Introduction
Neuroendocrine tumors (NETs) originate from the
diffuse neuroendocrine system, which is composed of cells
functioning as neurocytes, as well as endocrine cells. This type of
tumor most commonly occurs in the respiratory system and
gastrointestinal tract, while intracranial origin is rare (0.74%).
We herein present a case of a 40-year-old woman with a primary
intracranial NET, which was immunonegative for adrenocorticotropic
hormone (ACTH), resulting in ectopic ACTH syndrome.
Case report
A 40-year-old woman was admitted to Sanbo Brain
Hospital (Beijing, China) with a 6-year history of intermittent
bilateral rhinorrhea, a 2-month history of rapid weight gain,
polydipsia, polyuria and weakness, and a 2-week history of dimness.
On examination, the patient was found to have hypertension,
dimness, bilateral exophthalmus, diminution of vision in the left
eye, pigmentation of the skin of the face and trunk and grade 4
muscle weakness; other findings were unremarkable. Computed
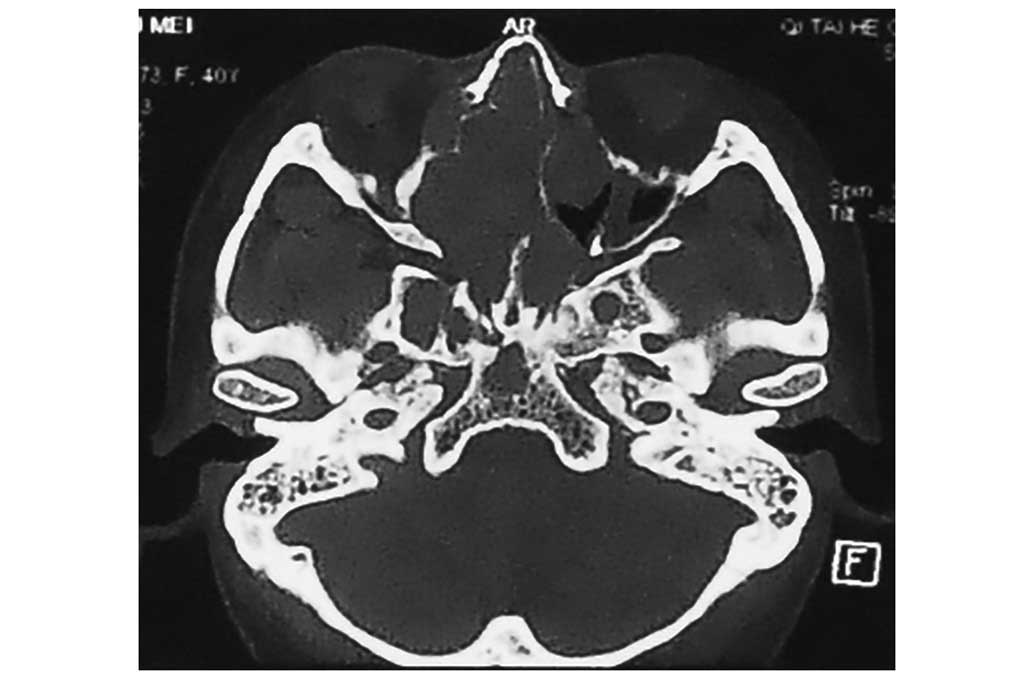
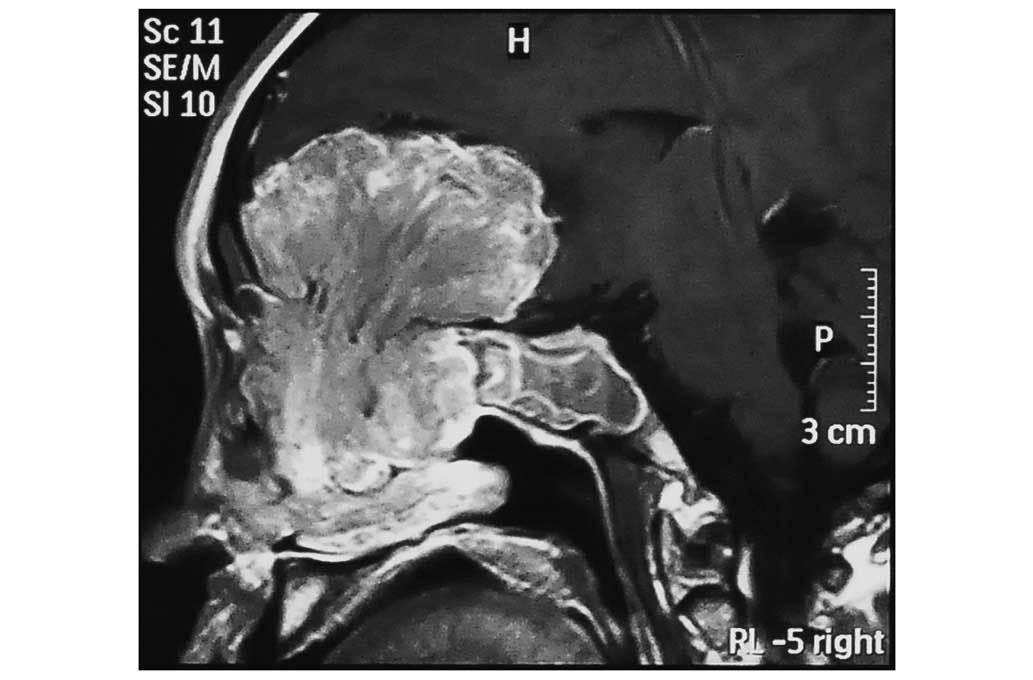
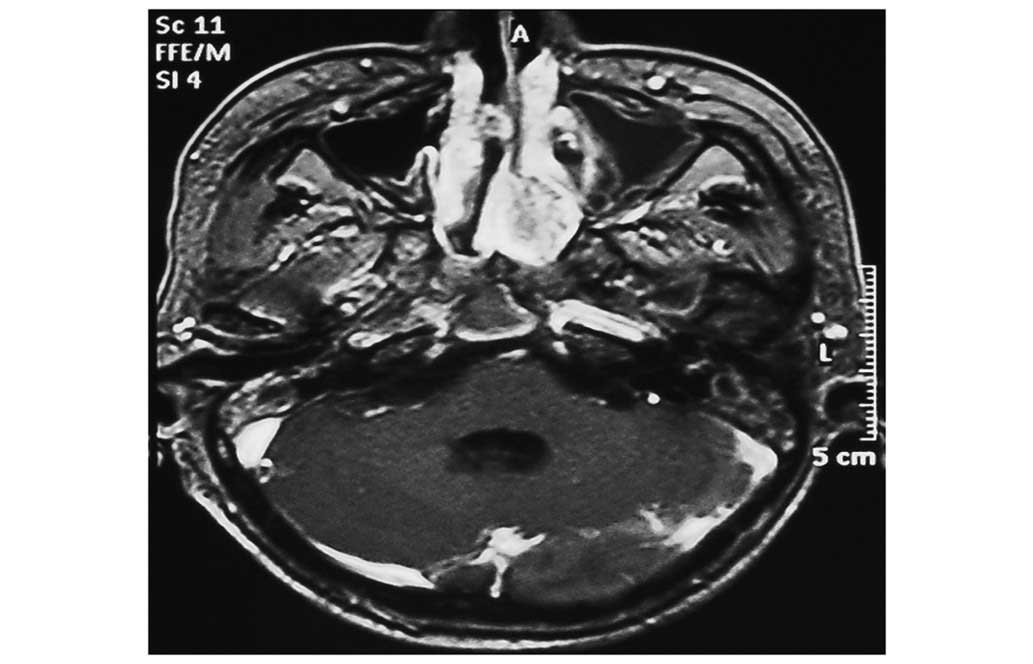
tomography (CT) and magnetic resonance imaging scans revealed a
sizeable contrast-enhanced tumor in the anterior cranial fossa,
which infiltrated the sphenoid and ethmoid sinuses bilaterally, the
left maxillary sinus and the nasal cavity (Figs. 1–4). The
abdominal CT scans revealed bilateral adrenal hyperplasia. On
laboratory biochemical tests, the serum potassium level was
1.92–2.82 mmol/l and the fasting blood glucose (FBG) level
14.9–20.0 mmol/l. The basal ACTH level was 33.72 pmol/l (normal,
<2.2 pmol/l), the 6 a.m. serum cortisol level was 1,779.78
nmol/l (normal, 83–359 nmol/l), the 24-h urinary free cortisol
level was ≤11,194.64 nmol/24 h (normal, 53.2–789.4 nmol/24 h), and
the testosterone level was 8.43 nmol/l (normal, 0.17–2.53
nmol/l).
