Introduction
cGMP-dependent protein kinase (PKG) is a
serine/threonine kinase. Mammalian cells have two PKG genes, which
encode the cytosolic PKG I and membrane-bound PKG II (1,2). PKG
I controls multiple physiological functions, including
proliferation, apoptosis, migration and the differentiation of
several cell types (3,4), and has been identified as a tumor
suppressor (5). A growing body of
evidence indicates that PKG II plays a role in inhibiting tumor
cell proliferation (6–8). However, more evidence is required to
identify PKG II as a tumor suppression factor. Previously, it has
been demonstrated that PKG II has a suppressive effect on
EGF-induced cell proliferation and cell cycle progression through
inhibiting the MAPK/ERK-mediated signal transduction pathway
(8,9). The current study provides direct
evidence linking increased PKG activity to the inhibition of
EGF-induced activation of the transcription factors AP-1 and Elk1
in gastric cancer cells.
AP-1 is a transcription factor and has been shown to
play a critical role in promoting carcinogenesis (10). AP-1 proteins consist of homodimers
of Jun family members (c-Jun, JunB, JunD) or heterodimers of
members of the Jun and Fos families (c-Fos, FosB, Fra-1 and Fra-2)
(11,12). It is well established that the
modulation of AP-1 activity is critical in the control of cell
proliferation and apoptosis (12–14).
Several studies have shown that AP-1 is activated by three MAPK
pathways (ERK, JNK and p38MAPK) in a selective manner and serves as
a common integrator of MAPK signaling to specific target gene
expression (12,15). However, the exact mechanism of EGF
treatment on AP-1 activation and the relative roles of various
MAPKs in these processes are diverse. Whether activated PKG II also
modulates the members of AP-1 family expression in response to the
stimulation of EGF in gastric cancer cells is unclear.
The transcription factor Elk1 is activated by a
variety of extracellular signals via the MAPK phosphorylation
cascades (16). Activated Elk1
binds to the serum response element of a variety of genes that
regulate cell growth, including c-Fos (17,18).
The molecular mechanism that results in Elk1 playing dominant roles
in regulating proliferation in gastric cancer has not yet been
elucidated.
We have previously demonstrated a potential role of
PKG II in regulating cancer cell proliferation, which is likely to
be associated with the activities of the MAPK-mediated pathway;
however, the downstream steps involved in this process remain to be
determined. The current study investigated the effect of PKG II on
the EGF-induced activation of AP-1 and Elk1 and the possible
association between the effect and the activities of MAPKs,
including ERK, JNK and p38MAPK, in order to provide further
evidence for indentifying PKG II as a cancer inhibitory factor.
Materials and methods
Cell lines and reagents
The human gastric cancer cell line BGC-823 was
provided by the Institute of Cell Biology (Shanghai, China).
Adenoviral vectors encoding the cDNA β-galactosidase (pAd-LacZ) and
PKG II (pAd-PKG II) were gifts from Dr Gerry Boss and Dr Renate
Pilz, University of California, San Diego, CA, USA. Dulbecco’s
modified Eagle’s medium (DMEM) and newborn calf serum (NBCS) were
purchased from Gibco (Grand Island, NY, USA). Antibodies against
JNK, phospho-JNK (Thr183/Tyr185), p-c-Jun (Ser73) and p-c-Jun
(Ser63) were obtained from Bioworld Technology (St. Louis Park, MN,
USA). Antibodies against Lamin A, β-actin, c-Fos and c-Jun were
purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
Antibodies against phospho-Elk1 (Ser383), p38MAPK, phospho-p38MAPK,
ERK and phospho-ERK were from Cell Signaling Technology (Danvers,
MA, USA). The horseradish peroxidase (HRP)-conjugated secondary
antibodies were from Jackson ImmunoResearch Laboratories (West
Grove, PA, USA). The cellular permeable cGMP analog 8-pCPT-cGMP was
from Calbiochem (San Diego, CA, USA). PD98059, SP600125, SB203580
and EGF were from Sigma (St. Louis, MO, USA).
Electrochemiluminescence (ECL) reagents were from Millipore
(Billerica, MA, USA). All other reagents used were of analytical
grade.
Cell culture and infection with
adenoviral vectors
BGC-823 cells were cultured in DMEM supplied with
10% NBCS and maintained at 37°C in a humidified incubator with 95%
air and 5% CO2. The medium was changed every two days
and the cells were subcultured at confluence. On the day prior to
infection with adenovirus, cells were freshly planted at 70–80%
confluence and infection was performed as reported previously
(21). All experiments were
approved by the ethics committee of Jiangsu University.
