1. Introduction
Lung cancer remains a significant global health
concern, affecting ~2.5 million individuals annually and exhibiting
the highest mortality rates among all cancer types, accounting for
~18.7% of cancer-related deaths (1,2). The
elevated mortality rate is primarily attributed to the disease's
tendency to present with atypical or asymptomatic manifestations,
as well as the limited participation of high-risk individuals in
systematic preventative screening programs. Consequently, lung
cancer is often diagnosed at advanced stages (3). In some cases, systemic signs and
symptoms unrelated to the primary tumor may arise as paraneoplastic
manifestations.
Paraneoplastic syndrome (PNS) encompasses a
heterogeneous group of disorders caused by either the secretion of
functional amines, peptides, hormones, or cytokines by the tumor or
an aberrant immune cross-reactivity between the tumor and normal
tissues (4). PNS has been
estimated to affect ~8% of all patients with cancer (5). Among malignancies, lung cancer
[particularly small-cell lung cancer (SCLC)] is strongly associated
with PNS. Notably, the presence and severity of PNS do not
correlate with the size of the primary tumor. In some cases, PNS
may manifest before the primary cancer is diagnosed, while in
others, it may appear later in the disease course or serve as an
early indication of cancer recurrence (6).
The present review aims to provide a comprehensive
overview of endocrine PNS in lung cancer, with a particular focus
on its pathophysiology, clinical presentation, diagnosis and
management. By highlighting the importance of early recognition and
timely intervention, the current review seeks to equip healthcare
professionals with practical insights to optimize patient care.
2. Pathophysiology of endocrine PNS
Endocrine PNS represents a distinct subset of
paraneoplastic disorders in which neoplastic cells produce
bioactive substances such as hormones, peptides and cytokines,
leading to endocrine dysfunction independent of direct tumor
invasion or metastasis. These endocrine abnormalities can often
precede the diagnosis of cancer and are not necessarily associated
with the primary organ or tissue of origin. While the pathogenesis
of endocrine PNS is well-characterized in endocrine tumors, its
mechanisms in non-endocrine malignancies remain less clearly
defined. In these cases, the neoplasm may either mimic endogenous
hormone activity or interfere with normal endocrine regulatory
pathways, contributing to complex and often misleading clinical
presentations (7).
3. Diagnostic criteria
Clinicians should maintain a high index of suspicion
for endocrine PNS in both oncologic and non-oncologic patients when
there is evidence of dysregulated hormone production. Key
indicators include abnormally elevated hormone levels, a
significant concentration gradient between the hormone levels in
the venous effluent from the tumor and the arterial circulation,
the presence of bioactive and/or immunoreactive hormone in tumor
extracts, and the identification of relevant hormone mRNA within
tumor tissue. Furthermore, tumor cells may exhibit the ability to
synthesize and secrete the hormone in vitro.
Additional clinical clues reinforcing the tumor's
role in hormone production include the presence of endocrine or
metabolic disturbances in a patient with a known malignancy,
remission of these abnormalities following successful treatment
(for example, surgery, radiotherapy, or chemotherapy), and the
recurrence of endocrine dysfunction in parallel with tumor relapse
(8).
4. Syndrome of inappropriate antidiuretic
hormone (SIADH)
The first suspicion of inappropriate antidiuretic
hormone (ADH) secretion was raised by Schwartz in 1957 when he
observed euvolemic, hypotonic hyponatremia in two patients with
bronchogenic lung cancer, despite normal renal and adrenal function
(9). This hypothesis was confirmed
several years later, around 1963, when a molecule exhibiting
ADH-like activity was isolated from cancer biopsy material
(10).
ADH is synthesized in the neurohypophysis and plays
a crucial role in fluid homeostasis by binding to renal receptors
to reduce free water excretion. Under normal physiological
conditions, when plasma osmolality exceeds 280 mOsm/kg, the
pituitary gland increases ADH secretion to enhance renal water
reabsorption, maintaining fluid and osmotic balance. In patients
with SCLC, ectopic ADH production leads to impaired free-water
excretion in the distal renal tubules, resulting in hyponatremia.
SCLC cells have been found to express ADH mRNA and actively
synthesize and release ADH, leading to elevated plasma ADH levels.
However, some patients with SCLC-associated hyponatremia exhibit no
detectable plasma ADH. In such cases, the tumor may express mRNA
for atrial natriuretic peptide (ANP), which is subsequently
secreted, leading to elevated plasma ANP concentrations and an
alternative mechanism contributing to hyponatremia (11,12).
The diagnosis of SIADH is based on specific
laboratory findings, including decreased plasma osmolality (<275
mOsm/kg), inappropriately concentrated urine (>100 mOsm/kg),
euvolemic status, elevated urine sodium (>20 mEq/l), and normal
thyroid and cortisol function, with no history of diuretic use
(13). SIADH is commonly
associated with disorders affecting the central nervous system and
the lungs. Although it is most frequently linked to SCLC, it has
also been reported in malignancies of the brain, prostate, bladder,
pancreas, adrenal glands and digestive tract, as well as in
mesothelioma, thymoma, sarcoma and hematologic malignancies
(14).
Management of SIADH in cases of severe symptomatic
hyponatremia requires immediate intervention with hypertonic
saline. Administration of 100 ml of 3% NaCl, up to three doses, is
recommended to achieve an initial increase in plasma sodium (pNa)
of 4 to 6 mmol/l, alleviating symptoms and reducing intracranial
pressure. In chronic SIADH, pNa correction should be gradual to
avoid osmotic demyelination syndrome, with an increase of 4 to 8
mmol/l per day, not exceeding 10 mmol/l per day. The primary
treatment for chronic SIADH is fluid restriction to <1,000
ml/day, though this approach is effective in only ~50% of patients.
Second-line therapies include tolvaptan, urea, sodium-glucose
cotransporter 2 inhibitors, or high-dose salt tablets. The
cornerstone of SIADH management remains the treatment of the
underlying malignancy, either through oncological or surgical
interventions (15).
5. Hypercalcemia
Hypercalcemia of malignancy (HCM) is a common
metabolic complication in patients with cancer, frequently observed
in hospitalized individuals. Its prevalence varies widely, ranging
from 2-30%, with an estimated annual incidence of 1-2%. This
variability is largely dependent on the type of malignancy and the
disease stage (16,17). HCM is predominantly associated with
solid tumors, with lung cancer (particularly squamous cell
carcinoma) being a common culprit. The two primary mechanisms
underlying hypercalcemia in patients with cancer are humoral
hypercalcemia of malignancy (HCM) and bone metastases.