The provisional diagnosis was anterior cranial base
tumor with ectopic ACTH syndrome. The neoplasm was exposed
intradurally and extradurally through a right frontal craniotomy,
while anterior skull base reconstruction was performed during
surgery. The intracranial surgery achieved gross removal of the
tumor; however, part of the tumor remained in the nasal cavity
(Fig. 5).
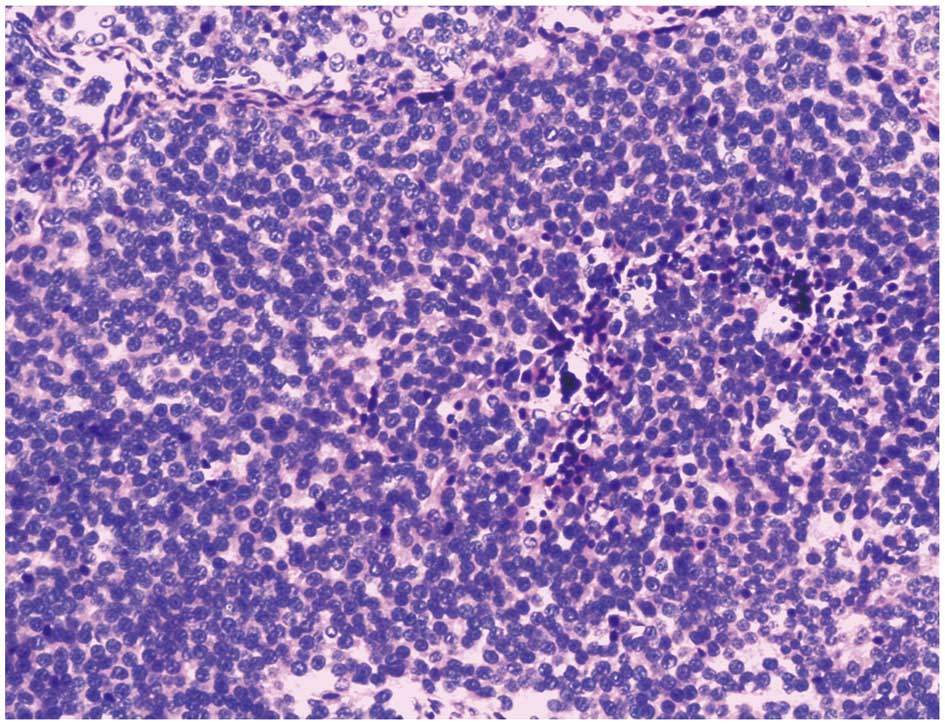
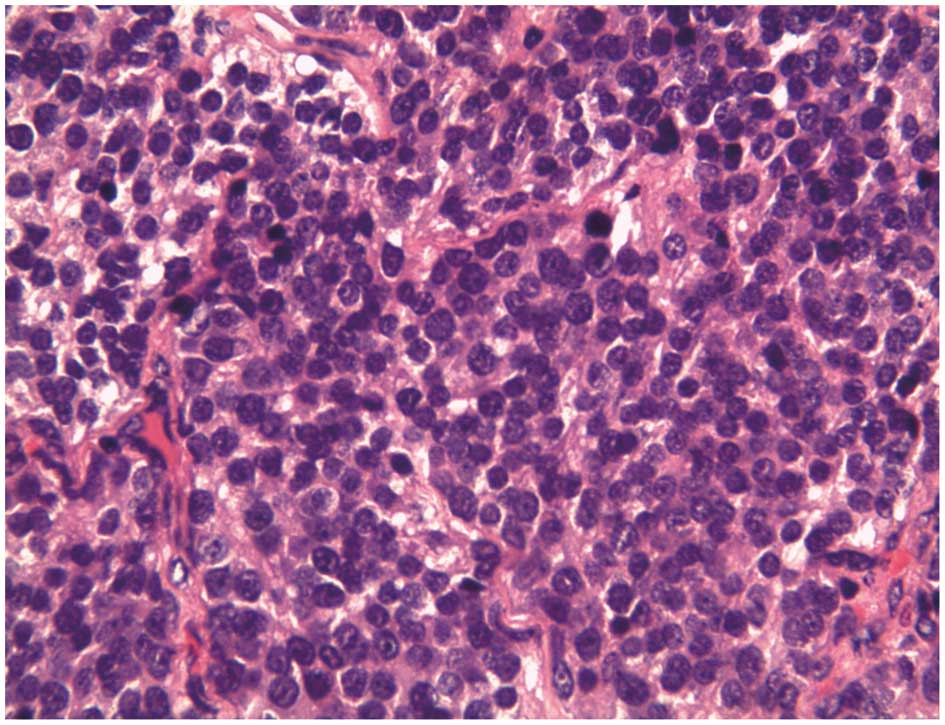
The histological examination revealed that the tumor
was composed of small round cells with uniform nuclei and scant
cytoplasm (Fig. 6).
