated
oncogene (GLI) signaling cascades, thus further illustrating the
complexity of autocrine and paracrine growth factor signaling
crosstalk during carcinogenesis and EMT (2,6,11).
For the therapeutic exploitation of these results,
different approaches are possible: at the first glance, the EMT
transcription factors, TWIST, Snail and the ZEB family, may be
targeted to inhibit the EMT process during tumor progression.
However, the pharmaceutical potency of available low molecular
weight drugs has not been sufficient until now (2,57).
RNA interference techniques may represent a promising approach to
repress these transcription factors on the mRNA level; however, the
in vivo stability and transfer efficiency of this drug
technology requires further investigation and optimization
(2).
For these reasons, another interesting therapeutic
approach may be to target the EMT inducers through small molecular
weight inhibitors which have already yielded promising results in
an in vitro setting (58).
The hierarchical regulatory role of EMT inducers depending on the
cancer type should be used as the basis for rational drug
selection. Based on the intertwined relationship between EMT
processes and cancer stem cells, direct targeting of the latter may
also manage the disease-related aspects of EMT in cancer. As an
example, a promising CSC-targeting drug, salinomycin, was isolated
by Gupta et al form a library of 16,000 small molecules
(59) and has yielded interesting
preclinical results in several tumor entities (60–64).
Additionally, a systematic approach to influence EMT
in cancer progression involves modulating the epigenetic regulation
of EMT: in the early 80s Jones et al demonstrated that the
differentiation status of cultured cells may effectively be
influenced by 5-azacytidine, a hypomethylating agent (65,66).
Additionally, it has been shown that the histone-associated
chromatin structure, as well as the DNA methylation pattern
influence the EMT transcriptional regulation of E-cadherin
(26,27,67,68)
(described in detail in the following chapter).
Epigenetic regulation of EMT
Epigenetic regulatory mechanisms refer to a series
of stable but reversible modifications not directly affecting the
DNA primary sequence but rather rely on dynamic transcriptional
programming effects. Such heritable regulations in the pattern of
gene expression are mediated by the DNA methylation of CpG
dinucleotides and several post-transcriptional covalent
modifications of the NH2 terminal of histone proteins, including
acetylation, biotinylation, methylation, phosphorylation and
SUMOylation (69). As a general
rule, DNA methylation, the di- and trimethylation of H3 lysine 9
(H3K9) and the trimethylation of H3K27 cause chromatin condensation
leading to gene silencing mediated by heterochromatin 1 (HP1) and
polycomb group (PcG) proteins (70). Several epigenetic events such as
global hypomethylation, specific hypermethylation at CpG islands
(71,72), as well as aberrations in the
histone modification landscape [‘histone onco-modifications’
(73)] have been specifically
associated with carcinogenesis. This chapter describes the
particular findings on how the EMT and EMT-related markers are
regulated via epigenetic events. An overview including some of the
epigenetic regulatory mechanisms involved in the control of EMT/MET
is presented in Fig. 2.
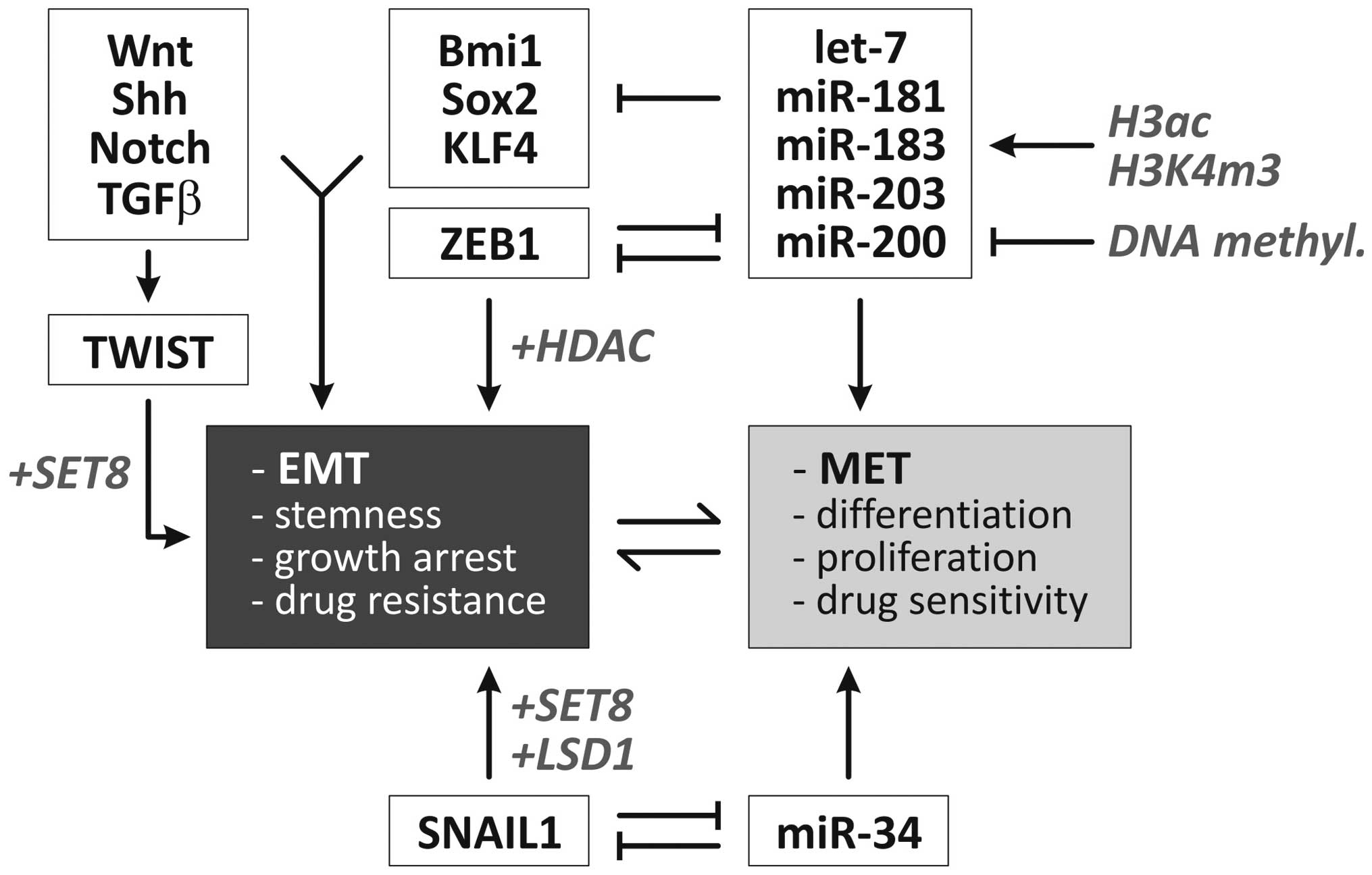 | Figure 2Overview of molecular regulators of
EMT. Two negative feedback loops are centrally involved in the
dynamic regulation of epithelial vs. mesenchymal cell phenotypes
(1,2). Additionally, specific microRNAs and
developmental signaling pathways are involved in the regulation of
EMT (6). Factors/processes
involved in epigenetic control of these pathways/factors are
highlighted in gray/italicised (6,16,126–131). Ac, acetylation; EMT,
epithelial-mesenchymal transition; H, histone; HDACi, histone
deacetylase inhibitor; MET, mesenchymal-epithelial transition; miR,
microRNA; m3, trimethylation; DNMTi, DNA methyltransferase
inhibitor; Shh, sonic hedgehog. |
Multiple epigenetic mechanisms have previously been
described that act during the EMT program in the repression of
epithelial markers and the conversion of epithelial cells into
aggressive, invasive tumor cells. In oral carcinoma cells,
hypermethylation at the CDH1 promoter inversely correlates with the
expression of E-cadherin and treatment with a demethylating agent
(5-azacytidine) causes the re-expression of E-cadherin in cell
lines which do not express the SIP1 E-cadherin repressor (74). Similar results have been found in