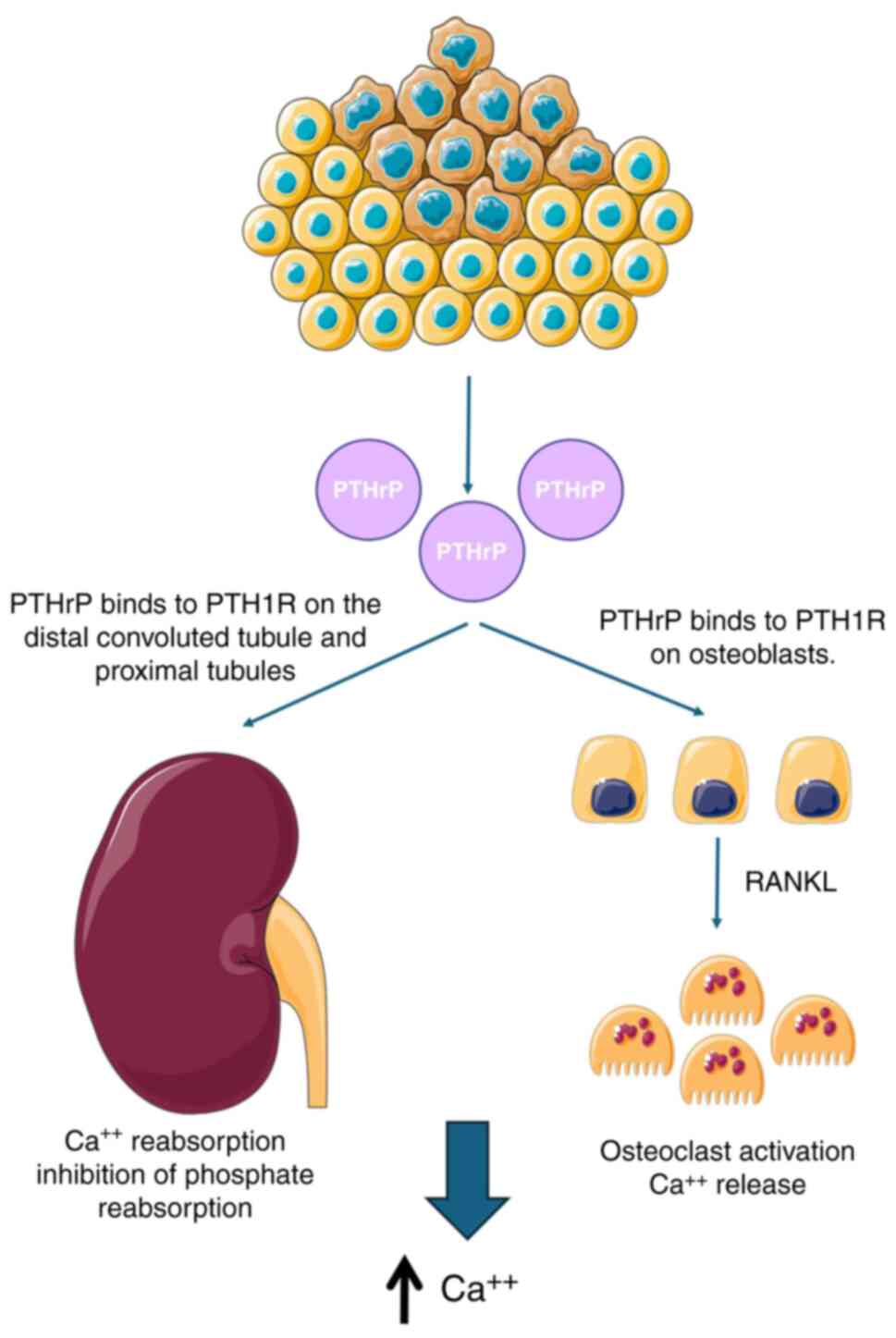
HCM, a PNS, arises when tumors secrete parathyroid
hormone (PTH)-related peptide (PTHrP), leading to increased calcium
levels. Though rare, lung cancer can also ectopically produce PTH.
Additionally, 1,25-dihydroxyvitamin D secretion is frequently
observed in hematologic malignancies. Granulocyte
colony-stimulating factor has also been implicated in HHM by
promoting osteoclast activation over time, further exacerbating
hypercalcemia (18).
Hypercalcemia disrupts normal cellular function and
manifests with a broad spectrum of symptoms. Neuromuscular effects
include confusion, fatigue and muscle weakness. Gastrointestinal
symptoms such as nausea, abdominal pain and constipation are
common. Renal complications may involve increased thirst and
urination, kidney stone formation and dehydration. Cardiovascular
manifestations include hypertension and a shortened QT interval on
ECG. Bone-related symptoms, including bone pain, osteoporosis and
osteitis fibrosa cystica, are also prevalent. The mnemonic ‘Stones,
bones, abdominal moans and psychic groans’ serves as a useful
reminder of the diverse clinical manifestations of hypercalcemia
(19).
Management of hypercalcemia begins with fluid
resuscitation using 0.9% NaCl, which reduces serum calcium levels
by expanding intravascular volume and enhancing renal calcium
excretion. Diuretics such as furosemide may be used following
volume expansion to further promote calcium elimination.
Corticosteroids such as prednisolone are particularly effective for
hypercalcemia associated with lymphoid malignancies but have
limited efficacy in solid tumors and may interfere with diagnostic
assessments. Bisphosphonates, which inhibit bone resorption, are
highly effective in managing hypercalcemia caused by multiple
myeloma and solid tumors with skeletal metastases, though they have
limited utility in HHM due to their minimal effect on
PTHrP-mediated hypercalcemia. Calcitonin provides rapid, short-term
relief by inhibiting bone resorption and increasing renal calcium
excretion, but its utility is limited by the rapid onset of
tachyphylaxis, necessitating frequent dosing (20).
Cinacalcet, a calcium-mimetic agent, effectively
lowers calcium levels by increasing the sensitivity of the
calcium-sensing receptor, making it particularly useful in cases of
humoral hypercalcemia that are resistant to conventional treatments
(21). The mechanisms of
PTHrP-induced hypercalcemia in lung cancer are demonstrated in
Fig. 1.
6. Cushing syndrome
Cushing syndrome is a well-recognized paraneoplastic
phenomenon associated primarily with SCLC and bronchial carcinoid
tumors. In total, ~5-10% of all Cushing syndrome (hypercortisolism)
cases are of paraneoplastic origin, with neuroendocrine lung tumors
accounting for ~50-60% of these cases. This condition arises due to
the ectopic production of adrenocorticotropic hormone (ACTH) by
tumor cells, leading to excessive cortisol production by the
adrenal glands. This phenomenon, known as ectopic Cushing syndrome,
results in a broad range of clinical manifestations. Unlike SIADH
and hypercalcemia, symptoms of paraneoplastic Cushing syndrome
frequently precede the diagnosis of cancer. Additionally,
recurrence of hypercortisolism may serve as an indicator of tumor
relapse.
The clinical presentation of ectopic Cushing
syndrome includes weight gain, hypertension, diabetes mellitus,
muscle weakness and characteristic skin changes such as easy
bruising and purple striae. Patients often exhibit classic
Cushingoid features, including moon facies, truncal obesity and
wide violaceous striae (22).
In lung cancer, ectopic ACTH production is most
commonly observed in SCLC, which accounts for ~10-15% of all lung
cancer cases. SCLC is a highly aggressive malignancy that is
frequently diagnosed at an advanced, metastatic stage.
