Introduction
Histiocytic sarcoma (HS) is a rare malignant
neoplasm originating from the hematopoietic system, composed of
cells exhibiting morphological and immunophenotypical
characteristics of mature tissue histiocytes, first described in
1939 as histiocytic medullary reticulosis, and as malignant
histiocytosis by Rappaport in 1966 (1). Current epidemiological data estimate
that <1% of tumors presenting in lymph nodes or soft tissue can
be defined as HS (2). Approximately
1/3 of the cases have been reported to occur in lymph nodes, while
the incidence of extranodular HS, such as in the gastrointestinal
tract, spleen and soft tissue, is relatively low (3).
Definitive morphological diagnosis of HS is
difficult, and it must be verified by immunohistochemistry. The
tumor cells express one or more tissue cell antigens, including
CD163, which is the most important antigen for detecting
macrophages (4), CD68 and
lysozyme.
The aim of the present report is to present in
detail the clinical and immunophenotypical characteristics of a
case of HS.
Case report
A 53-year-old female patient of Chinese descent
presented with a 3-month history of a rapidly enlarging right neck
mass. The mas was mobile and tender, and was associated with
symptoms including hoarseness, pharyngeal pain and occasionally
coughing. Imaging studies, including neck ultrasound and
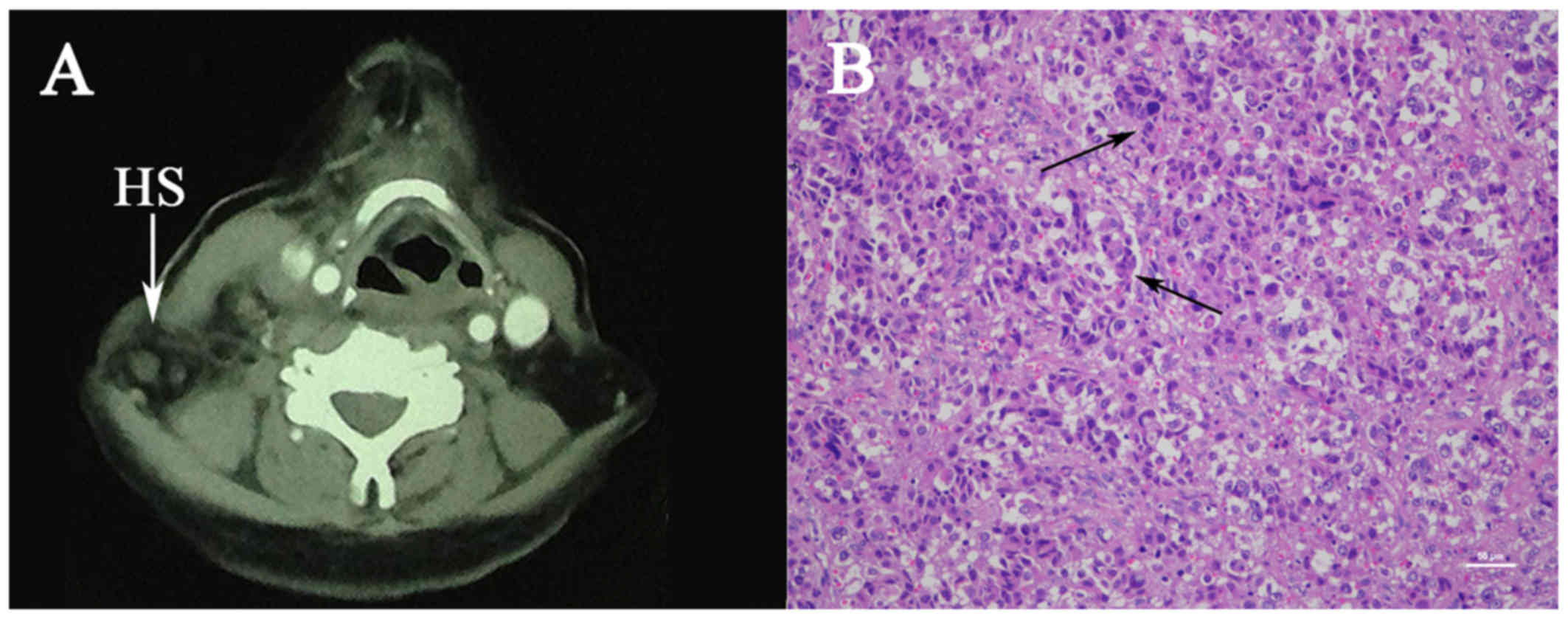
contrast-enhanced computed tomography (CT) revealed multiple right
cervical lymphadenectases with right jugular vein involvement
(Fig. 1A). Cervical biopsy of the
lesion was performed, followed by hematoxylin and eosin staining
