Introduction
Proteus syndrome is an extremely rare complex
disorder characterized by patchy or mosaic postnatal overgrowth of
different body parts (1). Proteus
syndrome was first described by Cohen and Hayden in 1979 (2), with an estimated prevalence of
<1/1,000,000 live births (3,4). The
onset may involve any site of the body and typically occurs during
infancy. The severity of Proteus syndrome has been found to vary
among different affected individuals, and commonly affected tissues
include the skin, connective tissue and bone, central nervous
system and eye (5). The affected
tissues vary widely regarding the extent of the involvement and the
severity in different patients, and they may be divided into four
main categories, namely soft tissue tumors, vascular anomalies,
macrodactyly and histopathological characteristics (6). The etiology of Proteus syndrome has not
been fully elucidated, but somatic genetic alterations, such as AKT
somatic mutations, in the affected tissue may be the major
causative factor. This is the case report of a Chinese male patient
with Proteus syndrome, with a review of the literature on the
molecular pathology underlying this disorder.
Case report
A 34-year-old man was admitted to Shenzhen People's
Hospital for treatment of progressive postnatal overgrowth and skin
problems mainly involving the limbs and hip. The patient's parents
had observed asymmetric overgrowth of the lower limbs during
childhood, but no systemic treatment was performed. On physical
examination, there was an obvious discrepancy in the length of the
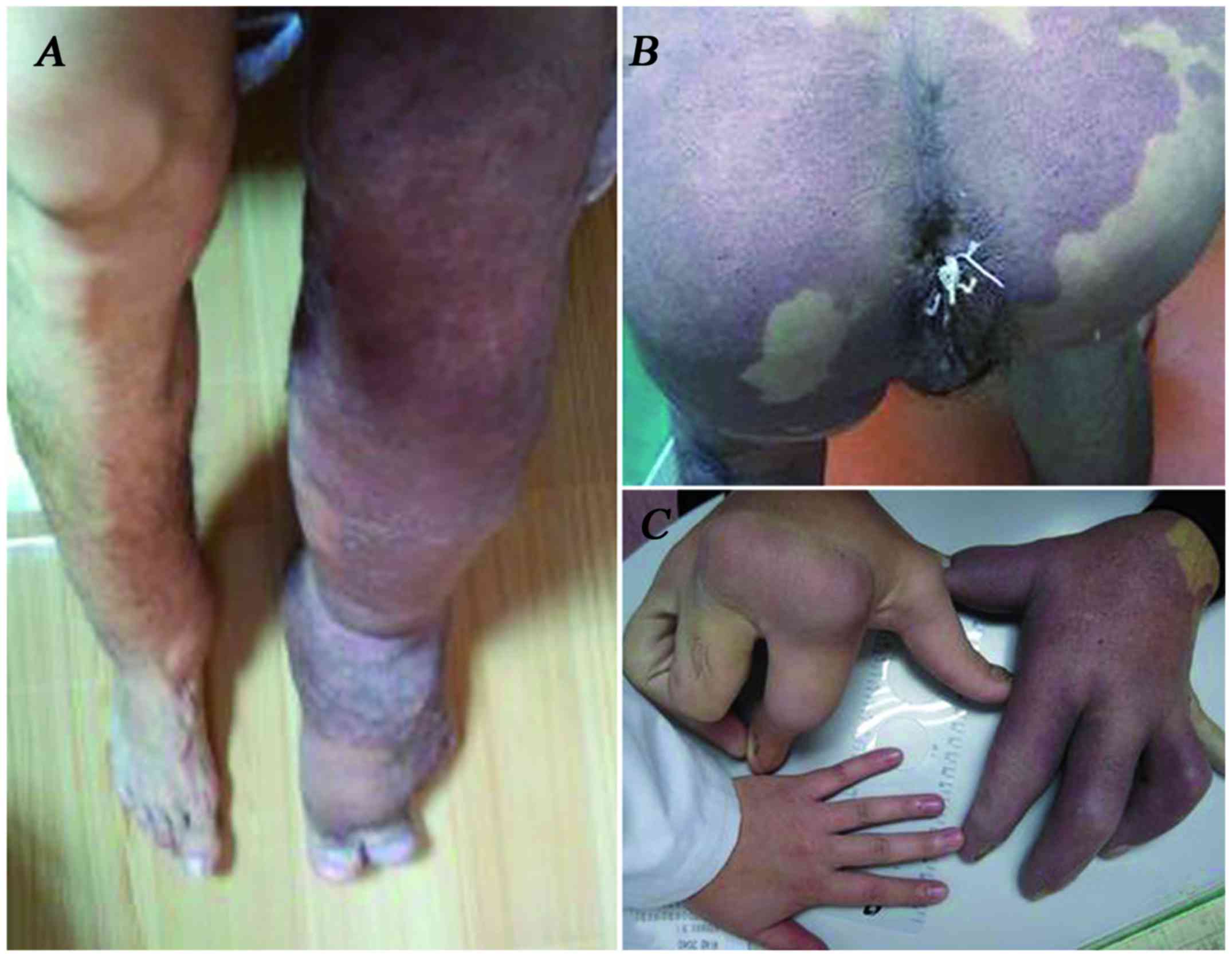
legs; in addition, the left lower limb was enlarged and covered by
an irregular red-brown plaque (Fig.
