Introduction
Granulosa cell tumors (GCTs) are rare,
hormone-producing ovarian malignancies, representing 80% of ovarian
sex cord-stromal tumors, and accounting for ~7–8% of all ovarian
neoplasms (1). Between 2008 and
2012, there were only 451 histologically confirmed cases of sex
cord-stromal cell tumors in 18 states of the USA, 67% of them
occurring in Caucasian female patients (2). A review of the Hungarian literature
(3–6)
revealed that the incidence of diagnosed GCTs is similar to that of
the international data; however, there is no relevant Hungarian
statistics database regarding sex cord-stromal tumors. The four
representative articles presented 120 cases of GCTs between 1960
and 2005, with patients between the ages of 14 and 86 (3–6). No
androgen-producing tumors were mentioned, and there was only one
article reporting an adolescent case (5).
Adult patients with GCTs usually present with a
palpable mass, or with symptoms due to hormone production,
including estrogens or androgens, leading to diagnosis at an early
stage with a better prognosis (7).
Hormone-producing malignancies are rare in children or adolescent
patients: Only 0.1% of all ovarian tumors and 4–5% of GCTs occur in
the sexually non-active ages (8). A
proper bimanual vaginal examination cannot be performed in the
majority of adolescent patients, thereby leading to a more
difficult differential diagnosis. Signs and symptoms of these
tumors are not as specific as the hormone-producing neoplasms in
adults; therefore, a more specific investigation is required in
such cases. In the present study, the case of an ovarian
juvenile-type GCT with androgenic manifestation in a 14-year-old
girl is reported, also including a review of the Hungarian and
international literature.
Case report
A 14-year-old girl presented in Zala County