Luciferase reporter experiments
BGC-823 cells were subcultured in 24-well plates in
triplicate 24 h prior to transfection. pAP-1-luciferase or
pElk1-luciferase was co-transfected with β-galactosidase reporter
plasmids (as an internal control) using LipofectamineTM
2000 in an antibiotic-free medium. Following 18 h incubation, the
cells were treated as designated. The cells were then lysed in
lysis buffer (25 mM Tris-phosphate, pH 7.8, 2 mM EDTA, 1% Triton
X-100 and 10% glycerol) and reporter gene activity was determined
with the Promega luciferase assay system using LUMI-ONE
luminometer. β-galactosidase activity was measured using the
Promega β-galactosidase enzyme assay system and used for
normalization of transfection efficiency.
Nuclear protein preparation
According to the method of Chen et
al(19), cells growing on
100-mm plates were harvested in HEM buffer (10 mM HEPES pH 7.5, 2
mM EDTA, 1 mM MgCl2) and homogenized with an ultrasonic
homogenizer. The homogenate was centrifuged at 500 × g at 4°C for 5
min to obtain the nuclei of the cells. Preheated SDS-PAGE loading
buffer was added to the pellet and boiled for 5 min to obtain
nuclear proteins.
Western blotting
Proteins from whole-cell and nuclear extracts were
separated by 10% SDS-PAGE. The primary antibodies were incubated
overnight at 4°C and the corresponding secondary antibodies were
incubated for 1 h at RT, with three washes following each
incubation. ECL reagents were used to show the positive bands on
the membrane.
Co-immunoprecipitation
The binding between c-Fos and c-Jun was detected by
co-immunoprecipitation. The cells were lysed with RIPA buffer (50
mM Tris-HCl pH 7.4, 1% Triton X-100, 1 mM EDTA, 1 mM leupeptin, 1
mM phenylmethylsulfonyl fluoride, 10 mM NaF, 1 mM
Na3VO4). Antibodies against c-Fos and c-Jun
and isotype-matched IgG were used for immunoprecipitation and
immunoblotting assay.
Statistical analysis
The data are shown as the mean ± standard error
(SE). Statistical significance was performed using ANOVA with SPSS
statistical software. P<0.05 was considered to indicate a
statistically significant difference.
Results
PKG II inhibits EGF-induced AP-1
transcriptional activity
The AP-1 transcription factor is a key regulatory
molecule that plays a central role in the control of cell
proliferation and transformation by converting MAPK signals into
the expression of specific target genes (20). In order to test the effect of PKG
II on AP-1 transcriptional activity, BGC-823 cells were transiently
transfected with a reporter plasmid containing a luciferase gene
driven by a minimal human collagenase gene promoter that contains a
single AP-1 site. To normalize transfection efficiency, a plasmid
containing a β-galactosidase gene was co-transfected as an internal
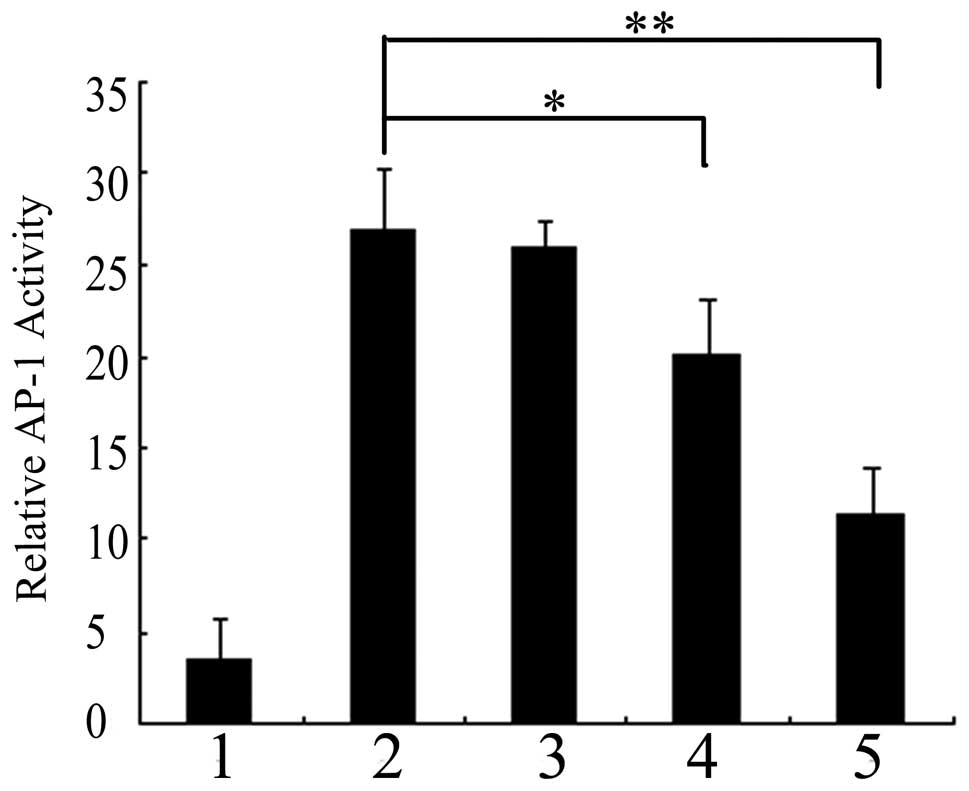
control. As shown in Fig. 1,
preinfecting pAd-LacZ or pAd-PKG II cells with EGF for 5 min
resulted in a 7.8-fold increase in AP-1 transcriptional activity.