breast tumor cells where CDH1 promoter hypermethylation rather than
mutational inactivation caused the reduced expression of
E-cadherin. The expression profile of the cell lines complied with
fibroblastic (mesenchymal) morphology and CDH1 promoter
hypermethylation (75).
A set of transcription factors has been
mechanistically linked to the induction of the EMT program,
including Twist, Snai1 (Snail), Snai2 (Slug) as mediators of the
molecular alterations occurring during EMT (76). In several model systems, epigenetic
modifications have been shown to contribute to the repressive
function of these transcription factors on epithelial genes. As
shown by Lin et al(77),
Snai1 recruits the histone demethylase lysine-specific demethylase
1 (LSD1) (KDM1A, AOF2) which removes dimethylation of Lys4 on
histone H3 (H2K4m2) and mediates the transcriptional repression of
Snai1 target genes, such as CDH1. The short-hairpin RNA-mediated
depletion of LSD1 results in partial re-expression of epithelial
genes associated with increased levels of H3K4m2 at the CDH1
promoter. These EMT-inducing transcription factors also interact
with HDAC1, HDAC2 and the co-repressor mSin3A (74) via their SNAG N-terminal domain as
well as polycomb protein repressive complex (PRC2) (77) and cause epigenetic silencing of the
CDH1 promoter. Additionally, Yang et al(78) demonstrated that the Twist
transcription factor interacts with the monomethyltransferase SET8
which can function both as a repressor (78) or inducer (80) of gene expression. Interestingly,
following the interaction of TWIST with SET8, the latter acts as a
dual epigenetic modifier on the promoters of E- and N-cadherin to
induce the expression of N-cadherin and the repression of
E-cadherin via its H4K20 monomethylation activity (78).
As Snail interacts with several repressor complexes
including HDAC, PRC2 and Ajuba-PRMT5 (74,77),
Snail causes bivalent histone modifications (e.g., coexistence of
H3K4m3 and H3K27m3) which render affected genes susceptible to
reactivation (81). This is of
particular interest as it explains the reversible nature of EMT
which, under certain circumstances, can be reversed via MET to
generate (e.g., metastasized) cells with epithelial characteristics
(1,13).
A large body of evidence demonstrates that the
miRNA-200 family and miRNA-205 play an important regulatory role in
EMT (82,83). In the context of the epigenetic
regulation of EMT, it was found that the CpG island near the
miRNA-200c and miRNA-141 transcription start is unmethylated in
miRNA-expressing tumor/normal cells and is heavily methylated in
miRNA-negative and invasive tumor cells. miRNA expression is
further facilitated by the enrichment of chromatin-permissive
histone modifications (H3 acetylation and H3K4 trimethylation)
(84). Likewise, Davalos et
al(85) demonstrated that in
epithelial cancer cell lines, the 5′-CpG islands of miRNA-200
family members are unmethylated, whereas the
hypermethylation-mediated silencing of these miRNAs was found in
transformed mesenchymal cells. The reversibility of this
methylation state mediates the shift between EMT and MET (85). Similar results were obtained in
bladder cancer (86) and breast
cancer cell lines (87). It was
further shown that ectopic miRNA-200b and -200c expression inhibits
ZEB1 translation and disrupts ZEB1-histone deacetylase repressor
complexes. This results in increased histone acetylation and
E-cadherin expression. Interestingly, the chemo- and
radiosensitivity of these breast cancer cells was increased by
enhanced p53-mediated apoptotic pathways (88).
Therapeutic options targeting epigenetics
and EMT
The overall aim of an epigenetic therapy is to
‘renew’ the epigenome of the cells by reconstituting the normal
expression level of epigenetically misregulated genes (89). Our understanding of the association
between modifications of DNA or histones via methylation or
acetylation and human diseases has increased over the years,
leading to the development of epigenetically functioning drugs,
some of which have been approved by the US Food and Drug
Administration for the treatment of human cancer (90). As recently reviewed by us, the
combination regimen of DNA methyltransferase inhibitors (DNMTi) and
histone deacetylase inhibitors (HDACi) yielded promising results in
the treatment of myelodysplastic syndrome, a clonal hematological
disease (91). Additionally,
clinical trials (up to phase IIb) have been performed for other
hematological diseases, such as non-Hodgkin’s lymphoma
(particularly T-cell lymphoma and diffuse large B-cell lymphoma)
and acute myeloid leukemia (90).
