Introduction
Sellar xanthogranuloma, also referred to as
cholesterol granuloma or xanthogranulomatous reaction, is a rare
granulomatous lesion consisting of cholesterol clefts, hemosiderin
deposits, macrophages, chronic inflammatory infiltrates and fibrous
proliferation (1,2). Sellar xanthogranuloma is most commonly
associated with craniopharyngioma or Rathke's cleft cyst, but may
also occur in isolation (2). The
xanthogranulomatous change was first described in 1988 (3). Xanthogranulomatous pituitary adenomas
are extremely rare, with only a few cases reported in the
literature to date (1,3,4).
Therefore, the etiology, diagnosis, management and prognosis of
this condition have yet to be fully elucidated. We herein report a
case of pituitary adenoma with concomitant xanthogranuloma in a
female patient who developed diabetes insipidus postoperatively.
The relevant literature is also reviewed and discussed.
Case report
A 56-year-old woman presented to The First Hospital
of Jilin University (Changchun, China) on August 30, 2014, with a
20-day history of intermittent headache, vomiting and distending
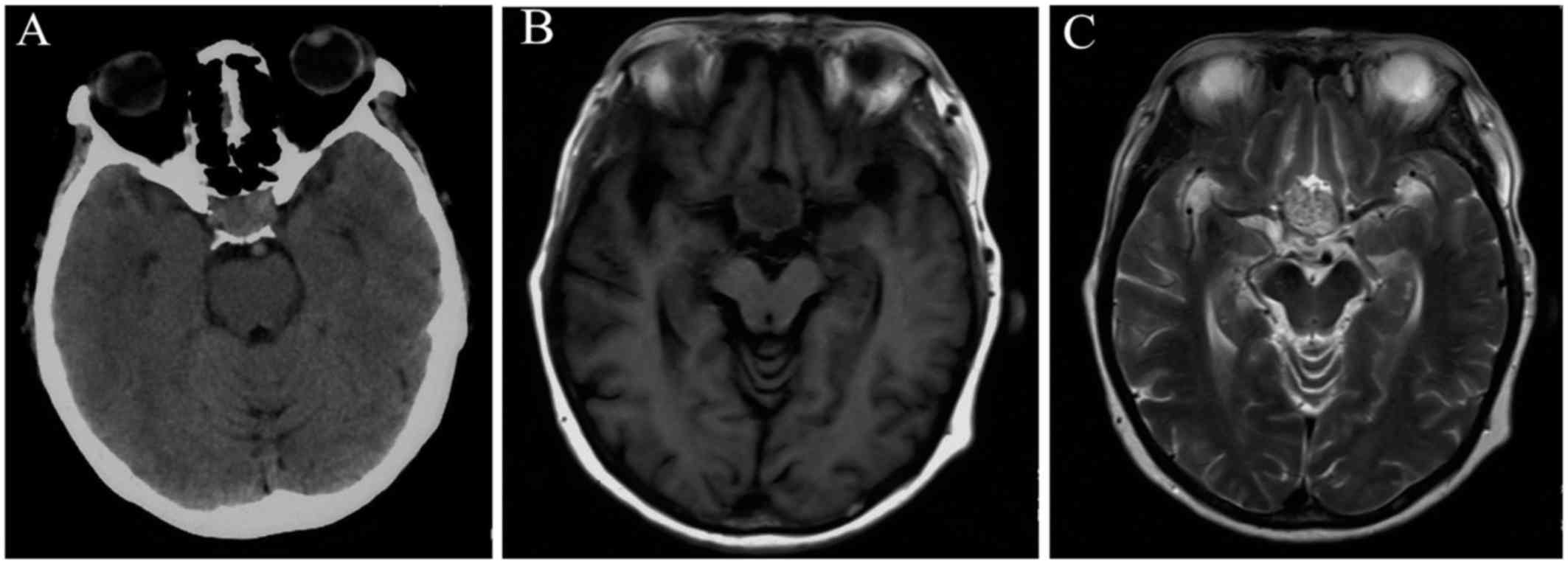
pain in the bilateral orbital regions. Brain computed tomography
(CT) scanning revealed a sellar mass exhibiting heterogeneous
hyperintensity (Fig. 1A).
