Introduction
Retinoblastoma is a common primary intraocular
malignant tumor in infants and young children. It is considered to
be the most common primary malignant intraocular tumor. According
to the World Health Organization, the reported incidence is 1 case
per 16,000-18,000 live births, with approximately 7,000-8,000 new
cases worldwide each year. It is caused by germline and somatic
mutations in the RB1 gene or, rarely, somatic amplification of
MYCN. Hereditary retinoblastomas are typically bilateral with
multiple tumor foci in one or two eyes (1).
Survivors of hereditary retinoblastoma generally
have a good prognosis but radiation therapy for hereditary
retinoblastoma increases the risk of secondary malignancy (2-4).
However, the precise molecular mechanism remains unknown. Here, we
report on a survivor of possible hereditary retinoblastoma,
diagnosed with secondary leiomyosarcoma 42 years after radiation
therapy. To elucidate the molecular mechanism of secondary
leiomyosarcoma, we performed an RNA panel sequencing and DNA panel
sequencing on her sarcoma tissue. We could not obtain this
patient's retinoblastoma tissue itself. However, we also performed
reverse transcription-polymerase chain reaction (RT-PCR) analysis
for the fusion genes detected by RNA sequencing to distinguish
whether the fusion genes were somatic or germline mutations on both
sarcoma and normal tissues, specifically lymph node and skin,
respectively. We also examined short variants found in the DNA
panel test by Sanger sequencing to distinguish between somatic and
germline mutations.
Case report
In April 2014, a 44-year-old woman with a residual
nasal cavity tumor was referred to our hospital. She had suffered
from left nasal bleeding in September 2013 and consulted a nearby
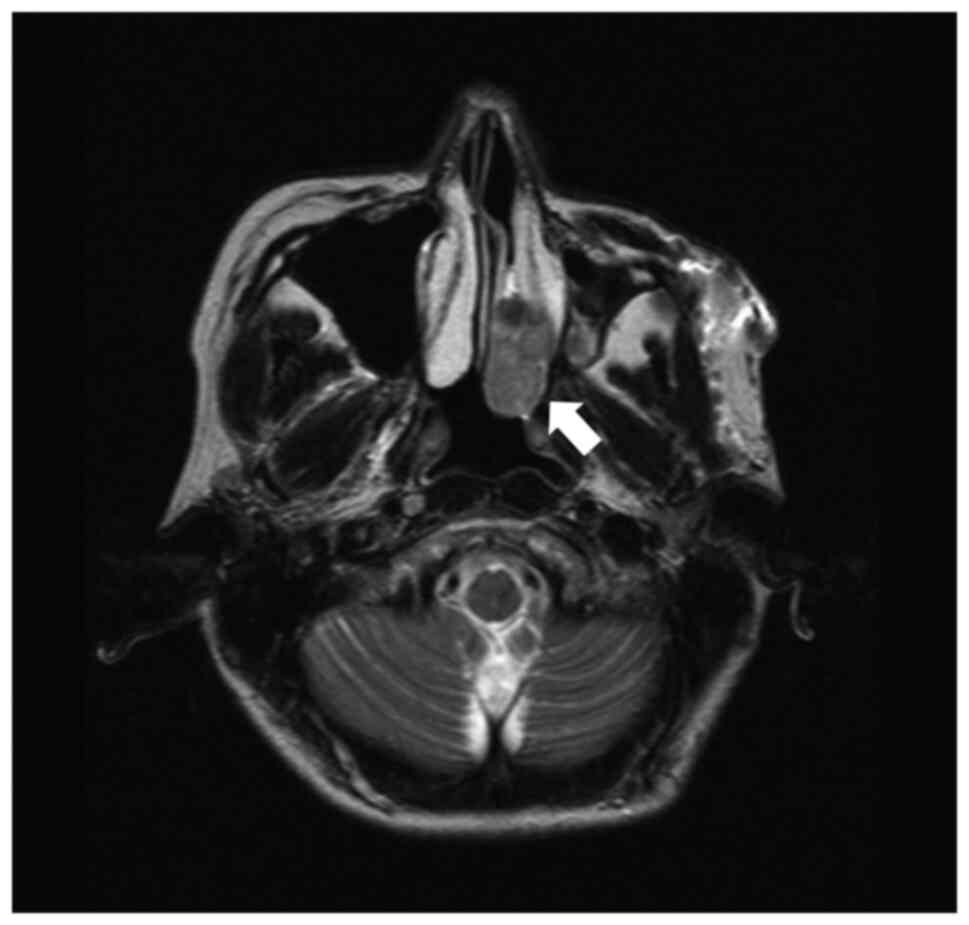
otolaryngologist. A tumor in her left nasal cavity was discovered
(Fig. 1), and she was referred for
treatment. After the embolization of the tumor, endoscopic
resection of the tumor was performed in March 2014, but the tumor
was located in the lateral wall of the nasal cavity. She was then
transferred to our hospital for future treatment.
The patient had a history of retinoblastoma of the
left eye at 2 years old. She received radiotherapy at the age of
two, and her left eyeball was removed at the age of 24. Her family
had no family history of retinoblastoma. Other information, such as
total radiation dose and chemotherapy, could not be obtained.
Our hospital pathologists re-examined the previously
resected nasal tumor and diagnosed leiomyosarcoma. An endoscopic
local examination did not identify the residual tumor, and imaging
examinations revealed no metastatic lesions. Her laboratory tests
yielded almost normal values except for lactate dehydrogenase was
263 U/l (normal range: 119-229 U/l).
In July 2014, partial removal of the left maxilla,
including the pharyngeal opening of the auditory tube and palatine
tonsils, was performed, where the location of the residual tumor
was suspected. Left submandibular lymph nodes were also dissected.