Nevertheless, the clinical application of epigenetic drugs for
solid tumors is still in the pilot phase for e.g., non small-cell
lung cancer or only used as experimental therapy in advanced,
recurrent or refractory malignancies (90,92).
With respect to the molecular effects of these
drugs, the detailed mechanisms of epigenetic therapies were
primarily focused on their anti-proliferative and pro-apoptotic
effects as well as anti-angiogenic potency as supported by many
experimental studies in vitro and in vivo(93,94).
Of particular interest, recent investigations revealed that
acetylation and de-acetylation are centrally integrated in a
cellular network of regulations: the ‘acetylome’ which affects RNA
splicing, DNA damage repair, cell cycle control, nuclear transport,
actin remodeling, ribosome and chaperone functions (91,95).
In our previous studies, we have shown that the cinnamic hydroxamic
acid pan-DACi panobinostat (LBH589), a novel potent inhibitor of
all HDAC enzymes, influences not only proliferation and apoptosis
(96), but also the expression of
markers of differentiation and EMT, particularly in an in
vivo xenograft model of human hepatoma (97) by upregulation of epithelial markers
(cytokeratins) and downregulation of mesenchymal markers
(vimentin). Additionally, we demonstrated that the combination of
the histone deacetylase inhibitor, SAHA, and the methyltransferase
inhibitor, Zebularine, altered the patterns of differentiation in
pancreatic cancer models (98).
Furthermore, treatment of myelodysplastic syndromes (MDS) and acute
myeloid leukemia (AML) with the DNA methylation inhibitor,
decitabine (trade name: Vidaza), induced different morphological
changes (such as colony forming capacity) and the expression of
hematopoietic differentiation markers (99).
These experimental findings are significant since
the differentiation status and the associated EMT/MET status of
tumor cells is modulated by classical chemotherapy, selecting
transitional, stem cell-like tumor cells which are possibly
chemotherapy resistant and are responsible for the clinical
recurrence as hypothesized by Todaro et al(100). Therefore, the tumor
differentiation status should be characterized in detail prior to,
during and after tumor treatment in order to obtain ‘personalized’
predictive, prognostic and therapeutic stratifications. For
example, Handra-Luca et al(50) showed that the expression of the
‘basic’ mesenchymal marker, vimentin, in classical pancreatic
ductal adenocarcinoma is associated with a worse outcome of
patients using immunohistochemistry on a tissue microarray of 387
patients. Additionally, the same group demonstrated that the loss
of E-cadherin protein expression was linked to a worse survival of
patients with resectable pancreatic adenocarcinomas (51). As clinicopathological
investigations of epigenetic treatment and its impact on EMT/MET in
cancer specimen are lacking, to date, only experimental data
support the theory that epigenetic treatment of cancer cell lines
in vitro and in vivo directly influences the
Twist-Snail/ZEB-E-cadherin axis and indirectly influences EMT
inducers such as Wnt-TGFβ-BMP or other classical pathways (as
described in chapter 4). Another interesting recent approach was
presented by Ivanova et al(101) who investigated the methylation
status of different gastric cancer cell lines, revealing that DNA
methylation predicts the responsiveness of these cell lines to
treatment with cisplatin, a standard chemotherapy for gastric
cancer. Additionally, one of the candidate genes, BMP-4, was
epigenetically upregulated in cisplatin-resistant gastric cancer
cell lines; therefore, the authors speculated that targeting BMP-4
may improve the sensitivity of such cancer cells to chemotherapy
(101). Taken together, several
tumorigenic properties initiated/driven by EMT such as invasion,
metastasis and drug resistance may be targeted by means of
epigenetic therapeutic approaches as illustrated in Fig. 3.
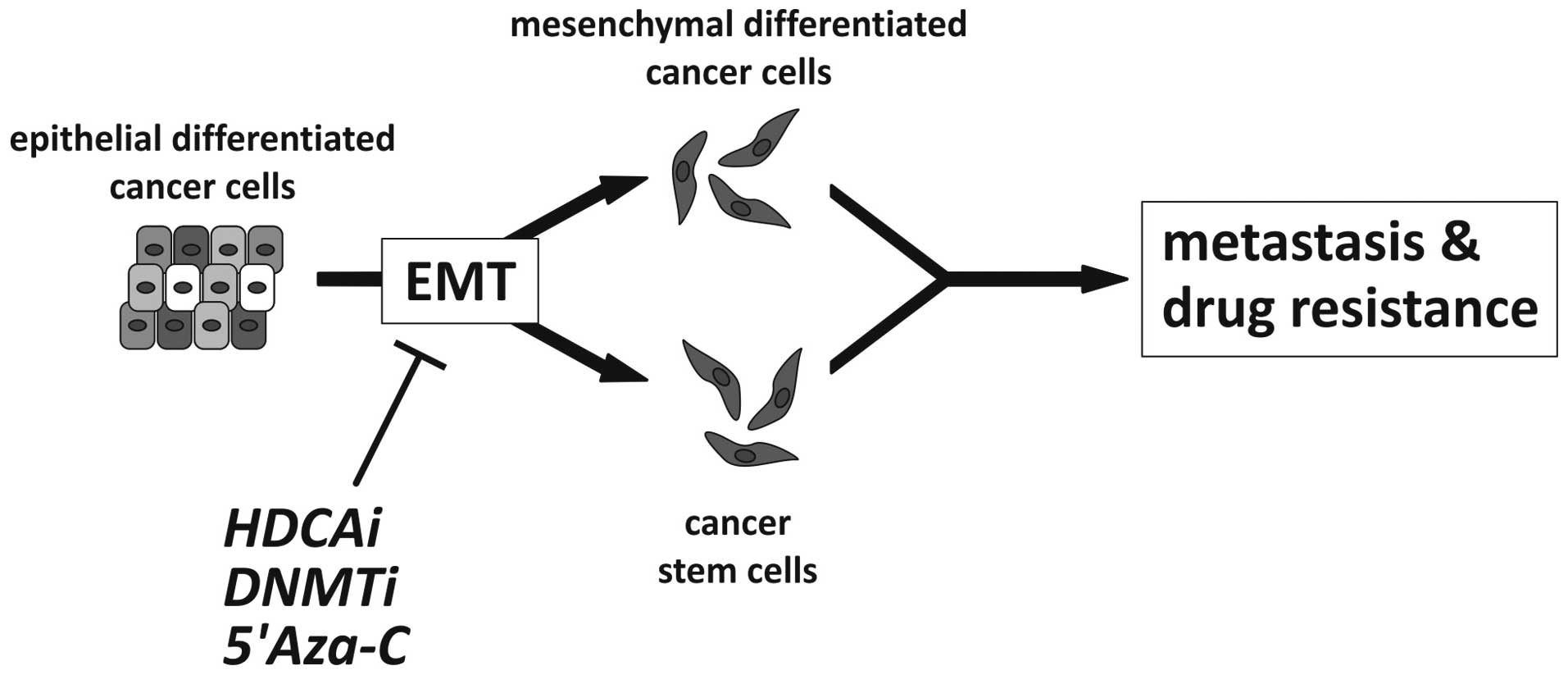 | Figure 3Possible effect of epigenetic cancer
therapy targeting EMT. Epithelial-mesenchymal transition
contributes to tumor progression by either generating migratory,
invasive mesenchymal cancer cells or by the induction of stemness
and generation of cancer stem cells, two processes that may involve
similar phenomena, i.e., acquisition of stemness and mesenchymal
characteristics. Possible epigenetic (classes of) drugs are
illustrated to inhibit EMT, including histone deacetylase inhibitor