The level of AP-1 luciferase activity in pAd-PKG II-infected cells
stimulated with cGMP decreased as compared with those infected with
pAd-LacZ along with EGF, indicating that activated PKG II
efficiently inhibits EGF-induced AP-1 transcriptional activity in
BGC-823 cells.
PKG II blocks the EGF-induced
transcriptional activity of Elk1
ERK is known to be activated by stress stimuli and
one of the downstream targets of the ERK pathway is the
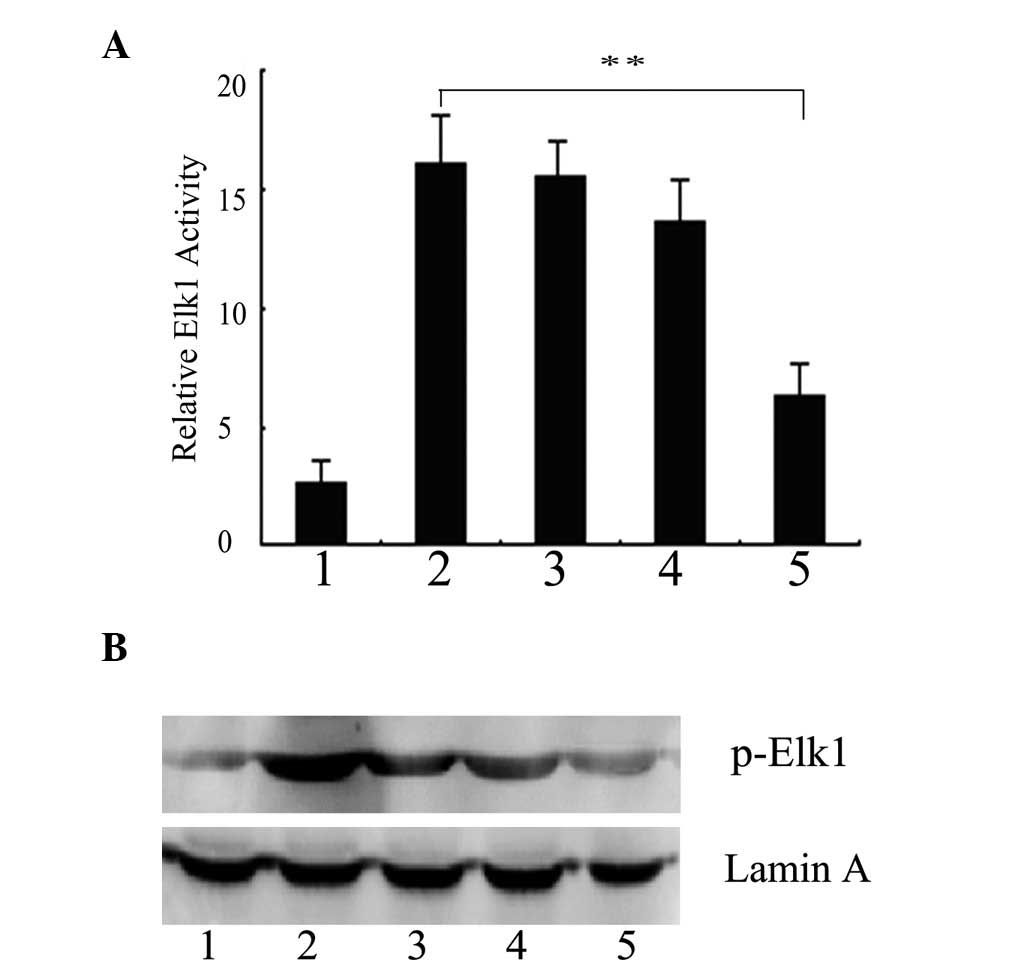
transcription factor Elk1. EGF increased the pElk1-luc activity
almost 6-fold compared with the cells infected with pAd-LacZ alone.