Subsequently, brain magnetic resonance imaging (MRI) revealed an
intra- and suprasellar oval mass sized 30×25×30 mm, involving the
hypothalamus and the foramen of Monro. The pituitary gland and the
pituitary stalk could not be clearly identified. The mass was
heterogeneously isointense on T1-weighted images (WI) and
heterogeneously hyperintense on T2WI, with significant enhancement
following contrast administration (Fig.
1B and D). The third ventricle was deformed due to compression,
and lateral ventricular dilation was observed. The laboratory
examinations revealed a decreased serum cortisol hormone level at 8
a.m. (69.25 nmol/l; normal range, 240–619 nmol/l), with normal
levels of other endocrine hormones: Luteinizing hormone (LH; 39.08
mIU/ml; normal range, 10.87–58.64 mIU/ml), prolactin (PRL; 191.3
mIU/l; normal range, 70.81–566.5 mIU/l) and growth hormone (GH;
0.206 ng/ml; normal range, 0.01–3.607 ng/ml). Preoperatively, the
suspected diagnosis was pituitary adenoma.
A craniotomy was performed 8 days after admission,
via the right pterional approach. Intraoperatively, a soft
pinkish-grey mass was identified, which was well-demarcated and
hypervascular. The bilateral optic nerves were surrounded by the
tumor and, after the optic nerve was isolated, the tumor was
partially resected in a piecemeal fashion due to poor exposure.
Cholesterol clefts were noted in the tumor. The posterior section
of the tumor infiltrated the third ventricle, which could not be
completely exposed, and gross total resection was not forced. The
pituitary stalk, bilateral optic nerves and internal carotid artery
were well-preserved.
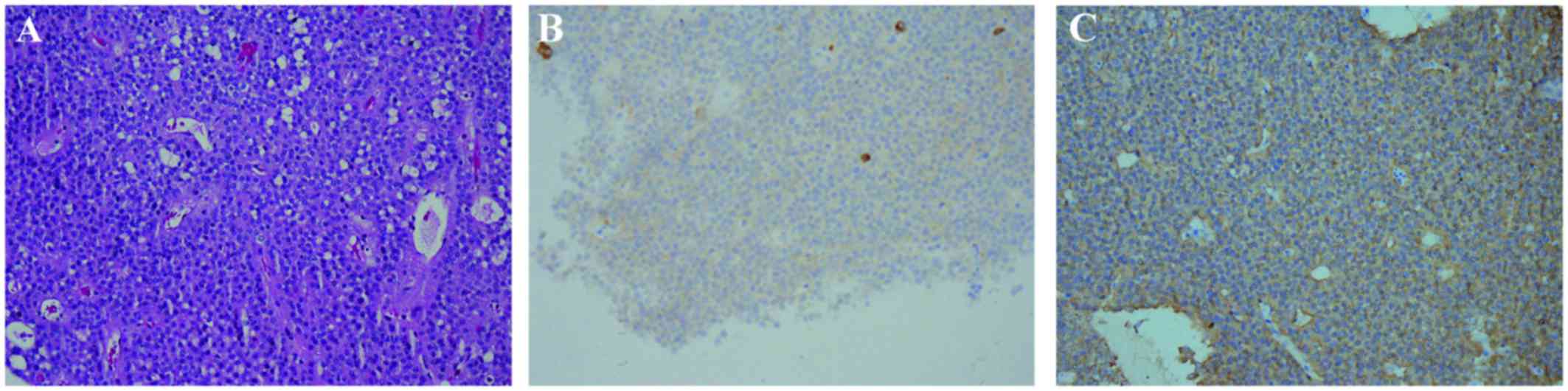
Histopathological examination confirmed the
diagnosis of plurihormonal xanthogranulomatous pituitary adenoma
(Fig. 2A). In the
xanthogranulomatous sections, cholesterol clefts, hemosiderin
deposits, macrophages, chronic inflammatory infiltrates and fibrous
proliferation were observed. Immunohistochemical staining revealed
that the tumor was positive for synaptophysin (Syn), chromogranin A
(CgA), GH, PRL, LH and thyroid-stimulating hormone (TSH) (Fig. 2B and C). The percentage of
Ki-67-positive tumor cells was ~1%.
