1. Introduction
The Epstein-Barr virus (EBV), also referred to as
human herpesvirus 4 (HHV-4), was the first human oncogenic virus to
be isolated 50 years ago by Epstein and co-workers from
lymphoblasts cultured from a Burkitt’s lymphoma (1). Thirty years later, herpesviral DNA
was identified in cells of Kaposi’s sarcoma (KS), giving rise to a
new virus, KS-associated herpesvirus (KSHV), also referred to as
HHV-8 (2). KSHV was shown to be
highly related to EBV and, therefore, represented the second human
γ-herpesvirus. To date, these are the only known oncogenic human
herpesviruses and are each associated with numerous malignancies
(Table I).
 | Table IThe human γ-herpesviruses and their
associated diseases. |
Table I
The human γ-herpesviruses and their
associated diseases.
| Virus | Associated
malignancy | Name |
|---|
| EBV | B-cell
lymphoma | Burkitt’s
lymphoma
Hodgkin’s lymphoma
PTLD |
| T-cell
lymphoma | Extranodal natural
killer/T-cell lymphoma |
| Carcinoma | NPC
Gastric carcinoma |
| KSHV | Sarcoma | KS |
| B-cell
lymphoma | MCD
PEL |
Over 90% of the global adult population is
seropositive for EBV and infection usually occurs asymptomatically
in early childhood (3,4). However, primary infection during
adolescence often results in infectious mononucleosis, which is
characterised by T-cell hyperproliferation with a strong
CD8+ response, with affected individuals suffering from
flu-like symptoms and severe fatigue. Furthermore, only a minute
proportion of those infected with EBV develop malignant diseases
(5) and these diseases are
invariably associated with immune suppression, either iatrogenic,
or due to human immunodeficiency virus (HIV) or malaria
co-infection (6).
The KSHV prevalence in northern Europe is
significantly lower compared to that of EBV, with approximately 3%
of the population testing seropositive (7,8).
However, several parts of Africa have an incidence rate of >80%,
whereas intermediate rates are found in the Mediterranean (9,10).
Concurrent with these findings, KS is one of the most common
cancers in sub-Saharan Africa, where individuals are often
co-infected with KSHV and HIV (11).
The two γ-herpesviruses readily infect B
lymphocytes, where they establish a lifelong reservoir (12,13).
However, initial infection and replication may also occur in oral
epithelial cells, allowing transmission of the viruses via
contaminated saliva (12–17). Similar to all members of the
herpesvirus family, EBV and KSHV have a biphasic life cycle,
consisting of a latent phase and a lytic replication cycle
(18). Following primary
infection, viral latency is established with the viral genome
maintained as a circularised episome in the nucleus of the host
cell. During this phase of infection, no infectious virions are
produced. A limited number of genes are expressed during latency,
encoding proteins that promote cell survival and proliferation, in
order to avoid loss of the viral episome. While this proliferation
is limited by the immune system in healthy individuals, individuals
with a compromised immune system, under conditions such as
iatrogenic immunosuppression, suffering from congenital
immunodeficiency or co-infected with HIV, may develop
lymphoproliferative disorders (19).
EBV latency is associated with at least four gene
expression programmes, termed latency 0, I, II and III. Latency
III, which is found in the B cells of post-transplant
lymphoproliferative disease (PTLD) patients and in in vitro
immortalised B cells, displays the broadest gene expression
profile, with expression of all the EBV-encoded nuclear antigens
(EBNA1, 2, 3A, 3B, 3C and leader protein), three latent membrane
proteins (LMP1, 2A and 2B) and several non-coding RNAs, such as the
Epstein-Barr virus-encoded small RNAs (EBERs) and microRNAs
(miRNAs) (5,20,21).
Latency II, which is observed in Hodgkin’s lymphoma and
EBV-associated carcinomas, is more restrictive, as only EBNA1 and
the LMPs are expressed (along with EBERs). Latency I, the most
restrictive latency programme associated with malignancy is found
in Burkitt’s lymphoma, where, along with the EBERs, EBNA1 is the
only EBV protein expressed. However, in resting memory B cells, the
latent reservoir in immunocompetent individuals, no viral proteins
are expressed, which is known as latency 0 (22,23).
It is considered that primary infection by EBV
initially requires the latency III transcriptional programme in
order to drive the proliferation and differentiation of B cells to
promote host colonisation. However, as several of the EBNAs are
immunodominant, a healthy immune system prevents this from evolving
into a lymphoproliferative disorder. Therefore, EBV possesses
various sophisticated mechanisms that promote the downregulation of
gene expression as latency is established. These mechanisms are
poorly understood, but it is hypothesized that EBV transcription is
sequentially restricted as it transits the germinal centre reaction
on its way to acquiring a memory B-cell phenotype. It is not
surprising, therefore, given the effect of latency-associated genes
on cellular function, that all EBV diseases are associated with
latency.
KSHV differs from EBV, as lytic replication is
essential for the most common malignancy associated with this
virus, i.e., KS. The lytic cycle may be activated by cellular
stimuli, such as hypoxia (24),
secretion of inflammatory factors (25) or co-infection with other viruses,
such as human cytomegalovirus or HIV (26,27).
Lytic replication results in the shedding of infectious virions
from the B-cell reservoir and subsequent infection of endothelial
cells, where KS tumours occur (28). Although the origin of these cells
(blood or lymphatic endothelium) remains to date undetermined
(29), it is a hallmark of KS
tumours that these cells adopt a spindle cell morphology. The
majority of tumour cells remain latently infected, but low levels
of virus sporadically enter the lytic cycle, meaning that there is
always a small percentage of cells undergoing lytic infection
(30,31). It is hypothesized that lytic
replication is essential for tumourigenesis, as KSHV latency is not
sufficient to maintain the pool of infected endothelial cells.
Furthermore, replicating KSHV expresses several oncogenic,
angiogenic and antiapoptotic gene products that contribute to
cancer formation (32–37), including the secretion of
tumour-promoting factors, such as interleukin (IL)-6 and IL-8
(38–40).
Other malignancies associated with KSHV include the
rare B-cell lymphomas primary effusion lymphoma (PEL) and
multicentric Castleman’s disease. Importantly, PEL cells, unlike
KS-derived cells, are able to maintain high copy numbers of the
latent KSHV genome when cultured and, therefore, provide an
essential research tool for studying KSHV in vitro (41).
The gene expression profile of KSHV in healthy,
immunocompetent individuals is currently unknown, as all our
knowledge is gleaned from studying cancer cell lines or
histological examination of malignant tissues. However, it is known
that KSHV also expresses a restricted gene expression profile
during latency, which is essential for the viability of these
cancer cells. The expressed genes include the latency-associated
nuclear antigen (LANA)1 (42), the
cyclin homologue v-cyclin, the viral homologue of the cellular
FLICE-inhibitory protein vFLIP and kaposin (43,44).
Of note, expression of vFLIP alone is sufficient to alter the
morphology of cultured endothelial cells to the characteristic
spindle cells of KS (45,46).
There is much yet to learn about EBV and KSHV; these
two viruses have been extensively investigated in recent years,
particularly using proteomic-based methods. These technologies have
proved to be powerful tools in aiding our understanding of these
important pathogens and may help elucidate new aspects of viral
biology as they become more widely used.
2. Approaches to proteomics
Gel electrophoresis-based methods
The first attempts to analyse the proteome occurred
in the early 1970s, making use of a new technique known as
polyacrylamide gel electrophoresis (PAGE) (47). Since then, there have been a number
of developments in proteomic techniques and a significant increase
in their utility. In this review, we provide a brief overview of
the most commonly used proteomic approaches.
Gel-based methods belong to the most established
techniques in proteomics and remain the preferred choice for
separation and semi-quantitative identification of various
proteins. The most commonly used method is one-dimensional
(1D)-PAGE, which may be used to separate denatured protein samples
according to their molecular weight in a polyacrylamide gel
(47). Although this technique is
used for western blot analyses in numerous laboratories, it has
limitations with regards to identification of single proteins
within large protein mixtures. Proteins of the same mass are
detected at the same position in the gel, leading to a mixed
signal.
This problem may be overcome by using
two-dimensional (2D)-PAGE. With this method, the proteins are
separated by their isoelectric point along a pH gradient in the
first dimension, a process termed isoelectric focusing (48). This is followed by separation
according to molecular mass. Due to the introduction of the second
dimension, separation of ≤10,000 proteins is possible (49). Furthermore, recent developments in
protein staining techniques, such as the introduction of
fluorescent dyes, have allowed not only a sensitivity of detection
in the femtomolar range, but also widened the linear dynamic range
by approximately three orders of magnitude (50–55).
However, direct comparison between different protein samples has
always been a weak point, as separate gels must be run for each
sample and methodical errors may intervene with the obtained
results. This has been overcome by introducing fluorescence 2D
difference gel electrophoresis (2D-DIGE), which allows comparison
of ≤3 protein samples in one gel (56,57).
In this approach, the respective protein mixes are labelled with
cyanine dyes (CyDyes), which bind covalently to the e-amino group
of the lysine residues of the proteins. Currently, three different
CyDyes are available, namely, Cy2, Cy3 and Cy5, which may be
separately detected (57).
Specific software, e.g., DeCyder, has been developed to help
analyse the gel images. Although this technique is highly
sensitive, its power is limited due to potential inconsistent
labelling of proteins (56).
Furthermore, 2D-DIGE does not overcome the limitations of 2D-PAGE,
as all the proteins must be in a molecular weight range between 5
and 150 kDa and a pH range of 3.5–10 in order to be detected
(56,58). Proteins that are successfully
separated by 2D- or 1D-PAGE, may be further defined by mass
spectrometry (MS).