for 30 min at 37°C, which revealed enlarged lymph nodes around the
anterior border of the sternocleidomastoid muscle, with a greatest
diameter of 3 cm, adhering to the surrounding tissue and fusing
with deep cervical lymph nodes. Microscopic examination revealed
diffusely distributed large non-cohesive tumor cells, round or oval
and focally spindle-shaped, with abundant eosinophilic cytoplasm
and large bizarre pleomorphic nuclei. Phagocytosis of red blood
cells, multinucleated giant cells and nuclear mitosis were readily
identified (Fig. 1B).
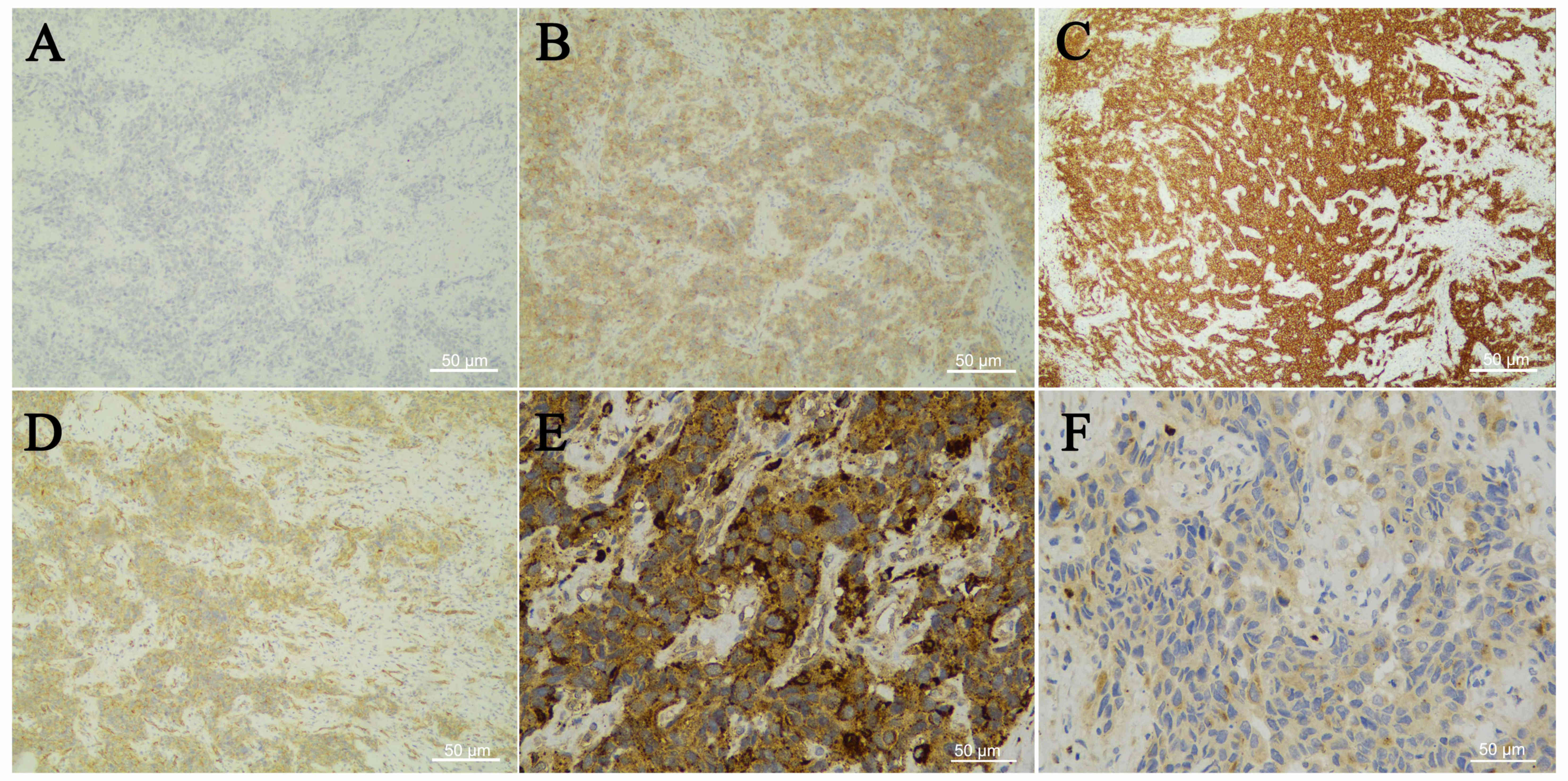
Immunohistochemical examination (antibody dilution, 1:100; OriGene
Technologies, Inc., Wuxi, China) was negative for cytokeratin (CK;
Fig. 2A), CD117, paired box (PAX)-5,
S-100, CD20, anaplastic lymphoma kinase (ALK), CD34 and epithelial
membrane antigen (EMA), ruling out epithelial, melanocytic or
myeloid origin, while it was positive for CD4, CD5, CD31 (Fig. 2B-D) and vimentin, which complicated
the diagnosis. However, the expression of histiocytic antigens,
CD68 (Fig. 2E) and lysozyme
(Fig. 2F), confirmed the diagnosis
of HS (5), with a Ki-67
proliferation labeling index of 90%. As it is a rare malignant
neoplasm originating from the lympho-hematopoietic system, HS
usually occurs in the lymph nodes and skin (6), it may affect multiple systems and
carries a dismal prognosis. An enhanced CT of the chest, abdomen
and pelvis revealed a right cervical space-occupying lesion and a
nodular lesion in the superior lobe of the right lung, multiple
mediastinal lymphadenectases, calcified lesions in the left lobe of