Hospital, Zalaegerszeg, Hungary with complaints of secondary
amenorrhea over the course of the past 18 months, followed by
masculinization. Her menarche was at the age of 10. After one year
of normal menstrual periods, the patient experienced irregular
menstruation, followed by amenorrhea. The patient's past medical
history was unremarkable. Her body mass index was 18.7
kg/m2, with 25–50% weight-for-age and 50–75%
height-for-age percentiles. A physical examination revealed
prominent hirsutism on the upper lip, thighs with a
Ferriman-Gallway score of 20, delayed thelarche and a deepened
voice. A pelvic examination revealed an anteflected, normal-sized
uterus, a palpable mass of 4 cm in the right ovarial area and an
enlarged clitoris of 5 cm. The patient's vital parameters and other
physical findings were normal.
Laboratory findings revealed an elevated plasma
total testosterone level of 8.84 nmol/l (normal: 0.17–2.81 nmol/l).
The serum levels of dehydroepiandrosterone sulfate,
follicle-stimulating hormone, luteinizing hormone, estradiol,
progesterone, thyroid-stimulating hormone, prolactin, α-fetoprotein
(AFP), cancer antigen-125 and cancer antigen-15-3 were within
normal limits. A pelvic ultrasonography revealed a well-defined
heterogeneous mass of 12×12 mm within the right ovary measuring
23×16 mm; other findings were normal. Pelvic, retroperitoneal and
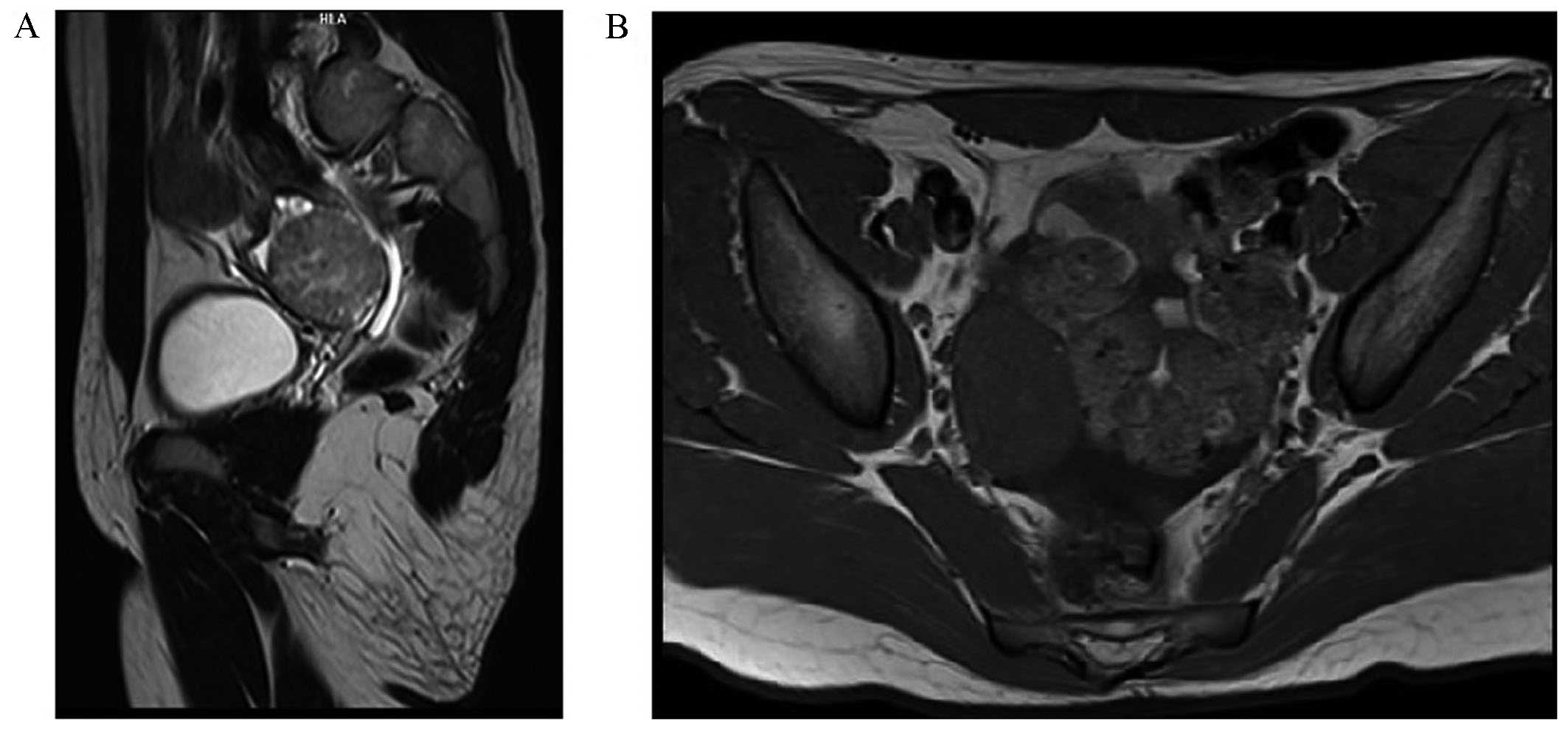
renal magnetic resonance imaging analyses made with a Siemens
Magnetom Avanto™ MRI scanner demonstrated the presence of a solid
lesion in the right ovary of 36×42×45 mm, minor grade
hepatosplenomegaly and ascites (Fig.
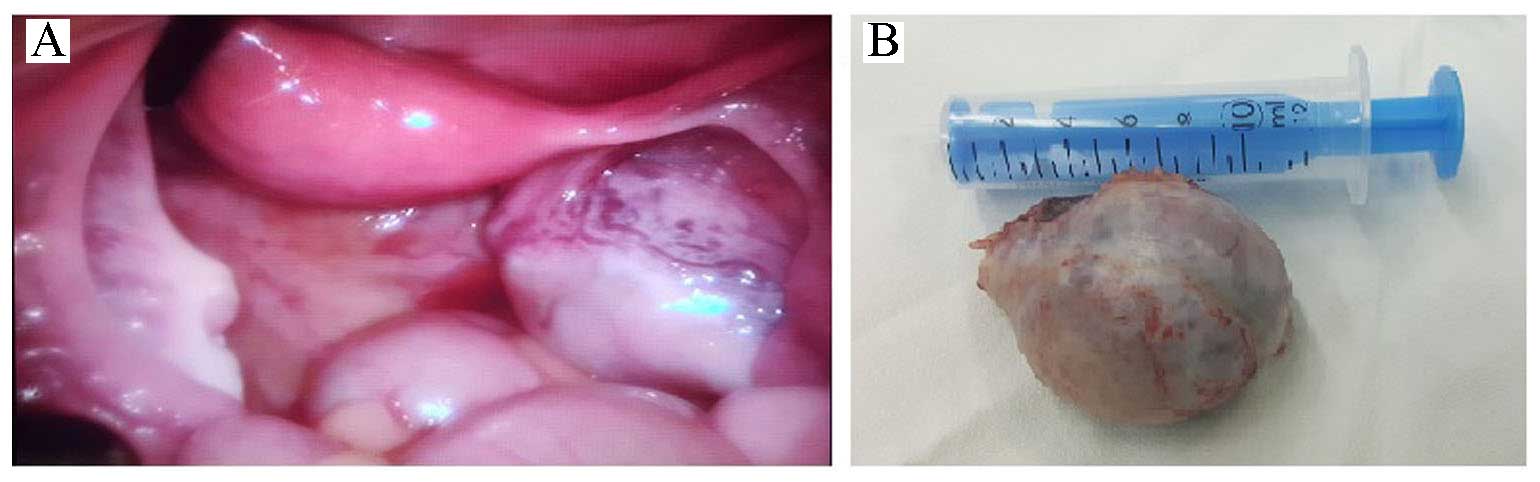
1). The patient underwent laparoscopic surgery, during which a
right-ovarian tumorous mass of 5×4 cm with abnormal vascularization
was encountered. The right and left Fallopian tubes, the left ovary