1A). Asymmetric growth of the upper limbs was also observed,
with the right forearm, which was covered by an irregular epidermal
nevus, being larger compared with the left forearm. The structure
of the right hand was also clearly affected and the middle and
index fingers could not be freely extended; in addition, certain
areas of the neck, abdomen and back were covered by a red-brown
irregular plaque with hyperpigmented borders (Fig. 1B and C); subcutaneous lipomas were
also present. Affected tissue samples were collected from the
patient's back for further molecular biological analysis by
whole-exome sequencing, as previously described (7). Briefly, the whole exome of affected and
control tissues was analyzed by high-throughput sequencing. Quality
control and basic filtering were performed, and the 90-bp
paired-end sequence reads were aligned and used for variant
calling. A total of 381 variants were found after the variants were
filtered. However, no mutation reportedly associated with the
Proteus syndrome was identified. The patient also presented with
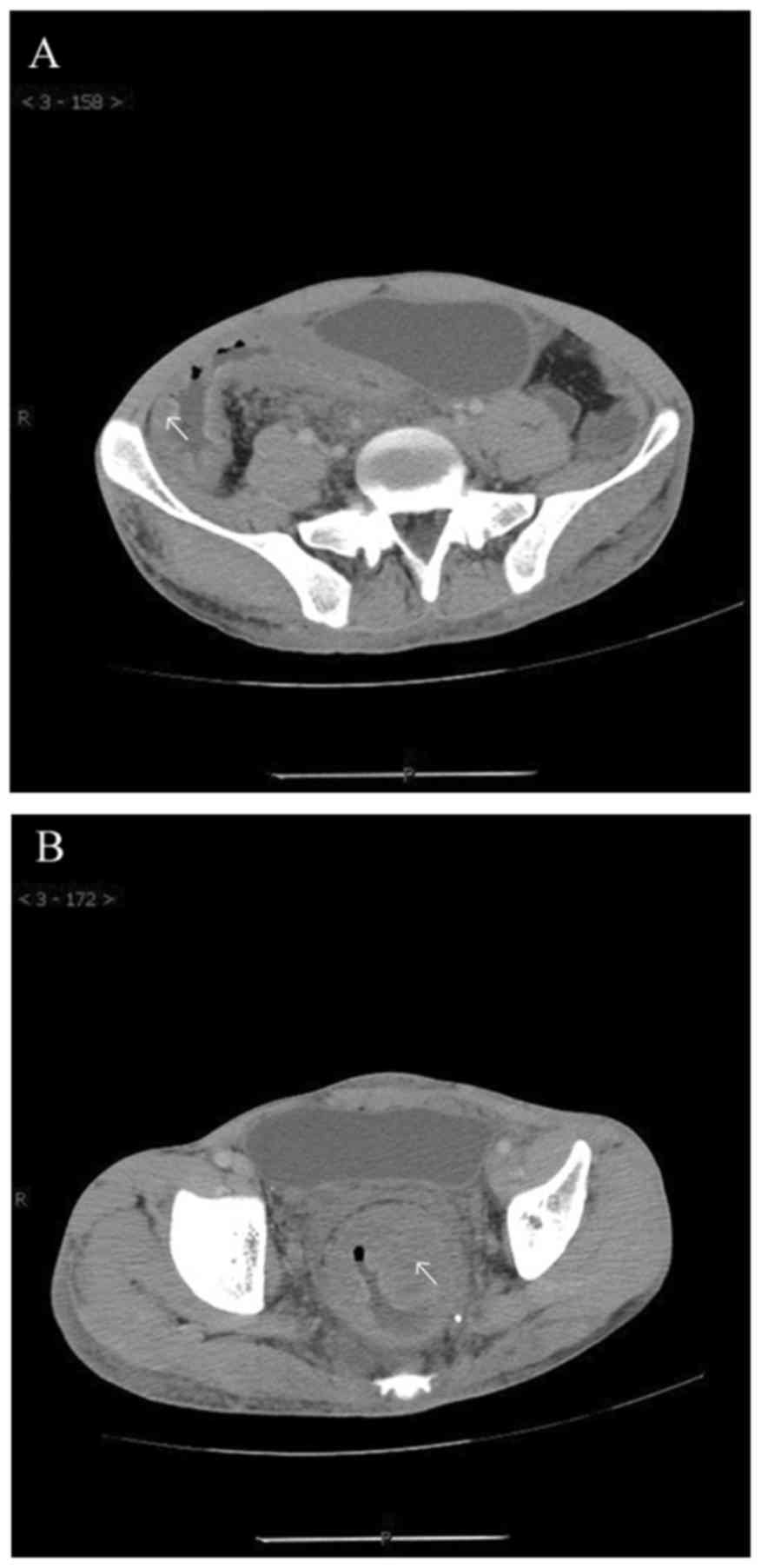
hematochezia, and a multislice spiral CT revealed multiple
hemangioma-like lesions of the large intestinal mucosa and systemic
vascular disease (Fig. 2). The
patient refused surgery due to the high cost of the treatment and
the associated surgical risk, and symptomatic treatment was instead
administered to control the skin and gastrointestinal symptoms. The
treatment included topical mupirocin for a concurrent infection of
the skin lesions on the left lower limb, and oral levofloxacin for
the gastrointestinal problems. On the last follow-up, in November
25, 2016, the condition of the patient remained stable; the skin
and gastrointestinal symptoms were partly controlled and there was
no obvious disease progression.
Written informed consent was obtained from the
patient regarding the publication of the case details and
associated images.
Discussion
The exact cause of Proteus syndrome remains unclear,
although AKT1 mutations were recently identified as an important
cause of this uncommon disease. The Proteus syndrome is a relative
rare and complex disease, which is characterized by partial
gigantism of the limbs, lipomas, varicosities and verrucous
epidermal nevi. After this type of disease was first reported in
1979 (2), similar cases were
described by Wiedemann et al in 1983 (8); however, only a limited number of cases
of the Proteus syndrome have been reported to date. The treatment