MS-based methods
Over the last three decades, developments in liquid
chromatography (LC), MS and protein labelling, e.g., the
introduction of electrospray ionisation (ESI) and stable isotope
labelling by amino acids in cell culture (SILAC), have enabled the
establishment of shotgun proteomics. MS is currently commonly used
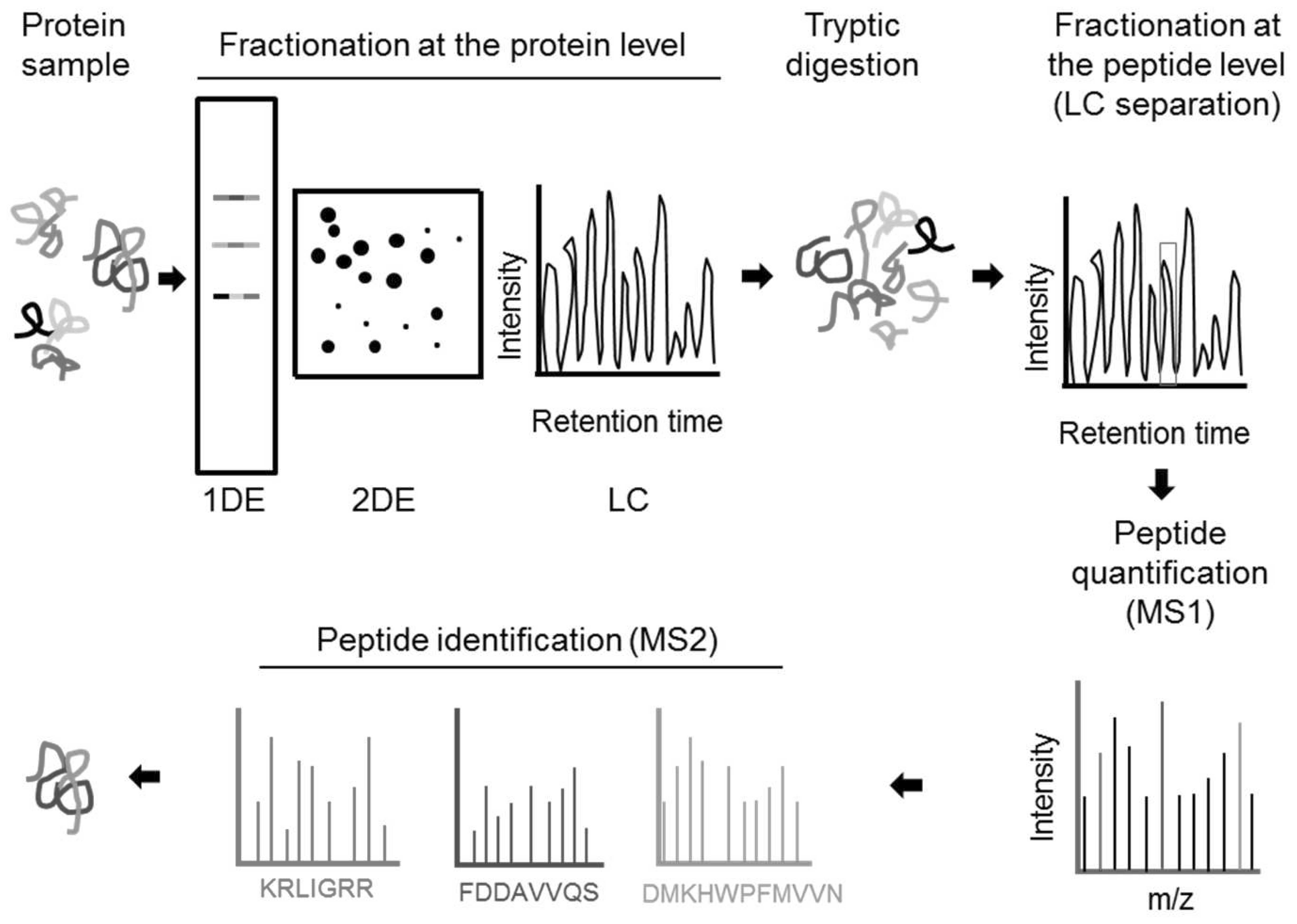
for accurate peptide identification and quantification.
Quantitative proteomics strategies rely on relative quantification,
comparing the levels of individual peptides identified from one or
more experimental samples to those levels from an untreated control
sample. This provides an invaluable and powerful tool for global
analysis of protein composition; however, it is important to
highlight that the proteomic coverage of the genome remains a major
technical challenge, as only ~10% of the proteins present in the
sample are identified and even fewer are quantified (59). MS-based proteomic approaches may be
used to identify and quantify proteins from a whole-cell lysate, an
isolated organelle/region of a cell population (e.g., nuclear
envelope), or a particular tissue or organism. As previously
alluded to, further protein purification may be achieved by means
of LC or gel electrophoresis. These strategies allow the removal of
high-abundance proteins, which would otherwise mask low-abundance
proteins, while also reducing sample complexity for increased
peptide identification and quantification. Following purification,
equal amounts of protein from each of the samples are
proteolytically digested into peptides, often using trypsin. The
digested peptides are separated and ionized by LC coupled to ESI
and are then subjected to tandem MS (MS/MS). The fractions undergo
a first round of MS (MS1), in which the mass-to-charge ratio (m/z)
of all ionised peptides is measured, generating a precursor ion
spectrum. The relative intensity of each spectrum peak correlates
proportionally to peptide concentration, allowing relative
quantification across different experimental conditions. Individual
peptide populations are then selected for collision-induced
dissociation (CID) with an inert gas and a second round of MS (MS2)
determines the m/z of the peptide fragments generating a CID
spectrum. This spectrum is then compared with a theoretical
database produced via an in silico digest for peptide
identification (Fig. 1) (60,61).
The majority of global proteomic analyses across different
experimental conditions are currently performed using stable
isotope labelling, mass tag labelling or label-free approaches
(62).
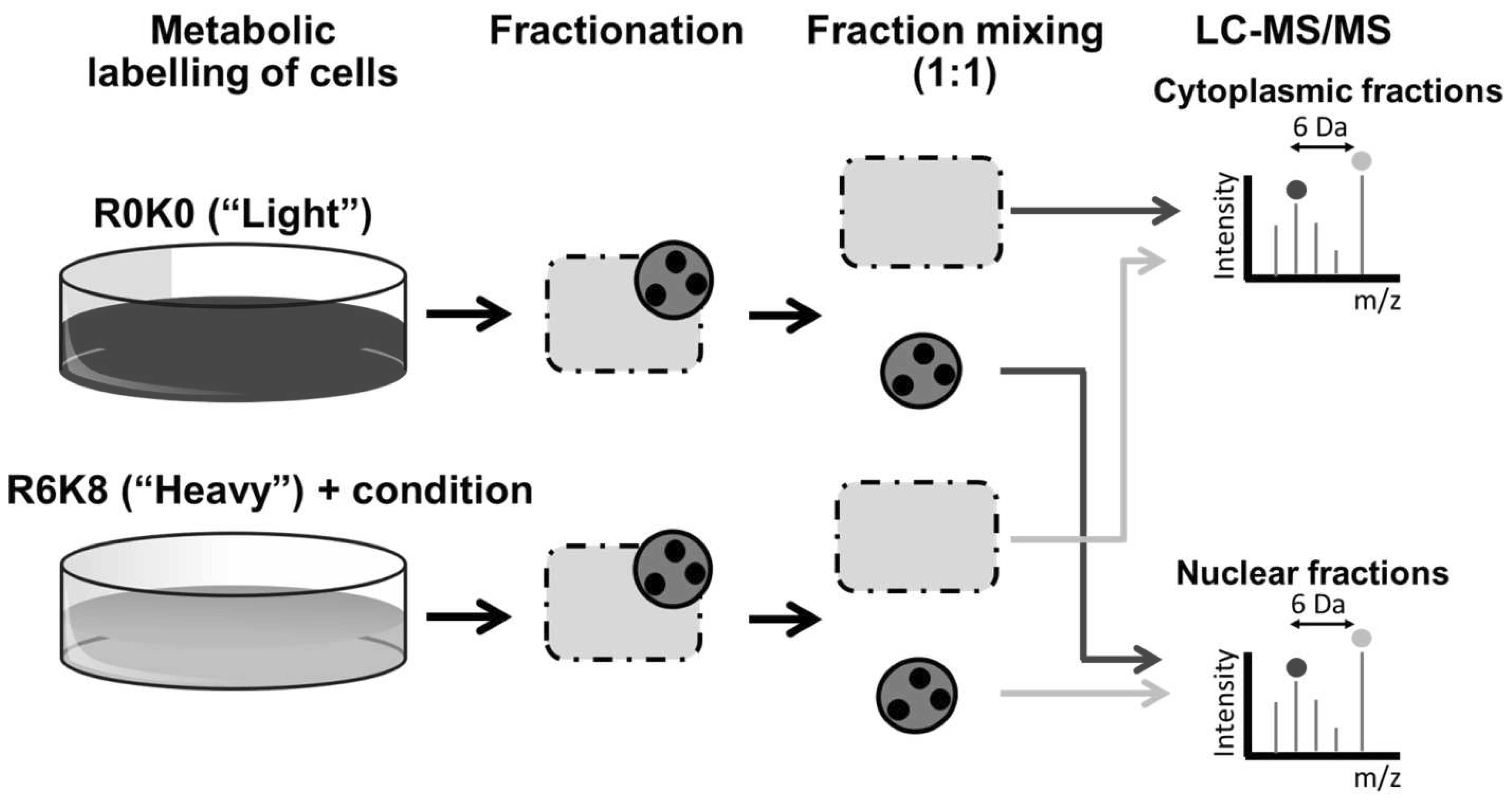
SILAC
SILAC, as first described by Ong et al
(63), is an in vivo
metabolic labelling technique, enabling the incorporation of
different atomic isotopes into proteins as they are being
synthesised in the living cell. SILAC is routinely performed using
cell culture systems, but it has also been applied to simple
organisms (e.g., C. elegans or Drosophila
melanogaster) (64) and even
mice (65). Stable
(non-radioactive) naturally occurring isotopes are variants of a
specific chemical element (commonly H, C, N and O), which contain
the same number of protons but different numbers of neutrons in
each atomic nucleus. Isotopes have identical chemical properties,
reducing the risk of introducing experimental bias between
differently labelled samples, but the isotopes have different
masses; therefore, they are easily identified by MS. The cells are
cultured in growth medium with specifically labelled amino acids
containing either a light isotope (e.g., 12C-lysine
and/or 12C-arginine) or a heavy isotope (e.g.,
13C-lysine and/or 13C-arginine).
13C has an extra neutron compared to 12C,
which gives 13C an increment in its mass number of 1 Da.
The substitution of light 12C-arginine with heavy
13C-arginine results in a 6-Da increment in the mass
spectrum of a peptide containing one arginine, as a single arginine
residue contains six carbons. Trypsin cleaves lysine and arginine
residues at the C-terminus; therefore, using these particular
labelled amino acids ensures that all tryptic peptides (except for
the C-terminal peptide) contain at least one labelled residue. For
SILAC to work effectively, the cells must be grown in labelled
growth medium for a number of passages in order to incorporate
isotopes into the majority of the proteins. A very high rate of
isotopic incorporation (>90%) is achieved in cells after 6–8
doublings (63). At this point,
cells that have been differentially labelled undergo the desired
treatment, for example, a viral infection. SILAC allows for a
maximum of three different samples in a single mass spec run with
the use of light (e.g., 12C-arginine and
12C-lysine; R0K0), medium (e.g., 13C-lysine
and 2D lysine; R6K4) and heavy (e.g., 13C
15N-arginine and 13C 15N lysine;
R10K8) isotope-containing media. A major advantage of SILAC is
that, once generated, labelled and unlabelled samples are mixed in
equal amounts into one pooled sample. This allows all the samples
to be processed together, reducing the risk of external
experimental error. Furthermore, due to its accuracy, SILAC is
particularly suitable for revealing relatively small changes in
protein levels or post-translational modifications. A limitation of
SILAC, however, is that certain cell lines may convert excess
arginine into proline and, therefore, titration of arginine into
the medium may be required (66).