and the uterus appeared to be normal. A right ovarian oophorectomy
was performed with a LigaSure™ device (5 mm blunt tip, ForceTriad™
energy platform; Covidien-Medtronic, Minneapolis, MN, USA), and the
mass was removed in an Endobag™ (ASID BONZ GmbH, Herrenberg,
Germany) to avoid spreading of the cancer cells.
A histopathological examination confirmed a
yellow-tan ovarian mass of 5.5×4×3 cm with a lobulated cut surface
containing a grey-white solid area of 15 mm (Fig. 2). Microscopic findings revealed the
presence of a heterogeneous, solid and cystic tumor with a
formation of lobules, nests and perivascular palisades of granulosa
tumor cells with scant cytoplasm, ovoid nuclei and nuclear grooving
in several zones. There was no lymph vascular invasion, and the
ovarial serosa was intact.
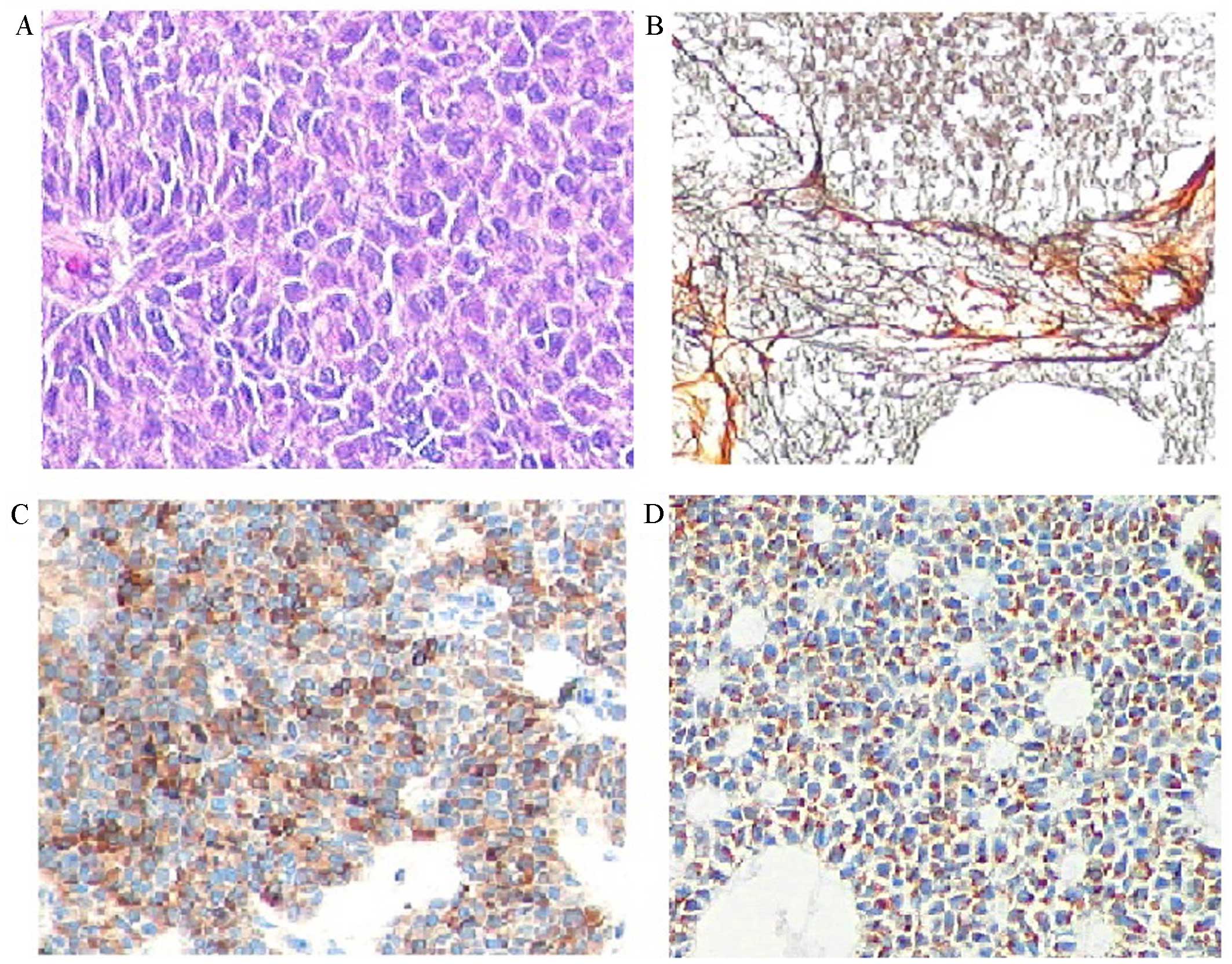
Immunohistochemical studies (Fig. 3) revealed positive cytoplasmic
staining for inhibin and Melan-A, nuclear staining for WT1, nuclear
and cytoplasmic staining for calretinin, and membrane staining for
CD99. The stain was negative for epithelial membrane antigen (EMA),
CD117, placental alkaline phosphatase (PLAP), AFP and CD30. A final
diagnosis of juvenile-type GCT was established.
The postoperative course was uncomplicated. Two days
after the surgery, the serum total testosterone level had
dramatically declined to 0.497 nmol/l. The patient was discharged
on the sixth postoperative day. During her follow-up, she got her
normal period again, and the 1-week, 1-month and 4-month serum
total testosterone levels were 0.50 nmol/l, 0.41 nmol/l and
<0.17 nmol/l, respectively.
Prior to the submission of this case study for
publication, written informed consent was obtained from the
patient's guardian.
Discussion
GCTs are divided into two histopathological
subtypes, classified as adult-type and juvenile-type GCTs. The