(HDACi), DNA methyltransferase inhibitor (DNMTi) and 5-azacytidine
(5-Aza-C) as an example of a demethylating agent (5,16,96,100,127,128). |
Summary and future directions
Our understanding of the role of EMT/MET-related
processes in different phases of human development, homoeostasis,
regeneration and reparation, as well as carcinogenesis has
dramatically increased. The central molecular pathways associated
with the downstream effects on the most important EMT phenotype,
i.e., loss of E-cadherin and vimentin expression have been well
described. The direct and indirect inducers of EMT/MET are known
and we are beginning to decipher their integrated regulatory
crosstalk and feedback mechanisms. As reviewed in this article, the
role of epigenetics in EMT is being increasingly strengthened by
recent experimental data. Nevertheless, further research is
required to fully uncover the whole spectrum of the epigenetic
regulation of EMT/MET in human cancer. Based on these insights,
novel epigenetic therapies that target the EMT-related processes in
tumor progression may become feasible.
With respect to basic cell culture experiments
showing the influence of epigenetics on therapy responsiveness or
resistance (101), recent data
demonstrate that the epigenetic pre-treatment of human cancer cells
induces differentiation and, therefore, presents us with a chance
to improve the efficiency of classical chemotherapies (102). Therefore, we hypothesize that
epigenetic therapy may stabilize the epithelial tumor phenotype or
induce MET which may subsequently improve tumor sensitivity to
conventional chemotherapy (Fig.
3). However, this hypothesis requires further confirmation in
appropriate pre-clinical studies and large prospective clinical
trials.
References
|
1.
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006.
|
|
2.
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelialmesenchymal transitions in development and disease.
Cell. 139:871–890. 2009.
|
|
3.
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial - mesenchymal
and mesenchymal - epithelial transitions in carcinoma progression.
J Cell Physiol. 213:374–383. 2007.
|
|
4.
|
Choi SS and Diehl AM:
Epithelial-to-mesenchymal transitions in the liver. Hepatology.
50:2007–2013. 2009.
|
|
5.
|
Brabletz T, Jung A, Spaderna S, Hlubek F
and Kirchner T: Opinion: migrating cancer stem cells - an
integrated concept of malignant tumour progression. Nat Rev Cancer.
5:744–749. 2005.
|
|
6.
|
Kiesslich T, Berr F, Alinger B, Kemmerling
R, Pichler M, Ocker M and Neureiter D: Current status of
therapeutic targeting of developmental signalling pathways in
oncology. Curr Pharm Biotechnol. 13:2184–2220. 2012.
|
|
7.
|
Kirchner T and Brabletz T: Patterning and
nuclear beta-catenin expression in the colonic adenoma-carcinoma
sequence. Analogies with embryonic gastrulation. Am J Pathol.
157:1113–1121. 2000.
|
|
8.
|
Brabletz T: To differentiate or not -
routes towards metastasis. Nat Rev Cancer. 12:425–436. 2012.
|
|
9.
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009.
|
|
10.
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007.
|
|
11.
|
Moustakas A and Heldin CH: Signaling
networks guiding epithelial-mesenchymal transitions during
embryogenesis and cancer progression. Cancer Sci. 98:1512–1520.
2007.
|
|
12.
|
Voulgari A and Pintzas A:
Epithelial-mesenchymal transition in cancer metastasis: mechanisms,
markers and strategies to overcome drug resistance in the clinic.
Biochim Biophys Acta. 1796:75–90. 2009.
|
|
13.
|
Scheel C and Weinberg RA: Cancer stem
cells and epithelialmesenchymal transition: Concepts and molecular
links. Semin Cancer Biol. Apr 23–2012, (E-pub ahead of print).
|
|
14.
|
McConkey DJ, Choi W, Marquis L, et al:
Role of epithelial-to-mesenchymal transition (EMT) in drug
sensitivity and metastasis in bladder cancer. Cancer Metastasis
Rev. 28:335–344. 2009.
|
|
15.
|
Ouyang G, Wang Z, Fang X, Liu J and Yang
CJ: Molecular signaling of the epithelial to mesenchymal transition
in generating and maintaining cancer stem cells. Cell Mol Life Sci.
67:2605–2618. 2010.
|
|
16.
|
Moll R, Divo M and Langbein L: The human
keratins: biology and pathology. Histochem Cell Biol. 129:705–733.
2008.
|
|
17.
|
Frixen UH, Behrens J, Sachs M, et al:
E-cadherin-mediated cell-cell adhesion prevents invasiveness of
human carcinoma cells. J Cell Biol. 113:173–185. 1991.
|
|
18.
|
Onder TT, Gupta PB, Mani SA, Yang J,
Lander ES and Weinberg RA: Loss of E-cadherin promotes metastasis
via multiple downstream transcriptional pathways. Cancer Res.