In addition, SILAC may not be applicable to cells that are
sensitive to changes in the medium composition. Combining SILAC
with subcellular fractionation may further reduce sample complexity
(Fig. 2).
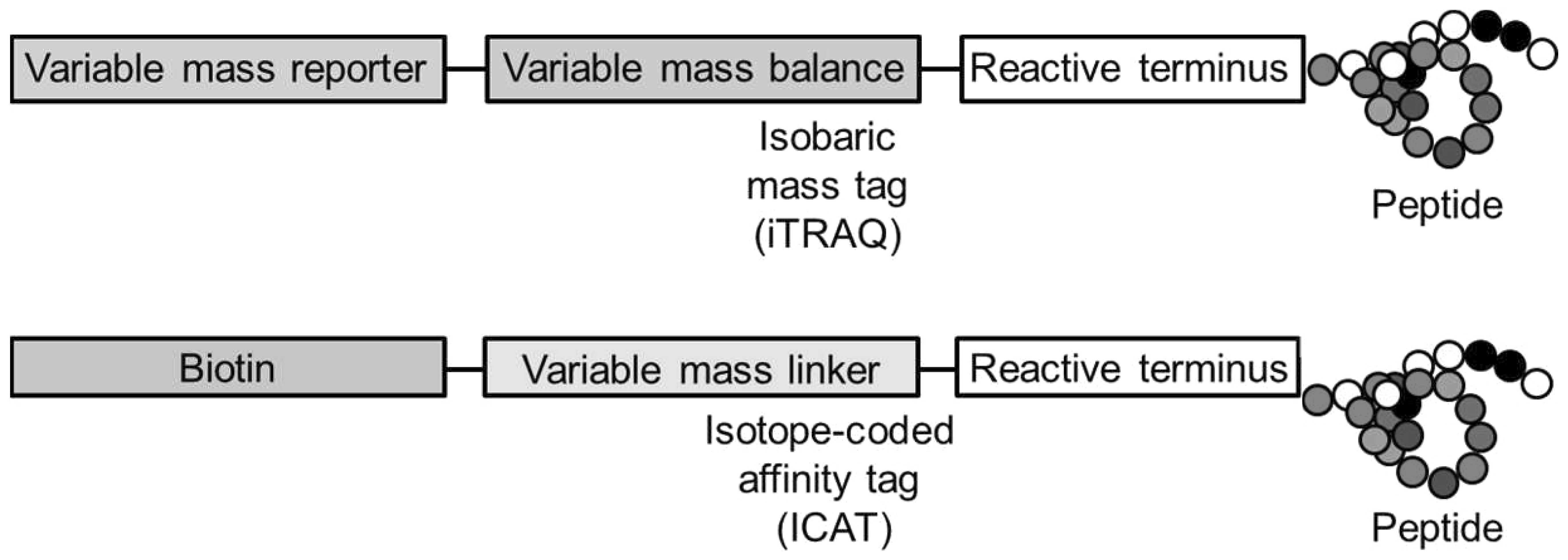
Isotope-coded affinity tagging (ICAT),
isobaric tags for relative and absolute quantification (iTRAQ) and
tandem mass tagging (TMT)
ICAT was the first method involving the addition of
isotopic tags to proteins for quantitative MS. This method was
reported in 1999 by Gygi et al (67) and allows labelling of proteins
present in any protein sample, including extracts from tissues or
body fluids. As opposed to SILAC, labelling occurs in vitro
following cell lysis. ICAT relies on the use of a reagent composed
of three elements: a biotin molecule to enable purification of the
labelled peptides with the use of avidin or streptavidin affinity
chromatography; a linker group, which contains stable isotopes
(light or heavy); and a reactive iodoacetyl group, which
specifically reacts with free thiols on cysteine residues.
Following labelling, light and heavy samples are combined into one
sample, which is subjected to proteolytic digestion, affinity
purification and LC-MS/MS. As labelling only occurs in proteins
containing cysteine, which is a rare amino acid, ICAT inherently
implies a considerable bias against proteins that lack or contain
few cysteine residues; however, for the same reason, this technique
is also advantageous when one wishes to reduce complexity in the
protein mixture. A modification of ICAT has been devised to
overcome this limitation, known as isotope-coded protein labelling
(68), in which all free amino
groups on intact proteins are isotopically labelled with either
light or heavy tags, resulting in a higher proteomic coverage
compared to ICAT.
Further techniques using tags of the same mass have
also been developed for use in MS-based quantitative proteomics,
most commonly known as iTRAQ and TMT. These techniques enable
examination of four, six or even eight different experimental
conditions in a single MS experiment with the use of up to eight
different isobaric mass tags. These tags are all of the same mass
and consist of three components: an N-hydroxysuccinimide ester
group, which reacts with primary amino groups; a mass tag
(reporter) group; and a balance (carbonyl) group. The reporter
group is based on N-methylpiperazine and contains a unique number
of heavy 13C and 15N substitutions. The
masses added by each tag are compensated with balance groups of
variable masses to make all tags equal in mass, meaning that the
same protein from different samples will behave in the same manner
during LC and MS1. Each experimental condition is enzymatically
digested individually, the peptides are then tagged and the samples
are pooled for LC-MS/MS analysis. Upon collision-induced
dissociation, the mass tags are cleaved from the balance groups.
During MS2, the intensity of the different tags are used for
relative peptide quantification, whereas the peptide fragment ions
are sequenced to determine protein identity. As isotopic labelling
occurs after enzymatic digestion, the sample complexity is
increased and more abundant proteins are overrepresented during MS
identification (69). iTRAQ and
TMT may also be used for absolute quantification, in which the
exact amount of a protein can be measured. The differences between
these isobaric mass tags and the previously mentioned isotope-coded
affinity tags are shown in Fig.
3.
Label-free MS analysis
As the labelling of proteins is always limited by
various factors, two label-free techniques have recently been
developed for use in quantitative proteomics.
Spectral counting is simply based on counting
mass spectra collected during a tandem MS run, as previous studies
demonstrated that the number of these mass spectra is proportional
to the concentration of a given peptide and, therefore, its
respective protein in the sample (58,70).
Prior to ionisation and MS/MS, the peptides resulting from the
initial protein trypsinisation are separated by LC. Elution of
highly concentrated peptides into the MS/MS ionisation chamber
occurs over a longer period compared to the elution of peptides
with lower concentrations; therefore, more spectra of the more
concentrated peptides may be acquired compared to the less
concentrated peptides (70).
Although modern LC systems are capable of very accurate separation,
there is always the possibility of co-eluting peptides when
analysing complex protein samples (58). This has resulted in critical
discussion regarding this label-free method, particularly as no
other physicochemical properties of the individual peptides are
measured (71). However, there
have been recent developments to improve spectral counting and
moving from relative quantification to absolute protein
measurement. The normalised spectral abundance factor (NSAF), the
distributed NSAF and the normalised spectral index take into
account not only the number of spectra, but also peptide count,
protein length, sample-to-sample variation for replicate analyses,
fragment-ion intensity and the distribution of spectral counts of
peptides shared by different proteins (72–74).
The absolute protein expression aims for absolute protein
measurement by calculating the proportionality of expected peptides
after trypsinisation and the observed peptides for a given protein
after MS/MS (75).
Measurement of chromatographic peak areas (or
MS signal intensities) is the second method used for label-free
quantitative proteomics. The peptides are separated by LC according
to their physical properties, e.g., charge or hydrophobicity,
followed by ionisation and detection of peptide precursor ions in a
mass spectrometer. Every precursor ion generates a specific
mono-isotopic peak, the intensity of which may be shown as function
of the LC retention time in an extracted ion chromatogram. The
chromatographic peak areas for each peptide, determined by the area
under the curve, correlate with the abundance of the respective
peptide. Therefore, a relative quantification between different
protein samples is possible (76–78).
However, this requires a high level of reproducibility between the
different samples (71,78). Moreover, due to identifying and
focusing on specific precursor ions at an early stage, there may be
a reduction in the peptides sequenced during MS/MS (79). To overcome this, two separate
analyses, one LC-MS and one LC-MS/MS, may be performed for each
sample. Nevertheless, newer and faster mass spectrometers are able
to collect data for MS as well as MS/MS scans at a satisfying rate
(71,79).
Although label-free is currently the least accurate
proteomic method, it is worth considering, as it displays distinct
advantages. First, the money- and time-consuming step of
introducing labels into proteins is not necessary, which also leads
to a reduction in experimental stages. Furthermore, although
multiplexing is not available for label-free samples, there is no
limit for parallel experiments if all the stages of sample
preparation and analyses are reproducible. Finally, label-free
quantifications exhibit a higher dynamic range compared to stable
isotope labelling. The merits of spectral counting, in particular,
have been demonstrated when seeking large changes in protein levels
compared to SILAC-labelled samples (70,71,80).
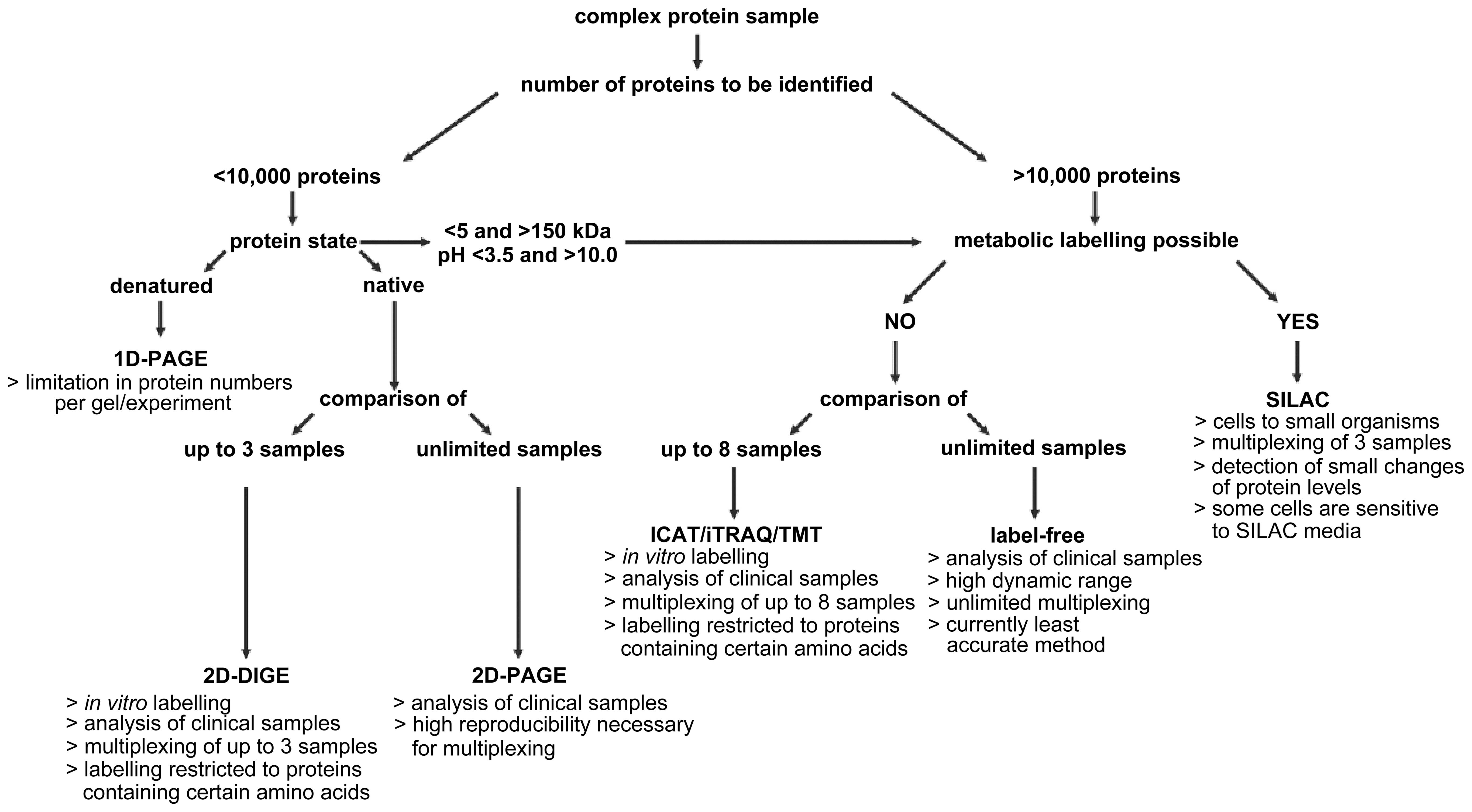
There are several factors to be considered when
selecting a particular method (Fig.
