Introduction
Juvenile polyposis syndrome (JPS) is a rare disease
characterized by multiple hamartomatous polyps in the
gastrointestinal tract (1). The most
frequently affected site is the colorectum (98%), followed by the
stomach (14%) and the small intestine (8.8%) (2). JPS is subdivided into three groups:
Juvenile polyposis of infancy, juvenile polyposis coli and
generalized juvenile polyposis (3,4). A fourth
category, juvenile polyposis of the stomach, has been reported,
which is used to describe polyps limited to the stomach at the time
of the initial diagnosis (5,6). JPS is generally recognized as consisting
of benign hyperplastic polyps; however, it has recently been
reported that these hyperplastic polyps have malignant potential.
The present study reported a case of juvenile polyposis of the
stomach with multiple early gastric cancers treated by
laparoscopy-assisted total gastrectomy.
Case report
A 24-year-old man with an unremarkable medical
history was referred to Onomichi General Hospital (Hiroshima,
Japan) as a result of melena and anemia in 2004. An upper
gastrointestinal endoscopy in 2004 revealed multiple edematous
polyps around the fundus of the stomach (Fig. 1). Histological examination revealed a
hyperplastic and disorganized foveolar epithelium, and an edematous
lamina propria with infiltration of inflammatory cells, and the
histological diagnosis was hyperplastic polyps. The patient began
taking a proton-pump inhibitor and iron supplements, and he
underwent an upper gastrointestinal endoscopy annually. The polyps
appeared around the fundus in 2004 and had gradually progressed to
the entire stomach in 2014 (Fig. 2).
The number and size of the polyps had also increased. Endoscopic
mucosal resection was performed frequently due to bleeding from the
polyps. It was difficult to control the progressing anemia by
non-surgical treatment, and, in 2014, histological examination
revealed for the first time that the benign polyps were
adenocarcinoma. Surgical treatment was therefore performed. At the
time of surgery, the patient was 171 cm tall and weighed 62.5 kg.
No skin lesions or loss of hair, which is characteristic of
Cronkhite-Canada syndrome, was exhibited. Laboratory data
demonstrated mild anemia (hemoglobin, 9.6 g/dl), but neither
hypoproteinemia nor elevation of tumor markers, including
carcinoembryonic antigen and carbohydrate antigen 19–9 was
observed. With respect to the patient's family history, the
patient's mother had succumbed to colon cancer in her 50s and his
cousin had succumbed to an unknown primary cancer in his 30s.
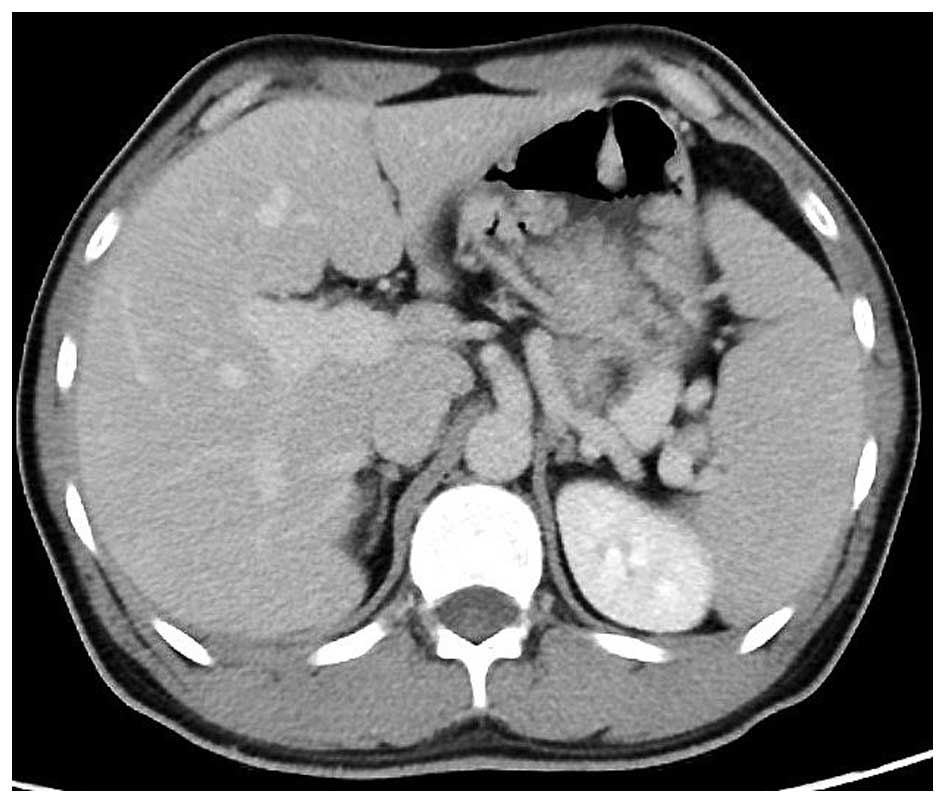
Colonoscopy revealed no specific findings. Enhanced computed
tomography revealed that the stomach wall was edematous with
enhanced thickening from multiple polyps (Fig. 3). An upper gastrointestinal series
revealed that multiple polypoid lesions were limited to the entire
stomach and were not present in the small intestine (Fig. 4). The patient underwent
laparoscopy-assisted total gastrectomy with Roux-en-Y
esophagojejunostomy. The resected specimen revealed numerous
diffuse polyps throughout the entire stomach (Fig. 5). Microscopic findings revealed a