Paraneoplastic ACTH secretion by SCLC results in bilateral adrenal
hyperplasia and excessive cortisol production. The biochemical
profile of ectopic Cushing syndrome typically includes elevated
plasma ACTH and cortisol levels, with a disruption of the normal
diurnal rhythm of cortisol secretion. Specific laboratory findings
associated with this condition include a serum cortisol level
>29 µg/dl, a urinary free cortisol level >47 µg/24 h, and a
midnight ACTH level >100 ng/l (23).
The management of Cushing syndrome in patients with
lung cancer is complex and requires a multidisciplinary approach.
The first-line treatment involves targeting the underlying
malignancy with chemotherapy, which can reduce tumor burden and
consequently lower ectopic hormone production. Medical therapies
such as ketoconazole, metyrapone, or mifepristone can be used to
control hypercortisolism by inhibiting adrenal steroidogenesis.
Additionally, somatostatin analogs (for example, octreotide) have
shown efficacy in reducing hormone secretion and controlling
symptoms. In refractory cases, bilateral adrenalectomy may be
considered to alleviate severe hypercortisolism; however, this is a
last-resort option due to significant surgical risks and the
complexities of postoperative adrenal insufficiency management
(4,24).
7. Carcinoid syndrome
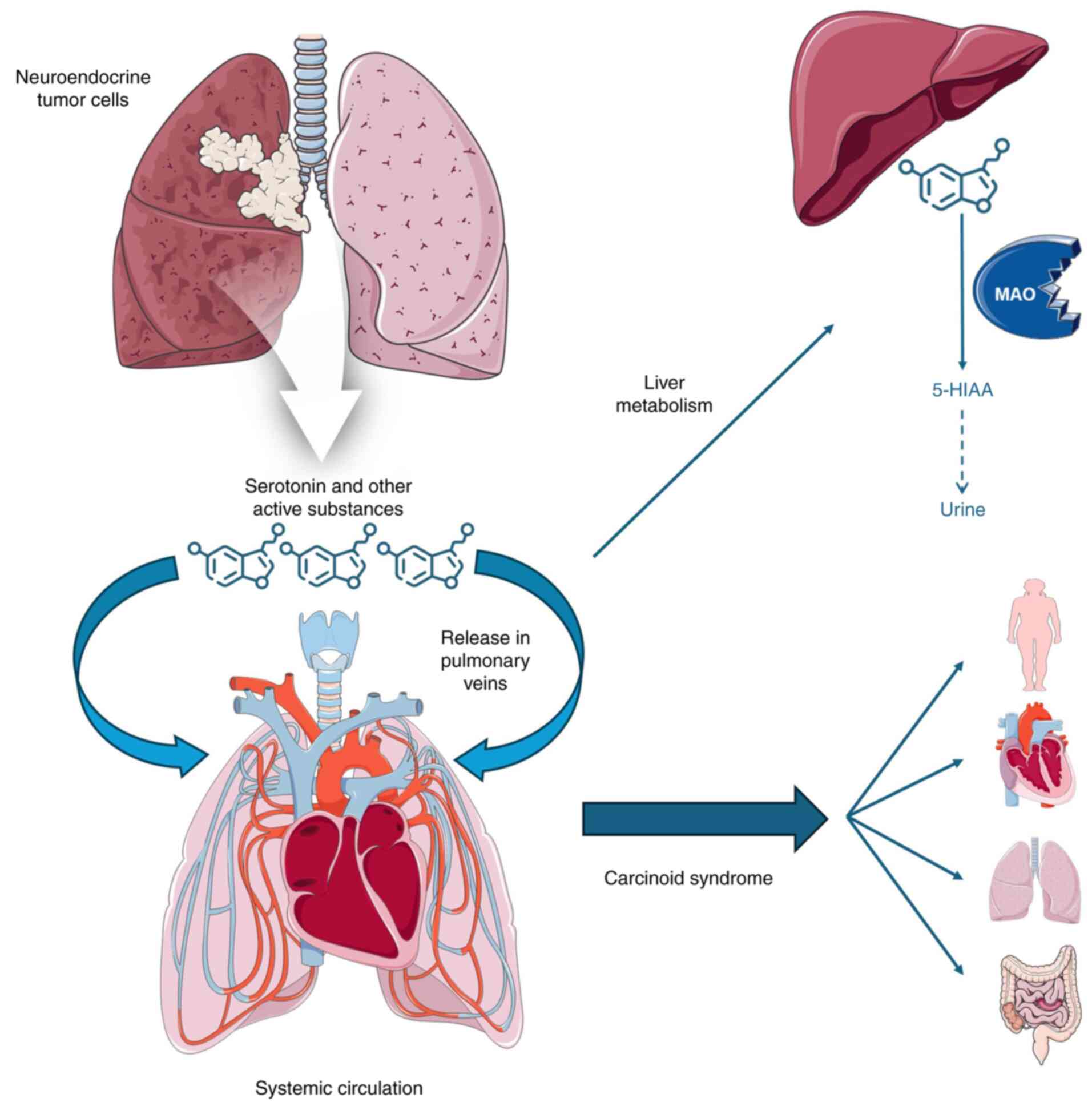
Carcinoid syndrome is a rare but significant
paraneoplastic phenomenon that can occur in patients with lung
cancer, particularly those with bronchial carcinoid tumors.
Bronchial carcinoids are a type of neuroendocrine tumor (NET) that
originates from neuroendocrine cells in the lung. These tumors can
secrete various bioactive amines and peptides, most notably
serotonin, which is responsible for the clinical manifestations of
carcinoid syndrome (25).
The classic symptoms of carcinoid syndrome include
flushing, diarrhea, bronchoconstriction, and, in advanced cases,
carcinoid heart disease characterized by fibrosis of the heart
valves. Flushing is typically episodic, affecting the face and
upper chest, and can be triggered by stress, alcohol, or specific
foods. Diarrhea can be severe and debilitating, leading to
significant weight loss and electrolyte imbalances.
Bronchoconstriction presents as wheezing and asthma-like symptoms,
often resulting in misdiagnosis as a primary pulmonary disorder
(26). In lung cancer, bronchial
carcinoids are less common than their gastrointestinal counterparts
but remain crucial to recognize due to their distinct clinical
behavior and therapeutic implications. These tumors are classified
as typical or atypical carcinoids based on histological criteria,
with atypical carcinoids exhibiting a more aggressive clinical
course and a higher likelihood of metastasis (25).
The diagnosis of carcinoid syndrome involves
detecting elevated levels of serotonin metabolites, such as
5-hydroxy-indole-acetic acid in urine, and serum chromogranin A, a
key marker for NETs. Imaging studies, including computed tomography
(CT) and somatostatin receptor scintigraphy (SRS), can help
localize the primary tumor and assess disease extent. Somatostatin
receptor imaging can identify nearly 80% of primary tumors and is
particularly useful for detecting metastatic disease (27).
The treatment of carcinoid syndrome in lung cancer
requires a combination of surgical, medical and supportive
approaches. Somatostatin analogs, such as octreotide and
lanreotide, are the first-line treatment for controlling symptoms.