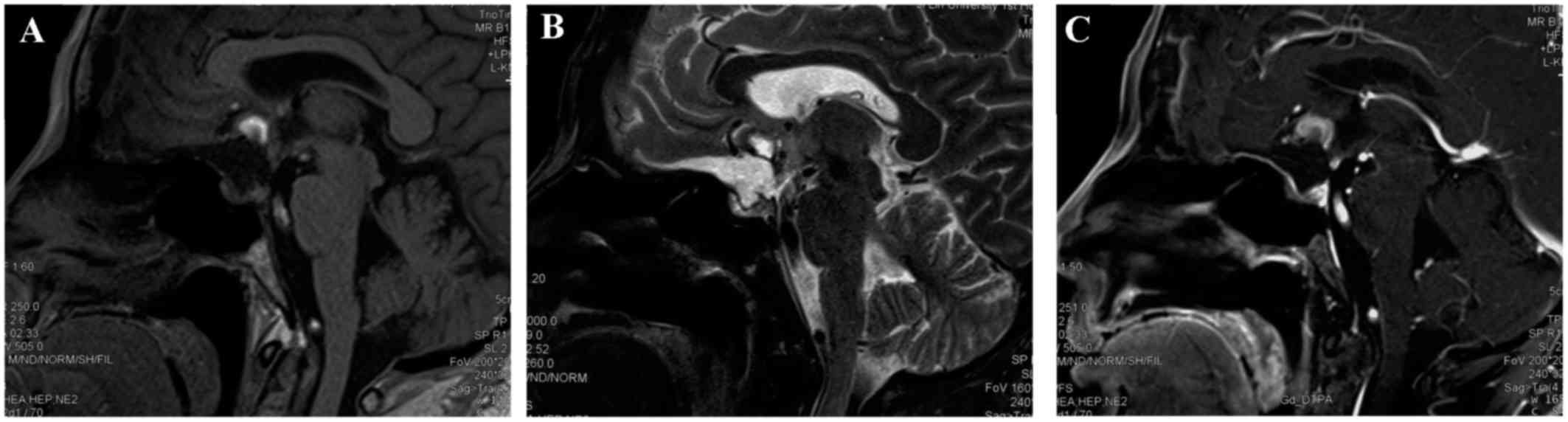
Postoperative MRI confirmed partial resection, with
residual tumor noted above the optic chiasma, which remained
heterogeneously isointense on T1WI and heterogeneously hyperintense
on T2WI, with significant contrast enhancement (Fig. 3). Postoperative endocrine
examinations revealed decreased serum levels of thyroid-related
hormones (TSH=0.08 µIU/ml, normal range: 0.27–4.2 µIU/ml; free
triiodothyronine=2.19 pmol/l, normal range: 3.1–6.8 pmol/l; and
free thyroxine=11.29 pmol/l, normal range: 12.0–22.0 pmol/l),
cortisol (69.25 nmol/l; normal range: 240–619 nmol/l) and
adrenocorticotrophic hormone (ACTH; 0.72 pmol/l; normal range:
1.6–13.9 pmol/l). The 24-h urine volume was 9.22 l. Hypopituitarism
was diagnosed and the patient was treated with hormone
replacement.
At the 3-month follow-up after surgery, the
patient's previous symptoms, including headache and vomiting, had
completely resolved, and her vision and visual fields were normal.
However, intractable hyponatremia and diabetes insipidus due to
hypopituitarism remained. No progression of the residual tumor was
observed on follow-up MRI. The last follow-up was performed in
November 30, 2016, and the patient remained on hormone replacement
therapy and recurrence-free. Written informed consent was obtained
from the patient regarding the publication of the case details and
accompanying images.
Discussion
A search the relevant literature yielded only 15
reported cases of pituitary adenoma with xanthogranulomatous change
(1,3,4). The
clinical profiles of the 15 patients are summarized as follows.
With respect to demographic characteristics, the male:female ratio
was 0.2:1, with an apparent female predominance. The average
patient age was 53 years (range, 33–67 years) and the tumors were
located in the intra-, supra-, or parasellar regions. The clinical
manifestations included hypopituitarism, visual impairment,
headache and vomiting. Radiologically, the tumors were usually
cystic, with heterogeneous intensity on MRI. The tumors appeared
iso- to hyperintense on T1WI, and the intensity varied from hypo-
to hyperintense on T2WI. Following gadolinium-DTPA
(diethylenetriaminepentaacetic acid) administration, enhancement
was usually significant and heterogeneous. The differential
diagnosis included craniopharyngioma and Rathke's cleft cyst.