4), but often different approaches yield comparable results;
therefore, most consideration should potentially be given to the
ease of application and how well equipped a lab is to attempt any
given approach.
Proteomic methodologies have been demonstrated to be
useful in a wide range of biological investigations, including
numerous studies of interactions between virus and host proteomes.
There are a number of examples of γ-herpesvirus studies that have
utilised these approaches, which have helped elucidate several
aspects of their biology.
3. Understanding γ-herpesviruses through
proteomics
EBV is an extremely successful oncovirus, infecting
a predicted 90% of the population; however, very few infected
individuals actually develop neoplasia. This demonstrates the
ability of EBV to limit its oncogenic potential, despite expressing
multiple proteins able to immortalise cells in vitro. It is
not surprising, therefore, that various studies have demonstrated
that EBV exerts a significant effect on the cellular transcriptome.
Less attention has been given to the effect of EBV of the cellular
proteome; however, this is an important consideration, as changes
in mRNA abundance do not always translate into alterations at the
protein level. Hence, a thorough investigation of the effect of EBV
on the cellular proteome is warranted.
As the most recently discovered human herpesvirus,
our knowledge of KSHV is behind that of other, more characterised
viruses, such as EBV. However, over the last decade, several
investigations into KSHV biology have been performed, utilising
advanced proteomic-based technologies, which have resulted in
significant advances in our understanding of this virus. As with
EBV, there remains much to learn regarding KSHV biology and large
systems biology approaches, such as proteomic-based studies, may
play a major role in elucidating the intricacies of these two
complicated pathogens.
Proteomic analysis of γ-herpesvirus
virions
Early analyses of viral proteomes often focus on the
composition of the mature viral particle, as the proteins found
within are often instrumental to a successful infection. As with
all herpesviruses, EBV and KSHV have complex virions, constructed
of an inner nucleoprotein core, a viral capsid, an inner tegument
layer and a lipid envelope (81).
Surprisingly, despite being the first true human oncovirus to be
discovered, only a single study has used proteomic analyses to
comprehensively study EBV virion composition (82). In that study, the EBV lytic cycle
was induced from latently infected B cells by the ectopic
expression of the EBV lytic switch protein, Zta. Mature virus
particles were purified using density gradients, de-enveloped and
subjected to in-gel trypsin digestion and LC-MS/MS. This analysis
largely demonstrated that EBV virion morphogenesis follows that of
other herpesviruses, such as the model herpesvirus, herpes simplex
virus (HSV). ICAT was also used to determine how likely a protein
was to be associated with the envelope or the EBV capsid. In this
example, 12C ‘light’ labelled enveloped virions were
mixed with 13C ‘heavy’ labelled virions that had been
treated with detergent to remove the envelope. Following
normalisation between samples on the major capsid protein, it was
observed that several cellular proteins were major components of
mature EBV virions, including β-actin (one of the most abundant
tegument proteins), cofilin, tubulin, heat shock protein (Hsp)70,
Hsp90 and other actin-associated proteins (ARPs), such as profilin,
ezrin, radixin, moesin and α-actinin. Hsp70, α-tubulin, β-tubulin
and actin were shown to be tightly associated with capsids,
suggesting that they may be involved in stabilising the capsids or
that they may be required immediately after de-envelopment during
de novo infections. Additional functions for the cellular
proteins may include EBV virion morphogenesis and egress.
Initial determinations of the KSHV virion components
were performed in a similar manner, subjecting mature virions
purified by density gradient to 1D-gel electrophoresis, in-gel
tryptic digest and MS (83). In
total, 24 proteins of structural, regulatory and signalling roles
were identified, including 5 capsid proteins, 8 tegument proteins,
8 glycoproteins found within the lipid envelope and a further 3
proteins associated with the virion. Of note, several of the
identified proteins were of cellular origin, including a number of
proteins also found in EBV virions, such as β-actin and the ARPs
ezrin and moesin. The roles of these proteins in the viral particle
have not yet been elucidated; however, their presence suggests that
they are required for virion stability or during the early stages
of infection.
The chaperones Hsp90 and Hsc70, identified in the
virions of a wide range of viral families, were also identified in
the tegument of KSHV. These proteins have been shown to play
numerous roles in viral processes, ranging from relocalization of
key viral factors to aiding in the reverse transcription of
retroviral genomes. Furthermore, the KSHV lytic reactivator protein
replication and transcription activator (RTA) (84) was identified as a component of the
KSHV tegument, suggesting that RTA drives a round of lytic gene
expression immediately after infection. These studies demonstrated
the efficacy of using reduced-complexity samples with MS-based
proteomics to identify a subset of viral and cellular proteins
involved in different aspects of viral biology.
Elucidating the effects of fully
competent γ-herpesvirus infection through proteomics
In an attempt to gain understanding of EBV
pathogenesis, Alsayed et al (85) performed a proteomic analysis of
PTLD tissues from solid organ transplant patients using a protein
microarray technique. The proteins that were dysregulated included
proteins of the phosphoinositide 3-kinase (PI3K)/mammalian target
of rapamycin, nuclear factor-κB and Hsp90 pathways, all of which
are entirely consistent with the latency III gene expression
profile in PTLD. Interestingly, when specific inhibitors of these
proteins were then tested in EBV+ and EBV-
cell lines, they were found to be specifically cytotoxic in the
EBV+ lines. Indeed, it was recently demonstrated that
the Hsp90 inhibitors geldanamycin,
17-allylamino-17-demethoxygeldanamycin and
17-dimethylaminoethylamino-17-demethoxygeldanamycin specifically
decreased EBNA1 translation, leading to cytotoxicity of
EBV+ cells and significantly inhibiting
lymphoproliferative disease in severe combined immunodeficiency
mice (86,87). Together, these early results
highlighted the value of proteomics in identifying novel
therapeutic targets.
Proteomic analyses have also been applied in order
to understand how the human γ-herpesviruses manipulate
intercellular communication. In addition to the secretion of
soluble factors that promote the establishment of latency (or
indeed, tumourigenesis), virally infected cells also release
modified exosomes. These are a class of endosomal-derived membrane
microvesicles from multivesicular bodies that, after release, may
fuse with recipient cells, into which they release their contents
and modulate biological activities. Although this is a normal
physiological process, there is evidence suggesting that altered
exosome function may be pathogenic, particularly during malignancy.
It was demonstrated that EBV-infected cells release exosomes
containing virally-encoded miRNAs and proteins and that these
factors are able to alter target cell function (88–90).
For example, EBV’s major oncoprotein LMP1 was highly enriched in
exosomes secreted from nasopharyngeal carcinoma (NPC) cells and the
transfer of these to recipient cells activated several
LMP1-associated signalling cascades, including PI3K/AKT and
mitogen-activated protein kinase/extracellular signal-regulated
kinase (91). LMP1 has also been
identified in B-cell-secreted exosomes and this may be important
for inducing proliferation and B-cell survival, a crucial step in
the establishment of EBV latency and colonisation.
However, there remains the major question whether
viral infections dictate the composition of secreted exosomes. This
was recently addressed by Meckes et al (92) using a combination of MS and 2D-DIGE
to determine the repertoire of proteins in EBV+,
KSHV+ and dually infected cell line exosomes, compared
to non-infected counterparts. In that study, a total of 871
proteins were identified from all exosome preparations and 449 of
these proteins were common among infected and non-infected cell
lines. Therefore, several hundred exosome proteins were enriched as
a result of γ-herpesvirus infections. Some of these proteins were
specific to EBV (n=93) and others to KSHV (n=22), suggesting that
these different viruses manipulate the exosome proteome in diverse
ways, possibly in order to optimally promote their particular life
cycles. Furthermore, a 2D-DIGE, coupled with a spectral count-based
quantitative proteomic approach, was employed to compare the
exosomes of LMP1+ and LMP1- cells, confirming
that several of the alterations in the EBV-associated exosome cargo
were due to LMP1. Moreover, the pathway analysis revealed that both
EBV- and KSHV-associated exosome components largely affect similar
cellular pathways, although KSHV exosomes were enriched in proteins
involved in cellular metabolism, whereas EBV-associated exosomes
were geared towards cellular signalling pathways.
The abovementioned findings demonstrate that
proteomic-based research is able to identify multiple protein
pathways that are altered by related viruses and put these into the
context of a viral infection. However, a potential caveat that
exists in a majority studies regarding the human γ-herpesviruses is
the lack of isogenic virus-negative counterparts to rule out the
possibility that some differences are purely cell type-specific.
Despite this caveat, however, these studies enhance our
understanding of how KSHV and EBV manipulate the tumour
microenvironment and provide potential new avenues for therapeutic
intervention. Interestingly, several of the proteins identified in
mature γ-herpesvirus virions have also been detected in their
exosomes (e.g., actin and ezrin), signifying the possibility that
virally induced exosomes may mimic viral infection. If this is
true, the extent to which it may benefit virus colonisation remains
to be determined. The next challenge will be to elucidate how
viruses dictate exosome proteomes and how this translates in
vivo.