These medications inhibit the release of serotonin and other
vasoactive substances, thereby managing flushing, diarrhea and
wheezing. While they are effective in symptom control, they may not
completely prevent long-term complications (27). In cases of metastatic disease,
additional therapies may be required, including peptide receptor
radionuclide therapy (PRRT), systemic chemotherapy, or targeted
therapies. Advances in genomic research have led to the exploration
of targeted therapies, particularly inhibitors of the mammalian
target of rapamycin (mTOR) pathway, such as everolimus, which have
shown promise in treating progressive NETs and controlling hormone
secretion (28). Surgical
resection of the primary tumor remains the cornerstone of treatment
and can be curative in localized disease. Removing the tumor
significantly reduces or eliminates hormone secretion, leading to
symptom resolution. In metastatic cases, debulking surgery may
still provide symptomatic relief (29). The mechanisms of carcinoid syndrome
in lung cancer are illustrated in Fig.
2.
8. Non-islet cell tumor hypoglycemia
(NITCH)
Hypoglycemia can be life-threatening and requires
accurate diagnosis. Cancer-related hypoglycemia arises through
three mechanisms: Insulin-secreting tumors (for example,
insulinomas), tumor-induced organ damage impairing glucose
regulation, and NITCH, where tumors secrete substances such as
‘big’ IGF-II, which mimic insulin (30). Insulin-like growth factor
(IGF)-2-mediated hypoglycemia, though rare (1 case/million
person-years), has significant morbidity and mortality, yet
literature on its course and treatment remains limited (31).
‘Big’-IGF-II, an abnormal IGF-2 variant (10-20 kDa),
forms a binary complex with IGFBP-3, enabling easier vascular
passage and enhanced glucose-lowering effects (32). It is commonly associated with
fibrous tumors, liver-originating tumors, hemangiopericytomas and
mesotheliomas (33). Although
reported in lung cancer cases, no strong correlation exists with
specific subtypes (30,34,35).
Diagnosing hypoglycemia in non-diabetic patients
requires evaluating medication effects, severe illness, organ
dysfunction and hormone deficiencies while also differentiating
endogenous hyperinsulinism. If a tumor is suspected, NITCH should
be considered. Diagnostic measures include serum glucose, insulin,
proinsulin, C-peptide, β-hydroxybutyrate and hypoglycemic drug
screening. If NITCH is suspected, additional IGF-I, IGF-II and
growth hormone (GH) assessments should be performed (36).
Endocrine Society guidelines suggest NITCH presents
with glucose <55 mg/dl, insulin ≤3 U/ml, proinsulin ≤5 pmol/l,
C-peptide ≤0.2 nmol/l and β-hydroxybutyrate ≤2.7 mmol/l, with no
hypoglycemic agents detected. Unlike transient hypoglycemia, GH
remains low due to negative feedback. While IGF-II levels may be
normal (275-750 ng/ml), IGF-I is often below 100 ng/ml, leading to
an elevated IGF-II/IGF-I ratio, occasionally exceeding 10:1-a
valuable NITCH marker (32).
Currently, no commercial test exists for big IGF-II,
necessitating specialized research labs (37). If the hypoglycemia etiology is
unclear, a glucagon stimulation test may help: A rise in glucose
indicates a hormonal origin, whereas an inadequate response
suggests insufficient liver glycogen reserves (38). Cross-sectional imaging of the
chest, abdomen, and pelvis is crucial for tumor identification
(32).
Immediate management of hypoglycemia involves oral
or intravenous glucose administration. While complete tumor
resection is the definitive treatment, reports of recurrence
following disease-relapse exist (32). Glucagon injections provide
short-term relief, while continuous glucagon infusions may be
effective in selective cases (39). Diazoxide and octreotide have shown
limited efficacy (40,41). Glucocorticoids remain the most
extensively studied treatment, effectively reducing IGF-II levels
and controlling hypoglycemia in 25-30% of cases. Combination
therapy with recombinant human GH may enhance outcomes, though the
absence of randomized controlled trials limits definitive treatment
guidelines (42,43).
9. Acromegaly
In 1959, Altman and Schutz described a case of
acromegaly in a patient who did not improve following pituitary
irradiation but showed significant clinical improvement after the
surgical removal of a lung carcinoid tumor. This case provided
early evidence that ectopic production of growth-hormone-releasing
hormone (GHRH) by a non-pituitary tumor can lead to acromegaly and
emphasized the importance of considering ectopic hormone secretion
when patients fail to respond to standard pituitary-directed
treatments (44). Although rare,
autonomous hormone secretion due to GHRH or, in exceptional cases,
direct secretion of GH occurs in <1% of acromegaly cases, with
bronchial carcinoid tumors being the most frequently implicated
ectopic source (45).
The diagnosis of acromegaly begins with clinical
suspicion and measurement of serum IGF-1, which is typically
elevated. To confirm the diagnosis, an Oral Glucose Tolerance Test
is performed, as glucose administration normally suppresses GH
levels, whereas in acromegaly, GH remains elevated. Imaging with
magnetic resonance imaging (MRI) of the pituitary gland is then
conducted to identify a pituitary tumor responsible for excess GH
production. Additional endocrine testing may include evaluating
other pituitary hormones and ruling out alternative conditions with
similar clinical manifestations. A definitive diagnosis is
established by integrating clinical symptoms, biochemical findings
and imaging results, which confirm persistently elevated IGF-1,
inadequate GH suppression following glucose intake, and the
presence of a pituitary tumor.
A patient with biochemically confirmed acromegaly
but a normal pituitary MRI presents a diagnostic and therapeutic
challenge, as the tumor may be too small to be detected. In such
cases, further evaluation is necessary to investigate the
possibility of ectopic hormone secretion. Additional tests should
include serum GHRH measurement and imaging techniques such as SRS
(octreoscan), which has a reported sensitivity of 97%, as well as
thoracic and abdominal imaging to identify potential ectopic
sources of hormone production (46,47).
The preferred treatment approach for ectopic
acromegaly is surgical resection of the tumor, as it offers the
highest chance of cure and symptom resolution. Whenever possible,
complete surgical removal of the ectopic source should be pursued
to achieve optimal clinical outcomes (48).
10. Zollinger-Ellison syndrome
Zollinger-Ellison syndrome is a rare paraneoplastic
disorder caused by the ectopic secretion of gastrin, typically by
gastrin-producing NETs known as gastrinomas. While the majority of
gastrinomas are located in the abdomen, particularly in the
pancreas and duodenum, ectopic gastrin production has been reported
in association with lung cancer, primarily in NETs such as atypical
carcinoids and SCLC (49).
The hallmark of Zollinger-Ellison syndrome is
hypergastrinemia, which leads to excessive gastric acid secretion
and severe peptic ulcer disease. Patients commonly present with
abdominal pain, chronic diarrhea, gastroesophageal reflux disease
and weight loss. The excessive acid production results in multiple
and recurrent peptic ulcers that are often resistant to standard
medical therapy. Chronic diarrhea in Zollinger-Ellison syndrome is
typically caused by the direct effects of excess gastric acid on
the intestinal mucosa, leading to malabsorption (50).