However, in the cells infected with pAd-PKG II and stimulated with
8-pCPT-cGMP, the increase was only ~2.3-fold by EGF treatment,
suggesting that PKG II markedly reduces the EGF-induced Elk1
transcriptional activity. The EGF-induced Elk1 luciferase activity
decreased gradually with the increasing concentrations of cGMP,
suggesting that the high expression and activity of PKG II results
in the dose-dependent reduction of the Elk1 activation induced by
EGF (Fig. 2A). Nuclear lysates
were prepared and the phosphorylation of Elk1 was detected by
western blot analysis. Fig. 2B
shows that EGF markedly induced the Ser383 phosphorylation of Elk1,
but such activation was blocked by activated PKG II.
PKG II inhibits the expression of c-Jun
and c-Fos, which are the predominant components of the EGF-induced
AP-1 complex
In order to investigate whether PKG II altered the
protein levels of AP-1 family members in BGC-823 cells, nuclear
lysates from pAd-PKG II-infected cells treated with various
concentrations of 8-pCPT-cGMP were subjected to western blot
analysis. Low levels of c-Jun and c-Fos proteins were detected in
the cells infected with pAd-LacZ. EGF treatment induced a
substantial increase in the nuclear protein levels of c-Jun and
c-Fos (Fig. 3A). In the cells
infected with pAd-PKG II and stimulated with cGMP, the increased
levels of EGF-induced c-Jun and c-Fos were significantly inhibited.
These data clearly reveal that c-Fos and c-Jun were components of
the EGF-induced AP-1 complex and that the expression of c-Jun was
more sensitive and more susceptible to EGF and PKG II than that of
c-Fos. c-Jun transcriptional activation, which is necessary for
tumor development, is regulated by a variety of post-translational
modifications. The results shown in Fig. 3A indicate that EGF markedly
enhanced the Ser73 phosphorylation of c-Jun and activated PKG II
almost completely blocked the increase in the phosphorylation of
c-Jun induced by EGF, without affecting the level of Ser63
phosphorylation of c-Jun. These results imply that PKG II inhibited
the phosphorylation of c-Jun at Ser73, which may impair the
formation of a heterodimer of the JNK substrate c-Jun and c-Fos to
form the AP-1 complex. To test this hypothesis and further
determine the pattern of the activated-AP-1 complex induced by EGF
in BGC-823 cells, co-immunoprecipitation experiments were
performed. As shown in Fig. 3B,
co-immunoprecipitation performed with anti-p-c-Jun antibody
revealed the coprecipitation with c-Fos and c-Jun from nuclear
extracts of BGC-823 cells. These results confirm that the
EGF-activated AP-1 complex was constituted by the p-c-Jun-c-Jun
homodimers and p-c-Jun-c-Fos heterodimers and the high expression
and activity of PKG II restrained the combination.