hyperplastic and disorganized foveolar epithelium with an atypical
nuclear shape. Enlargement of the nucleolus was also observed, and
the patient was diagnosed with well-differentiated adenocarcinoma.
A total of three lesions of early gastric cancer, and no lymph node
metastasis were detected. Immunohistochemical staining revealed
that the specimen was positive for p53 and carcinoembryonic antigen
and exhibited a high index of mindbomb E3 ubiquitin protein ligase
1 (Fig. 6). The postoperative course
was uneventful, hemoglobin level returned to the normal range, and
the patient experienced no recurrence over a 1 year follow-up.
Discussion
To the best of our knowledge, the present case is a
rare case of juvenile polyposis of the stomach, associated with
multiple early gastric cancers. In Japan, the incidence of juvenile
polyposis of the stomach is higher compared with that observed in
previous reports (1,7). The association of gastric cancer with
juvenile polyposis of the stomach is well-known in Japan, with a
frequency of ~50% (8). The present
strategy for symptomatic juvenile polyposis of the stomach is
surgical intervention. Notably, recurrence of gastric cancer and
polyps in the remnant stomach has been previously reported;
therefore, total gastrectomy is recommended as standard treatment
in cases of juvenile polyposis of the stomach (2). JPS is an autosomal dominant hereditary
syndrome with characteristic multiple hamartomatous polyps
throughout the entire intestine, and was first reported by McColl
et al (9) in 1964. Watanabe
et al (5) first reported JPS
limited to the stomach as juvenile polyposis of the stomach in
1979. Few reports exist in the English literature of polyps
predominantly occurring in the stomach. Certain hamartomatous
diseases of the stomach occur, for which differential diagnosis is
necessary, including Cronkhite-Canada syndrome and Peutz-Jeghers
syndrome (10). Skin lesions and
family history are helpful for distinguishing JPS from these
syndromes. It is known that 20–50% of patients have a family
history of JPS, and its mechanism of inheritance is autosomal
dominant with variable penetrance. Previously, mutations in
SMAD4 and BMPR1A, which are implicated in the
transforming growth factor-β pathway, have been identified as the
disease genes for this syndrome, and the probability of a mutation
in each in JPS is 20% (11,12). The incidence of gastrointestinal
cancer in JPS is ~20% (11). The most
frequently observed clinical feature is anemia (89%), followed by
hypoproteinemia (67%). It is difficult to control this condition
with non-surgical treatment. The histological findings of the
present study revealed a hyperplastic and disorganized foveolar
epithelium with atypia, and the patient was diagnosed with
well-differentiated adenocarcinoma. In the present case, three
lesions of mucosal adenocarcinoma were observed in the resected
specimen. It has been previously reported that colorectal lesions
have developed into adenocarcinoma during follow-up (2). Howe et al (13) reported that the risk of
gastrointestinal cancer in family members of patients with JPS
exceeded 50%.