In the setting of lung cancer, diagnosing ectopic
gastrin production can be challenging due to the overlap of
symptoms with other gastrointestinal and PNSs. The diagnosis is
based on clinical presentation, biochemical testing and imaging
studies. A high degree of suspicion is warranted in patients with
recurrent, severe peptic ulcers that fail to respond to
conventional treatment. Biochemical confirmation involves measuring
elevated fasting serum gastrin levels, which indicate the presence
of gastrin-secreting tumors. The secretin stimulation test is often
used to confirm the diagnosis; an abnormal rise in gastrin levels
following secretin administration strongly suggests
Zollinger-Ellison syndrome. Imaging studies such as CT, MRI and SRS
help localize the gastrin-producing tumors, which are most commonly
found in the pancreas or duodenum. Endoscopic ultrasound and biopsy
can further aid in precise localization and tissue diagnosis.
Accurate identification of the condition is essential for effective
management, which typically includes both pharmacologic control of
gastric acid secretion and surgical intervention (51,52).
The treatment of Zollinger-Ellison syndrome
associated with lung cancer requires a comprehensive approach that
targets both the excessive gastric acid secretion and the
underlying malignancy. Management usually begins with high-dose
proton pump inhibitors, such as omeprazole or esomeprazole, to
control the severe acid hypersecretion, often at doses higher than
those used for standard peptic ulcer disease (53,54).
In cases where the gastrinoma is localized, surgical resection is
considered; however, if the tumors are metastatic or inoperable,
somatostatin analogs such as octreotide or lanreotide can help
reduce gastrin secretion (55).
For patients with metastatic disease or those who are not surgical
candidates, somatostatin analogs remain an essential therapeutic
option (56,57).
Managing Zollinger-Ellison syndrome in the context
of lung cancer is particularly complex and requires a
multidisciplinary approach involving oncologists,
gastroenterologists and surgeons. Treatments directed at the lung
cancer, including chemotherapy, radiation therapy, or targeted
therapies, may influence the progression of gastrin-secreting
tumors, especially if they are part of a broader NET syndrome.
Emerging therapies such as everolimus, which targets tumors with
mTOR pathway activation, as well as immunotherapy and PRRT, are
being explored as potential treatment strategies for metastatic
disease (58-60).
11. Gynecomastia
Gynecomastia, the enlargement of male breast tissue
due to hormonal imbalance, can occur as a PNS in various cancers,
affecting ~2.4% of patients with lung cancer (61,62).
The therapeutic approach to gynecomastia depends on identifying the
underlying cause. The initial evaluation should exclude common
pathological factors, including medication use, chronic liver or
kidney disease, or androgen therapy in men with hypogonadism
(63). If no apparent cause is
found, pathological gynecomastia should be considered.
The first step in hormonal assessment involves
measuring levels of luteinizing hormone, testosterone,
dehydroepiandrosterone sulfate, thyroid function, estradiol, and
the beta subunit of human chorionic gonadotropin (β-hCG) (63). When β-hCG levels are elevated,
further investigation is warranted to identify potential testicular
tumors, which are the most common cause, as well as extragonadal
germ cell tumors or hCG-secreting trophoblastic tumors. Although
rare, non-trophoblastic tumors, including those of the lung,
kidney, liver and stomach, can also cause gynecomastia via ectopic
hCG production. Among these, lung cancer is the most frequently
implicated (64-66).
Only a few cases of gynecomastia associated with
lung cancer have been documented in the literature, predominantly
in non-small cell lung cancer, particularly adenocarcinoma, and
typically presenting as bilateral gynecomastia (67). The underlying mechanism often
involves ectopic β-hCG production by tumor cells, which has been
confirmed using immunohistochemical methods (7,68).
The treatment of gynecomastia in the context of lung
cancer is directly tied to managing the underlying malignancy,
typically through chemotherapy and/or surgical resection, depending
on the type and stage of the tumor (61,69).
12. Hyperprolactinemia
Ectopic production of prolactin associated with SCLC
remains a rare manifestation, with only a few cases documented in
the literature to date (70-72).
The clinical presentation of hyperprolactinemia results from
suppression of the hypothalamic-pituitary-gonadal axis and varies
depending on the patient's sex and age.
In women, hyperprolactinemia may present with
oligomenorrhea, amenorrhea, galactorrhea, decreased libido,
infertility, and, in prolonged cases, possible osteopenia. Chronic
hyperprolactinemia can also lead to hyperandrogenism, causing
hirsutism and acne, potentially due to increased secretion of
adrenal dehydroepiandrosterone sulfate and reduced levels of sex
hormone-binding globulin, resulting in elevated free testosterone
levels.
In men, hyperprolactinemia can manifest as erectile
dysfunction, decreased libido, infertility, gynecomastia, reduced
bone mass, and, in rare cases, galactorrhea. Over time, men may
also experience fatigue, reduced muscle mass and an increased risk
of osteopenia (73).
Persistent hyperprolactinemia, after excluding
common causes such as non-fasting blood samples, excessive
exercise, recent medication use, chest wall trauma or surgery,
renal disease, cirrhosis, or recent seizures, may indicate a
paraneoplastic etiology. In such cases, further imaging evaluation
is warranted to investigate underlying neoplasms (74).
The treatment of hyperprolactinemia includes
dopamine receptor agonists, such as cabergoline or bromocriptine,
which effectively suppress prolactin secretion. In cases where
medical therapy fails, surgical removal of the tumor may be
necessary (75).
Emerging research suggests that aberrant prolactin
activation could serve as a biomarker for lung cancer
aggressiveness, with elevated prolactin levels correlating with
rapid disease progression. Additionally, histone deacetylase
inhibitors have been proposed as a potential therapeutic strategy,
highlighting the role of prolactin in both diagnosis and treatment
(71). The endocrine PNSs in lung
cancer are summarized in Table
I.
 | Table IOverview of endocrine paraneoplastic
syndromes in lung cancer. |
Table I
Overview of endocrine paraneoplastic
syndromes in lung cancer.