Investigating the interactions and
effects of specific viral factors
Proteomic approaches have also been used to
investigate the function of individual viral proteins. The EBNA2
protein is considered to be the master transcriptional activator,
switching on the expression of all of EBV’s immortalising genes, as
well as various cellular genes known to be critical for EBV
infection, such as CD23. In an attempt to identify proteins altered
due to EBNA2 expression, 2D-PAGE was applied to a protein sample
from a conditionally immortalised B-cell line, in which EBNA2 is
activated by translocation to the nucleus, due to the addition of
oestrogen (93). In that study,
EBNA2 target proteins and the proteins that were induced during
primary B-cell infection were shown to correlate, highlighting the
major role of EBNA2 during the establishment of latency and B-cell
immortalisation. A total of 20 differentially expressed proteins
were identified, 12 of which were upregulated, including the
protein synthesis and degradation factors eIF5a, P0, PA28γ and
ubiquitin thiolesterase L3, as well as two proteins involved in
nucleotide metabolism, Nm23-H1 and pyrophosphatase. Some of the
best characterised EBNA2-responsive proteins were not identified,
however, suggesting incomplete coverage due to the nature of the
proteomic approach employed.
Similarly, the effect of individual KSHV protein
expression has been investigated using a gel-based approach.
2D-DIGE was successfully utilised to analyse the protein content of
endothelial cells expressing the KSHV vFLIP protein (94). This study identified 14 proteins
upregulated upon vFLIP expression, the most highly upregulated of
which was manganese superoxide dismutase (MnSOD). Interestingly,
this is an antiapoptotic enzyme that has also been shown to be
upregulated in KSHV-infected cells. Importantly, previous
transcriptome analyses also identified MnSOD to be upregulated at
the RNA level, demonstrating the possibility of combining
proteomics and transcriptomics approaches to validate and enhance
results (94,95) (see Section 4).
Another proteomic-based study of EBV investigated
the transformation potential of the EBV LMP1 oncoprotein in NPC
cell lines (96). In that study, a
total of 250 proteins were detected by iTRAQ-based analysis, 12 of
which were shown to be differentially expressed (although proteins
that changed as little as 1.33-fold were also considered),
including 6 proteins involved in signal transduction:
voltage-dependent anion channel 1, S100-A2, hormone-induced
protein-70, ubiquitin, tumour protein translationally-controlled
1-like protein and 4F2hc, as well as 3 proteins pertaining to
cyctoskeleton formation: keratin-75, TB8 and dynein light chain
1.
The first SILAC-based study designed to investigate
a small number of changes in specific subcellular compartments
utilised the expression of the KSHV K5 protein (97). Modulation of the host immune
response, particularly via major histocompatibility complex (MHC)
class I molecules, is a common trait amongst pathogens. The
KSHV-encoded K3 and K5 proteins, which function as membrane-bound
RING-CH ubiquitin ligases and target MHC I for lysosomal
degradation, are homologues of the K3 family of viral immune
modulators. Importantly, other viral downregulators of MHC I are
also known to target additional transmembrane proteins pertaining
to different cellular pathways. In order to identify any novel K5
targets, plasma membrane, endoplasmic reticulum (ER) and Golgi
apparatus were isolated from labelled control cells or cells
expressing K5. In addition to the expected downregulation of MHC I,
3 novel peptides were found to be downregulated in the plasma
membrane of cells expressing K5: activated leukocyte cell adhesion
molecule, bone marrow stromal cell antigen 2 (BST-2) and
syntaxin-4. The identification of these proteins marked a precedent
in SILAC proteomics, as it demonstrated a new approach for
identifying additional targets of viral proteins. BST-2 (also known
as tetherin) is of particular interest, as this protein was first
identified as a host restriction factor that prevents the release
of budding retroviruses from the plasma membrane. HIV is able to
counteract BST-2 via its auxiliary protein, viral protein unique,
by co-opting a cellular ubiquitin ligase, βTrCP, resulting in BST-2
degradation. Subsequent studies confirmed that BST-2 is indeed a
bona fide target of K5, although it was shown to be
subjected to proteasomal degradation as it left the ER, rather than
being degraded at the plasma membrane by the lysosome (as is true
of other K5 targets). Therefore, similar to HIV, KSHV has also
developed a mechanism of circumventing BST-2 function, although
this virus has evolved its own specialised ubiquitin ligase, namely
K5.
MS-based proteomics have also been used to identify
cellular proteins binding to the terminal repeat regions (TRs) of
the KSHV genome during latency. KSHV TRs contain the latent origin
of viral replication and play a major role in the maintenance and
spread of the viral episome among dividing cells. In order to
further characterise these important regulatory elements, TR
sequences were used to affinity capture proteins from KSHV-positive
cell lysates, which were subjected to MS-based proteomic analysis
(98). In total, 123 proteins were
found to bind the TRs of KSHV and further analysis on 4 of these
proteins, namely ataxia telangiectasia and Rad3-related protein,
transcription activator BRG1, nucleophosmin 1 and poly(ADP-ribose)
polymerase 1, demonstrated that they associate with the
KSHV-encoded LANA. This suggested that proteins binding TR
sequences may play a role in viral latency.
Furthermore, by employing affinity capture methods
coupled to 1D-PAGE and MS, glutathione S-transferase-tagged N- and
C-terminal regions of LANA were used to capture and identify
protein interaction partners of LANA (99). Several proteins were found to bind
to the N-terminus (n=53) and the C-terminus (n=56) and functional
analysis of the protein groups identified 27 different categories,
including ATPases, ribonucleoproteins and cytoskeletal proteins.
Whilst the majority of these novel interactions are yet to be fully
characterised, most of the proteins that bound LANA were found to
be transcription factors and DNA-binding proteins, suggesting that
LANA plays an important role in transcriptional control.
More recently, our laboratory has undertaken
SILAC-based proteomics to elucidate the effects of the essential
KSHV mRNA processing protein, ORF57 (100,101), on cellular pathways. To this end,
an inducible HEK293-based system expressing ORF57 was utilised. To
maximise the data obtained from the study, cellular fractionation
was performed to separate the major cellular compartments where
ORF57 is known to be present: the cytoplasm, nucleoplasm and
nucleoli (102). Predictably, as
ORF57 is a known viral RNA processing protein, its expression led
to an enrichment of proteins involved in RNA processing, RNA
post-transcriptional modification and gene expression, particularly
in the nucleolar fraction, where ORF57 is highly abundant (103,104). Importantly, the analysis also
identified an enrichment in DNA repair proteins that led to the
discovery of a novel role for ORF57 in genome instability (105). That study highlighted the
significance of large-scale proteomics studies for identifying
novel proteins or cellular pathways of interest. Moreover, cellular
fractionation and isolation of cellular compartments or organelles
linked to SILAC proteomics will substantially increase the
flexibility of this type of approach.
Combining protein immunoprecipitation with mass
spec-based quantitative proteomics provides another way to reduce
sample complexity and gain greater insight into the intricacies of
viral infection. Malik-Soni and Frappier (106) have used this method to elucidate
the interaction partners of the EBV EBNA1 protein in EBV-positive
and -negative NPC cell lines. In that study, EBNA1 was found to
have very similar interactomes in the two different cell
backgrounds and these interaction profiles largely agreed with
previous studies using HEK 293T cells. Furthermore, the EBNA1
interaction partners in AGS cells infected with latent EBV were
highly similar to those in lytically infected cells, demonstrating
that several of the EBNA1-host protein interactions occur
independent of EBV infection. Of note, EBNA1 was also shown to
associate with numerous heterogeneous nuclear ribonucleoproteins
and the La protein, which associated with the 3′ ends of newly
synthesized small RNAs and had previously been shown to bind EBV
EBERs (107). The interaction
between La and EBNA1 only occurred in the context of full EBV
infection and was ablated by RNase treatment, suggesting this
interaction is bridged by EBERs. EBNA1 also appeared to interact
with the nucleolar protein B23 (nucleophosmin), a protein that
frequently plays a role in infection with other viruses, such as
KSHV and HIV-1 (98,108).
Other large-scale studies have also added to our
knowledge of the KSHV proteome, although not strictly through a
proteomic approach. For example, a large study was performed by
Sander et al (109) with
the aim to map the subcellular localisation of every KSHV protein
in HeLa cells in order to elucidate potential roles of proteins
within the context of the viral infection with regard to their
location.
Greater understanding of a viral proteome may also
be used to create novel diagnostic tools. Recently, a 96-well
format multiplex bead-based assay was used to analyse the
serological response of 2 groups of individuals previously
diagnosed with KSHV-associated malignancies (n=36) or with a low
likelihood of KSHV infection (n=43) to 73 different recombinant
proteins encompassing most of the KSHV proteome (110). Historically, the KSHV ORF73 and
K8.1 proteins have been used to determine whether an individual is
infected; however, the patterns of reactivity in those infected
with KSHV were found to be highly variable. That study identified 6
more viral proteins that may be tested for to distinguish between
KSHV-positive and -negative individuals, providing a more powerful
and reliable tool for clinical KSHV serodiagnosis.
This selection of proteomic-based research
demonstrates the power of the numerous techniques that have been
developed in this field. Combining proteomic techniques with other
large-scale systems biology approaches may provide an even greater
insight into the various processes occurring within
populations.
4. Combining OMICS approaches to maximise
function
It is becoming increasingly apparent that utilising
multiple high-throughput systems techniques can create a synergy,
enabling greater insight into more biological processes, rather
than by relying on an individual technique. As the field of
large-scale proteomics is still in its infancy, it is likely that
the full potential of combining OMICS approaches has not yet been
realised, although there are numerous published examples of groups
combining two or more methods to answer newly arising
questions.
Currently, the majority of proteomic-based studies
focus on model organisms, for which there exists a database of
encoded proteins. A recently developed approach, known as
Proteomics Informed by Transcriptomics (PIT), combines deep
sequencing-based transcriptomics (RNA-seq) with MS-based
quantitative proteomics (111).
PIT allows the interpretation of the proteomes of multiple species
in the same sample, or of species without a complete reference
genome, using the transcriptome data to reveal any proteins that
may be present. This approach may also be used to identify changes
in mRNA levels that are not reflected at the protein level,
allowing the identification of potential post-transcriptional
degradation targets. Furthermore, combining these different OMICS
procedures allows researchers to overcome the limitations of an
individual technique, such as incomplete coverage of the
transciptome, due to high mRNA turnover rate, or of the proteome,
due to the issues previously mentioned with LC-MS/MS analysis.
Other attempts to apply large-scale OMICS techniques
to non-model species rely on the sequencing and annotation of a
novel reference genome. The initial interpretation of a novel
genome is often heavily guided by using sequence homology to
closely related species and in silico predictions of open
reading frames. This commonly results in incomplete genome
annotations, often ignoring non-canonical elements, such as
transcription start sites or genomic regions that lack sequence
homology to other previously characterised genes. Indeed, some of
the most interesting aspects of KSHV and EBV biology involve EBERs,
EBNA1 and KSHV LANA, all of which could be missed using these
traditional methods of genome annotation. However, the combination
of LC-MS/MS-based proteomics with custom tiling arrays has filled
in a number of gaps in the viral genome and identified several
novel genes (112). That previous
study demonstrated that combining two or more high-throughput
approaches may compensate for the limitations of each individual
approach and further demonstrated that combining transcriptome and
proteome data may also provide some insight into the stability of
the detected transcripts and proteins.