Pathological examination of the resected tissue revealed residual
lesions of leiomyosarcoma in the inferior nasal concha. There was
no metastases in the dissected lymph nodes (Sample 1).
After the operation, she received four courses of
adjuvant chemotherapy consisting of ifosfamide (2 g/m2
per day, infusions performed on days 1-5) and doxorubicin (30
mg/m2 per day, infusions performed on days 1-2) every
28-day cycle. However, several subcutaneous metastatic nodules
occurred in May 2016. In September 2016, a total of five
subcutaneous lesions were resected, two from the anterior chest,
and one from the right shoulder, left upper arm, and left lumbar
region (Sample 2). All of the lesions were metastatic
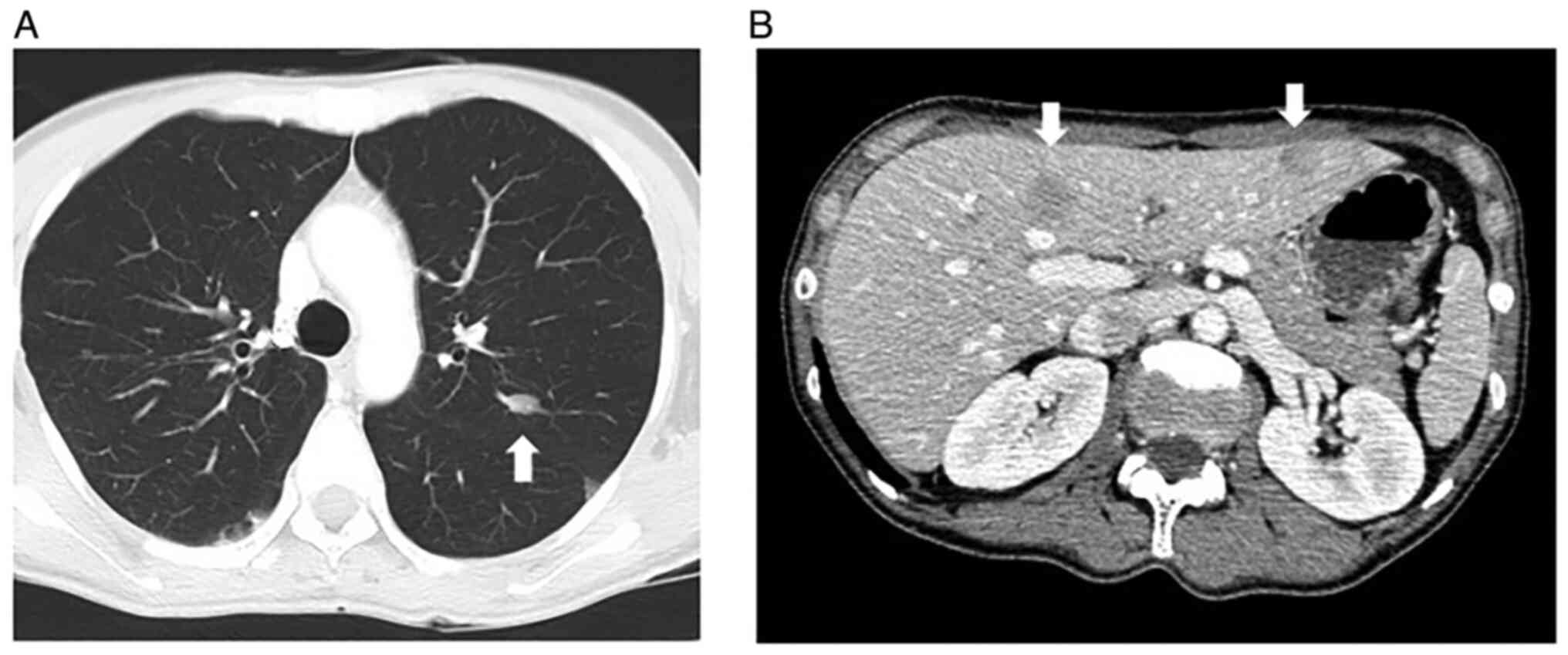
leiomyosarcomas. In November 2016, multiple metastases were
detected in her lung (Fig. 2A) and
liver (Fig. 2B) via computed
tomography. The first-line systemic chemotherapy, consisting of
gemcitabine (900 mg/m2/day; infusions performed on day 1
and 8) and docetaxel (70 mg/m2; infusions performed on
day 8) every 28-day cycle, was started, but it was discontinued
after two cycles due to the suspected pneumonitis caused by these
anticancer drugs. The second-line chemotherapy consisting of
eribulin mesylate (1.4 mg/m2/day; infusions performed on
day 1 and 8 of every 21-day cycle) was started, but it was
discontinued during the second cycle due to progressive disease.
The third-line therapy consisting of pazopanib, 600 mg/day orally,
was started in April 2017 but was discontinued due to high fever,
thrombocytopenia, and punctate erythema throughout the body. At
that time, a skin biopsy was performed on her left lower leg
(Sample 3), but no metastases of leiomyosarcoma were found. The
fourth line of chemotherapy, trabectedin (1.2 mg/m2),
was started, but after 2 cycles, there was evidence of progressive
disease. A reduced dose of pazopanib, 200-400 mg/day, was
re-introduced, and it was continued for approximately 2 years.
However, her leiomyosarcoma gradually increased, and she died in
January 2020.
Methods for pathological studies
Immunohistochemical staining was completed using a
Ventana BenchMark GX system (Ventana Medical Systems Inc.). These
evaluations used the following primary antibodies: pan Keratin
(AE1/AE3/PCK26 [prediluted], Ventana Medical Systems Inc.), smooth
muscle actin (SMA, 1A4 [prediluted] Ventana Medical Systems Inc.),
desmin (DE-R-11 [prediluted] Ventana Medical Systems Inc.), MyoD1
(EP212 [prediluted] Ventana Medical Systems Inc.), p53 (DO7
[prediluted], Ventana Medical Systems Inc.), and retinoblastoma
transcriptional corepressor 1 (Rb1, G3-245, 1:200; BD
Pharmingen).