Surgical resection was performed in all 15 patients.
In each case, the tumor was removed via the transsphenoidal
approach. Gross total resection was achieved in 11 of the 15 cases
(73.3%). Intraoperatively, the tumors were usually soft,
pinkish-grey and well-demarcated. Oily yellow liquid and fibrous
tissue were found within the tumors. Pathologically, pituitary
adenoma cells and xanthogranulomatous changes were observed,
including cholesterol clefts, hemosiderin deposits, macrophages,
chronic inflammatory infiltrates and fibrous proliferation.
Immunohistochemical staining showed positivity for Syn, CgA, ACTH,
GH, PRL, LH and TSH. In all 15 cases, the diagnosis was
plurihormonal pituitary adenoma.
Symptoms of headache and visual impairment may be
immediately relieved by surgical treatment; however, endocrine
abnormalities may be difficult to resolve.
Sellar xanthogranuloma is a rare granulomatous
lesion, which was first described by Shirataki et al in 1988
(3). Then in 1999, Paulus et
al suggested that sellar xanthogranuloma is clinically and
pathologically distinct from the classical adamantinomatous
craniopharyngioma (2). According to
a study published in 2011, the incidence of intracranial
xanthogranuloma is 1.6–7%, and lesions are only rarely found in the
sellar and parasellar regions (5).
Of the 643 patients with sellar or parasellar tumors
retrospectively reviewed by Rahmani et al only 4 patients
(0.6%) had histologically confirmed xanthogranulomas (6). Additionally, in our review of the
literature regarding sellar or parasellar xanthogranulomas, 160
cases were identified. Among those, 67 were pathologically
diagnosed as isolated xanthogranulomas (41.8%) (7–11), 52
(32.5%) as xanthogranulomatous craniopharyngiomas (2,3,6), 20 (12.5%) as xanthogranulomatous
Rathke's cleft cysts (5,6,12), 15
(9.4%) as xanthogranulomatous pituitary adenomas (1,3,4) and 6 cases (3.8%) as xanthogranulomatous
hypophysitis (13–15). To the best of our knowledge, the 15
cases of pituitary adenomas with xanthogranuloma are the only cases
reported in the literature to date, confirming the rarity of this
condition.
The etiology, the pathogenesis of sellar
xanthogranulomas remains incompletely understood. One study
proposed that xanthogranulomas may develop secondary to hemorrhage,
inflammation, or degeneration (6).
Amano et al analyzed the pathological characteristics of 7
cases of sellar xanthogranuloma and observed components of Rathke's
cleft cyst in 6/7 cases (12); thus,
they considered sellar xanthogranuloma as a terminal condition
resulting from a secondary reaction caused by repeated
inflammation, hemorrhage and degeneration of a Rathke's cleft cyst.
Other studies proposed that xanthogranulomas may occur as a
component of systemic autoimmune disease; secondary to a reactive
degenerative response to an epithelial lesion such as
craniopharyngioma, Rathke's cleft cyst, or pituitary adenoma; or
within the context of multiorgan involvement related to conditions
such as tuberculosis, sarcoidosis, or Erdheim-Chester disease
(16–18).
The most common symptoms of xanthogranulomatous
pituitary adenomas include vision and visual field disorders,
hypopituitarism and headache. Nishioka et al analyzed the
endocrinological and radiological characteristics of
xanthogranulomas associated with pituitary adenoma. They identified
5 patients (2.2%) with a remarkable xanthogranulomatous reaction
among 231 consecutive cases of pituitary adenoma, and all 5
patients exhibited anterior pituitary insufficiencies (1). However, in the present case, the
patient only presented with headache, vomiting and mild visual
impairment, whereas there was no endocrine dysfunction or
hypopituitarism. Thus, there may be no detectable connection
between endocrine dysfunction and the size or extension of a
pituitary adenoma. Sporadic cases have also manifested as
obstructive hydrocephalus with acute changes in consciousness
(19).
