Introduction
Plexiform schwannoma (PS) is a benign tumour of the
peripheral nerve sheath, in which Schwann cells exhibit a
multinodular growth pattern (plexiform) (1). PS represents a unique variant,
accounting for ~5% of all schwannomas (2). Berg et al (3) reported a series of 2,259 schwannomas
and found that PS represented only 4.3% (97 patients) of the total
cases.
PS is typically found in the skin, while visceral
localization is rare (2,4). The series from the Mayo Clinic
described 3 patients (3/97, 3%) with visceral PS, involving the
lung, sigmoid colon and parathyroid gland (3). To date, there have been fewer than 15
reported cases of intra-abdominal and visceral PS in both adults
and children (3,5). In particular, mesenteric PS has never
been reported, to the best of our knowledge.
PS is typically characterised by a benign course,
but it may be associated with neurofibromatosis (NF) and the
differential diagnosis from its malignant counterparts (e.g.,
neurofibroma and malignant peripheral nerve sheath tumour) may be
difficult (6). The aim of the
present study was to retrospectively describe two unique cases of
PS with a visceral location in young children, as well as one case
that has been previously reported by one of the authors, in order
to highlight the importance of considering this entity in the
differential diagnosis of abdominal masses and to perform a careful
investigation of associated abnormalities, particularly NF.
Case reports
Case 1
A 16-year-old female patient was admitted to Buzzi
Children's Hospital in July 2017 due to acute onset of abdominal
and back pain associated with fever and rectal bleeding. Over the
last few months, the patient had suffered from recurrent episodes
of abdominal pain that were attributed to constipation and treated
with stool softeners. The family history was unremarkable.
Upon admission, the patient was in good clinical
condition and the results of the laboratory tests were
unremarkable.
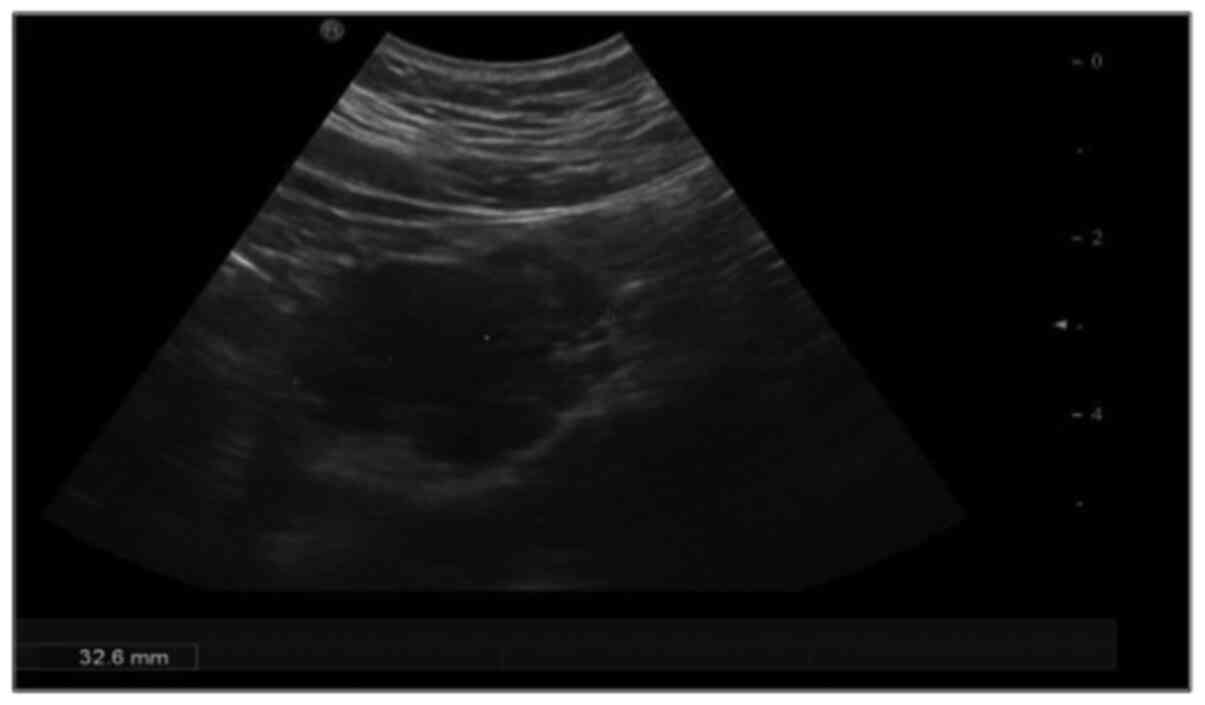
On abdominal ultrasound (Fig. 1), an epigastric mass sized 3.5 cm
was identified. Magnetic resonance imaging (MRI) examination
(Fig. 2) confirmed the presence of
an irregular solid inhomogeneous mass in the left upper abdomen
with contrast enhancement. The Tc-99m-octreotide scintigraphy was
negative.