Methods for genetic studies.
Extraction of RNA and DNA from formalin-fixed, paraffin-embedded
(FFPE) tissues
RNAs were extracted from slices of FFPE tissue using
Maxwell RSC Instrument and Maxwell RCS RNA FFPE Kit (Promega) and
their concentrations were measured with the QuantiFluor RNA System
(Promega). The DV200 score for quality assessment of each RNA was
determined using Bioanalyzer 2100 and RNA 6000 Nano/Pico Kit. DNA
was extracted from slices of FFPE tissue with QIAamp DNA FFPE
Advanced UNG Kit (Qiagen) and its concentration was measured using
the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Inc.). The
quality of DNA was confirmed using 1% agarose gel
electrophoresis.
RNA panel analysis. The extracted tumor RNA
was converted to a next-generation sequencing (NGS) library with
the TruSight RNA Pan-Cancer Panel (RS-303-1002, Illumina) following
their reference guide (Document # 1000000001632 v01). The quality
and quantity of the library were measured using Agilent DNA1000 Kit
(Agilent) and the Qubit dsDNA HS Assay Kit (Thermo Fisher
Scientific, Inc.). Libraries from 12 samples were equimolarly
pooled and diluted to a concentration of 1.5 pM. 1.3 ml of the
diluted library was sequenced on a NextSeq 550 instrument using the
NextSeq 500/550 Mid Output Kit v2.5 (20024905, Illumina). The data
were analyzed using RNA-Seq Alignment (version 2.0.2) of the
BaseSpace Sequence Hub (https://basespace.illumina.com/, Illumina).
RT-PCR analysis. RNAs were converted to cDNA
with a random hexamer and SuperScript IV Reverse Transcriptase
(Thermo Fisher Scientific, Inc.) as described in the manufacturer's
instructions. The cDNAs were used as templates for PCR reactions
with Platinum SuperFi DNA Polymerase and SuperFi GC Enhancer
(Thermo Fisher Scientific, Inc.). The primer sequences are as
follows: RB1exon8_F, 5'-GATACAAGAATTATTGAAGTTCTCTGTAAAGAAC-3';
DMXL1exon9_R, 5'-CAAGTGATCTCCTCCGACCTCT-3'; DMXL1exon9_F,
5'-GAACACTCCACTGCATGCC-3'; RB1exon9_R,
5'-AGTCCATTAGATGTTACAAGTCCA-3'; RAD51exon5_F,
5'-ACCCAGATCTGTCATACGCT-3'; KNL1utr5_R, 5'-TGCCGTCTTCTAACAGGTCT-3'.
The expected amplicon sizes are 97 bp (RB1exon8-DMXL1exon9), 139 bp
(DMXL1exon9-RB1exon9), 125 bp (RB1exon8-RB1exon9), 121 bp
(RAD51exon5-KNL1utr5). The cycling profile was as follows: 30 s at
98˚C for initial denaturation, followed by 40 or 45 cycles of 10 s
at 98˚C for denaturation, 10 s at 60˚C for annealing, and 30 s at
72˚C for extension. The PCR products were separated on a 2% agarose
gel electrophoresis and stained with GelGreen (Biotium). The low
molecular weight DNA ladder (cat. no. N3233S; New England BioLabs,
Inc.) was used.
DNA panel analysis. The extracted tumor DNA
was sheared using the Covaris M220 according to TruSight Tumor 170
DNA Shearing Quick Guide (Part Number: 010515 Rev B, Covaris) with
slight modification (Repeat/Iterations: 16) and checked the
distribution of the fragments using Bioanalyzer 2100 and High
Sensitivity DNA Kit (Agilent Technologies). The sheared DNA was
converted to an NGS library with the TruSight Oncology 500 Kit
(Illumina) following the reference guide (Document # 1000000067621
v04, Illumina). Sequencing was performed on a NextSeq 550
instrument, and the data were analyzed using the Local Run Manager
TruSight Oncology 500 Analysis Module on the sequencer. After
filtering out candidate germline variants using public databases,
1000 genomes (https://www.internationalgenome.org/) (5) and gnomAD exome/genom (https://gnomad.broadinstitute.org/) (6) according to the Workflow Guide of the
above analysis module (Document # 10000000099600 v00, Illumina), we
annotated the pathogenicity of the detected variants using
knowledge bases, OncoKB (v3.8, https://www.oncokb.org/) (7), Cancer Genome Interpreter (https://www.cancergenomeinterpreter.org/home)
(8), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/)
(9), COSMIC (https://cancer.sanger.ac.uk/cosmic) (10). We used ANNOVAR (https://annovar.openbioinformatics.org/en/latest/)
(11) to annotate the ClinVar data
(version 20210501).
Sanger sequencing. To determine whether the
short variants detected by DNA panel sequencing were somatic or
germline mutations, we performed PCR followed by Sanger sequencing
on the normal lymph node tissue (Sample 1). Each mutant gene was
amplified from DNA using appropriate primer sets (Table SI).