In conclusion, the present study described a rare
case of juvenile polyposis of the stomach with early gastric
cancers following curative resection. The patient has remained
disease-free for 1 year following surgery. Special attention should
be paid to the intestine and colorectum in patients with JPS.
Additionally, family members of the present patient should also
take gastrointestinal fiber and undergo genetic examination as a
result of the patient's strong family history.
Glossary
Abbreviation
Abbreviations:
|
JPS
|
juvenile polyposis syndrome
|
References
|
1
|
Larsen Haidle J and Howe JR: Juvenile
Polyposis Syndrome. GeneReviews®. Pagon RA, Adam MP,
Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong C,
Mefford HC, Smith RJH and Stephens K: University of Washington.
(Seattle). 2013.
|
|
2
|
Hizawa K, Iida M, Yao T, Aoyagi K and
Fujishima M: Juvenile polyposis of the stomach: Clinicopathological
features and its malignant potential. J Clin Pathol. 50:771–774.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sachatello CR, Hahn IS and Carrington CB:
Juvenile gastrointestinal polyposis in a female infant: Report of a
case and review of the literature of a recently recognized
syndrome. Surgery. 75:107–114. 1974.PubMed/NCBI
|
|
4
|
Agnifili A, Verzaro R, Gola P, Marino M,
Mancini E, Carducci G, Ibi I and Valenti M: Juvenile polyposis:
Case report and assessment of the neoplastic risk in 271 patients
reported in the literature. Dig Surg. 16:161–166. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Watanabe A, Nagashima H, Motoi M and Ogawa
K: Familial juvenile polyposis of the stomach. Gastroenterology.
77:148–151. 1979.PubMed/NCBI
|
|
6
|
Coburn MC, Pricolo VE, DeLuca FG and Bland
KI: Malignant potential in intestinal juvenile polyposis syndromes.
Ann Surg Oncol. 2:386–391. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma C, Giardiello FM and Montgomery EA:
Upper tract juvenile polyps in juvenile polyposis patients:
dysplasia and malignancy are associated with foveolar, intestinal,
and pyloric differentiation. Am J Surg Pathol. 38:1618–1626. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ozawa T, Wachi E and Yamashita N: A case
of juvenile polyposis limited to the stomach accompanied by double
gastric cancers and Ménétrier's disease. Nihon Shokakibyo Gakkai
Zasshi. 107:1641–1650. 2010.(In Japanese). PubMed/NCBI
|
|
9
|
McColl I, Busxey HJ, Veale AM and Morson
BC: Juvenile polyposis coli. Proc R Soc Med. 57:896–897.
1964.PubMed/NCBI
|
|
10
|
Zbuk KM and Eng C: Hamartomatous polyposis
syndromes. Nat Clin Pract Gastroenterol Hepatol. 4:492–502. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chow E and Macrae F: A review of juvenile
polyposis syndrome. J Gastroenterol Hepatol. 20:1634–1640. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Howe JR, Sayed MG, Ahmed AF, Ringold J,
Larsen-Haidle J, Merg A, Mitros FA, Vaccaro CA, Petersen GM,
Giardiello FM, et al: The prevalence of MADH4 and BMPR1A mutations
in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1
mutations. J Med Genet. 41:484–491. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Howe JR, Mitros FA and Summers RW: The
risk of gastrointestinal carcinoma in familial juvenile polyposis.
Ann Surg Oncol. 5:751–756. 1998. View Article : Google Scholar : PubMed/NCBI
|