of Proteus syndrome is challenging, and multiple orthopedic
procedures have been attempted in the clinical setting to control
abnormal overgrowth; however, patients with this syndrome may
suffer severe cosmetic and functional consequences, even with
aggressive treatment (4).
Although diagnostic criteria for the Proteus
syndrome have been established, the variable phenotype may be a
major cause of misdiagnosis. According to the diagnostic criteria
revised by Turner et al in 2004 (9), each of the general criteria and some of
the specific criteria should be present to establish the diagnosis
of Proteus syndrome. In the present case, the diagnosis of the
Proteus syndrome was based on the presence of 3 of the major
criteria, namely mosaic distribution of the lesions, sporadic
occurrence and progressive course; and 5 of the specific criteria,
namely disproportionate overgrowth of the left leg and epidermal
nevi on some areas of the neck, abdomen and back (category B),
lipomas, venous malformation and facial phenotype (category C).
Given the high complexity of the clinical
characteristics of the Proteus syndrome, the hypothesis of somatic
mosaicism underlying this syndrome is important. Based on this
hypothesis, Proteus syndrome may develop from certain postzygotic
mutations. It was recently indicated that mutations causing
dysfunction of the phosphoinositide 3 kinase (PI3K)-AKT pathway,
such as phosphatase and tensin homolog (PTEN) and AKT1 mutations,
may be important causes of the Proteus syndrome (10). However, there is a controversial
association between PTEN mutations and the clinical characteristics
of the Proteus syndrome, and certain affected individuals harboring
somatic PTEN mutations and clinical characteristics such as
segmental overgrowth, lipomatosis, arteriovenous malformation and
epidermal nevi, were diagnosed with the SOLAMEN or the Cowden
syndromes (11,12). Considering the clinical
characteristics and gene function of PTEN, these three syndromes
may belong to the same class of disorders. In 2011, Biesecker
(4) identified the genetic basis of
Proteus syndrome through analysis of 12 samples of exomes obtained
from 6 patients with Proteus syndrome using exome sequencing;
additional cases of validation confirmed that somatic mutations in
AKT1 were an important cause of Proteus syndrome (10). However, no mutation in the reported
Proteus syndrome-associated genes, including AKT1, PTEN or PIK3CA,
was found by exome sequencing in our case, which was similar to 3
of the 29 individuals with Proteus syndrome described by Biesecker.