Results from pathological studies
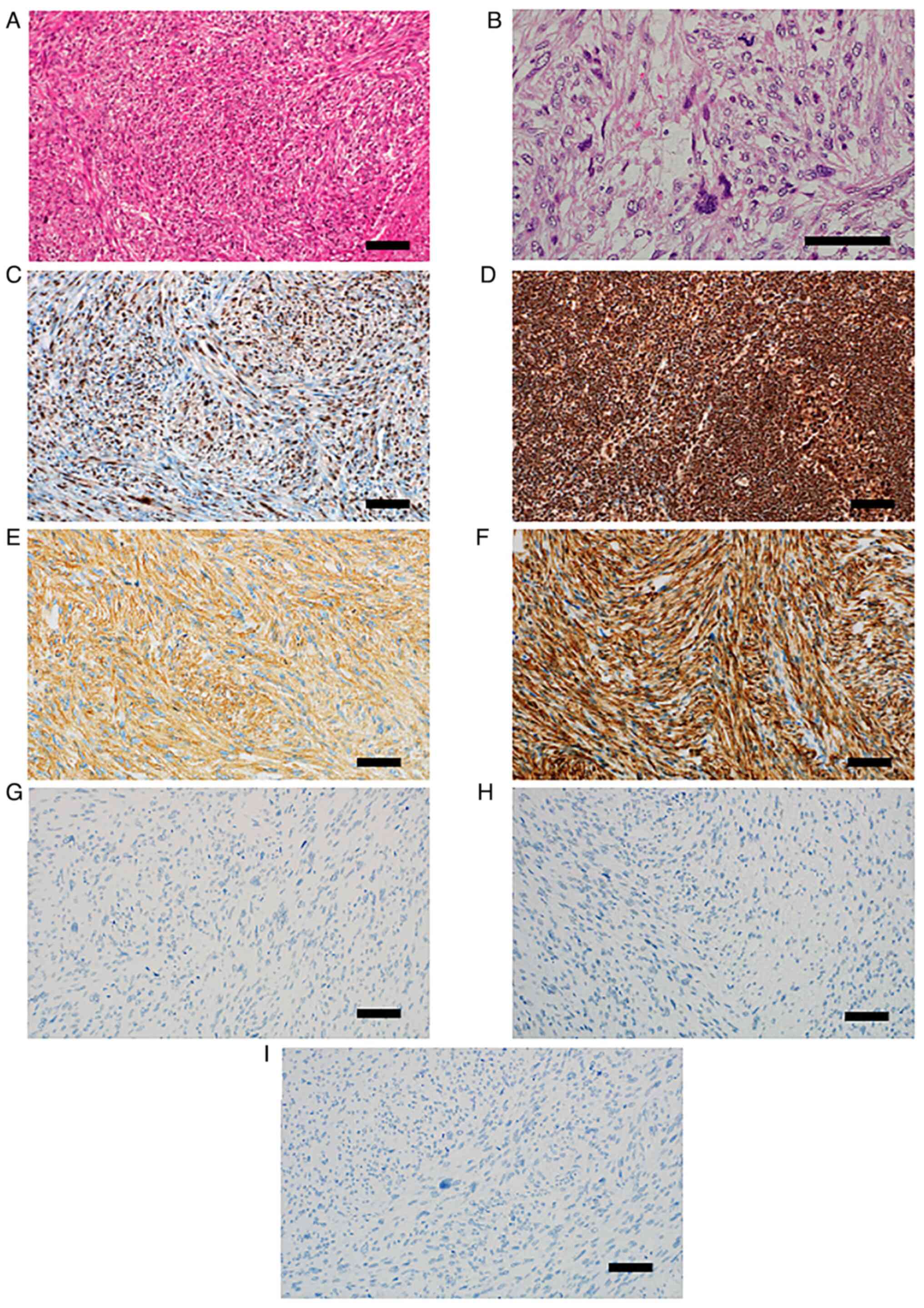
Pathological examination of the resected tissue
revealed residual lesions of leiomyosarcoma in the inferior nasal
concha, in which atypical spindle-shaped cells intermingle in
bundles proliferated (Fig. 3A).
These tumor cells have oval to spindle-shaped nuclei which contain
small nucleoli, and eosinophilic cytoplasm. The cellularity of the
tumor cells was moderate. Tumor cells often show strong nuclear
atypia and mitotic figures were observed with a frequency of 30/10
high-power field (HPF) (Fig. 3B).
The immunohistochemical staining identified that approximately 50%
of tumor cells lacked nuclear retinoblastoma transcriptional
corepressor 1 (Rb1) protein expression (Fig. 3C). In contrast, in normal lymph
node tissue (Sample 1), Rb1 protein expression was observed in more
than 90% of the component cell nuclei (Fig. 3D). Tumor cells were diffusely
positive for smooth muscle actin (Fig.
3E) and desmin (Fig. 3F). Pan
Keratin (Fig. 3G) and MyoD1
(Fig. 3H) were completely
negative. p53 also showed complete loss of expression (Fig. 3I).