 | Figure 3PKG II inhibits the expressions of
c-Jun and c-Fos and blocks the combination between c-Jun, c-Fos and
p-c-Jun (Ser73). BGC-823 cells were infected with either pAd-LacZ
or pAd-PKG II, serum-starved for 12 h, stimulated with 8-pCPT-cGMP
for 12 h and treated with EGF (100 ng/ml) for 12 h (lane 1, LacZ;
lane 2, LacZ+EGF; lane 3, PKG II+EGF; lane 4, PKG II+100 μM
cGMP+EGF; lane 5, PKG II+250 μM cGMP+EGF). (A) Cells were lysed and
the nuclear proteins were obtained. Western blotting with the
indicated antibodies was used to analyze the protein levels of
c-Jun, c-Fos and p-c-Jun (Ser73). (B) Cell nuclear lysates were
subjected to IP with an antibody against p-c-Jun followed by IB
with antibodies against c-Jun and c-Fos. Representative results
from three independent experiments are shown. IP,
immunoprecipitation; IB, immunoblotting. PKG II, type II
cGMP-dependent protein kinase. |
Blocking of EGF-induced activation of
AP-1 and Elk1 by PKG II was not associated with the inhibition of
activation of p38MAPK
The activation of AP-1 was regulated by MAPKs,
including ERK, p38MAPK and JNK, in various cell types. To elucidate
the correlation between MAPKs and AP-1/Elk1 transactivation in
BGC-823 cells, the effects of MAPK inhibitors on the
transcriptional activities of AP-1 and Elk1 in response to EGF was
investigated. Pretreatments with PD98059, SP600125 and SB203580
(inhibitors of ERK, JNK and p38MAPK, respectively) completely
blocked the increases in the luciferase activities of AP-1 and Elk1
induced by EGF (Fig. 4A), but
these pretreatments alone did not affect the activities of AP-1 and
Elk1 in the cells infected with pAd-LacZ alone (data not shown).
These results suggest that ERK, JNK and p38MAPK are necessary for
EGF-induced AP-1 and Elk1 transactivation in BGC-823 cells.
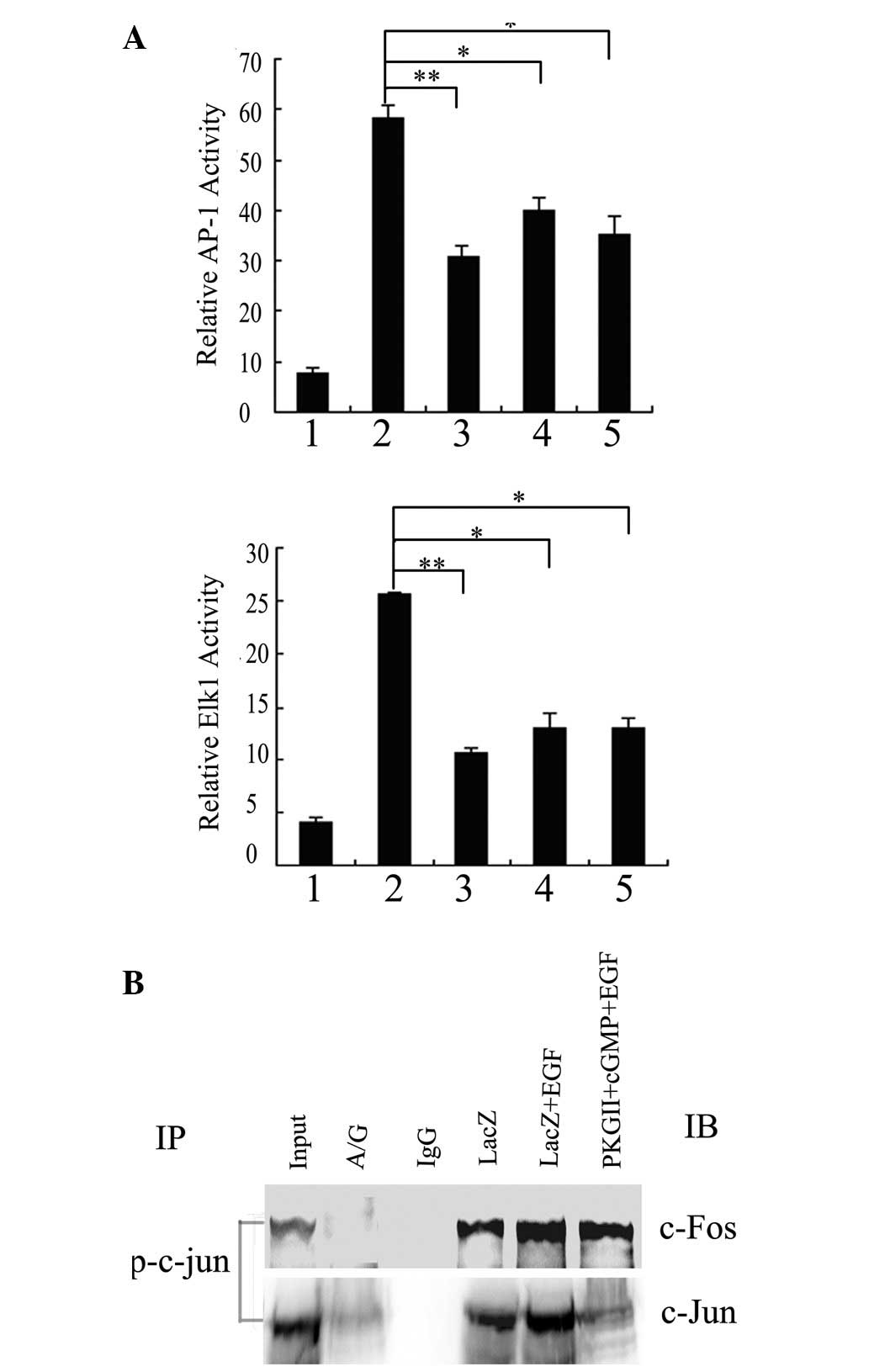 | Figure 4The inhibitory effect of PKG II on
AP-1 and Elk1 transactivation is dependent on the PKG II-inhibited
phosphorylation of JNK and ERK in BGC-823 cells exposed to EGF
treatment. (A) BGC-823 cells were transiently transfected with
pAP-1-Luc/pElk1-Luc and β-galactosidase plasmids. Cells were
infected with pAd-LacZ 8 h following transfection, serum-starved
for 12 h, pretreated with PD98059 (10 μM), SP600125 (10 μM) or
SB203580 (10 μM) for 2 h and then treated with EGF (100 ng/ml) for
5 min. 1, LacZ; 2, LacZ+EGF; 3, LacZ+PD98059+EGF; 4,
LacZ+SP600125+EGF; 5, LacZ+SB203580+EGF. Cell extracts were
prepared with luciferase reporter lysis buffer and analyzed for