68:3645–3654. 2008.
|
|
19.
|
Perl AK, Wilgenbus P, Dahl U, Semb H and
Christofori G: A causal role for E-cadherin in the transition from
adenoma to carcinoma. Nature. 392:190–193. 1998.
|
|
20.
|
Schipper JH, Frixen UH, Behrens J, Unger
A, Jahnke K and Birchmeier W: E-cadherin expression in squamous
cell carcinomas of head and neck: inverse correlation with tumor
dedifferentiation and lymph node metastasis. Cancer Res.
51:6328–6337. 1991.
|
|
21.
|
Umbas R, Isaacs WB, Bringuier PP, et al:
Decreased E-cadherin expression is associated with poor prognosis
in patients with prostate cancer. Cancer Res. 54:3929–3933.
1994.
|
|
22.
|
Cavallaro U, Schaffhauser B and
Christofori G: Cadherins and the tumour progression: is it all in a
switch? Cancer Lett. 176:123–128. 2002.
|
|
23.
|
Maeda M, Johnson KR and Wheelock MJ:
Cadherin switching: essential for behavioral but not morphological
changes during an epithelium-to-mesenchyme transition. J Cell Sci.
118:873–887. 2005.
|
|
24.
|
Gregory PA, Bert AG, Paterson EL, et al:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008.
|
|
25.
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008.
|
|
26.
|
Batlle E, Sancho E, Franci C, Dominguez D,
Monfar M, Baulida J and Garcia DH: The transcription factor snail
is a repressor of E-cadherin gene expression in epithelial tumour
cells. Nat Cell Biol. 2:84–89. 2000.
|
|
27.
|
Cano A, Perez-Moreno MA, Rodrigo I, et al:
The transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000.
|
|
28.
|
Bolos V, Peinado H, Perez-Moreno MA, Fraga
MF, Esteller M and Cano A: The transcription factor Slug represses
E-cadherin expression and induces epithelial to mesenchymal
transitions: a comparison with Snail and E47 repressors. J Cell
Sci. 116:499–511. 2003.
|
|
29.
|
Grooteclaes ML and Frisch SM: Evidence for
a function of CtBP in epithelial gene regulation and anoikis.
Oncogene. 19:3823–3828. 2000.
|
|
30.
|
Comijn J, Berx G, Vermassen P, et al: The
two-handed E box binding zinc finger protein SIP1 downregulates
E-cadherin and induces invasion. Mol Cell. 7:1267–1278. 2001.
|
|
31.
|
Perez-Moreno MA, Locascio A, Rodrigo I,
Dhondt G, Portillo F, Nieto MA and Cano A: A new role for E12/E47
in the repression of E-cadherin expression and
epithelial-mesenchymal transitions. J Biol Chem. 276:27424–27431.
2001.
|
|
32.
|
Nakaya Y and Sheng G: Epithelial to
mesenchymal transition during gastrulation: an embryological view.
Dev Growth Differ. 50:755–766. 2008.
|
|
33.
|
Qin Q, Xu Y, He T, Qin C and Xu J: Normal
and disease-related biological functions of Twist1 and underlying
molecular mechanisms. Cell Res. 22:90–106. 2012.
|
|
34.
|
Piera-Velazquez S, Li Z and Jimenez SA:
Role of endothelial-mesenchymal transition (EndoMT) in the
pathogenesis of fibrotic disorders. Am J Pathol. 179:1074–1080.
2011.
|
|
35.
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011.
|
|
36.
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000.
|
|
37.
|
Chaffer CL, Thompson EW and Williams ED:
Mesenchymal to epithelial transition in development and disease.
Cells Tissues Organs. 185:7–19. 2007.
|
|
38.
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008.
|
|
39.
|
Neureiter D, Herold C and Ocker M:
Gastrointestinal cancer - only a deregulation of stem cell
differentiation? (Review). Int J Mol Med. 17:483–489. 2006.
|
|
40.
|
Zeisberg M, Yang C, Martino M, Duncan MB,
Rieder F, Tanjore H and Kalluri R: Fibroblasts derive from
hepatocytes in liver fibrosis via epithelial to mesenchymal
transition. J Biol Chem. 282:23337–23347. 2007.
|
|
41.
|
Kim KK, Kugler MC, Wolters PJ, et al:
Alveolar epithelial cell mesenchymal transition develops in
vivo during pulmonary fibrosis and is regulated by the
extracellular matrix. Proc Natl Acad Sci USA. 103:13180–13185.
2006.
|
|
42.
|
Zeisberg EM, Tarnavski O, Zeisberg M, et
al: Endothelial-to-mesenchymal transition contributes to cardiac
fibrosis. Nat Med. 13:952–961. 2007.
|
|
43.
|
Zeisberg EM, Potenta SE, Sugimoto H,
Zeisberg M and Kalluri R: Fibroblasts in kidney fibrosis emerge via
endothelial-to-mesenchymal transition. J Am Soc Nephrol.
19:2282–2287. 2008.
|
|
44.
|
Yanez-Mo M, Lara-Pezzi E, Selgas R, et al:
Peritoneal dialysis and epithelial-to-mesenchymal transition of
mesothelial cells. N Engl J Med. 348:403–413. 2003.
|
|
45.
|
Stintzing S, Ocker M, Hartner A, Amann K,
Barbera L and Neureiter D: Differentiation patterning of vascular
smooth muscle cells (VSMC) in atherosclerosis. Virchows Arch.
455:171–185. 2009.
|
|
46.
|
Saika S, Ikeda K, Yamanaka O, et al:
Transient adenoviral gene transfer of Smad7 prevents injury-induced
epithelial-mesenchymal transition of lens epithelium in mice. Lab
Invest. 84:1259–1270. 2004.
|
|
47.
|
Tan X, Li Y and Liu Y: Paricalcitol
attenuates renal interstitial fibrosis in obstructive nephropathy.
J Am Soc Nephrol. 17:3382–3393. 2006.
|
|
48.
|
Zeisberg M, Bottiglio C, Kumar N, Maeshima
Y, Strutz F, Muller GA and Kalluri R: Bone morphogenic protein-7
inhibits progression of chronic renal fibrosis associated with two
genetic mouse models. Am J Physiol Renal Physiol. 285:F1060–F1067.