Results from genetic studies. RNA
panel sequencing and RT-PCR
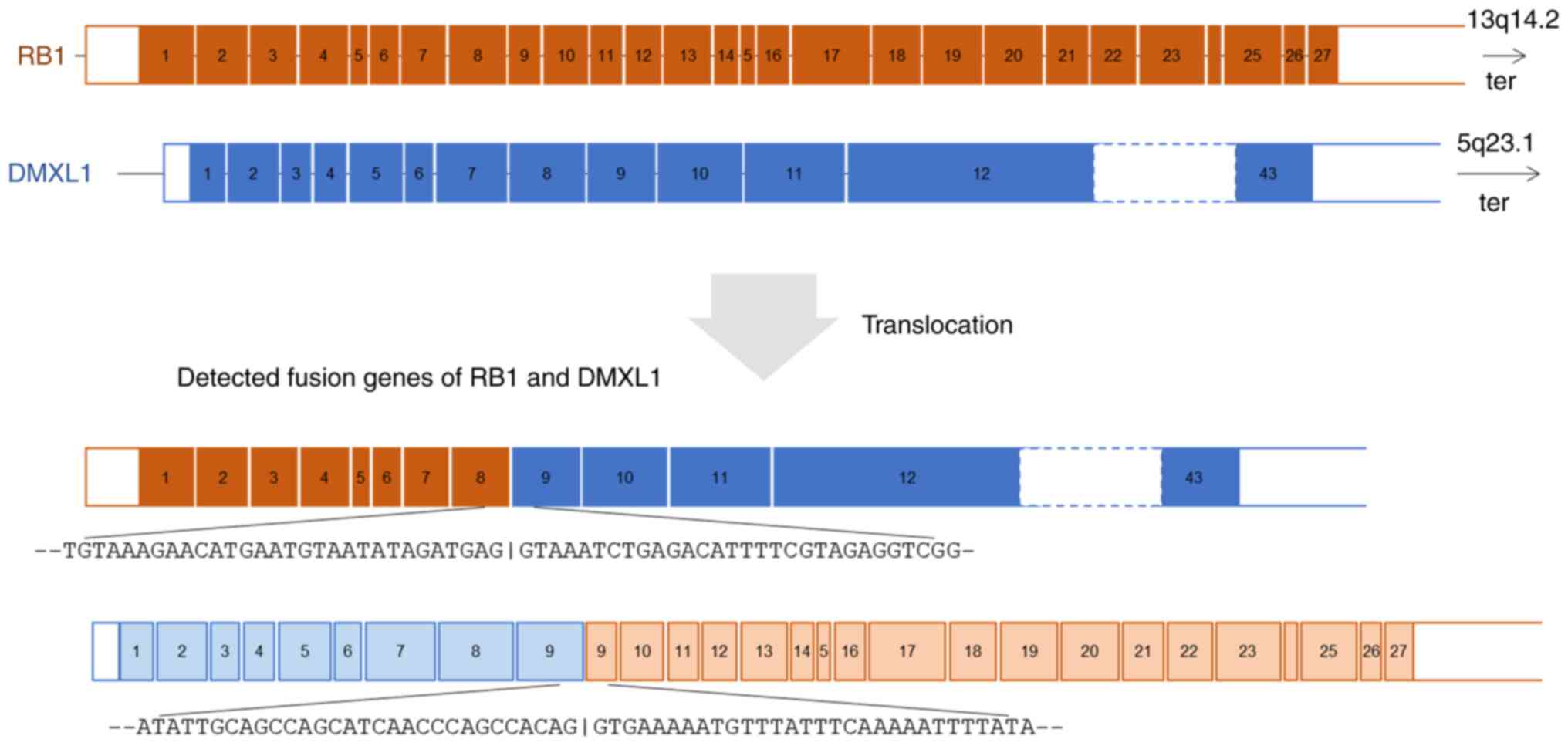
RNA panel sequencing of the metastatic
leiomyosarcoma (Sample 2) identified a novel reciprocal
translocation between 13q14.2 and 5q23.1 and a frameshift fusion
gene RAD51 (exon5; NM_002875.5)-KNL1 (exon2; NM_144508.5). This
reciprocal translocation resulted in two fusion genes: RB1(exon 8;
NM_000321.3)-DMXL1(exon 9; NM_001290321.3) and DMXL1(exon
9)-RB1(exon 9) (Fig. 4). The exon
9 of DMXL1 was included in both fusions. The fusion of RB1(exon
8)-DMXL1(exon 9) was validated by RT-PCR analysis revealing a
rearrangement between these loci. Similar rearrangement was
identified in Sample 1 (normal lymph node) and Sample 3 (skin)
(Fig. 5A). Wild-type RB1 genes
were proven by RT-PCR of RB1 exon 8-RB1 exon 9 in the
leiomyosarcoma, the lymph node, and the skin (Fig. 5B). On the other hand, the
RAD51-KNL1 fusion gene was identified only in the sarcoma tissue
and not in normal samples (Fig.
5C).
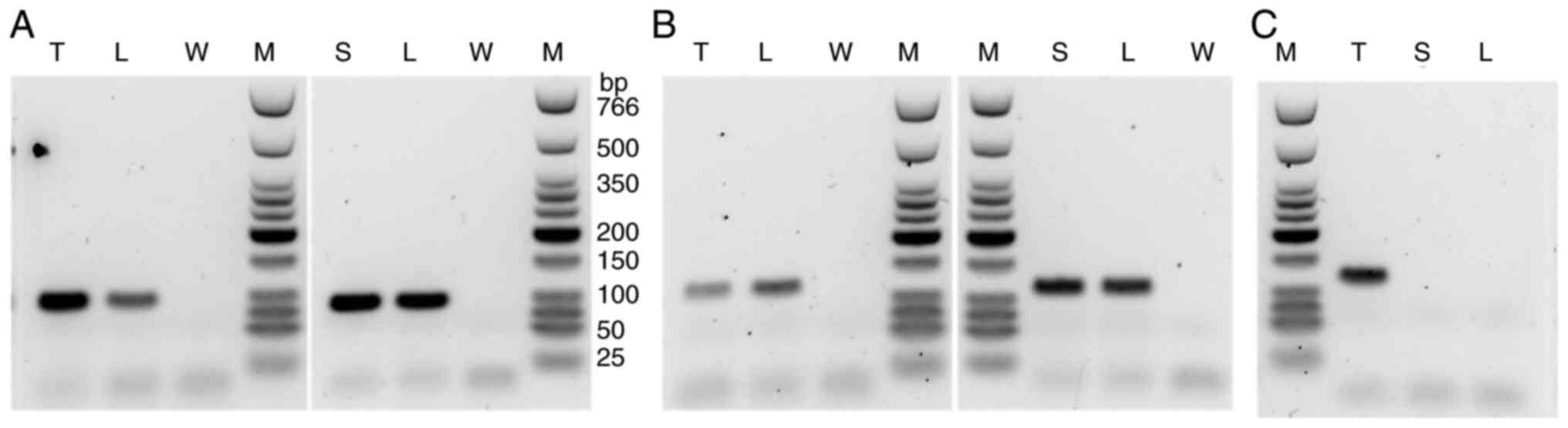 | Figure 5After reverse transcription of sample
RNAs, synthesized cDNAs were used as templates for PCR of (A) RB1
exon 8-DMXL1 exon 9 (amplicon size, 97 bp), (B) RB1 exon 8-RB1 exon
9 (amplicon size, 125 bp) and (C) RAD51 exon 5-KNL1 5' untranslated
region (amplicon size, 121 bp). The marker lane was run on the same
gel for the data shown in (B) (left panel) and (C). L, sample 1
(lymph node); T, sample 2 (tumor); S, sample 3 (skin); W, water; M,
low molecular weight DNA ladder. |
DNA panel sequencing and Sanger sequencing.