Another recently identified gap in the viral genome
annotations came in the form of small non-coding RNAs, named
miRNAs, which are primarily involved in the repression of
translation and have been hailed as one of the most important
breakthroughs in biology (113,114). The identification and
characterisation of functional miRNAs has proved to be a difficult
endeavour in recent years, with numerous predictive algorithms
being designed to elucidate target mRNA molecules. A number of
these algorithms, however, rely on assumptions about miRNAs binding
to target sequences at the 3′ end of the target mRNAs, despite
other mechanisms having been demonstrated (115). Even when this simplistic approach
is applied, it often results in a high rate of false-positives and
predicts a large list of potential targets that could not feasibly
be experimentally validated. Using deep sequencing, aberrantly
expressed miRNAs have been identified in osteopetrosis patients and
proposed targets were identified by their altered protein
expression using iTRAQ proteomics (116). This approach led to the
identification of 96 novel miRNAs in the osteopetrosis samples and
also provided a useful method for identifying miRNA-target pairs
with greater confidence compared to that which could be achieved by
relying on predictive algorithms alone. A similar approach has also
been used to identify targets of several viral miRNAs encoded by
KSHV, whereby pulsed SILAC was employed to identify host cell genes
repressed at the protein level upon expression of individual viral
miRNAs (117). While it may be
possible to identify some miRNA-mRNA target pairs using deep
sequencing alone, this may result in only identifying degraded mRNA
targets and not those for which translation is inhibited by other
means (118).
Until recently, ~32–39% of the predicted genes on
chromosome 1 alone lacked any protein identification according to
the Chinese Human Chromosome Proteome Consortium (119), despite the publishing of a
complete draft of the human genome over 10 years ago. In a recent
effort to characterize all the missing proteins, an integrative
OMICS approach was employed, combining MS-based proteomics, RNAseq
and ribosome profiling, an emerging technique that identifies the
translation potential of mRNAs present in a sample (120). This approach, utilising proteome,
transcriptome and translatome data, reduced the as yet
uncharacterised genes to ~15%, most of which are predicted to be
tissue-specific.
5. Future perspectives
Single-cell sequencing is now a reality and numerous
studies are currently being undertaken (121,122). This technology provides greater
insight into the biology of heterogeneous populations of cells,
e.g., a tumour, in which many different cell states contribute to
the overall function. Similarly, single-cell proteomic methods are
currently being devised, utilising an antibody-bound chip,
analogous to microarray technology (123). These approaches are expected to
take high-throughput systems biology approaches to new heights and
will also generate significantly larger datasets, for which new
strategies will be required.
Our ability to interpret the data produced via
high-throughput experimental technologies is continuously
improving. There are currently numerous online tools widely
available for use, such as PepTracker (124), DAVID (125) and STRING (126), that allow scientists with minimal
understanding of bioinformatics to obtain useful data from large
proteomic datasets. Many of these systems biology-based web
applications are available free of charge to the academic
community. The aim of the Proteomics Specification in Time and
Space project is to keep improving the utility of large datasets
through the development of analysis tools and the application of
new methodologies (127).
The increasing trend towards utilising large-scale
systems biology across the scientific community has led to an
increasing availability of OMICS datasets. Indeed, multiple
projects have been established in order to set up and maintain
large data repositories, such as the ProteomeXchange and RNA-seq
Atlas web applications, allowing the wider scientific community to
use data previously obtained by other groups to inform their own
research (128,129). This is highlighted by a recent
study using 42 previously published and 7 unpublished
Drosophila phenotypes inferred by RNAi screens (130). This approach enabled a ‘signed’
protein-protein interaction network to be constructed, consisting
of 6,125 interactions between 3,352 proteins. Subsequent testing of
the network revealed a high predictive power. As more OMICS
datasets become freely available, the power of such predictive
models will increase, as will our collective understanding of the
questions we can answer by using them. It is important to remain
critical, however, and not rely solely on computational prediction
biology. Despite the success of many of these projects, the data
require validation in a wet lab environment.
Regarding EBV and KSHV, further research using
large-scale systems biology approaches will help develop our
understanding of the intricacies of infection. Proteomic screens
are currently routinely performed and it will be of particular
interest to view the results of OMICS-based analysis of numerous
single cells infected with fully competent γ-herpesviruses. The
challenges arising from this will be validating these studies and
attempting to link them to clinically relevant applications for
these important pathogens.
Acknowledgements
The authors would like to thank all the members of
the Whitehouse Laboratory for the useful discussions. This study
was supported in part by BBSRC, YCR, Wellcome Trust and AICR.
References
|
1
|
Epstein MA, Achong BG and Barr YM: Virus
particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet.
1:702–703. 1964.
|
|
2
|
Chang Y, Cesarman E, Pessin MS, Lee F,
Culpepper J, Knowles DM and Moore PS: Identification of
herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma.
Science. 266:1865–1869. 1994.
|
|
3
|
Henle G, Henle W, Clifford P, et al:
Antibodies to Epstein-Barr virus in Burkitt’s lymphoma and control
groups. J Natl Cancer Inst. 43:1147–1157. 1969.
|
|
4
|
Kieff E and Rickinson AB: Fields’
Virology. Knipe DM and Howley PM: 2. 6th Edition. Lippincott
Williams and Wilkins; Philadelphia: pp. 2655–2700. 2007
|
|
5
|
Damania B and Pipas JM: DNA Tumour
Viruses. 1st Edition. Springer; New York, NY: pp. 205–216. 2009
|
|
6
|
Taylor GS and Blackbourn DJ: Infectious
agents in human cancers: lessons in immunity and immunomodulation
from gammaherpesviruses EBV and KSHV. Cancer Lett. 305:263–278.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Preiser W, Szép NI, Lang D, Doerr HW and
Rabenau HF: Kaposi’s sarcoma-associated herpesvirus seroprevalence
in selected German patients: evaluation by different test systems.
Med Microbiol Immun. 190:121–127. 2001.
|
|
8
|
Simpson GR, Schulz TF, Whitby D, et al:
Prevalence of Kaposi’s sarcoma associated herpesvirus infection
measured by antibodies to recombinant capsid protein and latent
immunofluorescence antigen. Lancet. 348:1133–1138. 1996.
|
|
9
|
Engels EA, Sinclair MD, Biggar RJ, Whitby
D, Ebbesen P, Goedert JJ and Gastwirth JL: Latent class analysis of
human herpesvirus 8 assay performance and infection prevalence in
sub-saharan Africa and Malta. Int J Cancer. 88:1003–1008. 2000.
View Article : Google Scholar
|
|
10
|
Mesri EA, Cesarman E and Boshoff C:
Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer.
10:707–719. 2010.
|
|
11
|
Parkin DM, Sitas F, Chirenje M, Stein L,
Abratt R and Wabinga H: Part I: Cancer in indigenous Africans -
burden, distribution, and trends. Lancet Oncol. 9:683–692. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ambroziak JA, Blackbourn DJ, Herndier BG,
et al: Herpes-like sequences in HIV-infected and uninfected
Kaposi’s sarcoma patients. Science. 268:582–583. 1995.PubMed/NCBI
|
|
13
|
Kurth J, Spieker T, Wustrow J, Strickler
GJ, Hansmann LM, Rajewsky K and Küppers R: EBV-infected B cells in
infectious mononucleosis: viral strategies for spreading in the B
cell compartment and establishing latency. Immunity. 13:485–495.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Webster-Cyriaque J, Duus K, Cooper C and
Duncan M: Oral EBV and KSHV infection in HIV. Adv Dent Res.
19:91–95. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tugizov SM, Berline JW and Palefsky JM:
Epstein-Barr virus infection of polarized tongue and nasopharyngeal
epithelial cells. Nat Med. 9:307–314. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Blackbourn DJ, Lennette ET, Ambroziak J,
Mourich DV and Levy JA: Human herpesvirus 8 detection in nasal
secretions and saliva. J Infect Dis. 177:213–216. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pauk J, Huang ML, Brodie SJ, et al:
Mucosal shedding of human herpesvirus 8 in men. New Engl J Med.
343:1369–1377. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goodwin DJ, Walters MS, Smith PG, Thurau
M, Fickenscher H and Whitehouse A: Herpesvirus Saimiri open reading
frame 50 (Rta) protein reactivates the lytic replication cycle in a
persistently infected A549 cell line. J Virol. 75:4008–4013. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cesarman E: Gammaherpesvirus and
lymphoproliferative disorders in immunocompromised patients. Cancer
Lett. 305:163–174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Klein E, Kis LL and Klein G: Epstein-Barr
virus infection in humans: from harmless to life endangering
virus-lymphocyte interactions. Oncogene. 26:1297–1305. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Speck SH and Ganem D: Viral latency and
its regulation: lessons from the gamma-herpesviruses. Cell Host
Microbe. 8:100–115. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Babcock GJ, Decker LL, Volk M and
Thorley-Lawson DA: EBV persistence in memory B cells in vivo.
Immunity. 9:395–404. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Babcock GJ, Hochberg D and Thorley-Lawson
AD: The expression pattern of Epstein-Barr virus latent genes in
vivo is dependent upon the differentiation stage of the infected B
cell. Immunity. 13:497–506. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Davis DA, Rinderknecht AS, Zoeteweij JP,
et al: Hypoxia induces lytic replication of Kaposi
sarcoma-associated herpesvirus. Blood. 97:3244–3250. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Blackbourn DJ, Fujimura S, Kutzkey T and
Levy JA: Induction of human herpesvirus-8 gene expression by
recombinant interferon gamma. AIDS. 14:98–99. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vieira J, O’Hearn P, Kimball L, Chandran B
and Corey L: Activation of Kaposi’s sarcoma-associated herpesvirus
(human herpesvirus 8) lytic replication by human cytomegalovirus. J
Virol. 75:1378–1386. 2001.
|
|
27
|
Zeng Y, Zhang X, Huang Z, et al:
Intracellular Tat of human immunodeficiency virus type 1 activates
lytic cycle replication of Kaposi’s sarcoma-associated herpesvirus:
role of JAK/STAT signaling. J Virol. 81:2401–2417. 2007.PubMed/NCBI
|
|
28
|
Wilson SJ, Tsao EH, Webb BL, et al: X box
binding protein XBP-1s transactivates the Kaposi’s
sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking
plasma cell differentiation to KSHV reactivation from latency. J
Virol. 81:13578–13586. 2007.PubMed/NCBI
|
|
29
|
Roth WK, Brandstetter H and Sturzl M:
Cellular and molecular features of HIV-associated Kaposi’s sarcoma.
