Introduction
DNA molecular weight (mw) standard controls of
nucleic acids, also known as DNA ladders, are widely used in
molecular biology studies to determine the mw or the base pair (bp)
length of nucleic acids. The DNA ladder is also used to
quantitatively analyze target DNA fragments. At present, there are
two ways to prepare a DNA ladder: i) amplification by polymerase
chain reaction (PCR) (1,2); and ii) digestion of plasmid DNA by
restriction endonucleases (3,4).
However, each of these methods have advantages and disadvantages.
The former achieves regular bands, but it is difficult to amplify
DNA fragments of a high mw. The latter involves the preparation of
the DNA ladder mostly from bacteriophages or plasmids, with
digestion of the purified DNA by restriction endonucleases, but
this process is complex and produces unevenly distributed DNA
fragments, particularly for the preparation of large quantities of
DNA ladders. The present study describes a method that is based on
the combination of PCR amplification and plasmid digestion by
restriction endonucleases to prepare a high mw DNA ladder. The
results show that the prepared DNA ladder bands were clear,
accurate and cheap and may be used as a standard in future
molecular biology experiments.
Materials and methods
PCR primers
According to the λ phage DNA sequence (GenBank
accession no. J02459), pairs of 1,000-, 2,000-, 3,000- and 4,000-bp
primers were designed with Primer 5.0 software and were obtained
from Shanghai Sangon Co., Ltd. (Shanghai, China). The primers are
shown in Table I.
 | Table IPrimers used in this study. |
Table I
Primers used in this study.
| Length (bp) | Primers | Melting temperature
(°C) |
|---|
| 1,000 |
5′-GCGGCACGGAGTGGAGCAAG-3′ | 66.00 |
|
5′-GTTATCGAAATCAGCCACAGGGC-3′ | 63.68 |
| 2,000 |
5′-GCAGTGACACTGCGCTGGATC-3′ | 61.90 |
|
5′-GTTATCGAAATCAGCCACAGGGC-3′ | 63.68 |
| 3,000 |
5′-CAGGCCCGCAGTTATCAGGTC-3′ | 63.87 |
|
5′-GTTATCGAAATCAGCCACAGGGC-3′ | 63.68 |
| 4,000 |
5′-CAGCATGCCACGTAAGCGAAACAAAAA-3′ | 62.00 |
|
5′-CACGGAAAAAGAGACGCAGAAACAGC-3′ | 63.52 |
PCR amplification
DNA fragments of 1,000–4,000-bp were amplified by
PCR. The reaction mixture (100 μl) for each quantitative PCR
contained 100 ng template DNA, 0.2 mM dNTP, 10 mM Tris-Cl (pH 8.8),
1.5 mM MgCl2, 50 mM KCl, 0.1 mM of each primer and 2.5
units Taq DNA polymerase. All PCR procedures of these DNA
fragments were adopted by the improved touch-down PCR method
(5) and DNA fragments were
amplified using various PCR procedures. The specific PCR procedures
were as follows: the PCR conditions of 1,000- and 2,000-bp included
a temperature profile of 30 cycles, which included 2 cycles at 95°C
for 40 sec, 60°C for 50 sec and 72°C for 50 sec, 2 cycles 95°C for
40 sec, 59°C for 50 sec and 72°C for 50 sec, 2 cycles for every
annealing temperature interval one temperature, until 56°C
polishing 20 cycles. The PCR conditions of 3,000- and 4,000-bp
included a temperature profile of 30 cycles, which contained 2
cycles at 95°C for 40 sec, 65°C for 1 min and 72°C for 2 min, 2
cycles 95°C for 40 sec, 64°C for 1 min and 72°C for 2 min, 2 cycles
for every annealing temperature interval one temperature, until
61°C polishing 20 cycles.
Construction, purification and
identification of plasmids
The construction of high mw plasmids, including 5-,
6-, 8- and 10-kb was carried out as described previously (6), purified with a plasmid DNA
purification kit and identified with endonuclease HindIII.