| Syndrome | Primary
hormone/chemical involved |
Pathophysiology | Clinical
presentation | Diagnosis | Management |
|---|
| Syndrome of
inappropriate ADH | ADH | Ectopic production
of ADH leading to impaired water excretion and hyponatremia | Hyponatremia,
headache, nausea, confusion, seizures | Low plasma
osmolality, concentrated urine, euvolemia, and elevated urine
sodium | Fluid restriction,
hypertonic saline, tolvaptan, and addressing underlying
malignancy |
| Cushing
syndrome | ACTH | Ectopic ACTH
production causing excess cortisol secretion and
hypercortisolism | Weight gain,
hypertension, diabetes, muscle weakness, moon face | Elevated ACTH and
cortisol, loss of cortisol diurnal rhythm | Chemotherapy,
metyrapone, ketoconazole, octreotide, or bilateral
adrenalectomy |
| Hypercalcemia | PTHrP | Secretion of PTHrP
or osteolytic activity of bone metastases, leading to increased
calcium levels | Bone pain,
confusion, muscle weakness, constipation | Elevated serum
calcium, suppressed PTH, and increased PTHrP levels | Hydration,
bisphosphonates, calcitonin, cinacalcet, and addressing
malignancy |
| Carcinoid
syndrome | Serotonin | Secretion of
serotonin and other bioactive substances by neuroendocrine
tumors | Flushing, diarrhea,
bronchospasm, heart valve fibrosis | Elevated urinary
5-hydroxy-indole-acetic acid, elevated plasma chromogranin A,
somatostatin receptor scintigraphy | Octreotide,
lanreotide, peptide receptor radionuclide therapy, surgical
resection |
| Non-islet cell
Tumor Hypoglycemia (NITCH) | IGF-II | Ectopic production
of ‘big’ IGF-II mimicking insulin and lowering glucose levels | Hypoglycemia,
confusion, sweating, weakness | Low glucose, low
insulin, low C-peptide, elevated IGF-II/IGF-I ratio | Glucocorticoids,
surgery, glucagon infusion, addressing underlying tumor |
| Acromegaly | GHRH | Ectopic GHRH
production leading to excess growth hormone and IGF-1 | Enlarged
hands/feet, coarse facial features, headache | Elevated IGF-1,
lack of GH suppression after glucose load, pituitary magnetic
resonance imaging | Surgery,
somatostatin analogs, GH receptor antagonists, addressing ectopic
source |
| Zollinger-Ellison
syndrome | Gastrin | Ectopic production
of gastrin causing hyper-secretion of gastric acid | Recurrent peptic
ulcers, GERD, diarrhea, abdominal pain | Elevated fasting
gastrin, secretin stimulation test | Proton pump
inhibitors, somatostatin analogs, surgery, chemotherapy for
underlying malignancy |
| Gynecomastia | β-HCG | Ectopic β-HCG
secretion leading to an imbalance in testosterone and estrogen | Breast enlargement,
tenderness | Elevated β-HCG,
imaging to rule out testicular and other tumors | Treat underlying
malignancy, consider hormonal therapy |
|
Hyperprolactinemia | Prolactin | Ectopic production
of prolactin leading to suppression of the
hypothalamic-pituitary-gonadal axis | Galactorrhea,
amenorrhea, infertility, decreased libido | Elevated prolactin,
imaging to exclude pituitary source | Dopamine agonists
(cabergoline, bromocriptine), surgery for non-responsive cases |
13. Conclusions
Endocrine PNSs in lung cancer are rare but
clinically significant, often serving as early indicators of
malignancy or disease recurrence. The present review highlights the
pathophysiology, diagnostic challenges and management strategies of
these syndromes, emphasizing the importance of early recognition
and a multidisciplinary approach. Given their impact on patient
outcomes, heightened clinical vigilance and systematic evaluation
are essentisal to improving early diagnosis and optimizing
treatment.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
MEK and VEG conceptualized the study. MEK, VEG, DAS,
DA, MPY and MS made a substantial contribution to data
interpretation and analysis and wrote and prepared the draft of the
manuscript. MEK and VEG analyzed the data and provided critical
revisions. All authors contributed to manuscript revision, read and
approved the final version of the manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, artificial
intelligence tools were used to improve the readability and
language of the manuscript, and subsequently, the authors revised
and edited the content produced by the artificial intelligence
tools as necessary, taking full responsibility for the ultimate
content of the present manuscript.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ferlay J, Colombet M, Soerjomataram I,
Parkin DM, Piñeros M, Znaor A and Bray F: Cancer statistics for the
year 2020: An overview. Int J Cancer: Apr 5, 2021 (Epub ahead of
print).
|
|
3
|
Soomro Z, Youssef M, Yust-Katz S, Jalali
A, Patel AJ and Mandel J: Paraneoplastic syndromes in small cell
lung cancer. J Thorac Dis. 12:6253–6263. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Henry K: Paraneoplastic syndromes:
Definitions, classification, pathophysiology and principles of
treatment. Semin Diagn Pathol. 36:204–210. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Thapa B, Mahendraker N and Ramphul K:
Paraneoplastic syndromes. [Updated 2023 Mar 31]. In: StatPearls
[Internet]. Treasure Island (FL): StatPearls Publishing, 2025.
|
|
6
|
Anwar A, Jafri F, Ashraf S, Jafri MAS and
Fanucchi M: Paraneoplastic syndromes in lung cancer and their
management. Ann Transl Med. 7(359)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dimitriadis GK, Angelousi A, Weickert MO,
Randeva HS, Kaltsas G and Grossman A: Paraneoplastic endocrine
syndromes. Endocr Relat Cancer. 24:R173–R190. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Onyema MC, Drakou EE and Dimitriadis GK:
Endocrine abnormality in paraneoplastic syndrome. Best Pract Res
Clin Endocrinol Metab. 36(101621)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Schwartz WB, Bennett W, Curelop S and
Bartter FC: A syndrome of renal sodium loss and hyponatremia
probably resulting from inappropriate secretion of antidiuretic
hormone. Am J Med. 23:529–542. 1975.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Amatruda TT Jr, Mulrow PJ, Gallagher JC
and Sawyer WH: Carcinoma of the lung with inappropriate
antidiuresis. Demonstration of antidiuretic-hormone-like activity
in tumor extract. N Engl J Med. 269:544–549. 1963.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Adrogué HJ, Tucker BM and Madias NE:
Diagnosis and management of hyponatremia: A review. JAMA.
328:280–291. 2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sun NH, Wang SH, Liu JN, Liu A, Gong WJ,
Liu Y, Sun P and Li H: The productions of atrial natriuretic
peptide and arginine vasopressin in small cell lung cancer with
brain metastases and their associations with hyponatremia. Eur Rev
Med Pharmacol Sci. 21:4104–4112. 2017.PubMed/NCBI
|
|
13
|
Ghosal A, Qadeer HA, Nekkanti SK, Pradhan
P, Okoye C and Waqar D: A conspectus of euvolemic hyponatremia, its
various etiologies, and treatment modalities: A comprehensive
review of the literature. Cureus. 15(e43390)2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sørensen JB, Andersen MK and Hansen HH:
Syndrome of inappropriate secretion of antidiuretic hormone (SIADH)
in malignant disease. J Intern Med. 238:97–110. 1995.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Warren AM, Grossmann M, Christ-Crain M and
Russell N: Syndrome of inappropriate antidiuresis: From
pathophysiology to management. Endocr Rev. 44:819–861.
2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Mc Donald D, Drake MT and Crowley RK:
Treatment of hypercalcaemia of malignancy in adults. Clin Med
(Lond). 23:503–507. 2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Feldenzer KL and Sarno J: Hypercalcemia of
malignancy. J Adv Pract Oncol. 9:496–504. 2018.PubMed/NCBI
|
|
18
|
Giannetta E, Sesti F, Modica R,
Grossrubatscher EM, Ragni A, Zanata I, Colao A and Faggiano A: What
lies behind paraneoplastic hypercalcemia secondary to
well-differentiated neuroendocrine neoplasms? A systematic review
of the literature. J Pers Med. 12(1553)2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Goltzman D: Approach to hypercalcemia.