luciferase activity. (B) BGC-823 cells were infected with either
pAd-LacZ or pAd-PKG II, serum-starved for 12 h, stimulated with
8-pCPT-cGMP for 1 h and treated with EGF (100 ng/ml) for 5 min
(lane 1, LacZ; lane 2-LacZ+EGF; lane 3, PKGII+EGF; lane 4, PKG
II+100 μM cGMP+EGF; lane 5, PKG II+250 μM cGMP+EGF). Cell lysates
were analyzed by western blotting with anti-JNK/phospho-JNK,
anti-ERK/phospho-ERK and anti-p38MAPK/phospho-p38MAPK. (A) The
relative values of AP-1/Elk1 luciferase activity to β-galactosidase
are shown as the mean ± SE of three independent experiments
(*P<0.05 and **P<0.01 vs.
pAd-LacZ+EGF). (B) Representative results from three independent
experiments are shown. IP, immunoprecipitation; IB, immunoblotting.
PKG II, type II cGMP-dependent protein kinase. |
To test the effect of PKG II on these signaling
cascades, the phosphorylation and activation of ERK, JNK and
p38MAPK were examined. As shown in Fig. 4B, the high level of expression and
activity of PKG II markedly inhibited the EGF-induced
phosphorylation of ERK and JNK in a concentration-dependent manner,
but had no effect on the phosphorylation of p38MAPK. Activated PKG
II hardly affected the total protein level of ERK, JNK and p38MAPK.
Combining with the results of Fig.
4A, these data suggest that the inhibition of ERK and JNK
phosphorylation by activated PKG II is one of the underlying
mechanisms involved in its suppressive effect on EGF-induced AP-1
and Elk1 transactivation.
Discussion
AP-1 complexes induce cellular proliferation and
transformation and may also promote differentiation and trigger
apoptosis, depending upon their composition (21). Thus, the effect of AP-1 activation
on cell proliferation may be temporally and spatially restricted.
c-Jun and c-Fos are typical immediate early genes induced by a wide
variety of growth factors and trigger downstream events relevant to
cell cycle progression and proliferation (10). In the present study, PKG II
suppressed the EGF-induced AP-1 activation through inhibiting the
expression of c-Jun and c-Fos, which have positive effects on cell
proliferation (22,23). In accordance with these results, it
has previously been confirmed that increasing PKG II activity
suppresses the proliferation of BGC-823 cells by inducing G0/G1
phase arrest (9). The inhibitory
pattern of c-Jun expression by cGMP in a dose-dependent manner was
extremely similar to that of AP-1 transcriptional activity,
suggesting that the inhibition of c-Jun expression may be
correlated with AP-1 transcriptional activity inhibition.