DNA panel sequencing identified eight short variants:
CDK12(Arg271Lys), CDK6(Val62Gly), HGF(Asp330Val),
KDM5A(Thr1343Ile), LRP1B(Cys138Gly), MSH2(Met688Ile),
SNCAIP(Glu87Lys), and TP53 (splice acceptor of intron 8). Only the
variants in HGF, KDM5A, LRP1B, and MSH2 were detected in the normal
lymph node (Table I). The panel
analysis identified fourteen copy number changed genes. Nine of
these genes, AR, FGF23, FGF4, FGF6, FGF7, FGFR1, KRAS, NRG1, and
RAF1, were amplified. The other five genes, ATM, FGF5, FGF8, MDM4,
and PTEN, were lost (Table
II).
 | Table IShort variants detected by DNA panel
sequencing. |
Table I
Short variants detected by DNA panel
sequencing.
| Gene | Variant | VAF | OncoKB | CGI | COSMIC | COSMIC_id | ClinVar |
|---|
| CDK12 | Arg271Lys | 0.418 | | Passenger | | | |
| CDK6 | Val62Gly | 0.408 | | Driver | | | |
| HGF | Asp330Val | 0.416 | | Driver | | | |
| KDM5A | Thr1343Ile | 0.333 | | Passenger | | | |
| LRP1B | Cys138Gly | 0.613 | | Passenger | | | |
| MSH2 | Met688Ile | 0.611 | | Driver | Pathogenic | COSV 51886508 | US |
| SNCAIP | Glu87Lys | 0.035 | | Passenger | Pathogenic | COSV 54401827 | |
| TP53 | Splice acceptor of
intron 8 | 0.849 | | Driver | | | |
| Table IICopy number changes. |
Table II
Copy number changes.
| Gene | Amp/Loss | OncoKB | CGI |
|---|
| AR | Amp | LO | Known |
| ATM | Loss | LO | Known |
| FGF23 | Amp | | Predicted
passenger |
| FGF4 | Amp | Inconclusive | Known |
| FGF5 | Loss | | Predicted
passenger |
| FGF6 | Amp | | Predicted
passenger |
| FGF7 | Amp | | Predicted
passenger |
| FGF8 | Loss | | Predicted
passenger |
| FGFR1 | Amp | O | Known |
| KRAS | Amp | LO | Predicted
driver |
| MDM4 | Loss | | Predicted
passenger |
| NRG1 | Amp | | Predicted
passenger |
| PTEN | Loss | O | Known |
| RAF1 | Amp | O | Known |
Discussion
Retinoblastoma is a common primary intraocular
malignant tumor in infants and young children. Knudson's ‘two-hit
hypothesis’ accurately explains the development mechanism of
retinoblastoma (12,13). Briefly, in hereditary
retinoblastoma, individuals carry a germline heterozygous
alteration in RB1. Somatic inactivation of the second RB1 allele
results in the development of retinoblastoma.
To diagnose hereditary retinoblastoma in clinical
practice, various sequencing methods are used to identify a
heterozygous germline pathogenic variant in RB1. Pathogenic
variants in RB1 show various types of mutations, including single
nucleotide variations, small indels, and large
deletions/duplications (14).
Schieffer et al (15)
performed RNA sequencing on the tumor tissue of an infant with
sporadic and intracranial retinoblastoma and reported multiple
fusion genes, including RB1-SIAH3. However, the reports of fusion
genes, including RB1, are rare.
We did not have this patient's retinoblastoma
tissue, so the secondary malignancy (leiomyosarcoma) and normal
tissues (lymph node and skin) were used for genetic testing. RNA
sequencing is a useful tool for detecting fusion genes. Using this
method on the leiomyosarcoma tissue, reciprocal translocations
RB1(exon 8)-DMXL1(exon 9) and DMXL1(exon 9)-RB1(exon 9), and fusion
gene of RAD51 (exon5)-KNL1 (exon2), were identified. To the best of
our knowledge, these are novel fusion genes. RT-PCR confirmed the
presence of RB1(exon 8)-DMXL1(exon9) in the leiomyosarcoma, lymph
node, and skin samples, identifying this fusion gene as a germline
mutation. DMXL1(exon 9)-RB1(exon 9) was confirmed in leiomyosarcoma
and lymph node but not in the skin. However, this fusion gene is
also probably a germline mutation. Because RNA quality was poorest
in the skin (DV200 score: 35 for tumor, 36 for lymph node, 15 for
skin), that RNA was highly fragmented, which may explain the
false-negative results. Since wild-type RB1 genes were proven by
RT-PCR of RB1 exon 8-RB1 exon 9 in the leiomyosarcoma, the lymph
node, and the skin, the reciprocal translocation between RB1 and
DMXL1 is regarded as heterozygous alteration, and, thus, we
considered that her retinoblastoma was likely hereditary. On the
other hand, RAD51-KNL1 was identified in leiomyosarcoma but not
lymph node or skin. Therefore, this fusion gene is regarded as a
somatic mutation.