The imaging characteristics of xanthogranulomatous
pituitary adenomas are non-specific. The tumors usually appear as
isointense to hyperintense on T1WI and hypointense to hyperintense
on T2WI, and contrast enhancement may be significant and
heterogeneous (1,4). In the present case, the sellar and
suprasellar tumor extended into the third ventricle and exhibited
heterogeneous isointensity on T1WI, with significant enhancement
and heterogeneous hyperintensity on T2WI. The MRI characteristics
may be associated with the multiple components of the tumor, with
cholesterol clefts showing characteristic signals, with
hyperintensity on T1WI and hypointensity on T2WI.
Xanthogranulomatous pituitary adenomas may also exhibit atypical
characteristics on MRI, including a dural tail, vascular encasement
and intra-axial lesions in the posterior fossa (10).
The differential diagnosis of xanthogranulomatous
pituitary adenoma is challenging and should include
craniopharyngioma, Rathke's cleft cyst and granulomatous
hypophysitis. Craniopharyngiomas most commonly occur in the
suprasellar regions in young patients who present with visual
impairment, intracranial hypertension and endocrine dysfunction.
Moreover, calcifications are usually found on CT scans. Patients
with Rathke's cleft cysts usually present with headache and
endocrine dysfunction, and MRI of the tumors usually shows
homogenous intensity with nodules in the cyst and circular
enhancement. The main symptoms of granulomatous hypophysitis are
hypopituitarism, diabetes insipidus, headache and visual
impairment.
Accurate diagnosis of xanthogranulomatous pituitary
adenoma is based on histopathological criteria. Microscopically,
the typical histological characteristics of xanthogranuloma consist
of cholesterol clefts, hemosiderin deposits, macrophages, chronic
inflammatory infiltrates and fibrous proliferation. The
histological findings in our patient were in accordance with the
literature, supporting the diagnosis of xanthogranulomatous
pituitary adenoma. Although xanthogranuloma may occur as an
isolated entity, it usually coexists with other sellar tumors. In
the present case, the presence of pituitary adenoma cells and the
immunohistological staining results confirmed the diagnosis of
associated plurihormonal pituitary adenoma. With regard to the
differential diagnosis, enamel epithelial cells and cytokeratin may
be found in adamantinomatous craniopharyngiomas; Rathke's cleft
cysts usually display columnar epithelial cells, ciliated cells,
hemorrhage and necrosis; and xanthogranulomatous hypophysitis
mainly consists of foamy macrophages and small lymphocytes.
Xanthogranulomatous adenomas are benign,
slow-growing entities. According to previous reports, surgical
resection via the transsphenoidal approach is the preferred
treatment, and gross total resection has been achieved in 73.3% of
the cases (1,6). In the present case, the tumor was
resected via the right pterional approach; however, only the
posterior portion of the tumor could be removed due to poor
exposure. Previous reports indicated that headache and visual
impairment may be completely relieved postoperatively, whereas
endocrine dysfunction is difficult to treat. Our patient developed
hypopituitarism and diabetes insipidus, which has not been
previously reported. The specific pathogenic mechanisms responsible
for these conditions remain unclear, and we hypothesized that these
postoperative complications may be associated with dysfunction of
the hypothalamus-pituitary axis due to inflammatory cell
infiltration (20). Although
recurrence was not observed in previously reported cases, close MRI
follow-up is recommended given the rarity of xanthogranulomatous
pituitary adenoma.
In conclusion, we herein reported a case of
pituitary adenoma and concomitant xanthogranuloma in a female
patient who developed diabetes insipidus postoperatively.
Previously reported cases of this condition were reviewed, and
definitive diagnosis of this condition relies on pathological
examination. Xanthogranulomatous change should be carefully
assessed in adenomas.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nishioka H, Shibuya M, Ohtsuka K, Ikeda Y
and Haraoka J: Endocrinological and MRI features of pituitary
adenomas with marked xanthogranulomatous reaction. Neuroradiology.
52:997–1002. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paulus W, Honegger J, Keyvani K and
Fahlbusch R: Xanthogranuloma of the sellar region: A
clinicopathological entity different from adamantinomatous
craniopharyngioma. Acta Neuropathol. 97:377–382. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shirataki K, Okada S and Matsumoto S:
Histopathological study of the ‘cholesterol granuloma reaction’ in
the sellar and juxta-sellar tumors. No To Shinkei. 40:133–139.