Therefore, the expression of the c-Jun protein may be crucial for
the activity of AP-1-regulated genes. Since c-Jun activity is
directly modulated at the protein level via regulatory
phosphorylation occurring on Ser63, Ser73 and Thr91 within the
transactivation domain (24), the
results clearly suggest that the inhibitory effect of PKG II on
AP-1 transcriptional activity acts by suppressing the
Ser73-phosphorylated-c-Jun. Furthermore, it was revealed that
activated AP-1 consisted of p-c-Jun-c-Jun homodimers and
p-c-Jun-c-Fos heterodimers following EGF treatment, but this
induction was blocked in cells infected with pAd-PKG II and
following 8-pCPT-cGMP exposure. The present study used only
antibodies against c-Jun and c-Fos to detect the formation of AP-1,
therefore, other dimeric forms of the AP-1 transcription factor
involved in regulating the AP-1 activity in BGC-823 cells cannot be
excluded from this process.
It is well established that the activation of JNK,
p38MAPK and/or ERK induces cancer proliferation through the AP-1
signaling pathway. Data in this study revealed that all three MAPK
inhibitors blocked the activation of AP-1 and Elk1, suggesting that
the JNK and p38MAPK pathways are also involved in EGF-induced
transcriptional activities of AP-1 and Elk1. However, western blot
analysis revealed that activated PKG II had a selective inhibitory
effect on ERK/JNK activation but not p38MAPK activation. Therefore,
it is believed that PKG II inhibits EGF-induced AP-1 and Elk1
transactivation in gastric cancer cells through suppressing ERK and
JNK signaling pathways.
It has been reported that the critical step in the
EGF/ERK signaling cascade is the change in cellular localization of
phosphorylated ERK from the cytoplasm to the nucleus, which results
in the activation of the transcription factor of Elk1 (25). The phosphorylation at Ser383 and
Ser389 of Elk1 is critical for its transcriptional activity and is
accomplished by all three types of MAPKs (26,27).
A recent study has shown that PKG II significantly inhibited the
EGF-induced nuclear translocation of phospho-ERK (8). This study demonstrated that
activated-PKG II suppressed the EGF-induced
Ser383-phosphorylated-Elk1, a crucial downstream molecule of ERK,
in the nuclei of BGC-823 cancer cells.
In summary, we have shown that PKG II inhibits the
EGF-stimulated ERK/JNK-dependent activation of AP-1 and Elk1 in
gastric cancer cells. Since AP-1/Elk1-associated transcriptional
activity is crucial in mediating gastric cancer cell proliferation
and growth, it is further confirmed that PKG II inhibited the
proliferation of gastric cancer cells through blocking the signal
transduction of MAPK (ERK/JNK)-mediated pathways. This is likely to
provide direct evidence for the therapeutic value of PKG II as a
cancer suppression factor.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (nos. 81001100, 31040002 and 31100974)
and by the Specialized Research Fund for Senior Personnel Program
of Jiangsu University (no. 08JDG033). We thank Dr Gerry Boss and Dr
Renate Pilz, University of California, San Diego, CA, USA, for the
kind gifts of adenoviral constructs.
References
|
1
|
Orstavik S, Natarajan V, Taskén K, Jahnsen
T and Sandberg M: Characterization of the human gene encoding the
type I alpha and type I beta cGMP-dependent protein kinase (PRKG1).
Genomics. 42:311–318. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Orstavik S, Solberg R, Taskén K, Nordahl
M, Altherr MR, Hansson V, Jahnsen T and Sandberg M: Molecular
cloning, cDNA structure and chromosomal localization of the human
type II cGMP-dependent protein kinase. Biochem Biophys Res Commun.
220:759–765. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Feil R, Hofmann F and Kleppisch T:
Function of cGMP-dependent protein kinases in the nervous system.
Rev Neurosci. 16:23–41. 2005.PubMed/NCBI
|
|
4
|
Pilz RB and Broderick KE: Role of cyclic
GMP in gene regulation. Front Biosci. 10:1239–1268. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hou Y, Gupta N, Schoenlein P, Wong E,
Martindale R, Ganapathy V and Browning D: An anti-tumor role for
cGMP-dependent protein kinase. Cancer Lett. 240:60–68. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cook AL and Haynes JM: Protein kinase G
II-mediated proliferative effects in human cultured prostatic
stromal cells. Cell Signal. 16:253–261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cook AL and Haynes JM: Phosphorylation of
the PKG substrate, vasodilator-stimulated phosphoprotein (VASP), in
human cultured prostatic stromal cells. Nitric Oxide. 16:10–17.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu Y, Chen YC, Qu R, Lan T and Sang JR:
Type II cGMP-dependent protein kinase inhibits EGF-triggered signal
transduction of the MAPK/ERK-mediated pathway in gastric cancer
cells. Oncol Rep. 27:553–558. 2012.PubMed/NCBI
|
|
9
|
Chen YC, Ren F, Sang JR, Tao Y and Xu WR:
Type II cGMP-dependent protein kinase inhibits proliferation of the
gastric cancer cell line BGC-823. Mol Med Rep. 3:361–366.