adult-subtype tumor, representing 95% of all GCTs, occurs in
perimenopausal or postmenopausal women, at a peak age frequency
between 50 and 55 years. The juvenile-type GCT is represented in 5%
of cases, mostly recognized in the prepubertal age, at a peak age
of 13 (9). The two subtypes may be
hormonally active and occur in children, adolescents and adults;
therefore, diagnosis is based on a histopathological evaluation
(10).
The symptoms of the tumor occur due to its hormone
production: Hyperestrogenism in 97–98% of the cases, and
hyperandrogenism in 2–3% of the cases. Clinical manifestations of
estrogen-producing tumors are amenorrhea, dysfunctional menstrual
bleeding, growth of uterine leiomyomas, hyperplasia of the
endometrium, or endometrial cancer. The symptoms and signs of the
rare virilizing GCTs are primary or secondary amenorrhea,
hirsutism, clitoris hypertrophy, deepening of the voice, muscular
development and acne due to elevated testosterone levels (11).
Diagnosis is based on laboratory and
histopathological findings. A total of 94% of the GCTs are
unilateral and diagnosed at an early stage; therefore, unilateral
oophorectomy or adnexectomy as the surgical treatment is the method
of choice (12). Following surgery,
the majority of the symptoms resulting from hormone production may
disappear. Based on the histopathological staging, adjuvant
chemotherapy or radiotherapy should be considered (13). The prognosis is excellent, with
90–95% 5-year survival in the early stages, and this correlates
with tumor stage, grade and mitotic index (14). Follow-up must be performed every two
to three months during the first few years following the operation,
but since late recurrences have been reported 20 years after the
initial treatment, long-term follow-up should also be considered
(15).