AIDS. 6:895–913. 1992.
|
|
30
|
Staskus KA, Zhong W, Gebhard K, et al:
Kaposi’s sarcoma-associated herpesvirus gene expression in
endothelial (spindle) tumor cells. J Virol. 71:715–719. 1997.
|
|
31
|
Orenstein JM, Alkan S, Blauvelt A, Jeang
KT, Weinstein MD, Ganem D and Herndier B: Visualization of human
herpesvirus type 8 in Kaposi’s sarcoma by light and transmission
electron microscopy. AIDS. 11:F35–F45. 1997.PubMed/NCBI
|
|
32
|
Arvanitakis L, Geras-Raaka E, Varma A,
Gershengorn MC and Cesarman E: Human herpesvirus KSHV encodes a
constitutively active G-protein-coupled receptor linked to cell
proliferation. Nature. 385:347–350. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bais C, Santomasso B, Coso O, et al:
G-protein-coupled receptor of Kaposi’s sarcoma-associated
herpesvirus is a viral oncogene and angiogenesis activator. Nature.
391:86–89. 1998.
|
|
34
|
Boshoff C, Endo Y, Collins PD, et al:
Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines.
Science. 278:290–294. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gao SJ, Boshoff C, Jayachandra S, Weiss
RA, Chang Y and Moore PS: KSHV ORF K9 (vIRF) is an oncogene which
inhibits the interferon signaling pathway. Oncogene. 15:1979–1985.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee H, Guo J, Li M, Choi JK, DeMaria M,
Rosenzweig M and Jung JU: Identification of an immunoreceptor
tyrosine-based activation motif of K1 transforming protein of
Kaposi’s sarcoma-associated herpesvirus. Mol Cell Biol.
18:5219–5228. 1998.PubMed/NCBI
|
|
37
|
Sarid R, Sato T, Bohenzky RA, Russo JJ and
Chang Y: Kaposi’s sarcoma-associated herpesvirus encodes a
functional bcl-2 homologue. Nat Med. 3:293–298. 1997.
|
|
38
|
Moore PS, Boshoff C, Weiss RA and Chang Y:
Molecular mimicry of human cytokine and cytokine response pathway
genes by KSHV. Science. 274:1739–1744. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun Q, Matta H, Lu G and Chaudhary PM:
Induction of IL-8 expression by human herpesvirus 8 encoded vFLIP
K13 via NF-kappaB activation. Oncogene. 25:2717–2726. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Osborne J, Moore PS and Chang Y:
KSHV-encoded viral IL-6 activates multiple human IL-6 signaling
pathways. Hum Immunol. 60:921–927. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cesarman E, Moore PS, Rao PH, Inghirami G,
Knowles DM and Chang Y: In vitro establishment and characterization
of two acquired immunodeficiency syndrome-related lymphoma cell
lines (BC-1 and BC-2) containing Kaposi’s sarcoma-associated
herpesvirus-like (KSHV) DNA sequences. Blood. 86:2708–2714.
1995.PubMed/NCBI
|
|
42
|
Hu J, Garber AC and Renne R: The
latency-associated nuclear antigen of Kaposi’s sarcoma-associated
herpesvirus supports latent DNA replication in dividing cells. J
Virol. 76:11677–11687. 2002.
|
|
43
|
Jenner RG, Alba MM, Boshoff C and Kellam
P: Kaposi’s sarcoma-associated herpesvirus latent and lytic gene
expression as revealed by DNA arrays. J Virol. 75:891–902.
2001.
|
|
44
|
Pearce M, Matsumura S and Wilson AC:
Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA
cluster of Kaposi’s sarcoma-associated herpesvirus originate from a
common promoter. J Virol. 79:14457–14464. 2005.PubMed/NCBI
|
|
45
|
Alkharsah KR, Singh VV, Bosco R, et al:
Deletion of Kaposi’s sarcoma-associated herpesvirus FLICE
inhibitory protein, vFLIP, from the viral genome compromises the
activation of STAT1-responsive cellular genes and spindle cell
formation in endothelial cells. J Virol. 85:10375–10388. 2011.
|
|
46
|
Grossmann C, Podgrabinska S, Skobe M and
Ganem D: Activation of NF-κB by the latent vFLIP gene of Kaposi’s
sarcoma-associated herpesvirus is required for the spindle shape of
virus-infected endothelial cells and contributes to their
proinflammatory phenotype. J Virol. 80:7179–7185. 2006.
|
|
47
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
O’Farrell PH: High resolution
two-dimensional electrophoresis of proteins. J Biol Chem.
250:4007–4021. 1975.
|
|
49
|
Klose J and Kobalz U: Two-dimensional
electrophoresis of proteins: an updated protocol and implications
for a functional analysis of the genome. Electrophoresis.
16:1034–1059. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gorg A, Weiss W and Dunn MJ: Current
two-dimensional electrophoresis technology for proteomics.
Proteomics. 4:3665–3685. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Miller I, Crawford J and Gianazza E:
Protein stains for proteomic applications: which, when, why?
Proteomics. 6:5385–5408. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Neuhoff V, Arold N, Taube D and Ehrhardt
W: Improved staining of proteins in polyacrylamide gels including
isoelectric focusing gels with clear background at nanogram
sensitivity using Coomassie Brilliant Blue G-250 and R-250.
Electrophoresis. 9:255–262. 1988. View Article : Google Scholar
|
|
53
|
Berggren K, Chernokalskaya E, Steinberg
TH, et al: Background-free, high sensitivity staining of proteins
in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide
gels using a luminescent ruthenium complex. Electrophoresis.
21:2509–2521. 2000. View Article : Google Scholar
|
|
54
|
Berggren KN, Schulenberg B, Lopez MF, et
al: An improved formulation of SYPRO Ruby protein gel stain:
comparison with the original formulation and with a ruthenium II
tris (bathophenanthroline disulfonate) formulation. Proteomics.
2:486–498. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Patton WF: Detection technologies in
proteome analysis. J Chromatogr B Analyt Technol Biomed Life Sci.
771:3–31. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chandramouli K and Qian PY: Proteomics:
challenges, techniques and possibilities to overcome biological
sample complexity. Hum Genom Proteomics. 1:pii: 239204.
2009.PubMed/NCBI
|
|
57
|
Unlu M, Morgan ME and Minden JS:
Difference gel electrophoresis: a single gel method for detecting
changes in protein extracts. Electrophoresis. 18:2071–2077. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
May C, Brosseron F, Chartowski P,
Schumbrutzki C, Schoenebeck B and Marcus K: Instruments and methods
in proteomics. Methods Mol Biol. 696:3–26. 2011. View Article : Google Scholar
|
|
59
|
Michalski A, Cox J and Mann M: More than
100,000 detectable peptide species elute in single shotgun
proteomics runs but the majority is inaccessible to data-dependent
LC-MS/MS. J Proteome Res. 10:1785–1793. 2011. View Article : Google Scholar
|
|
60
|
Washburn MP, Wolters D and Yates JR III:
Large-scale analysis of the yeast proteome by multidimensional
protein identification technology. Nat Biotechnol. 19:242–247.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yates JR III, Eng JK, McCormack AL and
Schieltz D: Method to correlate tandem mass spectra of modified
peptides to amino acid sequences in the protein database. Anal
Chem. 67:1426–1436. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Munday DC, Surtees R, Emmott E, et al:
Using SILAC and quantitative proteomics to investigate the
interactions between viral and host proteomes. Proteomics.
12:666–672. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ong SE, Blagoev B, Kratchmarova I,
Kristensen DB, Steen H, Pandey A and Mann M: Stable isotope
labeling by amino acids in cell culture, SILAC, as a simple and
accurate approach to expression proteomics. Mol Cell Proteomics.
1:376–386. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Krijgsveld J, Ketting RF, Mahmoudi T, et
al: Metabolic labeling of C. elegans and D.
melanogaster for quantitative proteomics. Nat Biotechnol.
21:927–931. 2003.
|
|
65
|
Kruger M, Moser M, Ussar S, et al: SILAC
mouse for quantitative proteomics uncovers kindlin-3 as an
essential factor for red blood cell function. Cell. 134:353–364.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ong SE, Kratchmarova I and Mann M:
Properties of 13C-substituted arginine in stable isotope
labeling by amino acids in cell culture (SILAC). J Proteome Res.
2:173–181. 2003.
|
|
67
|
Gygi SP, Rist B, Gerber SA, Turecek F,
Gelb MH and Aebersold R: Quantitative analysis of complex protein
mixtures using isotope-coded affinity tags. Nat Biotechnol.
17:994–999. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
68
|
Schmidt A, Kellermann J and Lottspeich F:
A novel strategy for quantitative proteomics using isotope-coded
protein labels. Proteomics. 5:4–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
DeSouza L, Diehl G, Rodrigues MJ, Guo J,
Romaschin AD, Colgan TJ and Siu KW: Search for cancer markers from
endometrial tissues using differentially labeled tags iTRAQ and
cICAT with multidimensional liquid chromatography and tandem mass
spectrometry. J Proteome Res. 4:377–386. 2005. View Article : Google Scholar
|
|
70
|
Liu H, Sadygov RG and Yates JR III: A
model for random sampling and estimation of relative protein
abundance in shotgun proteomics. Anal Chem. 76:4193–4201. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bantscheff M, Schirle M, Sweetman G, Rick
J and Kuster B: Quantitative mass spectrometry in proteomics: a
critical review. Anal Bioanal Chem. 389:1017–1031. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang Y, Wen Z, Washburn MP and Florens L:
Refinements to label free proteome quantitation: how to deal with
peptides shared by multiple proteins. Anal Chem. 82:2272–2281.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Florens L, Carozza MJ, Swanson SK,
Fournier M, Coleman MK, Workman JL and Washburn MP: Analyzing
chromatin remodeling complexes using shotgun proteomics and
normalized spectral abundance factors. Methods. 40:303–311. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zybailov B, Mosley AL, Sardiu ME, Coleman
MK, Florens L and Washburn MP: Statistical analysis of membrane
proteome expression changes in Saccharomyces cerevisiae. J
Proteome Res. 5:2339–2347. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Lu P, Vogel C, Wang R, Yao X and Marcotte
EM: Absolute protein expression profiling estimates the relative
contributions of transcriptional and translational regulation. Nat
Biotechnol. 25:117–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bondarenko PV, Chelius D and Shaler TA:
Identification and relative quantitation of protein mixtures by
enzymatic digestion followed by capillary reversed-phase liquid
chromatography-tandem mass spectrometry. Anal Chem. 74:4741–4749.
2002. View Article : Google Scholar
|
|
77
|
Chelius D and Bondarenko PV: Quantitative
profiling of proteins in complex mixtures using liquid
chromatography and mass spectrometry. J Proteome Res. 1:317–323.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wong JW and Cagney G: An overview of
label-free quantitation methods in proteomics by mass spectrometry.
Methods Mol Biol. 604:273–283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Tate S, Larsen B, Bonner R and Gingras AC:
Label-free quantitative proteomics trends for protein-protein
interactions. J Proteomics. 81:91–101. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Li Z, Adams RM, Chourey K, Hurst GB,
Hettich RL and Pan C: Systematic comparison of label-free,
metabolic labeling, and isobaric chemical labeling for quantitative
proteomics on LTQ Orbitrap Velos. J Proteome Res. 11:1582–1590.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lippe R: Deciphering novel
host-herpesvirus interactions by virion proteomics. Front
Microbiol. 3:1812012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Johannsen E, Luftig M, Chase MR, et al:
Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci USA.