2010.PubMed/NCBI
|
|
10
|
Angel P and Karin M: The role of Jun, Fos
and the AP-1 complex in cell-proliferation and transformation.
Biochim Biophys Acta. 1072:129–157. 1991.PubMed/NCBI
|
|
11
|
Chiariello M, Marinissen MJ and Gutkind
JS: Multiple mitogen-activated protein kinase signaling pathways
connect the cot oncoprotein to the c-jun promoter and to cellular
transformation. Mol Cell Biol. 20:1747–1758. 2000. View Article : Google Scholar
|
|
12
|
Shaulian E and Karin M: AP-1 in cell
proliferation and survival. Oncogene. 20:2390–2400. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shaulian E and Karin M: AP-1 as a
regulator of cell life and death. Nat Cell Biol. 4:E131–E136. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eferl R and Wagner EF: AP-1: a
double-edged sword in tumorigenesis. Nat Rev Cancer. 3:859–868.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Whitmarsh AJ, Shore P, Sharrocks AD and
Davis RJ: Integration of MAP kinase signal transduction pathways at
the serum response element. Science. 269:403–407. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Buchwalter G, Gross C and Wasylyk B: Ets
ternary complex transcription factors. Gene. 324:1–14. 2004.
View Article : Google Scholar
|
|
17
|
Hao D, Gao P, Liu P, Zhao J, Wang Y, Yang
W, Lu Y, Shi T and Zhang X: AC3–33, a novel secretory protein,
inhibits Elk1 transcriptional activity via ERK pathway. Mol Biol
Rep. 38:1375–1382. 2011.
|
|
18
|
Ely HA, Mellon PL and Coss D: GnRH induces
the c-Fos gene via phosphorylation of SRF by the calcium/calmodulin
kinase II pathway. Mol Endocrinol. 25:669–680. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen JC, Zhuang S, Nguyen TH, Boss GR and
Pilz RB: Oncogenic Ras leads to Rho activation by activating the
mitogen-activated protein kinase pathway and decreasing
Rho-GTPase-activating protein activity. J Biol Chem. 278:2807–2818.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ono K and Han J: The p38 signal
transduction pathway: activation and function. Cell Signal.
12:1–13. 2000. View Article : Google Scholar
|
|
21
|
Milde-Langosch K: The Fos family of
transcription factors and their role in tumourigenesis. Eur J
Cancer. 41:2449–2461. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brown JR, Nigh E, Lee RJ, Ye H, Thompson
MA, Saudou F, Pestell RG and Greenberg ME: Fos family members
induce cell cycle entry by activating cyclin D1. Mol Cell Biol.
18:5609–5619. 1998.PubMed/NCBI
|
|
23
|
Szabowski A, Maas-Szabowski N, Andrecht S,
Kolbus A, Schorpp-Kistner M, Fusenig NE and Angel P: c-Jun and JunB
antagonistically control cytokine-regulated mesenchymal-epidermal
interaction in skin. Cell. 103:745–755. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Karin M: The regulation of AP-1 activity
by mitogen-activated protein kinases. J Biol Chem. 270:16483–16486.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gille H, Kortenjann M, Thomae O, Moomaw C,
Slaughter C, Cobb MH and Shaw PE: ERK phosphorylation potentiates
Elk-1-mediated ternary complex formation and transactivation. EMBO
J. 14:951–962. 1995.PubMed/NCBI
|
|
26
|
Ricote M, García-Tuñón I, Fraile B,
Fernández C, Aller P, Paniagua R and Royuela M: p38 MAPK protects
against TNF-alpha-provoked apoptosis in LNCaP prostatic cancer
cells. Apoptosis. 11:1969–1975. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vikman P, Ansar S and Edvinsson L:
Transcriptional regulation of inflammatory and extracellular
matrix-regulating genes in cerebral arteries following experimental
subarachnoid hemorrhage in rats. Laboratory investigation. J
Neurosurg. 107:1015–1022. 2007. View Article : Google Scholar
|