The protein encoded by RB1 is a tumor suppressor
that is a regulator of the G1/S transition of the cell cycle
(https://www.uniprot.org/uniprot/P06400) (16). On the other hand, the protein
encoded by DMXL1 is a member of the WD repeat superfamily of
proteins with regulatory functions. DMXL1 is expressed in various
tissue types, including several types of eye tissue, and it has
been associated with ocular phenotypes (https://genome-asia.ucsc.edu/cgi-bin/hgGene?hgg_gene=ENST00000311085.8&hgg_chrom=chr5&hgg_start=119071489&hgg_end=119249127&hgg_type=knownGene&db=hg38)
(17). The role of chimeric
RB1-DMXL1 genes in the formation of retinoblastoma or secondary
leiomyosarcoma is difficult to understand, but this fusion gene
predicts the possibility of dysfunction of the RB1 gene. Dommering
et al (18) investigated
RB1 mutations in 44 hereditary retinoblastoma patients with a
second primary malignancy and found an increased risk of second
primary malignancy among carriers of one of the 11 recurrent
CGA>TGA nonsense RB1 mutations. In their samples, one case had a
deletion of exon 9-27, which was associated with an epithelial
second primary malignancy. In our case, we may have had a similar
mechanism of RB1 gene dysfunction. The protein coded by RAD51 plays
an important role in homologous strand exchange, a key step in DNA
repair through homologous recombination (https://www.uniprot.org/uniprot/Q06609) (16). The protein coded by KNL1 is
essential for spindle-assembly checkpoint signaling and correct
chromosome alignment (https://www.uniprot.org/uniprot/Q8NG31) (16). RAD51-KNL1 fusion causes a
frameshift and may result in the loss of both proteins.
We identified eight short variants and fourteen copy
number changed genes using DNA panel sequencing. To predict the
involvement of these mutations in carcinogenesis of secondary
leiomyosarcoma, we used cancer knowledge databases: OncoKB
(7), Cancer Genome Interpreter
(8), ClinVar (9), and COSMIC (10) (Table
I). Based on these results, we speculated that short variants
of CDK6, HGF, MSH2, SNCAIP, and TP53 might be involved in malignant
transformation. In the copy number change, amplification of AR,
FGFR1, KRAS, RAF1, and loss of ATM and PTEN were suspected.
To determine whether the eight short variants
detected by DNA panel sequencing were somatic or germline
mutations, Sanger sequencing was performed on the normal lymph node
(Sample 1). Four variants, TP53 (splice acceptor of intron 8),
CDK12(Arg271Lys), CDK6(Val62Gly), and SNCAIP(Glu87Lys), were
somatic mutations. The other four variants, HGF(Asp330Val),
KDM5A(Thr1343Ile), LRP1B(Cys138Gly), and MSH2(Met688Ile), were
germline mutations detected by DNA panel sequencing. The variant
allele fraction of TP53 was 0.85, which is a very high value. This
mutation was not detected in the normal lymph node by Sanger
sequencing. Therefore, we concluded that it is a somatic mutation
and speculated that there was a loss of heterozygosity. This
mutation is in the splice acceptor site and is expected to cause an
exon skipping or intron retention of mRNA, resulting in dysfunction
of TP53 protein.
Schaefer et al (19) reported a high frequency of
co-inactivation of TP53 and RB1 in local recurrent or distant
metastatic leiomyosarcomas using immunohistochemistry. Using
whole-exome and transcriptome sequencing, Chudasama et al
(20) reported that leiomyosarcoma
tumors are characterized by substantial mutational heterogeneity,
near-universal inactivation of TP53, as well as RB1, widespread DNA
copy number alterations including chromothripsis, and frequent
whole-genome duplication. As a molecular mechanism of our patient's
secondary leiomyosarcoma tumorigenesis, we speculate that the
patient had a reciprocal translocation of the RB1 and DMXL1 as a
germline mutation and various somatic mutations containing the
splice acceptor site mutation of TP53 accumulated, resulting in the
secondary leiomyosarcoma. Radiation therapy is thought to trigger
somatic mutations. However, her sarcoma developed 42 years after
the radiotherapy, and the involvement of age-related exposure to
environmental carcinogens cannot be ruled out.
This study has limitations. First, this study is
based on only one case report. Therefore, further prospective
investigations are needed to elucidate the role of reciprocal
translocation of the RB1 and DMXL1 or other mutations in
tumorigenesis of second malignancy in patients with hereditary
retinoblastoma. Second, after adjuvant chemotherapy, the
leiomyosarcoma tissue (Sample 2) was analyzed for mutation
analysis. Therefore, it cannot be ruled out that anticancer drugs
may have affected the somatic mutations in leiomyosarcoma. Third, a
detailed family history could not be confirmed due to the small
number of her relatives, no family history of retinoblastoma could
be found, and no genetic test was performed on her relatives.
Therefore, it is possible that the reciprocal translocation of the
RB1 and DMXL1 is a de novo mutation rather than hereditary. Fourth,
we did not perform the fluorescence in situ hybridization
(FISH) of RB1-DMXL1 fusion gene. Therefore, this fusion gene could
not be visualized in the tissues.
In summary, we report the case of a woman with a
history of possible hereditary retinoblastoma diagnosed with
secondary leiomyosarcoma 42 years after radiotherapy. She had
several germline mutations, including a reciprocal translocation of
RB1 and DMXL1. It is speculated that the addition of some somatic
mutations, such as the splice acceptor site mutation of TP53, might
have precipitated the development of the second malignant
leiomyosarcoma. Further prospective investigations are needed to
elucidate the role of reciprocal translocation of the RB1 and DMXL1
or other mutations in the tumorigenesis of second malignancy in
patients with hereditary retinoblastoma.