[Updated 2023 Apr 17]. In: Feingold KR, Anawalt B, Blackman MR,
Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K,
Hofland L et al (eds). In: Endotext [Internet]. South
Dartmouth (MA): MDText.com, Inc., 2000.
|
|
20
|
Almuradova E and Cicin I: Cancer-related
hypercalcemia and potential treatments. Front Endocrinol
(Lausanne). 14(1039490)2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
O'Callaghan S and Yau H: Treatment of
malignancy-associated hypercalcemia with cinacalcet: A paradigm
shift. Endocr Connect. 10:R13–R24. 2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li WY, Liu XD, Li WN, Dong SY, Qu XH, Gong
SL, Shao MR and Zhang L: Paraneoplastic Cushing's syndrome
associated with bronchopulmonary carcinoid tumor in youth: A case
report and review of the literature. Oncol Lett. 12:69–72.
2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Galli J and Greenlee J: Paraneoplastic
diseases of the central nervous system. F1000Res: F1000 Faculty
Rev-167, 2020.
|
|
24
|
Castinetti F: Medical management of
Cushing's disease: When and how? J Neuroendocrinol.
34(e13120)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Granberg D, Juhlin CC, Falhammar H and
Hedayati E: Lung carcinoids: A comprehensive review for clinicians.
Cancers (Basel). 15(5440)2023.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hofland J, Herrera-Martinez AD, Zandee WT
and de Herder WW: Management of carcinoid syndrome: A systematic
review and meta-analysis. Endocr Relat Cancer. 26:R145–R156.
2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Jacob N, Dasharathy SS, Bui V, Benhammou
JN, Grody WW, Singh RR and Pisegna JR: Generalized cytokine
increase in the setting of a multisystem clinical disorder and
carcinoid syndrome associated with a novel NLRP12 variant. Dig Dis
Sci. 64:2140–2146. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mota JM, Sousa LG and Riechelmann RP:
Complications from carcinoid syndrome: review of the current
evidence. Ecancermedicalscience. 10(662)2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Caplin ME, Baudin E, Ferolla P, Filosso P,
Garcia-Yuste M, Lim E, Oberg K, Pelosi G, Perren A, Rossi RE, et
al: Pulmonary neuroendocrine (carcinoid) tumors: European
neuroendocrine tumor society expert consensus and recommendations
for best practice for typical and atypical pulmonary carcinoids.
Ann Oncol. 26:1604–1620. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Legare TB, Hamilton O, Dhannoon S and Ali
S: Non-islet cell tumor hypoglycemia: A rare cause of hypoglycemia
in pulmonary sarcomatoid cancer. Cureus. 9(e1972)2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yamasaki H, Itawaki A, Morita M, Miyake H,
Yamamoto M, Sonoyama H, Tanaka S, Notsu M, Yamauchi M, Fujii Y, et
al: A case of insulin-like growth factor 2-producing
gastrointestinal stromal tumor with severe hypoglycemia. BMC Endocr
Disord. 20(60)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Bodnar TW, Acevedo MJ and Pietropaolo M:
Management of non-islet-cell tumor hypoglycemia: A clinical review.
J Clin Endocrinol Metab. 99:713–722. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ata F, Choudry H, Khan AA, Anum Khamees I,
Al-Sadi A, Mohamed A, Malkawi L and Aljaloudi E: A systematic
review of literature on Insulin-like growth factor-2-mediated
hypoglycaemia in non-islet cell tumours. Endocrinol Diabetes Metab.
7(e00471)2024.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Nauck MA, Reinecke M, Perren A, Frystyk J,
Berishvili G, Zwimpfer C, Figge AM, Flyvbjerg A, Lankisch PG, Blum
WF, et al: Hypoglycemia due to paraneoplastic secretion of
insulin-like growth factor-I in a patient with metastasizing
large-cell carcinoma of the lung. J Clin Endocrinol Metab.
92:1600–1605. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Gherbon A, Frandes M, Nicula M, Avram A
and Timar R: Igf-2 induced hypoglycemia associated with lung
sarcoma. Acta Endocrinol (Buchar). 18:232–237. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
McCall AL, Lieb DC, Gianchandani R,
MacMaster H, Maynard GA, Murad MH, Seaquist E, Wolfsdorf JI, Wright
RF and Wiercioch W: Management of individuals with diabetes at high
risk for hypoglycemia: An endocrine society clinical practice
guideline. J Clin Endocrinol Metab. 108:529–562. 2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wei LF, Weng XF, Huang XC, Peng YH, Guo HP
and Xu YW: IGFBP2 in cancer: Pathological role and clinical
significance (Review). Oncol Rep. 45:427–438. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Palani G, Stortz E and Moheet A: Clinical
presentation and diagnostic approach to hypoglycemia in adults
without diabetes mellitus. Endocr Pract. 29:286–294.
2023.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Isaacs D, Clements J, Turco N and Hartman
R: Glucagon: Its evolving role in the management of hypoglycemia.
Pharmacotherapy. 41:623–633. 2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Abell SK, Teng J, Dowling A, Hofman MS,
MacIsaac RJ and Sachithanandan N: Prolonged life-threatening
hypoglycaemia following dose escalation of octreotide LAR in a
patient with malignant polysecreting pancreatic neuroendocrine
tumour. Endocrinol Diabetes Metab Case Rep.
2015(140097)2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Iglesias P and Díez JJ: Management of
endocrine disease: A clinical update on tumor-induced hypoglycemia.
Eur J Endocrinol. 170:R147–R157. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
He D, Gong H, Pan J, Zhu F, Jiang X and Su
H: Recurrent Non-islet cell tumor hypoglycemia secondary to
hepatocellular carcinoma: Case report and literature review. Z
Gastroenterol. 62:752–758. 2024.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Karamanolis NN, Kounatidis D, Vallianou
NG, Alexandropoulos K, Kovlakidi E, Kaparou P, Karampela I,
Stratigou T and Dalamaga M: Paraneoplastic hypoglycemia: An
overview for optimal clinical guidance. Metabol Open.
23(100305)2024.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Ghazi AA, Amirbaigloo A, Dezfooli AA,
Saadat N, Ghazi S, Pourafkari M, Tirgari F, Dhall D, Bannykh S,
Melmed S and Cooper O: Ectopic acromegaly due to growth hormone
releasing hormone. Endocrine. 43:293–302. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sohail S, Shafiq W, Sajjad K, Azmat U and
Naveed MA: Ectopic acromegaly secondary to bronchial tumour: A case
report of rare occurrence. J Cancer Allied Spec.
7(e397)2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Katznelson L, Laws ER Jr, Melmed S,
Molitch ME, Murad MH, Utz A and Wass JA: Endocrine Society.
Acromegaly: An endocrine society clinical practice guideline. J
Clin Endocrinol Metab. 99:3933–3951. 2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Elenius H and Nieman LK: Recognition and
management of ectopic ACTH secreting tumors. J Endocr Soc.
9(bvae194)2025.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zendran I, Gut G, Kałużny M, Zawadzka K
and Bolanowski M: Acromegaly caused by ectopic growth hormone
releasing hormone secretion: A review. Front Endocrinol (Lausanne).
13(867965)2022.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Rosiek V, Kogut A and Kos-Kudła B:
Pro-gastrin-releasing peptide as a biomarker in lung neuroendocrine
neoplasm. Cancers (Basel). 15(3282)2023.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Rossi RE, Elvevi A, Citterio D, Coppa J,
Invernizzi P, Mazzaferro V and Massironi S: Gastrinoma and
Zollinger Ellison syndrome: A roadmap for the management between
new and old therapies. World J Gastroenterol. 27:5890–5907.