the liver and fibroids. As there are currently no consensus
guidelines for the management of HS due to its rarity, following an
extensive discussion with several clinical departments, it was
decided to commence chemoradiotherapy, as surgery would be
associated with a high risk of incomplete clearance of the tumor
and poor prognosis. However, the patient and her family asked to be
discharged to recuperate prior to further treatment. The patient
was discharged on July 15, 2016 and was lost to follow-up.
Due to its rapid invasiveness and high mortality
rate, HS remains a medical challenge for which there is yet no
effective treatment (7,8), which calls for more efforts and further
research.
Discussion
HS is an extremely rare lymphoid hematopoietic
malignancy with a morbidity of <0.5% among lymphoid
hematopoietic tumors. In the 2016 revision of the World Health
Organization classification of lymphoid neoplasms, HS is listed
under the major categories of histiocyte and dendritic cell tumors,
defined as malignant hyperplasia of cells exhibiting morphological
and immunophenotypical characteristics similar to those of mature
cells, with expression of one or more tissue cell markers,
excluding acute monocytic leukemia and primitive monocytic sarcoma.
HS may occur at any age, but the majority of the cases are
encountered in adults, with a median age of 52 years and a slight
male predominance. Approximately 1/3 of the cases occur in lymph
nodes, while the incidence of extranodular HS, such as in the
gastrointestinal tract, spleen and soft tissue, is relatively
low.