1988.(In Japanese). PubMed/NCBI
|
|
4
|
Yokoyama S, Goto M, Hirano H, Hirakawa W,
Noguchi S, Hirahara K, Kadota K and Asakura T: Pituitary adenoma
with cholesterol clefts. Endocr Pathol. 9:91–95. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miyajima Y, Oka H, Utsuki S and Fujii K:
Rathke's cleft cyst with xanthogranulomatous change-case report.
Neurol Med Chir. 51:740–742. 2011. View Article : Google Scholar
|
|
6
|
Rahmani R, Sukumaran M, Donaldson AM,
Akselrod O, Lavi E and Schwartz TH: Parasellar xanthogranulomas. J
Neurosurg. 122:812–817. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bao SS and Rapp R: Xanthogranuloma as an
unsuspected cause of idiopathic central diabetes insipidus. Endocr
Pract. 20:e42–e46. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ben Nsir A, Thai QA, Chaieb L and Jemel H:
Calcified suprasellar xanthogranuloma presenting with primary
amenorrhea in a 17-year-old girl: Case report and literature
review. World Neurosurg. 84:866.e11–e14. 2015. View Article : Google Scholar
|
|
9
|
Kamoshima Y, Sawamura Y, Motegi H, Kubota
K and Houkin K: Xanthogranuloma of the sellar region of children:
Series of five cases and literature review. Neurol Med Chir
(Tokyo). 51:689–693. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Madan Mohan B, Mohamed E, Jain SK, Jain M
and Jaiswal AK: Serial MR imaging in suprasellar xanthogranuloma:
Growth pattern and new lesions. J Neuroimaging. 25:677–679. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vajtai I, Kopniczky Z, Buza Z, Kovács J,
Kovács Z, Varga Z, Bodosi M and Paulus W: Cholesterol granuloma at
the sella region: A new method of the differential diagnosis of
craniopharyngioma. Orv Hetil. 142:451–457. 2001.(In Hungarian).
PubMed/NCBI
|
|
12
|
Amano K, Kubo O, Komori T, Tanaka M,
Kawamata T, Hori T and Okada Y: Clinicopathological features of
sellar region xanthogranuloma: Correlation with Rathke's cleft
cyst. Brain Tumor Pathol. 30:233–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Burt MG, Morey AL, Turner JJ, Pell M,
Sheehy JP and Ho KK: Xanthomatous pituitary lesions: A report of
two cases and review of the literature. Pituitary. 6:161–168. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gopal-Kothandapani JS, Bagga V, Wharton
SB, Connolly DJ, Sinha S and Dimitri PJ: Xanthogranulomatous
hypophysitis: A rare and often mistaken pituitary lesion.
Endocrinol Diabetes Metab Case Rep. 2015:1400892015.PubMed/NCBI
|
|
15
|
Yokoyama S, Sano T, Tajitsu K and Kusumoto
K: Xanthogranulomatous hypophysitis mimicking a pituitary neoplasm.
Endocr Pathol. 15:351–357. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abla AA, Wilson DA, Eschbacher JM and
Spetzler RF: Neurosurgical biopsy as the initial diagnosis of
xanthogranuloma of the Erdheim-Chester disease variety of the
infundibulum and optic apparatus: Letter to the editor. Acta
Neurochir (Wien). 152:925–927. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Reithmeier T, Trost HA, Wolf S, Stölzle A,
Feiden W and Lumenta CB: Xanthogranuloma of the Erdheim-Chester
type within the sellar region: Case report. Clin Neuropathol.
21:24–28. 2002.PubMed/NCBI
|
|
18
|
Sulentić P, Cupić H, Cerina V and Vrkljan
M: Xanthogranuloma of the sellar region in a patient with
sarcoidosis. Acta Clin Croat. 49:61–65. 2010.PubMed/NCBI
|
|
19
|
Liu ZH, Tzaan WC, Wu YY and Chen HC:
Sellar xanthogranuloma manifesting as obstructive hydrocephalus. J
Clin Neurosci. 15:929–933. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sugata S, Hirano H, Yatsushiro K, Yunoue
S, Nakamura K and Arita K: Xanthogranuloma in the suprasellar
region. Neurol Med Chir. 49:124–127. 2009. View Article : Google Scholar
|