Supplementary Material
Oligonucleotide sequences for
validation of detected variants.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not publicly available due to containing
information that could compromise the privacy of the research
participant and her relatives but are available from the
corresponding author on reasonable request.
Authors' contributions
TY conceived the present study. Data acquisition,
analysis and interpretation were performed by HN and YK. TW, HiT,
SN, MW, SK, HaT, RS, ST and YH collected clinical data. TY and HN
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
The tissues of the patient were collected after
written informed consent was obtained from the patient according to
the protocol approved by the Osaka International Cancer Institute
(Osaka, Japan).
Patient consent for publication
Written informed consent was obtained from the
patient's brother to publish her data and associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Eagle RC Jr, Chévez-Barrios P, Li B, Al
Hussaini M and Wilson M: Retinoblastoma. In: WHO classification of
tumours of the eye. Grossniklaus HE, Eberhart CG and Kivelä TT
(eds). 4th edition. International Agency for Research on Cancer,
Lyon, pp111-117, 2018.
|
|
2
|
Kleinerman RA, Schonfeld SJ, Sigel BS,
Wong-Siegel JR, Gilbert ES, Abramson DH, Seddon JM, Tucker MA and
Morton LM: Bone and soft-tissue sarcoma risk in long-term survivors
of hereditary retinoblastoma treated with radiation. J Clin Oncol.
37:3436–3445. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Temming P, Arendt M, Viehmann A, Eisele L,
Le Guin CH, Schündeln MM, Biewald E, Astrahantseff K, Wieland R,
Bornfeld N, et al: Incidence of second cancers after radiotherapy
and systemic chemotherapy in heritable retinoblastoma survivors: A
report from the German reference center. Pediatr Blood Cancer.
64:71–80. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wong FL, Boice JD Jr, Abramson DH, Tarone
RE, Kleinerman RA, Stovall M, Goldman MB, Seddon JM, Tarbell N,
Fraumeni JF Jr and Li FP: Cancer incidence after retinoblastoma.
Radiation dose and sarcoma risk. JAMA. 278:1262–1267.
1997.PubMed/NCBI View Article : Google Scholar
|
|
5
|
1000 Genomes Project Consortium. Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA and Abecasis GR: A global reference for human
genetic variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Karczewski KJ, Francioli LC, Tiao G,
Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A,
Birnbaum DP, et al: The mutational constraint spectrum quantified
from variation in 141,456 humans. Nature. 581:434–443.
2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chakravarty D, Gao J, Phillips SM, Kundra
R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, et
al: OncoKB: A precision oncology knowledge base. JCO Precis Oncol.
2017(PO.17.00011)2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tamborero D, Rubio-Perez C, Deu-Pons J,
Schroeder MP, Vivancos A, Rovira A, Tusquets I, Albanell J, Rodon
J, Tabernero J, et al: Cancer Genome Interpreter annotates the
biological and clinical relevance of tumor alterations. Genome Med.
10(25)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Landrum MJ, Lee JM, Benson M, Brown GR,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al:
ClinVar: Improving access to variant interpretations and supporting
evidence. Nucleic Acids Res. 46:D1062–D1067. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tate JG, Bamford S, Jubb HC, Sondka Z,
Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E,
et al: COSMIC: The catalogue of somatic mutations in cancer.
Nucleic Acids Res. 47:D941–D947. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38(e164)2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Knudson AG Jr: Mutation and cancer:
Statistical study of retinoblastoma. Proc Natl Acad Sci USA.
68:820–823. 1971.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Knudson AG: Two genetic hits (more or
less) to cancer. Nat Rev Cancer. 1:157–162. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Singh J, Mishra A, Pandian AJ, Mallipatna
AC, Khetan V, Sripriya S, Kapoor S, Agarwal S, Sankaran S,
Katragadda S, et al: Next-generation sequencing-based method shows
increased mutation detection sensitivity in an Indian
retinoblastoma cohort. Mol Vis. 22:1036–1047. 2016.PubMed/NCBI
|
|
15
|
Schieffer KM, Feldman AZ, Kautto EA,
McGrath S, Miller AR, Hernandez-Gonzalez ME, Lahaye S, Miller KE,
Koboldt DC, Brennan P, et al: Molecular classification of a complex
structural rearrangement of the RB1 locus in an infant with
sporadic, isolated, intracranial, sellar region retinoblastoma.
Acta Neuropathol Commun. 9(61)2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
UniProt Consortium: UniProt: A worldwide
hub of protein knowledge. Nucleic Acids Res. 47:D506–D515.
2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Navarro Gonzalez J, Zweig AS, Speir ML,
Schmelter D, Rosenbloom KR, Raney BJ, Powell CC, Nassar LR,
Maulding ND, Lee CM, et al: The UCSC genome browser database: 2021
update. Nucleic Acids Res. 49:D1046–D1057. 2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Dommering CJ, Marees T, van der Hout AH,
Imhof SM, Meijers-Heijboer H, Ringens PJ, van Leeuwen FE and Moll
AC: RB1 mutations and second primary malignancies after hereditary
retinoblastoma. Fam Cancer. 11:225–233. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Schaefer IM, Lundberg MZ, Demicco EG,
Przybyl J, Matusiak M, Chibon F, Ingram DR, Hornick JL, Wang WL,
Bauer S, et al: Relationships between highly recurrent tumor
suppressor alterations in 489 leiomyosarcomas. Cancer.
127:2666–2673. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chudasama P, Mughal SS, Sanders MA,
Hübschmann D, Chung I, Deeg KI, Wong SH, Rabe S, Hlevnjak M,
Zapatka M, et al: Integrative genomic and transcriptomic analysis
of leiomyosarcoma. Nat Commun. 9(144)2018.PubMed/NCBI View Article : Google Scholar
|