2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Metz DC and Jensen RT: Zollinger-Ellison
syndrome. Yamada's Textbook of Gastroenterology, pp977-1003,
2022.
|
|
52
|
Cho MS and Kasi A: Zollinger-Ellison
syndrome. In: StatPearls. StatPearls Publishing, Treasure Island,
FL, 2025. https://www.ncbi.nlm.nih.gov/books/NBK537344/.
|
|
53
|
Scarpignato C, Gatta L, Zullo A and
Blandizzi C: SIF-AIGO-FIMMG Group; Italian Society of Pharmacology,
the Italian Association of Hospital Gastroenterologists, the
Italian Federation of General Practitioners. Effective and safe
proton pump inhibitor therapy in acid-related diseases-A position
paper addressing benefits and potential harms of acid suppression.
BMC Med. 14(179)2016.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Jin XF, Spampatti MP, Spitzweg C and
Auernhammer CJ: Supportive therapy in gastroenteropancreatic
neuroendocrine tumors: Often forgotten but important. Rev Endocr
Metab Disord. 19:145–158. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Shao QQ, Zhao BB, Dong LB, Cao HT and Wang
WB: Surgical management of Zollinger-Ellison syndrome: Classical
considerations and current controversies. World J Gastroenterol.
25:4673–4681. 2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Verdugo MA, Soto RM, Salgado DL, Medrano
ME and León WA: Clinical and surgical management of
Zollinger-Ellison syndrome: A literature review. Int J Med Sci Clin
Res Stud. 2:1508–1511. 2022.
|
|
57
|
Guarnotta V, Martini C, Davì MV, Pizza G,
Colao A and Faggiano A: NIKE group. The Zollinger-Ellison syndrome:
Is there a role for somatostatin analogues in the treatment of the
gastrinoma? Endocrine. 60:15–27. 2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Ito T, Ramos-Alvarez I and Jensen RT:
Successful lifetime/long-term medical treatment of acid
hypersecretion in Zollinger-Ellison syndrome (ZES): Myth or fact?
Insights from an analysis of results of NIH long-term prospective
studies of ZES. Cancers (Basel). 15(1377)2023.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Metelski J, Metelska A, Sereda D, Nieścior
H and Szwed M: Zollinger-Ellison syndrome-review. J Educ Health
Sport. 12:523–532. 2022.
|
|
60
|
Yao JC, Fazio N, Singh S, Buzzoni R,
Carnaghi C, Wolin E, Tomasek J, Raderer M, Lahner H, Voi M, et al:
Everolimus for the treatment of advanced, non-functional
neuroendocrine tumours of the lung or gastrointestinal tract
(RADIANT-4): A randomised, placebo-controlled, phase 3 study.
Lancet. 387:968–977. 2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Swerdloff RS and Ng JCM: Gynecomastia:
Etiology, diagnosis, and treatment. [Updated 2023 Jan 6]. In:
Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E,
de Herder WW, Dhatariya K, Dungan K, Hofland J et al (eds).
Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc,
2000.
|
|
62
|
Gowda H, Phatak SV, Manoj M and Ghanta P:
Paraneoplastic syndrome presenting with unilateral gynecomastia:
Ultrasonography, mammography, and strain elastography evaluation. J
Datta Meghe Inst Med Sci Univ. 17:421–423. 2022.
|
|
63
|
Cuhaci N, Polat SB, Evranos B, Ersoy R and
Cakir B: Gynecomastia: Clinical evaluation and management. Indian J
Endocrinol Metab. 18:150–158. 2014.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Peng J, Lv S, Liu L, Feng S and Xing N:
Lung neoplasm mimicking as ectopic pregnancy due to paraneoplastic
secretion of human chorionic gonadotropin: A case report and
literature review. Arch Gynecol Obstet. 303:607–614.
2021.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Matsukuma S, Obara K, Utsumi Y, Miyai K,
Takeo H, Oshika Y and Sensaki K: Focal positivity of
immunohistochemical markers for pulmonary squamous cell carcinoma
in primary pulmonary choriocarcinoma: A histopathological study.
Oncol Lett. 16:7256–7263. 2018.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Yang RH, Ting CH and Chu YK: Cannonball
lung metastases as a presenting feature of ectopic hCG expression.
J Oncol Sci. 2:58–62. 2016.
|
|
67
|
Sanjan G, Banerjee S, Dua R and Sharma P:
A lung cancer patient presenting with gynecomastia: An uncommon
paraneoplastic syndrome. Cureus. 16(e54758)2024.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Singh J, Swaminathan U, Sharada P, Alur
JB, Chowdhury P and Mrinal U: Estimation of expression of
beta-human chorionic gonadotropin levels through progression of
disease from normal to epithelial dysplasia to malignancy. J Oral
Maxillofac Pathol. 23:108–113. 2019.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Lazopoulos A, Krimiotis D, Schizas NC,
Rallis T, Gogakos AS, Chatzinikolaou F, Tsiouda T, Zarogoulidis P,
Sarafis P, Kamparoudi P, et al: Galactorrhea, mastodynia and
gynecomastia as the first manifestation of lung adenocarcinoma. A
case report. Respir Med Case Rep. 26:146–149. 2018.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Yao H, Rui W, Zhang Y, Liu Y, Lin S, Tang
H, Zhao W and Wu Z: Prolactin-secreting lung adenocarcinoma
metastatic to the pituitary mimicking a prolactinoma: A case
report. Neurosurgery. 85:E773–E778. 2019.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Le Bescont A, Vitte AL, Debernardi A,
Curtet S, Buchou T, Vayr J, de Reyniès A, Ito A, Guardiola P,
Brambilla C, et al: Receptor-independent ectopic activity of
prolactin predicts aggressive lung tumors and indicates HDACi-based
therapeutic strategies. Antioxid Redox Signal. 23:1–14.
2015.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Legostaev VM, Ayrapetova TG, Kit OI,
Frantsiyants EM, Bandovkina VA, Babenkov OM, Chubaryan AV, Ezhova M
and Duritskiy MN: Ectopic prolactin production by lung cancer cells
as an indicator of aggressive nature of tumors. J Clin Oncol. 37
(15 Suppl)(e20021)2019.
|
|
73
|
Chen TY, Lee CH, Yang MY, Shen CC, Yang
YP, Chien Y, Huang YF, Lai CM and Cheng WY: Treatment of
hyperprolactinemia: A single-institute experience. J Chin Med
Assoc. 84:1019–1022. 2021.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Melmed S, Casanueva FF, Hoffman AR,
Kleinberg DL, Montori VM, Schlechte JA and Wass JA: Endocrine
Society. Diagnosis and treatment of hyperprolactinemia: An
endocrine society clinical practice guideline. J Clin Endocrinol
Metab. 96:273–288. 2011.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Barry L, Pather S, Gargya A and Marren A:
Prolactin-secreting leiomyoma causing hyperprolactinaemia
unresponsive to dopamine agonist therapy and resolution following
myomectomy. Case Rep Endocrinol. 2021(5553187)2021.PubMed/NCBI View Article : Google Scholar
|