The undigested plasmid of corresponding mw was used as the control
and then electrophoresed.
Electrophoresis, purification and
sequencing
The PCR products of 1,000- and 2,000-bp were
detected by 1% agarose gel electrophoresis at 120 V for 30 min. The
PCR products of 3,000- and 4,000-bp, the undigested plasmids and
the restriction endonuclease were detected by 0.8% agarose gel at
120 V for 30 min. The PCR products and plasmid DNA were recovered
using the agarose gel purification kit according to the
manufacturer’s instructions, followed by sequencing by Shanghai
Sangon Co., Ltd. The generated sequences were compared with BLAST
(http://www.ncbi.nlm.nih.gov/blast/Blast.cgi).
Preparation of a high mw DNA ladder
The PCR products and linear plasmids were extracted
by phenol/chloroform and precipitated with ethanol, then dissolved
in TE (10 mM Tris-HCl, 1 mM EDTA) buffers and had their UV
absorbance analyzed at 260 nm. According to the special
proportions, various sizes of DNA fragments were mixed and added to
10X loading buffer and were then ready for use. The prepared DNA
ladder was then frozen at −20°C.
Results
PCR amplification
All DNA fragments of 1,000–4,000-bp were amplified
successfully by PCR. As shown in Fig.
1, DNA fragments had high specificity and the sizes of the
amplified PCR products were consistent with those expected.
 | Figure 1DNA fragments (1,000–4,000-bp) were
amplified by PCR. (A) The electrophoresis results of 1,000-bp (lane
1), 2,000-bp (lane 2), DL200 (M). (B) The electrophoresis results
of 3,000-bp (lane 1), 4,000-bp (lane 2), 1-kb DNA ladder (M) in
(B). PCR, polymerase chain reaction. |
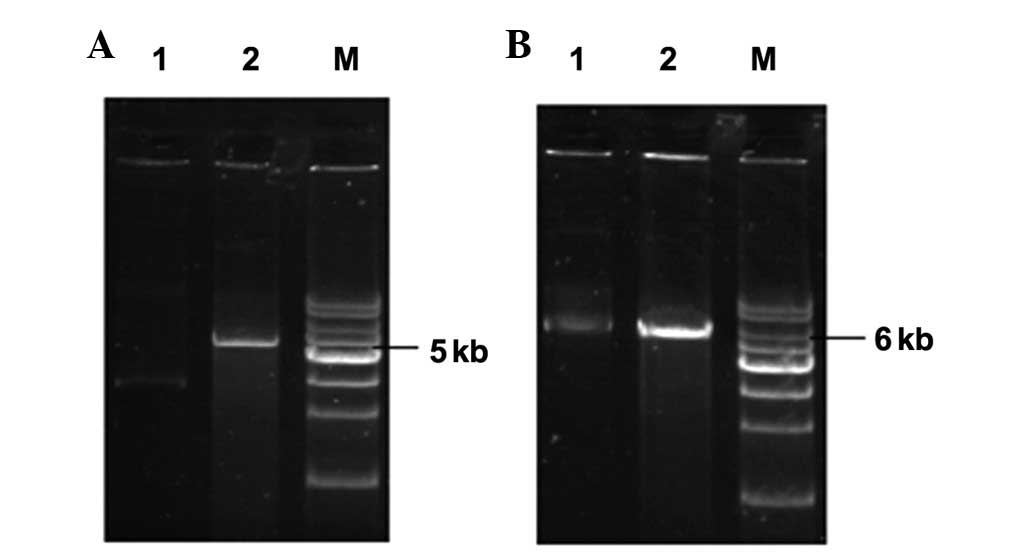
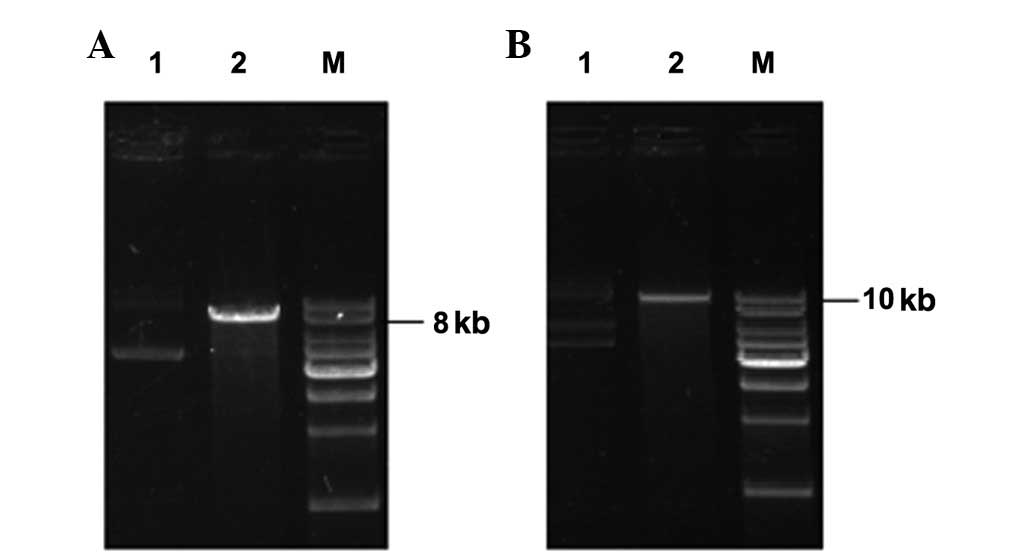