2003.
|
|
49.
|
Jung A, Brabletz T and Kirchner T: The
migrating cancer stem cells model - a conceptual explanation of
malignant tumour progression. Ernst Schering Found Symp Proc.
109–124. 2006.
|
|
50.
|
Handra-Luca A, Hong SM, Walter K, Wolfgang
C, Hruban R and Goggins M: Tumour epithelial vimentin expression
and outcome of pancreatic ductal adenocarcinomas. Br J Cancer.
104:1296–1302. 2011.
|
|
51.
|
Hong SM, Li A, Olino K, et al: Loss of
E-cadherin expression and outcome among patients with resectable
pancreatic adenocarcinomas. Mod Pathol. 24:1237–1247. 2011.
|
|
52.
|
Jouppila-Matto A, Tuhkanen H, Soini Y, et
al: Transcription factor snail1 expression and poor survival in
pharyngeal squamous cell carcinoma. Histol Histopathol. 26:443–449.
2011.
|
|
53.
|
Franci C, Gallen M, Alameda F, Baro T,
Iglesias M, Virtanen I and Garcia DH: Snail1 protein in the stroma
as a new putative prognosis marker for colon tumours. PLoS One.
4:e55952009.
|
|
54.
|
Bieche I, Lerebours F, Tozlu S, Espie M,
Marty M and Lidereau R: Molecular profiling of inflammatory breast
cancer: identification of a poor-prognosis gene expression
signature. Clin Cancer Res. 10:6789–6795. 2004.
|
|
55.
|
Sarkar FH, Li Y, Wang Z and Kong D:
Pancreatic cancer stem cells and EMT in drug resistance and
metastasis. Minerva Chir. 64:489–500. 2009.
|
|
56.
|
van Zijl F, Zulehner G, Petz M, et al:
Epithelial-mesenchymal transition in hepatocellular carcinoma.
Future Oncol. 5:1169–1179. 2009.
|
|
57.
|
Sabbah M, Emami S, Redeuilh G, et al:
Molecular signature and therapeutic perspective of the
epithelial-to-mesenchymal transitions in epithelial cancers. Drug
Resist Updat. 11:123–151. 2008.
|
|
58.
|
Thiery JP, Chua K, Sim WJ and Huang R:
Epithelial mesenchymal transition during development in fibrosis
and in the progression of carcinoma. Bull Cancer. 97:1285–1295.
2010.(In French).
|
|
59.
|
Gupta PB, Onder TT, Jiang G, Tao K,
Kuperwasser C, Weinberg RA and Lander ES: Identification of
selective inhibitors of cancer stem cells by high-throughput
screening. Cell. 138:645–659. 2009.
|
|
60.
|
Ketola K, Hilvo M, Hyotylainen T, et al:
Salinomycin inhibits prostate cancer growth and migration via
induction of oxidative stress. Br J Cancer. 106:99–106. 2012.
|
|
61.
|
Wang Y: Effects of salinomycin on cancer
stem cell in human lung adenocarcinoma A549 cells. Med Chem.
7:106–111. 2011.
|
|
62.
|
Gong C, Yao H, Liu Q, Chen J, Shi J, Su F
and Song E: Markers of tumor-initiating cells predict
chemoresistance in breast cancer. PLoS One. 5:e156302010.
|
|
63.
|
Bardsley MR, Horvath VJ, Asuzu DT, et al:
Kitlow stem cells cause resistance to Kit/platelet-derived growth
factor alpha inhibitors in murine gastrointestinal stromal tumors.
Gastroenterology. 139:942–952. 2010.
|
|
64.
|
Fuchs D, Daniel V, Sadeghi M, Opelz G and
Naujokat C: Salinomycin overcomes ABC transporter-mediated
multidrug and apoptosis resistance in human leukemia stem cell-like
KG-1a cells. Biochem Biophys Res Commun. 394:1098–1104. 2010.
|
|
65.
|
Jones PA and Taylor SM: Cellular
differentiation, cytidine analogs and DNA methylation. Cell.
20:85–93. 1980.
|
|
66.
|
Jones PA, Taylor SM and Wilson V: DNA
modification, differentiation, and transformation. J Exp Zool.
228:287–295. 1983.
|
|
67.
|
Fraga MF, Herranz M, Espada J, et al: A
mouse skin multistage carcinogenesis model reflects the aberrant
DNA methylation patterns of human tumors. Cancer Res. 64:5527–5534.
2004.
|
|
68.
|
Herranz N, Pasini D, Diaz VM, et al:
Polycomb complex 2 is required for E-cadherin repression by the
Snail1 transcription factor. Mol Cell Biol. 28:4772–4781. 2008.
|
|
69.
|
Vincent A and Van SI: On the epigenetic
origin of cancer stem cells. Biochim Biophys Acta. 1826:83–88.
2012.
|
|
70.
|
Rodriguez-Paredes M and Esteller M: Cancer
epigenetics reaches mainstream oncology. Nat Med. 17:330–339.
2011.
|
|
71.
|
Ehrlich M: DNA hypomethylation in cancer
cells. Epigenomics. 1:239–259. 2009.
|
|
72.
|
Ehrlich M: DNA methylation in cancer: too
much, but also too little. Oncogene. 21:5400–5413. 2002.
|
|
73.
|
Fullgrabe J, Kavanagh E and Joseph B:
Histone onco-modifications. Oncogene. 30:3391–3403. 2011.
|
|
74.
|
Maeda G, Chiba T, Aoba T and Imai K:
Epigenetic inactivation of E-cadherin by promoter hypermethylation
in oral carcinoma cells. Odontology. 95:24–29. 2007.
|
|
75.
|
Lombaerts M, van Wezel T, Philippo K, et
al: E-cadherin transcriptional downregulation by promoter
methylation but not mutation is related to
epithelial-to-mesenchymal transition in breast cancer cell lines.
Br J Cancer. 94:661–671. 2006.
|
|
76.
|
Savagner P: The epithelial-mesenchymal
transition (EMT) phenomenon. Ann Oncol. 21(Suppl 7): vii89–vii92.