During laparoscopy (Fig.
3), a hard polylobulated mesenteric mass was identified close
to the first jejunal loops. The mass and the affected bowel loops
were exteriorized through the umbilical wound and resected along
with the adjacent intestine (total length, 10 cm). Intestinal
continuity was restored by termino-terminal anastomosis and the
mesenteric gap was closed.
Histological examination (Fig. 4) following hematoxylin and eosin
staining revealed a PS with free margins. Immunohistochemical
staining for S100, CD34, epithelial membrane antigen (EMA) and
α-smooth muscle actin (α-SMA) was also performed (negative α-SMA
and CD34; positive S100 and EMA). The screening for NF, including
dermatological evaluation and cerebral MRI, was normal.
Two years after the surgery the patient remained
asymptomatic, and an abdominal MRI revealed no abnormalities (last
follow-up visit, December 2019).
Case 2
A 10-year-old boy was referred to New Delhi All
India Institute of Medical Sciences in May 2013 with gross
abdominal distension and pain. On palpation, there was a lump in
the abdomen. The computed tomography scan revealed a mass with a
whorled appearance. On exploratory laparotomy, a sizeable mass
(greatest diameter, 14 cm) arising from the retroperitoneum and
engulfing the aorta and inferior vena cava was identified. Frozen
section biopsy revealed a benign lesion. Near-total excision of the
mass was performed, shaving it off but leaving a part attached to
the great vessels. Histopathological examination was suggestive of
PS. The immunohistochemical staining for S100 was positive.
Screening for NF was performed, including dermatological evaluation
and cerebral and cervical MRI, and the findings were normal. The
patient was asymptomatic after 4 years of follow-up (last follow-up
visit, January 2018).
Case 3
This case was previously reported by one of the
authors (7). In April 2003, a
presacral schwannoma with intraspinal extension was diagnosed in a
10-year-old boy with type 2 NF and multiple neurofibromas, who was
admitted to the New Delhi All India Institute of Medical Sciences
with left sciatic pain and difficulty walking. MRI examination
revealed a unilateral vestibular schwannoma associated with an
intrathoracic schwannoma. Surgery was completed in two steps: The
intraspinal portion of the mass was first removed, followed by
removal of the presacral portion after 15 days, achieving complete
removal. Histological examination revealed S-100 positivity. Our
experience with this case enabled us to correctly diagnose case 2.
The patient was asymptomatic and disease-free at the last follow-up
visit (February 2008).
Discussion
In 1978, Harkin et al first described six
cases of PS, a rare Schwann cell tumour variant (4.3-5% of all
schwannomas) with intraneural, plexiform and possible multinodular
growth (1).
PS is frequently located in superficial soft tissues
or the cutaneous region (3).
Visceral involvement is extremely rare (3%) and only a few cases
involving the abdomen/digestive tract have been reported to date
(2,8). The first case of visceral PS
(ascending colon) was reported by Hirose et al in
1997(9).
In 2016, Kudose et al identified 10 cases of
PS arising in the digestive tract (5 in the colon, 3 in the
oesophagus and 2 in the small intestine) in a review of the English
medical literature and reported the first case of gastric PS
associated with NF2(2). Only 2
patients were aged ≤18 years (age range, 11-77 years). The majority
of the patients (6/10 cases) presented with acute symptoms,
including abdominal pain and rectal bleeding, as the patients in
the present study.
To the best of our knowledge, case 1 presented
herein is the first reported case of a mesenteric PS.
PS is generally associated with benign
characteristics, slow growth, and no metastatic potential. It
usually affects young adults and presents as a solitary lesion
(2,7).
On histological evaluation, PS has several elements
found in conventional schwannoma, such as being composed by compact
Schwann cells (but arranged as multinodular/plexiform rather than
in a globular configuration), Verocay bodies, hyalinized and/or
ectatic vessels and collagenous residues (3). Recent studies also demonstrated that
PS may not always display the Antoni A and B patterns (but when it
does, Antoni A is predominant) and is often positive for S100
expression (3-5).
A precise definition of the histopathological characteristics is
extremely important, as it is the best method for differentiating
PS from other benign or malignant lesions. Indeed, preoperative
diagnosis is extremely difficult, as the endoscopic and
radiological findings are non-specific (5).
On imaging evaluation of case 1, a mass that was
isointense relative to skeletal muscle was identified on MRI
T1-weighted images, displaying multinodularity, as previously
reported (2,4,7,10), but
this was not enough to confirm the diagnosis. A preoperative or
intraoperative biopsy was considered; however, the mass was
well-defined and appeared to be benign macroscopically. Moreover,
the resection also included the normal adjacent bowel, with a total
length of 10 cm of intestine removed. As there is the possibility
of other neuronal tumours with different natural histories and
behaviours, a preoperative or intraoperative biopsy should be
considered to optimise the treatment approach (11). The differential diagnosis from other
entities, such as plexiform neurinoma, which is associated with a
high risk of malignant transformation, and malignant cell tumours,
is crucial in order to avoid delay in treatment and, potentially,
the need for a more aggressive approach (7).