Histologically, HS is characterized by destruction
of normal tissue structure by diffuse, non-aggregated proliferative
tumor cells, which appear as monoclonal or pleomorphic. The tumor
cells are usually large, round or oval and focally spindle-shaped,
with abundant eosinophilic cytoplasm surrounding large bizarre
pleomorphic nuclei. Phagocytosis of red blood cells, multinucleated
giant cells and nuclear mitoses were readily identified.
A definitive morphological diagnosis of HS is very
difficult and it must be confirmed by immunohistochemistry. The
tumor cells may express one or more tissue cell antigens, including
CD163, CD68, lysozyme, CD11c and CD14, while CD45, leukocyte common
antigen RO subtype, CD4, macrophage antibody 387 and
α1-antichymotrypsin may also be positive in HS.
Langerhans cell marker (CD1a, langerin),
myeloperoxidase (MPO), CD1, CD33, CD34, dendritic cell markers
(CD21, CD35 etc.), specific B-cell and T-cell antigens, CD30, human
melanoma black (HMB)-45 and CK are negative in HS. S-100 may be
positive or negative. The Ki-67 proliferation index ranges from 10
to 90% (median, 20%), but whether the percentage reflects the
prognosis remains uncertain. Electron microscopy for the diagnosis
of HS is of auxiliary value, and can demonstrate tumor cells with
abundant cytoplasm, containing a large number of lysosomes, but
without Birbeck bodies or intercellular junctions.
Histologically, HS may be misdiagnosed as several
diseases: i) Large-cell lymphoma, mainly Hodgkin's lymphoma
(expression of CD30, CD15, PAX-5), diffuse large B-cell lymphoma
(expressing B cell markers) and anaplastic large-cell lymphoma
(CD30, ALK and EMA positivity are considered as identification
markers in addition to T-cell markers); ii) poorly differentiated
carcinoma expresses epithelial markers such as CK and EMA; iii)
malignant melanoma also expresses HMB-45 and melan A in addition to
S-100; iv) granulocytic sarcoma is positive for the
granulocyte-specific markers MPO and CD33, which may help in the
differentiation from HS; v) follicular dendritic cell sarcoma
(FDCS); the cells of FDCS are mostly spindle-shaped and oval with
large nuclei, forming unequally sized nodules, and are positive for
CD21, CD23 and CD35, while they are negative for CD68.
HS is an invasive tumor that responds poorly to
treatment and for which there is yet no widely accepted effective
therapy (9). Due to the fact that
the vast majority of patients are at an advanced stage when they
seek medical advice, the survival time is usually <2 years due
to the poor response to chemotherapy (10). In recent years, it has been
demonstrated that the use of radiotherapy and chemotherapy combined
with autologous hematopoietic stem cell transplantation may improve
treatment efficacy. In general, efficacy depends on a variety of
factors, the most important being the location and stage of the
tumor. For example, central nervous system and disseminated lesions
are associated with an aggressive clinical course. The size of the
tumor is also an important factor affecting the prognosis, and the
survival time of patients with a primary tumor diameter of >3.5
cm is short.
Despite the low incidence of HS, a number of cases
(11–13) have been reported. Compared with
previous reports, the tumor cells in the present case formed nests,
with a cord-like distribution, which is generally not
characteristic of HS, increasing the difficulty of morphological
diagnosis. In addition, the tumor cells not only expressed the
histiocytic antigens CD68 and lysozyme, but also expressed
lymphocyte-associated antigens, such as CD4, CD5 and CD31,
reflecting the complicated antigen expression pattern of HS.
Unfortunately, our patient was unable to tolerate chemotherapy due
to her poor physical condition; therefore, the patient underwent no
intervention other than cervical lymph node biopsy and we were
unable to collect more information on the treatment or outcome of
HS, as she was lost to follow-up.