In conclusion, virilizing GCTs are rare causes of
hyperandrogenism in adolescents. The diagnosis is based on signs
and symptoms of elevated testosterone levels, clinical and imaging
findings; however, a definitive diagnosis can only be made
following histopathology. The majority of the cases are discovered
at an early stage; therefore, 5-year and 10-year survival rates are
excellent. The tumor may be treated surgically; in the majority of
the cases, without a need for postoperative adjuvant therapy;
however, long-term follow up should be considered, as late
recurrences are mentioned in the relevant literature.
References
|
1
|
Thrall MM, Paley P, Pizer E, Garcia R and
Goff BA: Patterns of spread and recurrence of sex cord-stromal
tumors of the ovary. Gynecol Oncol. 122:242–245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Howlader N, Noone AM, Krapcho M, Garshell
J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, et al: SEER
Cancer Statistics Review, 1975–2012. National Cancer Institute;
Bethesda, MD: http://seer.cancer.gov/csr/1975_2012
|
|
3
|
Horányi D, Koiss R, Babarczi E and Siklós
P: A petefészek ivarléc-stroma eredetű daganatainak kezelésével
szerzett tapasztalataink. Nőgyógyászati Onkológia. 16:40–42.
2011.
|
|
4
|
Csapó ZS, Szirmai K, Nagy GyR and Papp Z:
Granulosa cell tumor (Retrospective study of 15 cases occuring
during 15 years). Magyar Nőorvosok Lapja. 69:471–474. 2006.
|
|
5
|
Göcze P, Krommer K, Csermely T, Cziráky K,
Garamvölgyi Z, Kovács K and Szabó I: Ovulation induction therapy
and ovarian cancer. Orvo Hetil. 141:71–75. 2000.(In Hungarin).
|
|
6
|
Tanyi J, Rigó JR, Kis Csitári I and Csapó
ZS: Juvenile granulosa cell tumor complicating pregnancy: Report of
2 cases. Magyar Nőorvosok Lapja. 61:451–454. 1998.
|
|
7
|
Haroon S, Idrees R, Zia A, Memon A, Fatima
S and Kayani N: Ovarian sex cord stromal tumours in children and
young girls-a more than two decade clinicopathological experience
in a developing country, Pakistan. Asian Pac J Cancer Prev.
15:1351–1355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hashemipour M, Moaddab MH, Nazem M,
Mahzouni P and Salek M: Granulosa cell tumor in a six-year-old girl
presented as precocious puberty. J Res Med Sci. 15:240–242.
2010.PubMed/NCBI
|
|
9
|
Kabaca C, Karateke A, Gurbuz A and Cesur
S: Androgenic adult granulosa cell tumor in a teenager: A case
report and review of the literature. Int J Gynecol Cancer.
16:(Suppl 1). S368–S374. 2006. View Article : Google Scholar
|
|
10
|
François Y, Berlier P, Chatelain P and
François R: Virilizing ovarian tumor in an adolescent. Pediatrie.
45:105–107. 1990.(In French). PubMed/NCBI
|
|
11
|
Patel SS, Carrick KS and Carr BR:
Virilization persists in a woman with an androgen-secreting
granulosa cell tumor. Fertil Steril. 91:933.e13–e15. 2009.
View Article : Google Scholar
|
|
12
|
Ayhan A, Salman MC, Velipasaoglu M,
Sakinci M and Yuce K: Prognostic factors in adult granulosa cell
tumors of the ovary: A retrospective analysis of 80 cases. J
Gynecol Oncol. 20:158–163. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tai YJ, Chang WC, Kuo KT and Sheu BC:
Ovarian steroid cell tumor, not otherwise specified, with
virilization symptoms. Taiwan J Obstet Gynecol. 53:260–262. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haroon NN, Agarwal G, Pandey R and
Dabadghao P: Juvenile granulosa cell tumor presenting as isosexual
precocious puberty: A case report and review of literature. Indian
J Endocrinol Metab. 17:157–159. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kota SK, Gayatri K, Pani JP, Meher LK,
Kota SK and Modi KD: Ovarian granulosa cell tumor: An uncommon
presentation with primary amenorrhea and virilization in a pubertal
girl. Indian J Endocrinol Metab. 16:836–839. 2012. View Article : Google Scholar : PubMed/NCBI
|