Purification and identification of
plasmids
The 4 plasmids and their restriction endonuclease
analyses are shown in Figs. 2 and
3. Following the digestion of
these constructed vectors by HindIII, revealing the
linearized DNA strands, the results of the restriction were
consistent with those expected, the above results indicate that 4
plasmids had been successfully constructed.
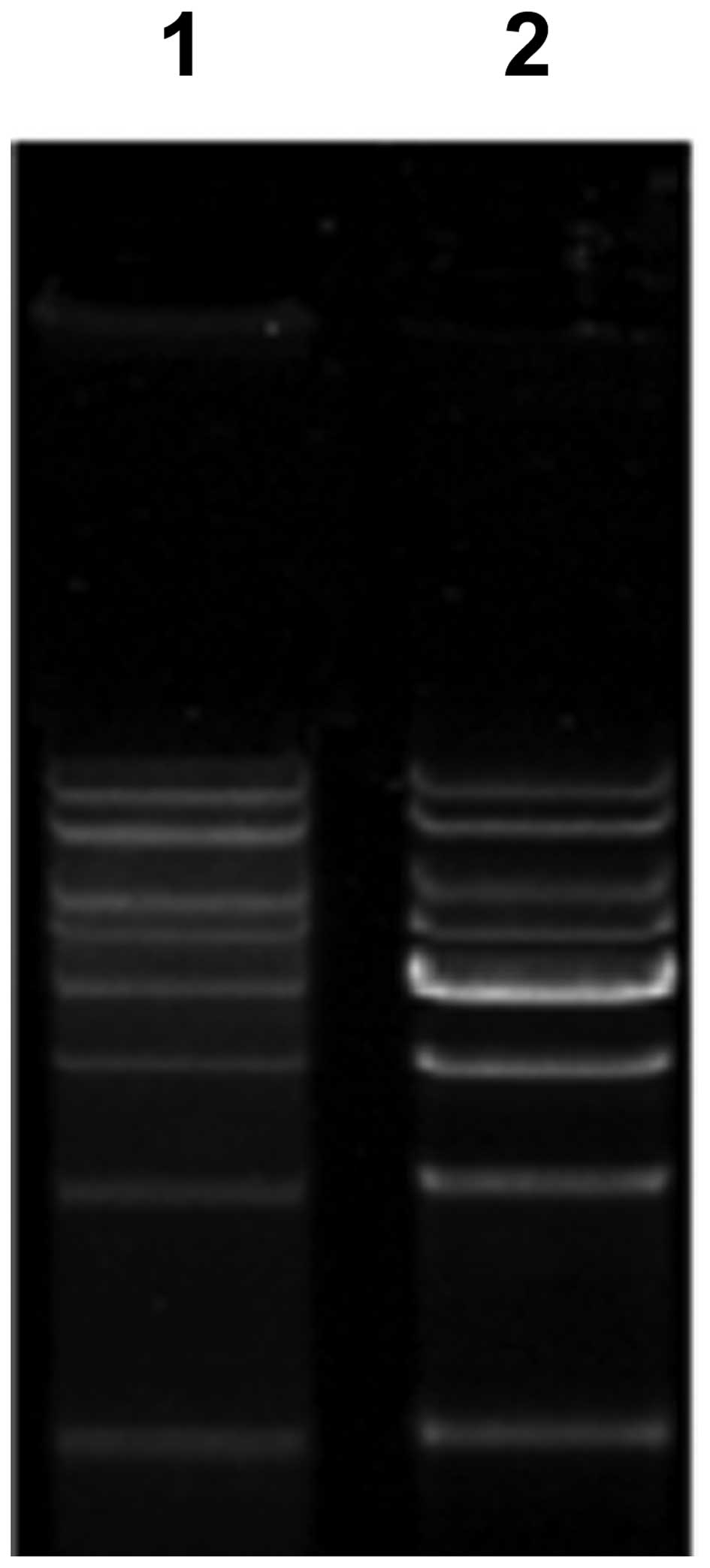
Preparation of high mw DNA ladder
Following extraction, purification and quantity
analysis, the PCR products and linear plasmids were mixed according
to the special proportions, while the 4,000-bp band was doubled to
increase the discrimination effect, then added to 10X loading
buffer and was then ready for use. The result of the agarose gel
electrophoresis revealed that all the DNA bands were clear and that
the ladder was comparable with the commercial version (Fig. 4).
Discussion
The DNA ladder is a widely applied method in
electrophoresis and is valuable in molecular biology experiments.
It is used to mark the mw of unknown samples in nucleic acid
electrophoresis and further aids the judgement of the properties of
DNA samples. DNA ladders have been widely applied in such fields as
biotechnology, medicine and agriculture, among others, and the
market demand is extremely high. PCR was initiated in 1983 by
Mullis et al(7) as a
specific DNA amplification technology and has the advantages of
easy operation, high sensitivity and specificity and good
selectivity, and has been widely applied in the fields of molecular
biology, genetics, biochemistry, genetic engineering and forensics
(8–10). Although PCR amplification is
convenient for preparing the DNA ladder, conventional PCR is