2010.
|
|
77.
|
Lin T, Ponn A, Hu X, Law BK and Lu J:
Requirement of the histone demethylase LSD1 in Snai1-mediated
transcriptional repression during epithelial-mesenchymal
transition. Oncogene. 29:4896–4904. 2010.
|
|
78.
|
Yang F, Sun L, Li Q, Han X, Lei L, Zhang H
and Shang Y: SET8 promotes epithelial-mesenchymal transition and
confers TWIST dual transcriptional activities. EMBO J. 31:110–123.
2011.
|
|
79.
|
Kalakonda N, Fischle W, Boccuni P, et al:
Histone H4 lysine 20 monomethylation promotes transcriptional
repression by L3MBTL1. Oncogene. 27:4293–4304. 2008.
|
|
80.
|
Li Z, Nie F, Wang S and Li L: Histone H4
Lys 20 monomethylation by histone methylase SET8 mediates Wnt
target gene activation. Proc Natl Acad Sci USA. 108:3116–3123.
2011.
|
|
81.
|
Bernstein BE, Mikkelsen TS, Xie X, et al:
A bivalent chromatin structure marks key developmental genes in
embryonic stem cells. Cell. 125:315–326. 2006.
|
|
82.
|
Mongroo PS and Rustgi AK: The role of the
miR-200 family in epithelial-mesenchymal transition. Cancer Biol
Ther. 10:219–222. 2010.
|
|
83.
|
Bullock MD, Sayan AE, Packham GK and
Mirnezami AH: MicroRNAs: critical regulators of epithelial to
mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in
cancer progression. Biol Cell. 104:3–12. 2012.
|
|
84.
|
Vrba L, Jensen TJ, Garbe JC, et al: Role
for DNA methylation in the regulation of miR-200c and miR-141
expression in normal and cancer cells. PLoS One. 5:e86972010.
|
|
85.
|
Davalos V, Moutinho C, Villanueva A, Boque
R, Silva P, Carneiro F and Esteller M: Dynamic epigenetic
regulation of the microRNA-200 family mediates epithelial and
mesenchymal transitions in human tumorigenesis. Oncogene.
31:2062–2074. 2012.
|
|
86.
|
Wiklund ED, Bramsen JB, Hulf T, et al:
Coordinated epigenetic repression of the miR-200 family and miR-205
in invasive bladder cancer. Int J Cancer. 128:1327–1334. 2011.
|
|
87.
|
Neves R, Scheel C, Weinhold S, et al: Role
of DNA methylation in miR-200c/141 cluster silencing in invasive
breast cancer cells. BMC Res Notes. 3:2192010.
|
|
88.
|
Tryndyak VP, Beland FA and Pogribny IP:
E-cadherin transcriptional down-regulation by epigenetic and
microRNA-200 family alterations is related to mesenchymal and
drug-resistant phenotypes in human breast cancer cells. Int J
Cancer. 126:2575–2583. 2010.
|
|
89.
|
Kelly TK, De Carvalho DD and Jones PA:
Epigenetic modifications as therapeutic targets. Nat Biotechnol.
28:1069–1078. 2010.
|
|
90.
|
Lane AA and Chabner BA: Histone
deacetylase inhibitors in cancer therapy. J Clin Oncol.
27:5459–5468. 2009.
|
|
91.
|
Stintzing S, Kemmerling R, Kiesslich T,
Alinger B, Ocker M and Neureiter D: Myelodysplastic syndrome and
histone deacetylase inhibitors: “to be or not to be acetylated”? J
Biomed Biotechnol. 2011:2141432011.
|
|
92.
|
Batty N, Malouf GG and Issa JP: Histone
deacetylase inhibitors as anti-neoplastic agents. Cancer Lett.
280:192–200. 2009.
|
|
93.
|
Ocker M: Deacetylase inhibitors - focus on
non-histone targets and effects. World J Biol Chem. 1:55–61.
2010.
|
|
94.
|
Ocker M and Schneider-Stock R: Histone
deacetylase inhibitors: signalling towards p21cip1/waf1. Int J
Biochem Cell Biol. 39:1367–1374. 2007.
|
|
95.
|
Spange S, Wagner T, Heinzel T and Kramer
OH: Acetylation of non-histone proteins modulates cellular
signalling at multiple levels. Int J Biochem Cell Biol. 41:185–198.
2009.
|
|
96.
|
Di Fazio P, Schneider-Stock R, Neureiter
D, et al: The pan-deacetylase inhibitor panobinostat inhibits
growth of hepatocellular carcinoma models by alternative pathways
of apoptosis. Cell Oncol. 32:285–300. 2010.
|
|
97.
|
Di Fazio P, Montalbano R, Quint K, et al:
The pan-deacetylase inhibitor panobinostat modulates expression of
epithelialmesenchymal transition markers in hepatocellular
carcinoma models. Oncol Lett. (In press).
|
|
98.
|
Neureiter D, Zopf S, Leu T, et al:
Apoptosis, proliferation and differentiation patterns are
influenced by Zebularine and SAHA in pancreatic cancer models.
Scand J Gastroenterol. 42:103–116. 2007.
|
|
99.
|
Ryningen A, Stapnes C and Bruserud O:
Clonogenic acute myelogenous leukemia cells are heterogeneous with
regard to regulation of differentiation and effect of epigenetic
pharmacological targeting. Leuk Res. 31:1303–1313. 2007.
|
|
100.
|
Todaro M, Francipane MG, Medema JP and
Stassi G: Colon cancer stem cells: promise of targeted therapy.
Gastroenterology. 138:2151–2162. 2010.
|
|
101.
|
Ivanova T, Zouridis H, Wu Y, et al:
Integrated epigenomics identifies BMP4 as a modulator of cisplatin
sensitivity in gastric cancer. Gut. Apr 25–2012.(E-pub ahead of
print).
|
|
102.
|
Bhatla T, Wang J, Morrison DJ, Raetz EA,
Burke MJ, Brown P and Carroll WL: Epigenetic reprogramming reverses
the relapse-specific gene expression signature and restores
chemo-sensitivity in childhood B-lymphoblastic leukemia. Blood.
119:5201–5210. 2012.
|
|
103.
|
Omenetti A, Bass LM, Anders RA, et al:
Hedgehog activity, epithelial-mesenchymal transitions, and biliary
dysmorphogenesis in biliary atresia. Hepatology. 53:1246–1258.