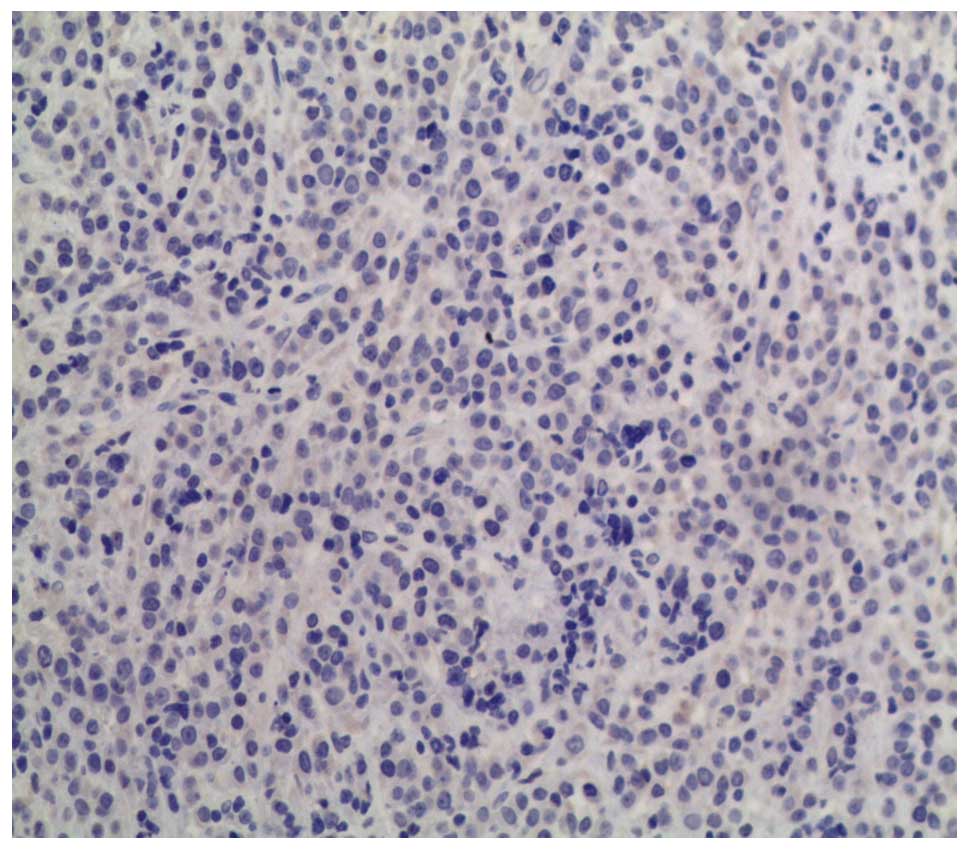
Immunohistochemically, the cells were positive for epithelial
markers, including cytokeratin (CK)8, CK18 and cytokeratin A (endo
A), and for neuroendocrine differentiation markers, including
synaptophysin (Syn), chromogranin A (ChrA), and neuron-specific
enolase, but negative for carcinoembryonic antigen, epithelial
membrane antigen, thyroid transcription factor 1 and ACTH (Figs. 7 and 8).