It is likely that some mutations in unknown genes may contribute to
the development of Proteus syndrome, but further investigation is
required.
Considering the severe complications of the Proteus
syndrome, it is necessary to diagnose this disorder earlier in
childhood, and remain alert regarding potential tumor development
in such patients, in order to improve their quality of life. In
addition, in the majority of patients, Proteus syndrome may be
caused by mutations in certain known genes, and genetic examination
of the family members of patients with the syndrome may be
performed, which may prove to be valuable for variant filtering and
identification of disease-related mutations.
In summary, Proteus syndrome is a relatively
recently described and complex disease with a variable phenotype,
and its diagnosis is challenging. Although AKT1 mutations have been
identified as a cause of Proteus syndrome, the precise pathogenesis
and etiology of this syndrome require further investigation.
Acknowledgements
The present study was funded by the Science and
Technology Innovation Commission of Shenzhen Municipality (grant
nos. JCYJ20130401093116730 and JCYJ20150403101146277), and the
Guangxi Natural Science Foundation (grant no.
2015GXNSFBA139176).
References
|
1
|
Cohen MM Jr: Proteus syndrome review:
Molecular, clinical, and pathologic features. Clin Genet.
85:111–119. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cohen MM Jr and Hayden PW: A newly
recognized hamartomatous syndrome. Birth Defects Orig Artic Ser
15(5B). 291–296. 1979.
|
|
3
|
Furquim I, Honjo R, Bae R, Andrade W,
Santos M, Tannuri U and Kim C: Proteus syndrome: Report of a case
with recurrent abdominal lipomatosis. J Pediatr Surg. 44:E1–E3.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Biesecker L: The challenges of Proteus
syndrome: Diagnosis and management. Eur J Hum Genet. 14:1151–1157.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alves C, Acosta AX and Toralles MB:
Proteus syndrome: Clinical diagnosis of a series of cases. Indian J
Endocrinol Metab. 17:1053–1056. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hoey SE, Eastwood D, Monsell F, Kangesu L,
Harper JI and Sebire NJ: Histopathological features of Proteus
syndrome. Clin Exp Dermatol. 33:234–238. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sui W, Ou M, Liang J, Ding M, Chen J, Liu
W, Xiao R, Meng X, Wang L, Pan X, et al: Rapid gene identification
in a Chinese osteopetrosis family by whole exome sequencing. Gene.
516:311–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wiedemann HR, Burgio GR, Aldenhoff P,
Kunze J, Kaufmann HJ and Schirg E: The proteus syndrome. Partial
gigantism of the hands and/or feet, nevi, hemihypertrophy,
subuutaneos tumors, macrocephaly or other skull anomalies and
possible accelerated growth and visceral affections. Eur J Pediatr.
140:5–12. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Turner JT, Cohen MM Jr and Biesecker LG:
Reassessment of the Proteus syndrome literature: Application of
diagnostic criteria to published cases. Am J Med Genet A.
130:111–122. 2004. View Article : Google Scholar
|
|
10
|
Lindhurst MJ, Sapp JC, Teer JK, Johnston
JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L,
et al: A mosaic activating mutation in AKT1 associated with the
Proteus syndrome. N Engl J Med. 365:611–619. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caux F, Plauchu H, Chibon F, Faivre L,
Fain O, Vabres P, Bonnet F, Selma ZB, Laroche L, Gérard M and Longy
M: Segmental overgrowth, lipomatosis, arteriovenous malformation
and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN
nullizygosity. Eur J Hum Genet. 15:767–773. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Loffeld A, McLellan NJ, Cole T and Moss C:
Type 2 segmental Cowden disease vs. Proteus syndrome: Reply from
authors. Br J Dermatol. 158:410–411. 2008.PubMed/NCBI
|