designed to amplify one type of DNA fragment, one tube at a time,
while the DNA ladder is an integration of several DNA fragments,
which makes the technological process more complex and cumbersome
(11). In addition, the technology
is only suitable for amplifying small DNA fragments, while the
corresponding fragments of a high mw DNA ladder are difficult to
obtain. Despite certain large fragments being amplified using
high-fidelity DNA polymerase, the specificity is poor and
production cost is high and cannot meet the experimental demand.
Another routine method of preparing a DNA ladder is using
bacteriophages or plasmids which are digested by restriction
endonucleases (12,13). This process requires the
construction of a series of vectors for preparing the DNA ladder,
further digesting the purified DNA with certain restriction
endonucleases and subsequently the combinations of DNA fragments
that were required are obtained (14,15).
This process is laborious, time consuming and material- and
equipment-intensive, involving the propagation of the virus or
plasmid in the appropriate host organism and the purification and
digestion of the genomic or plasmid DNA from the nucleic acids of
the host. Since the corresponding restriction enzyme sites of
bacteriophages or plasmid DNA are not well balanced, the
distribution of the prepared DNA ladder is irregular and the gaps
of the DNA bands are variable in size and not convenient (16).
Based on the analysis and research of the advantages
and disadvantages of the above two methods, the methods of PCR
amplification and plasmid digestion by restriction endonucleases
were adopted in the current study to prepare a high mw DNA ladder.