Another important aspect is the possible association
of PS with NF2 and schwannomatosis that has been reported in up to
10% of cases (5,9). The presence of multiple lesions
represents a high risk of having NF2 and is associated with
mutations of the NF2 gene on chromosome 22q12.2 (adjacent to the
SMARCB1/INI1 suppressor gene) (12). Therefore, it is important to exclude
the presence of associated dermal lesions and neurinoma in patients
with PS, particularly in paediatric patients in whom inherited
syndromes are more frequent, by performing thorough intracranial
and spinal MRI examinations (11,13).
Genetic studies are useful when there is a positive family history
or in case of a certain association with NF-schwannomatosis. In
these selected patients, attempts have been made to use DNA
next-generation sequencing to detect copy number alterations and
mosaicisms and to correlate genotype with clinical phenotype
(14). This may lead to a better
understanding of the underlying pathology and to the development of
new clinical models (e.g., germline mutations of SMARCB1 predispose
to schwannomas) (15). Moreover,
genetic studies are the base of new therapeutic strategies, such as
the inhibition of the tyrosine kinase receptor MET or EGFR/ErbB2
and FAK1 (PTK2) to suppress tumorigenesis and achieve a
growth-inhibitory effect (16). It
is unclear whether there is a correlation between these aspects and
the isolated form of PS; SMARCB1/INI1 immunoreactivity appears to
be uninformative, particularly in solitary schwannomas (17). On the other hand, recent studies
focused on the possibility to perform histological and genetic
characterization of Schwann cells from cutaneous PS, correlating
them to the presence of NF2 and allowing an early diagnosis of the
associated syndrome (18). The same
concept may be applied to cells obtained from visceral PS.
PS is treated with radical surgical excision to
prevent local recurrence (19). The
literature also reports endoscopic submucosal resection in case of
rectal PS confined to the submucosa (4,20).
Radical excision is associated with an excellent outcome; however,
if the mass involves vital structures, it may be possible to
perform limited resection. Indeed, the tumour in case 2 engulfed
the great vessels in the abdomen; once the diagnosis was confirmed
by frozen section biopsies, partial removal was deemed a safer
option. Our choice of treatment was also supported by the reported
absence of malignant characteristics and metastatic spread of
visceral PS (21). In 2017, a
series of paediatric non-vestibular schwannoma cases indicated the
presence of >4 mitoses/10 high-power fields as a risk factor for
recurrence (17). Unfortunately,
the rare visceral location of PS prevents drawing definitive
conclusions regarding partial surgical removal.
Retrosi et al suggest collecting data from
patients with PS in unusual locations to advance the overall
knowledge and plan adequate management and follow-up (11). Planning follow-up represents another
uncertain and critical aspect of patient management. According to
Kawaguchi et al, surveillance is not necessary following
complete surgical resection (5),
but there are no specific indications in case of incomplete
removal.
Considering the cases reported in the literature as
well as our experience, it appears that the histological and
biological characteristics of the tumour affect its natural history
more than its location: PS has a benign nature, regardless of its
localisation. The difficulties in preoperative diagnosis and the
asymptomatic pattern in younger patients may explain the relatively
late identification and diagnosis of visceral and abdominal PS.
However, this does not appear to affect postoperative outcomes,
particularly when complete surgical excision is achieved.
In conclusion, although benign schwannomas rarely
occur in the gastrointestinal tract, PS should be considered in the
differential diagnosis of all patients with an abdominal mass. As
the lesion is benign, subtotal resection may be a viable option if
the tumour involves vital structures.
Given its possible association with NF, the
diagnosis of PS should prompt an investigation for other
manifestations of this disorder.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
FD and SS conceptualized and co-wrote the
manuscript. FD and LM performed the literature search. LM was
actively involved in drafting the manuscript. PC performed the
histological examination and provided the related image. CV and GR
acted as supervisors and critically revised the manuscript for
important intellectual content. All the authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
This article does not contain any studies with human
participants performed by any of the authors.
Patient consent for publication
Verbal (case 1) and/or written (cases 2 and 3)
informed consent was obtained from all individual participants
included in the study or their parents/legal guardians.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Harkin JC, Arrington JH and Reed RJ:
Benign plexiform schwannoma, a lesion distinct from plexiform
neurofibroma (abstract). J Neuropathol Exp Neurol. 37(622)1978.
|
|
2
|
Kudose S, Kyriakos M and Awad MM: Gastric
plexiform schwannoma in association with neurofibromatosis type 2.