101:16286–16291. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhu FX, Chong JM, Wu L and Yuan Y: Virion
proteins of Kaposi’s sarcoma-associated herpesvirus. J Virol.
79:800–811. 2005.
|
|
84
|
Gould F, Harrison SM, Hewitt EW and
Whitehouse A: Kaposi’s sarcoma-associated herpesvirus RTA promotes
degradation of the Hey1 repressor protein through the ubiquitin
proteasome pathway. J Virol. 83:6727–6738. 2009.
|
|
85
|
Alsayed Y, Leleu X, Leontovich A, Oton AB,
Malhem M, George D and Ghobrial IM: Proteomics analysis in
post-transplant lymphoproliferative disorders. Eur J Haematol.
81:298–303. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Sun X, Barlow EA, Ma S, et al: Hsp90
inhibitors block outgrowth of EBV-infected malignant cells in vitro
and in vivo through an EBNA1-dependent mechanism. Proc Natl Acad
Sci USA. 107:3146–3151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Sun X and Kenney SC: Hsp90 inhibitors: a
potential treatment for latent EBV infection? Cell Cycle.
9:1665–1666. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Meckes DG Jr and Raab-Traub N:
Microvesicles and viral infection. J Virol. 85:12844–12854. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Pegtel DM, Cosmopoulos K, Thorley-Lawson
DA, et al: Functional delivery of viral miRNAs via exosomes. Proc
Natl Acad Sci USA. 107:6328–6333. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Raimondo F, Morosi L, Chinello C, Magni F
and Pitto M: Advances in membranous vesicle and exosome proteomics
improving biological understanding and biomarker discovery.
Proteomics. 11:709–720. 2011. View Article : Google Scholar
|
|
91
|
Meckes DG Jr, Shair KH, Marquitz AR, Kung
CP, Edwards RH and Raab-Traub N: Human tumor virus utilizes
exosomes for intercellular communication. Proc Natl Acad Sci USA.
107:20370–20375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Meckes DG Jr, Gunawardena HP, Dekroon RM,
et al: Modulation of B-cell exosome proteins by gamma herpesvirus
infection. Proc Natl Acad Sci USA. 110:E2925–E2933. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Schlee M, Krug T, Gires O, et al:
Identification of Epstein-Barr virus (EBV) nuclear antigen 2
(EBNA2) target proteins by proteome analysis: activation of EBNA2
in conditionally immortalized B cells reflects early events after
infection of primary B cells by EBV. J Virol. 78:3941–3952. 2004.
View Article : Google Scholar
|
|
94
|
Thurau M, Marquardt G, Gonin-Laurent N, et
al: Viral inhibitor of apoptosis vFLIP/K13 protects endothelial
cells against superoxide-induced cell death. J Virol. 83:598–611.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Sakakibara S, Pise-Masison CA, Brady JN
and Tosato G: Gene regulation and functional alterations induced by
Kaposi’s sarcoma-associated herpesvirus-encoded ORFK13/vFLIP in
endothelial cells. J Virol. 83:2140–2153. 2009.PubMed/NCBI
|
|
96
|
Feng X, Zhang J, Chen WN and Ching CB:
Proteome profiling of Epstein-Barr virus infected nasopharyngeal
carcinoma cell line: identification of potential biomarkers by
comparative iTRAQ-coupled 2D LC/MS-MS analysis. J Proteomics.
74:567–576. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Bartee E, McCormack A and Früh K:
Quantitative membrane proteomics reveals new cellular targets of
viral immune modulators. PLoS Pathog. 2:e1072006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Si H, Verma SC and Robertson ES: Proteomic
analysis of the Kaposi’s sarcoma-associated herpesvirus terminal
repeat element binding proteins. J Virol. 80:9017–9030. 2006.
|
|
99
|
Kaul R, Verma SC and Robertson ES: Protein
complexes associated with the Kaposi’s sarcoma-associated
herpesvirus-encoded LANA. Virology. 364:317–329. 2007.
|
|
100
|
Boyne JR, Jackson BR, Taylor A, Macnab SA
and Whitehouse A: Kaposi’s sarcoma-associated herpesvirus ORF57
protein interacts with PYM to enhance translation of viral
intronless mRNAs. EMBO J. 29:1851–1864. 2010.
|
|
101
|
Jackson BR, Boyne JR, Noerenberg M, et al:
An interaction between KSHV ORF57 and UIF provides mRNA-adaptor
redundancy in Herpesvirus intronless mRNA export. PLoS Pathog.
7:e10021382011. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Hiscox JA, Whitehouse A and Matthews DA:
Nucleolar proteomics and viral infection. Proteomics. 10:4077–4086.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Boyne JR and Whitehouse A: Nucleolar
disruption impairs Kaposi’s sarcoma-associated herpesvirus
ORF57-mediated nuclear export of intronless viral mRNAs. FEBS Lett.
583:3549–3556. 2009.PubMed/NCBI
|
|
104
|
Taylor A, Jackson BR, Noerenberg M, et al:
Mutation of a C-terminal motif affects Kaposi’s sarcoma-associated
herpesvirus ORF57 RNA binding, nuclear trafficking, and
multimerization. J Virol. 85:7881–7891. 2011.PubMed/NCBI
|
|
105
|
Jackson BR, Noerenberg M and Whitehouse A:
A novel mechanism inducing genome instability in Kaposi’s
sarcoma-associated herpesvirus infected cells. PLoS Pathog.
10:e10040982014.PubMed/NCBI
|
|
106
|
Malik-Soni N and Frappier L: Proteomic
profiling of EBNA1-host protein interactions in latent and lytic
Epstein-Barr virus infections. J Virol. 86:6999–7002. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Howe JG and Shu MD: Isolation and
characterization of the genes for two small RNAs of herpesvirus
papio and their comparison with Epstein-Barr virus-encoded EBER
RNAs. J Virol. 62:2790–2798. 1988.PubMed/NCBI
|
|
108
|
Szebeni A, Mehrotra B, Baumann A, Adam SA,
Wingfield PT and Olson MO: Nucleolar protein B23 stimulates nuclear
import of the HIV-1 Rev protein and NLS-conjugated albumin.
Biochemistry. 36:3941–3949. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Sander G, Konrad A, Thurau M, et al:
Intracellular localization map of human herpesvirus 8 proteins. J
Virol. 82:1908–1922. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Labo N, Miley W, Marshall V, et al:
Heterogeneity and breadth of host antibody response to KSHV
infection demonstrated by systematic analysis of the KSHV proteome.
PLoS Pathog. 10:e10040462014. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Evans VC, Barker G, Heesom KJ, Fan J,
Bessant C and Matthews DA: De novo derivation of proteomes from
transcriptomes for transcript and protein identification. Nat
Methods. 9:1207–1211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Dresang LR, Teuton JR, Feng H, et al:
Coupled transcriptome and proteome analysis of human lymphotropic
tumor viruses: insights on the detection and discovery of viral
genes. BMC Genomics. 12:6252011. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Couzin J: Breakthrough of the year. Small
RNAs make big splash. Science. 298:2296–2297. 2002.PubMed/NCBI
|
|
114
|
Dennis C: Small RNAs: the genome’s guiding
hand? Nature. 420:7322002.
|
|
115
|
Jopling CL: Regulation of hepatitis C
virus by microRNA-122. Biochem Soc Trans. 36:1220–1223. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Ou M, Zhang X, Dai Y, et al:
Identification of potential microRNA-target pairs associated with
osteopetrosis by deep sequencing, iTRAQ proteomics and
bioinformatics. Eur J Hum Genet. 5:625–632. 2013.PubMed/NCBI
|
|
117
|
Gallaher AM, Das S, Xiao Z, Andresson T,
Kieffer-Kwon P, Happel C and Ziegelbauer J: Proteomic screening of
human targets of viral microRNAs reveals functions associated with
immune evasion and angiogenesis. PLoS Pathog. 9:e10035842013.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Huang TC, Pinto SM and Pandey A:
Proteomics for understanding miRNA biology. Proteomics. 13:558–567.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Wu S, Li N, Ma J, et al: First proteomic
exploration of protein-encoding genes on chromosome 1 in human
liver, stomach, and colon. J Proteome Res. 12:67–80. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhang C, Li N, Zhai L, et al: Systematic
analysis of missing proteins provides clues to help define all of
the protein-coding genes on human chromosome 1. J Proteome Res.
13:114–125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Tang F, Barbacioru C, Wang Y, et al:
mRNA-Seq whole-transcriptome analysis of a single cell. Nat
Methods. 6:377–382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Islam S, Kjallquist U, Moliner A, Zajac P,
Fan JB, Lönnerberg P and Linnarsson S: Highly multiplexed and
strand-specific single-cell RNA 5′ end sequencing. Nat Protoc.
7:813–828. 2012.PubMed/NCBI
|
|
123
|
Salehi-Reyhani A, Kaplinsky J, Burgin E,
et al: A first step towards practical single cell proteomics: a
microfluidic antibody capture chip with TIRF detection. Lab Chip.
11:1256–1261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lamond A: http://www.peptracker.com/dm/.
Accessed June 1, 2014
|
|
125
|
Boisvert FM, Ahmad Y, Gierliński M, et al:
A quantitative spatial proteomics analysis of proteome turnover in
human cells. Mol Cell Proteomics. 11:M111.011429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
von Mering C, Huynen M, Jaeggi D, Schmidt
S, Bork P and Snel B: STRING: a database of predicted functional
associations between proteins. Nucleic Acids Res. 31:258–261.
2003.PubMed/NCBI
|
|
127
|
Mann M, Aebersold R, Robinson CV, et al:
http://www.propsects-fp7.eu/resources/index.html.
Accessed June 1, 2014
|
|
128
|
Krupp M, Marquardt JU, Sahin U, Galle PR,
Castle J and Teufel A: RNA-Seq Atlas - a reference database for
gene expression profiling in normal tissue by next-generation
sequencing. Bioinformatics. 28:1184–1185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Vizcaino JA, Deutsch EW, Wang R, et al:
ProteomeXchange provides globally coordinated proteomics data
submission and dissemination. Nat Biotechnol. 32:223–226. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Vinayagam A, Zirin J, Roesel C, et al:
Integrating protein-protein interaction networks with phenotypes
reveals signs of interactions. Nat Methods. 11:94–99. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Jones JF, Shurin S, Abramowsky C, et al:
T-cell lymphomas containing Epstein-Barr viral DNA in patients with
chronic Epstein-Barr virus infections. New Engl J Med. 318:733–741.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Old LJ, Boyse EA, Oettgen HF, De Harven E,
Geering G, Williamson B and Clifford P: Precipitating antibody in
human serum to an antigen present in cultured Burkitt’s lymphoma
cells. Proc Natl Acad Sci USA. 56:1699–1704. 1966.PubMed/NCBI
|