2011.
|
|
104.
|
Fabris L and Strazzabosco M:
Epithelial-mesenchymal interactions in biliary diseases. Semin
Liver Dis. 31:11–32. 2011.
|
|
105.
|
Bailey JM, Singh PK and Hollingsworth MA:
Cancer metastasis facilitated by developmental pathways: Sonic
hedgehog, Notch, and bone morphogenic proteins. J Cell Biochem.
102:829–839. 2007.
|
|
106.
|
Dasgupta P, Rizwani W, Pillai S, et al:
Nicotine induces cell proliferation, invasion and
epithelial-mesenchymal transition in a variety of human cancer cell
lines. Int J Cancer. 124:36–45. 2009.
|
|
107.
|
Li Y, Liu Y, Xu Y, Voorhees JJ and Fisher
GJ: UV irradiation induces Snail expression by AP-1 dependent
mechanism in human skin keratinocytes. J Dermatol Sci. 60:105–113.
2010.
|
|
108.
|
Hardy KM, Booth BW, Hendrix MJ, Salomon DS
and Strizzi L: ErbB/EGF signaling and EMT in mammary development
and breast cancer. J Mammary Gland Biol Neoplasia. 15:191–199.
2010.
|
|
109.
|
Guttilla IK, Adams BD and White BA:
ERalpha, microRNAs, and the epithelial-mesenchymal transition in
breast cancer. Trends Endocrinol Metab. 23:73–82. 2012.
|
|
110.
|
Gallo D, Ferlini C and Scambia G: The
epithelial-mesenchymal transition and the estrogen-signaling in
ovarian cancer. Curr Drug Targets. 11:474–481. 2010.
|
|
111.
|
Katoh Y and Katoh M: FGFR2-related
pathogenesis and FGFR2-targeted therapeutics (Review). Int J Mol
Med. 23:307–311. 2009.
|
|
112.
|
Ding W, You H, Dang H, et al:
Epithelial-to-mesenchymal transition of murine liver tumor cells
promotes invasion. Hepatology. 52:945–953. 2010.
|
|
113.
|
Semenza GL: Hypoxia-inducible factors:
mediators of cancer progression and targets for cancer therapy.
Trends Pharmacol Sci. 33:207–214. 2012.
|
|
114.
|
Jiang J, Tang YL and Liang XH: EMT: a new
vision of hypoxia promoting cancer progression. Cancer Biol Ther.
11:714–723. 2011.
|
|
115.
|
Yang SY, Miah A, Pabari A and Winslet M:
Growth Factors and their receptors in cancer metastases. Front
Biosci. 16:531–538. 2011.
|
|
116.
|
Mamuya FA and Duncan MK: aV integrins and
TGF-β-induced EMT: a circle of regulation. J Cell Mol Med.
16:445–455. 2012.
|
|
117.
|
Eble JA and Haier J: Integrins in cancer
treatment. Curr Cancer Drug Targets. 6:89–105. 2006.
|
|
118.
|
Lahsnig C, Mikula M, Petz M, et al: ILEI
requires oncogenic Ras for the epithelial to mesenchymal transition
of hepatocytes and liver carcinoma progression. Oncogene.
28:638–650. 2009.
|
|
119.
|
Yadav A, Kumar B, Datta J, Teknos TN and
Kumar P: IL-6 promotes head and neck tumor metastasis by inducing
epithelialmesenchymal transition via the JAK-STAT3-SNAIL signaling
pathway. Mol Cancer Res. 9:1658–1667. 2011.
|
|
120.
|
Andrae J, Gallini R and Betsholtz C: Role
of platelet-derived growth factors in physiology and medicine.
Genes Dev. 22:1276–1312. 2008.
|
|
121.
|
Peebles KA, Lee JM, Mao JT, et al:
Inflammation and lung carcinogenesis: applying findings in
prevention and treatment. Expert Rev Anticancer Ther. 7:1405–1421.
2007.
|
|
122.
|
Lee JM, Yanagawa J, Peebles KA, Sharma S,
Mao JT and Dubinett SM: Inflammation in lung carcinogenesis: new
targets for lung cancer chemoprevention and treatment. Crit Rev
Oncol Hematol. 66:208–217. 2008.
|
|
123.
|
Gherardi E, Birchmeier W, Birchmeier C and
Vande WG: Targeting MET in cancer: rationale and progress. Nat Rev
Cancer. 12:89–103. 2012.
|
|
124.
|
Tolnay E, Kuhnen C, Wiethege T, Konig JE,
Voss B and Muller KM: Hepatocyte growth factor/scatter factor and
its receptor c-Met are overexpressed and associated with an
increased microvessel density in malignant pleural mesothelioma. J
Cancer Res Clin Oncol. 124:291–296. 1998.
|
|
125.
|
Heuberger J and Birchmeier W: Interplay of
cadherin-mediated cell adhesion and canonical Wnt signaling. Cold
Spring Harb Perspect Biol. 2:a0029152010.
|
|
126.
|
Hollier BG, Evans K and Mani SA: The
epithelial-to-mesenchymal transition and cancer stem cells: a
coalition against cancer therapies. J Mammary Gland Biol Neoplasia.
14:29–43. 2009.
|
|
127.
|
Mimeault M and Batra SK: New advances on
critical implications of tumor- and metastasis-initiating cells in
cancer progression, treatment resistance and disease recurrence.
Histol Histopathol. 25:1057–1073. 2010.
|
|
128.
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: an emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010.
|
|
129.
|
Wang Z, Li Y, Ahmad A, Azmi AS, Kong D,
Banerjee S and Sarkar FH: Targeting miRNAs involved in cancer stem
cell and EMT regulation: an emerging concept in overcoming drug
resistance. Drug Resist Updat. 13:109–118. 2010.
|
|
130.
|
Wellner U, Schubert J, Burk UC, et al: The
EMT-activator ZEB1 promotes tumorigenicity by repressing
stemness-inhibiting microRNAs. Nat Cell Biol. 11:1487–1495.
2009.
|
|
131.
|
Wendt MK, Tian M and Schiemann WP:
Deconstructing the mechanisms and consequences of TGF-beta-induced
EMT during cancer progression. Cell Tissue Res. 347:85–101.
2012.
|