Clin J Gastroenterol. 9:352–357. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Berg JC, Scheithauer BW, Spinner RJ, Allen
CM and Koutlas IG: Plexiform schwannoma: A clinicopathologic
overview with emphasis on the head and neck region. Hum Pathol.
39:633–640. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Iida A, Imamura Y, Katayama K, Hirose K
and Yamaguchi A: Plexiform schwannoma of the small intestine:
Report of a case. Surg Today. 33:940–943. 2003.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kawaguchi S, Yamamoto R, Yamamura M,
Oyamada J, Sato H, Fuke H and Yabana T: Plexiform schwannoma of the
rectum. Dig Endosc. 26:113–116. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gkekas C, Kalyvas V, Symeonidis EN,
Malioris A, Papathanasiou M, Kalinderis N, Moisidis K and
Hatzimouratidis K: Plexiform schwannoma of the penis: A rare
subtype of genital Schwannoma. Case Rep Urol.
2019(1752314)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gangopadhyay AN, Sharma S, Kumar M and
Sharma SP: Presacral schwannoma with intraspinal extension in a
child with neurofibromatosis type 2-A case report and review of
literature. J Indian Association Pediatric Surgeons. 9:116–119.
2004.
|
|
8
|
Aktekın A, Özkara S, Merıç K, Günay
Gürleyık M, Aker F and Sağlam A: Plexiform schwannoma of the
duodenum accompanying pyloric stenosis: Report of a case. Turk J
Gastroenterol. 23:385–389. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hirose T, Scheithauer BW and Sano T: Giant
plexiform schwannoma: A report of two cases with soft tissue and
visceral involvement. Mod Pathol. 10:1075–1081. 1997.PubMed/NCBI
|
|
10
|
Shishiba T, Niimura M, Ohtsuka F and Tsuru
N: Multiple cutaneous neurilemmomas as a skin manifestation of
neurilemmomatosis. J Am Acad Dermatol. 10:744–754. 1984.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Retrosi G, Nanni L, Ricci R, Manzoni C and
Pintus C: Plexiform schwannoma of the esophagus in a child with
neurofibromatosis type 2. J Pediatr Surg. 44:1458–1461.
2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ishida T, Kuroda M, Motoi T, Oka T,
Imamura T and Machinami R: Phenotypic diversity of
neurofibromatosis 2: Association with plexiform schwannoma.
Histopathology. 32:264–270. 1998.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Matsuoka Y, Kakudo N, Fukui M and Kusumoto
K: Giant plexiform schwannoma in the plantar aspect of the foot: A
case report. J Surg Case Rep. 2019(rjz352)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Fisher MJ, Belzberg AJ, de Blank P, De
Raedt T, Elefteriou F, Ferner RE, Giovannini M, Harris GJ,
Kalamarides M, Karajannis MA, et al: 2016 Children's Tumor
Foundation conference on neurofibromatosis type 1,
neurofibromatosis type 2, and schwannomatosis. Am J Med Genet A.
176:1258–1269. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Karajannis MA, Legault G, Hagiwara M,
Ballas MS, Brown K, Nusbaum AO, Hochman T, Goldberg JD, Koch KM,
Golfinos JG, et al: Phase II trial of lapatinib in adult and
pediatric patients with neurofibromatosis type 2 and progressive
vestibular schwannomas. Neuro Oncol. 14:1163–1170. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Vitte J, Gao F, Coppola G, Judkins AR and
Giovannini M: Timing of Smarcb1 and Nf2 inactivation determines
schwannoma versus rhabdoid tumor development. Nat Commun.
8(300)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Broehm C, Al-Ibraheemi A and Fritchie KJ:
Pediatric Non-vestibular Schwannoma. Pediatr Dev Pathol.
20:232–239. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Castellanos E, Plana A, Carrato C, Carrió
M, Rosas I, Amilibia E, Roca-Ribas F, Hostalot C, Castillo A, Ros
A, et al: Early genetic diagnosis of neurofibromatosis type 2 from
skin plaque plexiform schwannomas in childhood. JAMA Dermatol.
154:341–346. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ijichi K, Muto M, Masaki A and Murakami S:
Recurrent plexiform schwannoma involving the carotid canal. Auris
Nasus Larynx. 45:358–361. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Sivak MV Jr, Sullivan BH Jr and Farmer RG:
Neurogenic tumors of the small intestine. Review of the literature
and report of a case with endoscopic removal. Gastroenterology.
68:374–380. 1975.PubMed/NCBI
|
|
21
|
Agaram NP, Prakash S and Antonescu CR:
Deep-seated plexiform schwannoma: A pathologic study of 16 cases
and comparative analysis with the superficial variety. Am J Surg
Pathol. 29:1042–1048. 2005.PubMed/NCBI
|