In the current study, 1,000–4,000-bp DNA bands were amplified by
PCR and the larger DNA bands, including 5-, 6-, 8- and 10-kb, were
obtained by restriction endonuclease digestion of the purified
plasmids. These DNA fragments were extracted with phenol/chloroform
and precipitated with ethanol, further mixed according to the
special proportions and added to 10X loading buffer to obtain the
high mw DNA ladder. Compared with commercial DNA ladders, this
method was simple, practical and low cost, and the DNA bands were
clear. It may also be adjusted to any size of mw standard in a
certain range according to the experimental requirement and may be
used as a standard in molecular experiments. In short, a new method
of preparing high mw DNA ladders was developed in this study.
Acknowledgements
This work was supported by grants from the National
Natural Science Foundation of China (30970055), Henan Province
(122102310194; 12A310005) and the key research areas bidding
subject of Xinxiang Medical University (ZD2011-36).
References
|
1
|
Wu J and Ye C: Tandem PCR: a novel and
efficient unit amplification model for the preparation of small DNA
fragments. Mol Biol Rep. 38:2729–2731. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang TY, Guo L and Zhang JH: Preparation
of DNA ladder based on multiplex PCR technique. J Nucleic Acids.
25:4218032010.PubMed/NCBI
|
|
3
|
Dodgson JB, Cheng HH and Okemoto R: DNA
marker technology: a revolution in animal genetics. Poult Sci.
76:1108–1114. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Z, Wu J, Li X, Ye C and Wenxing H:
Novel strategies to construct complex synthetic vectors to produce
DNA molecular weight standards. Mol Biotechnol. 42:128–133. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schunck B, Kraft W and Truyen U: A simple
touch-down polymerase chain reaction for the detection of canine
parvovirus and feline panleukopenia virus in feces. J Virol
Methods. 55:427–433. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sambrook J and Russell DW: Molecular
Cloning: A Laboratory Manual. 3rd edition. Cold Spring Harbor
Laboratory Press; New York, NY: 2001
|
|
7
|
Mullis K, Faloona F, Scharf S, Saiki R,
Horn G and Erlich H: Specific enzymatic amplification of DNA in
vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant
Biol. 51:263–273. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ting NC, Zaki NM, Rosli R, et al: SSR
mining in oil palm EST database: application in oil palm germplasm
diversity studies. J Genet. 89:135–145. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee JH, Shin SR and Cho JH: Evaluation of
direct immunofluorescence test with PCR for detection of novel
influenza A (H1N1) virus during 2009 pandemic. Yonsei Med J.
52:680–682. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bellstedt DU, Pirie MD, Visser JC, de
Villiers MJ and Gehrke B: A rapid and inexpensive method for the
direct PCR amplification of DNA from plants. Am J Bot. 97:e65–e68.
2010.PubMed/NCBI
|
|
11
|
Dawson EP: Method for the multiplexed
preparation of nucleic acid molecular weight markers and resultant
products US Patent 5,714,326. Filed January 3, 1996; issued
February 3, 1998.
|
|
12
|
Ye C, Gu J, Chen S, Deng A, Li YZ and Li
D: Unit cloning and amplification as novel and universal strategies
for complex vector construction and small DNA fragment preparation.
Electrophoresis. 31:2929–2935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu A-li W, Hartley JL and Jordan HJ:
Nucleic acid ladders US Patent 6,924,098. Filed December 3, 1999;
issued August 2, 2005.
|
|
14
|
Barvish Z, Davis C and Gitelman I: A
wide-range, low-cost 150 bp ladder for sizing DNA fragments between
150 and 4500 bp. Electrophoresis. 28:900–902. 2007.PubMed/NCBI
|
|
15
|
Cole KD: Preparative separation of plasmid
and bacterial artificial chromosome DNA by density gradient
electrophoresis in the presence of linear polymers.
Electrophoresis. 19:3062–3068. 1998. View Article : Google Scholar
|
|
16
|
Hyman ED: Method of making DNA ladders US
Patent 5,939,293. Filed June 5, 1998; issued August 17, 1999.
|