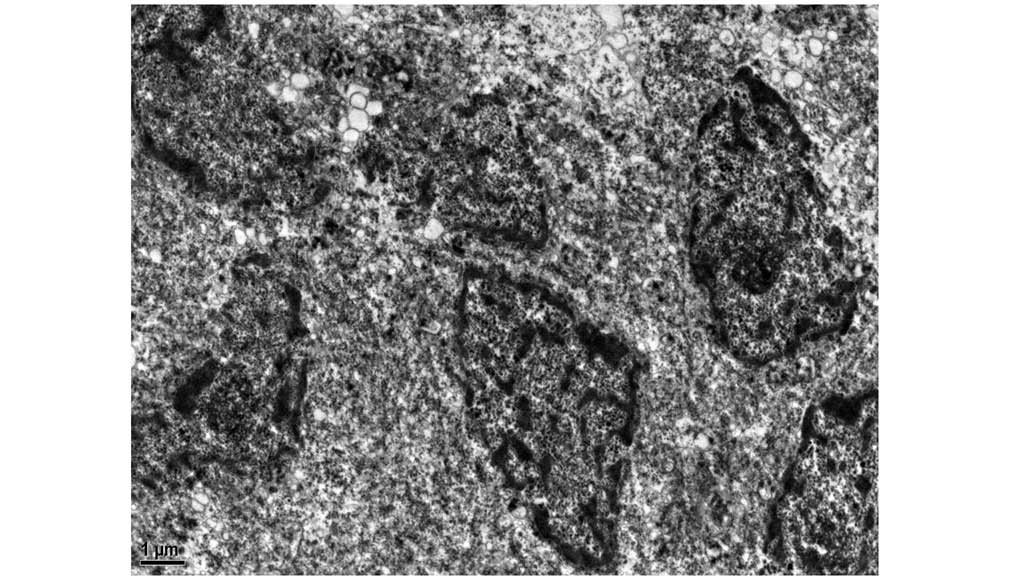
The Ki-67 labeling index was <1%. Electron microscopy revealed
the presence of occasional neuroendocrine granules (NEGs) (Fig. 9). These findings suggested low-grade
small-cell NET.
The postoperative course was relatively uneventful.
Abdominal CT scans revealed bilateral regression of the adrenal
gland hyperplasia and the biochemical results revealed a potassium
level of 4.25 mmol/l, FBG level of 6.2–6.5 mmol/l, basal ACTH level
of 1.42 pmol/l and 6 a.m. serum cortisol level of 272 nmol/l.
Discussion
NETs originate from amine precursor uptake and
decarboxylation cells, which are referred to as the diffuse
neuroendocrine system (1–3), whereas intracranial origin is rare
(0.74%) (4). To the best of our
knowledge, this is the first case of primary intracranial NET,
immunonegative for ACTH, resulting in Cushing's syndrome. In
biochemical measurements, ChrA may be used as a tumor marker, as it
increases in 70–90% of patients with NETs (5). On histological examination, the tumor is
composed of large cells with uniform nuclei, scant cytoplasm,
basophil granules and irregular mitoses (6). ChrA and Syn are selected as common
immunohistochemical markers, which are sensitive and non-specific
(7). According to the recent
literature, testing for peptide hormones and bioamines is no longer
recommended, as no correlation has been confirmed between hormone
levels and clinical presentation, and the level of hormone is not a
valuable prognostic indicator (8).
The presence of intracytoplasmic NEGs on electron micrographs,
whose compact cores show high electron density, is of value in the
differential diagnosis.
Immunohistochemistry is a significant differential
approach to the diagnosis of the neuroendocrine component,
epithelial component and detection of neuropeptides. In this case,
the tumor had to be differentiated from esthesioneuroblastoma and
neuroendocrine carcinoma of the nasal cavity and paranasal sinuses.
Klimstra (6) reported that
esthesioneuroblstoma was composed of neuroblasts with uniform
circular nuclei and longitudinal cerebromedullary tubes in the
dendrites. NETs mainly originate from the epithelium, lacking a
neural component. Immunohistochemistry of neuroendocrine carcinoma
of the nasal cavity and paranasal sinuses is similar to that of the
intracranial counterpart, but the Ki-67 labeling index is often
>3%. As determined by imaging in this case, the bulk of tumor
situated intracranially, with the skull base limiting tumor
growth.
The NEGs were rare and immunohistochemistry was
negative for ACTH, contradicting the preoperative changes in the
serum ACTH level. The main reasons were as follows: i) ACTH
consists of 39 amino acids, and is divided into the bioactive part,
located in the first 24 amino acid residues, and the immunoactive
part, located between the 22nd and 39th amino acid residues
(9). Thus, the antibody used in the
Pathology Department of our institute may not recognize the
secreted hormone. ii) Ectopic tumors may secrete ACTH or an ACTH
analogue and a simple anti-ACTH antibody is insufficient for
detecting ectopic secretion. iii) Some lesions may secrete ACTH and
corticotropin-releasing hormone (CRH) simultaneously (10). Moreover, Al-Gahtany et al
(11) reported three cases of ectopic
CRH tumors, with pituitary hyperplasia leading to high
corticotrophin hyperlipidemia; they also reported that,
postoperatively, the ACTH level immediately returned to normal if
the tumor produced ACTH, but it decreased gradually if the tumor
produced CRH. This patient's postoperative ACTH level returned to
normal after 16 days. iv) The tumor had been present for 6 years,
but Cushing's syndrome had been present for 2 months. During a
follow-up of ~6 months due to the residual tumor in the nasal
cavity, the patient did not present with signs or symptoms of
hypercortisolism. The reason may be that the ectopic hormone
secretion by the residual NET was not sufficient to cause typical
Cushing's syndrome.