In conclusion, HS is a rare malignant tumor with
poor prognosis. Due to its rapid invasiveness and high mortality
rate, HS remains a medical challenge without effective
treatment.
Acknowledgements
Not applicable.
Competing interests
The authors wish to declare that there is no known
conflict of interests associated with this publication.
Funding
This study was supported by the Central South
University Sports Medicine Scholarship and National College
Students' Innovation and Entrepreneurship Training Program (grant
no. 201710422116).
Authors' contributions
JZ and YL cared for the patient and contributed to
the concept and design. JZ, WCW and FLX drafted the report.
Ethics approval and consent to
participate
Not applicable.
Availability of data and materials
Not applicable.
Consent for publication
A signed written consent form was obtained from the
patient regarding the publication of the case details and
associated images.
Glossary
Abbreviations
Abbreviations:
|
HS
|
histiocytic sarcoma
|
|
CD
|
cluster of differentiation
|
|
CT
|
computed tomography
|
|
CK
|
cytokeratin
|
|
ALK
|
anaplastic lymphoma kinase
|
|
EMA
|
epithelial membrane antigen
|
|
FDCS
|
follicular dendritic cell sarcoma
|
References
|
1
|
Laviv Y, Zagzag D, Fichman-Horn S and
Michowitz S: Primary central nervous system histiocytic sarcoma.
Brain Tumor Pathol. 30:192–195. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pollen M, El Jamal S, Lewin J and Manucha
V: Histiocytic sarcoma in a kidney transplant patient: A case
report and review of the literature. Case Rep Pathol.
2016:35910502016.PubMed/NCBI
|
|
3
|
Hornick JL, Jaffe ES and Fletcher CD:
Extranodal histiocytic sarcoma: Clinicopathologic analysis of 14
cases of a rare epithelioid malignancy. Am J Surg Pathol.
28:1133–1144. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu L and Yang SJ: A case of primary
histiocytic sarcoma arising from thyroid gland. Pathol Oncol Res.
16:127–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Swerdlow SH, Campo E, Pileri SA, Harris
NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz
AD, et al: The 2016 revision of the World Health Organization
classification of lymphoid neoplasms. Blood. 127:2375–2390. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takahashi E and Nakamura S: Histiocytic
sarcoma: An updated literature review based on the 2008 WHO
classification. J Clin Exp Hematop. 53:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Munoz J, Sanchez BE and Wang D:
Histiocytic sarcoma of the thyroid. Am J Hematol. 87:5312012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pollen M, El Jamal S, Lewin J and Manucha
V: Histiocytic Sarcoma in a Kidney Transplant Patient: A Case
Report and Review of the Literature. Case Rep Pathol.
2016:35910502016.PubMed/NCBI
|
|
9
|
Shen XZ, Liu F, Ni RJ and Wang BY: Primary
histiocytic sarcoma of the stomach: A case report with imaging
findings. World J Gastroenterol. 19:422–425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao J, Niu X, Wang Z, Lu H, Lin X and Lu
Q: Histiocytic sarcoma combined with acute monocytic leukemia: A
case report. Diagn Pathol. 10:1102015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Munoz J, Sanchez BE and Wang D:
Histiocytic sarcoma of the thyroid. Am J Hematol. 87:5312012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pakravan A, Bhatia R, Oshima K, Chen G,
Fesler M, Prather C and Taylor JR: Histiocytic sarcoma: The first
reported case of primary esophageal involvement. Am J
Gastroenterol. 109:291–292. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nieuwenhuis MB, van der Salm SM, Verhoeff
JJ, van der Kooi AJ, Slavujecvic-Letic I, Pals ST and Vos JMA: A
43-Year-Old Female with Multifocal Cerebral Lesions. Histiocytic
Sarcoma. Brain Pathol. 25:371–372. 2015. View Article : Google Scholar : PubMed/NCBI
|