The optimal treatment of NETs is surgical resection.
Patients who receive comprehensive treatment, including completion
of chemoradiotherapy, are expected to have longer tumor-free
survival, compared with those receiving surgery alone (10). Drugs focusing on the epithelial
component may also be selected.
Anterior skull base reconstruction was performed
during surgery in this case. It was previously suggested that bony
reconstruction should be performed when the deficit is >3 cm
(12). The ‘sandwich’ technique is
currently used, involving tight suturing of the galea aponeurotica
or temporalis fascia and the skull base dura, bony rehabilitation
and covering with periosteum.
In summary, to the best of our knowledge, this is
the first case of primary intracranial NET with Cushing's syndrome.
The tumor was immunonegative for ACTH and may have displayed
ectopic secretion of CRH, or an ACTH analogue. The ‘sandwich’ skull
base reconstruction technique was performed, using the quadriceps
femoris, plastic skull flaps and periosteum, following tumor
removal.
References
|
1
|
Godwin JD II: Carcinoid tumors. A analysis
of 2837 cases. Cancer. 36:560–569. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Volante M, Rindi G and Papotti M: The grey
zone between pure (neuro)endocrine and non-(neuro)endocrine tumors:
A comment on concepts and classification of mixed
exocrine-endocrine neoplasms. Virchows Arch. 449:499–506. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi J, Xu Z, Wang ZD, et al: Pathological
category and diagnosis of neuroendocrine tumor in
non-neuroendocrine system. J Clin Exp Pathol. 25:548–550. 2009.(In
Chinese).
|
|
4
|
Faggiano A, Mansueto G, Ferolla P, Milone
F, del Basso de Caro ML, Lombardi G, Colao A and De Rosa G:
Diagnosis and prognostic implication of the World Health
Organization classification of neuroendocrine tumors. J Endocrinol
Invest. 31:216–223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lambert SW, Hofland LJ and Nobels FR:
Neuroendocrine tumor markers. Front Neuroendocrinol. 22:309–339.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klimstra DS: Pathology reporting of
neuroendocrine tumors: Essential elements for accurate diagnosis,
classification and staging. Semin Oncol. 40:23–36. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Travis WD, Rush W, Flieder DB, Falk R,
Fleming MV, Gal AA and Koss MN: Survival analysis of 200 pulmonary
neuroendocrine tumors with classification of criteria for atypical
carcinoid and its separation from typical carcinoid. Am J Surg
Pathol. 22:934–944. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hochwald SN, Zee S, Conlon KC, Colleoni R,
Louie O, Brennan MF and Klimstra DS: Prognostic factors in
pancreatic endocrine neoplasms: An analysis of 136 cases with a
proposal for low-grade and intermediate-grade groups. J Clin Oncol.
20:2633–2642. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith EM, Galin FS, LeBoeuf RD,
Coppenhaver DH, Harbour DV and Blalock JE: Nucleotide and amino
acid sequence of lymphocyte-derived corticotropin: Endotoxin
induction of a truncated peptide. Pro Natl Sci USA. 87:1057–1064.
1990. View Article : Google Scholar
|
|
10
|
Kolesnikova GS, Lapshina AM, Voronkova IA,
Marova EI, Arapova SD, Goncharov NP and Dedov II: Comparative
analysis of clinical, hormonal and morphological studies in
patients with neuroendocrine ACTH-producing tumors. Int J
Endocrinol. 2013:6592322013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Al-Gahtany M, Horvath E and Kovacs K:
Pituitary hyperplasia. Hormones (Athens). 2:149–158. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu H, Zhang Q and Yang Z: Skull base
reconstruction and rehabilitation. J Clin Otorhinolaryngol.
18:755–757. 2003.(In Chinese).
|